

Abstract
Amino acids metabolism, especially aspartate metabolism, is often altered in human cancers including hepatocellular carcinoma (HCC) and this metabolic remodeling is required for supporting cancer cell malignant activities. Argininosuccinate synthase 1 (ASS1), as a crucial rate-limiting enzyme in aspartate metabolism, participates in repressing tumor progression. However, the roles of long noncoding RNAs (lncRNAs) in aspartate metabolism remodeling and the underlying mechanisms remain unclear. Here, we screen LINC01234 as an aspartate metabolism-related lncRNA in HCC. Clinically, LINC01234 was highly expressed in HCC, and a high LINC01234 expression level was correlated with a poor prognosis of patients with HCC. LINC01234 promoted cell proliferation, migration, and drug resistance by orchestrating aspartate metabolic reprogramming in HCC cells. Mechanistically, LINC01234 downregulated the expression of ASS1, leading to am increased aspartate level and activation of the mammalian target of rapamycin pathway. LINC01234 bound to the promoter of ASS1 and inhibited transcriptional activation of ASS1 by transcriptional factors, including p53. Finally, inhibiting LINC01234 dramatically impaired tumor growth in nude mice and sensitized HCC cells to sorafenib. These findings demonstrate that LINC01234 promotes HCC progression by modulating aspartate metabolic reprogramming and might be a prognostic or therapeutic target for HCC.
Key Words: LINC01234, metabolic reprogramming, ASS1, p53, hepatocellular carcinoma
Graphical abstract
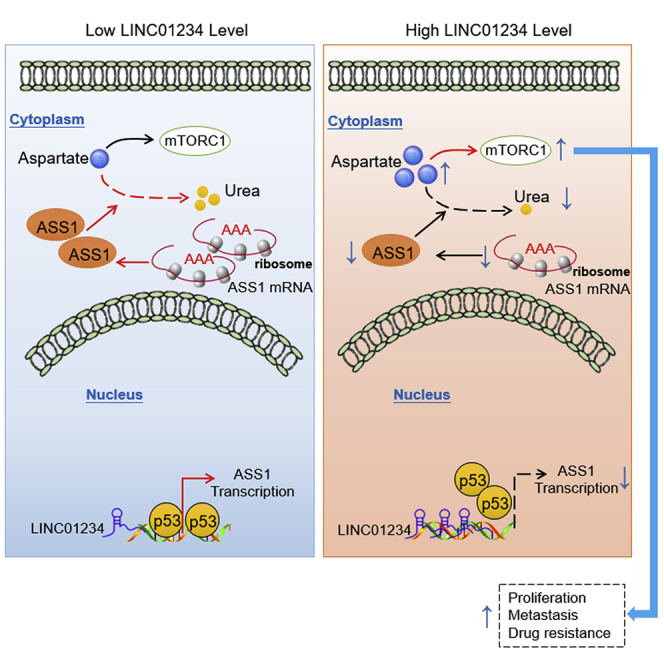
Chen et al. demonstrate that LINC01234 inhibits the transcription of ASS1 and decreases ASS1 activity, causing enhanced mammalian target of rapamycin complex 1 (mTORC1) activation to promote HCC progression. Depletion of LINC01234 attenuates growth, migration and drug-resistance of HCC cells, indicating a potential RNA interference-based strategy that targets lncRNAs and cancer metabolism for cancer treatment.
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer. Although various management strategies have shown considerable effects, the overall prognosis of HCC is still poor owing to high rates of metastasis and drug resistance.1,2 Metabolic remodeling and malignant activities are hallmarks of cancers including HCC. Recently, the roles of amino acid metabolism are emerging as an attractive topic in cancer therapeutics. Cancer cells reprogram their amino acid metabolism to support the biosynthetic pathways that are upregulated in cancer cells and provide specific adaptive traits to the anti-cancer drugs the cells are exposed to.3 Because arginine, asparagine, serine, and leucine are essential for cancer cells growth, deprivation of these amino acids has emerged as a potential strategy for cancer treatment.4, 5, 6 Characterizing the cooperative mechanisms underlying amino acid metabolism and tumor malignancy will give us a better understanding of human cancer progression.
Cellular aspartate is crucial for cell growth because it is essential for both protein synthesis and nucleotide biosynthesis.7,8 Since the circulating levels of aspartate are lower compared with other amino acids, cancer cells largely reprogram their intracellular aspartate metabolism to support cellular growth.9 The conversion of nitrogen from aspartate to urea decreases cellular aspartate through the urea cycle.10 Argininosuccinate synthase 1 (ASS1) is essential for catalyzing argininosuccinate formation from citrulline and aspartate, the rate-limiting step of de novo arginine synthesis in the urea cycle. Notably, ASS1 is downregulated in many cancers, and this low expression of ASS1 is associated with a poor prognosis for patients.11,12 ASS1 deficiency in cancer increases cytosolic aspartate level, which further enhances mammalian target of rapamycin (mTOR) pathway to support biomass synthesis and tumor malignancies.13 The ASS1 enzymatic activators, spinosyn A and its derivative, display the capabilities for inhibiting breast cancer.14 Nevertheless, the mechanism underlying how ASS1 is downregulated in cancer remains elusive. The tumor suppressor p53 directly promotes ASS1 expression, causing an increase in ASS1 activity.15
Long noncoding RNAs (lncRNAs) are a class of noncoding RNA transcripts with a length of more than 200 nucleotides. Numerous studies have demonstrated that lncRNAs play pivotal roles in cancer development and progression such as tumor cell proliferation, growth, migration, invasion and chemoresistance.16, 17, 18 Targeting lncRNAs using small interfering RNAs (siRNAs), antisense oligonucleotides, and the CRISPR/Cas9 system is a potential cancer treatment.19 In recent years, some lncRNAs have been shown to regulate cancer metabolism, but the roles of lncRNAs in aspartate metabolism remodeling are unknown. Thus, this topic attracts our interest. Here, we identified that LINC01234 is associated with aspartate metabolism in human HCC. Recent studies have reported that LINC01234 is upregulated in cancers, such as non-small-cell lung cancer, colorectal cancer, and breast cancer.20, 21, 22, 23, 24, 25, 26, 27 Moreover, LINC01234 is involved in the progression of these cancers. LINC01234 mainly regulates the biological activity of miRNAs as a competing endogenous RNA (ceRNA) and regulates the expression of messenger RNAs (mRNAs) targeted by these miRNAs, such as miR-340-5p, miR-31-5p. and miR-124-3p.23,28, 29, 30, 31 LINC01234 also regulates miR-106b biogenesis by interacting with HNRNPA2B1 to increase c-Myc activity.27 However, the roles of LINC01234 in cancer metabolism and the underlying mechanisms are unknown.
In our study, we found that LINC01234 promotes HCC progression. We further uncovered that LINC01234 bound to the promoter of ASS1 and impaired the activation of ASS1 by transcriptional factor (TFs), including p53. By downregulating the expression of ASS1, LINC01234 elevates cellular aspartate, resulting in enhanced mTORC1 activity to support tumor malignancies. Finally, the inhibition of LINC01234 is helpful for attenuating tumor growth and sensitizing HCC cells to sorafenib, suggesting that LINC01234 may be a biomarker and therapeutic target for HCC treatment.
Results
LINC01234 is identified as a potential metabolism-related lncRNA
It is often observed that ASS1 is severely reduced or absent in some types of aggressive cancers.14,32,33 We first investigated the expression of ASS1 in HCC. Using TCGA HCC cohort, we also found that ASS1 was significantly downregulated in HCC tissues compared with adjacent non-cancerous tissues (Figure 1A). Moreover, ASS1 was significantly decreased at clinical stage IV related to stage I and stage III. Importantly, Kaplan-Meier plots showed that a low expression of ASS1 was associated with a poor prognosis, indicating that the loss of ASS1 promotes HCC progression (Figure 1B). However, the mechanism of ASS1 downregulation in tumors has not been fully elucidated. It is unknown whether lncRNAs are involved in regulating ASS1-mediated tumor suppression. Thus, we identified upregulated lncRNAs in HCC and analyzed the lncRNAs associated with ASS1 expression level from TCGA database. Among these lncRNAs, LINC01234 was more highly expressed in HCC tissues than in normal tissues from the TCGA HCC database (Figure 1C). The correlation analysis revealed that the ASS1 mRNA level was negatively associated with LINC01234 level in the TCGA HCC database (Figure 1D). Furthermore, we verified the relationship between ASS1 and LINC01234 in patients with HCC in an independent cohort (Peking University Cancer Hospital, PKUCancer). As shown in Figure 1E, the expression of ASS1 was negatively correlated with the LINC01234 level. These results suggest that LINC01234 relates to the expression of ASS1.
Figure 1.

Identification of LINC01234 as a metabolism-related lncRNA
(A) The expression level of ASS1 in cancerous tissues and non-cancerous tissues derived from patients with HCC by UALCAN (http://ualcan.path.uab.edu/).
(B) Overall survival analysis based on ASS1 levels by Kaplan-Meier Plotter (http://kmplot.com/analysis/) (n = 364, log rank test, two-sided).
(C) Venn diagrams of overlapped lncRNA between ASS1-associated lncRNAs and top30 differentially expressed lncRNAs in TCGA LIHC.
(D) The correlation analysis between LINC01234 expression level and ASS1 expression level according to TCGA dataset.
(E) The LINC01234 expression and ASS1 expression were evaluated by RT-qPCR in a cohort of patients with HCC (PKUCancer cohort).
(F) Expression level of LINC01234 in HCC tissues and paired adjacent non-tumoral tissues based on the TCGA dataset.
(G) The expression level of LINC01234 and ASS1 in HCC tissues and paired adjacent non-tumoral tissues was detected by RT-qPCR.
(H) Overall survival and disease-free survival analyses based on LINC01234 level were analyzed using data from PKUCancer cohort.
(I) Overall survival and disease-free survival analyses based on LINC01234 levels were analyzed using data from TCGA-LIHC cohort.
(J) RT-qPCR detection of LINC01234 expression in the cytoplasmic and nuclear fractions.
(K) The subcellular localization of LINC01234 was determined by FISH.
(L) The expression of LINC01234 was evaluated by RT-qPCR and the expression of ASS1 was detected by western blot in multiple HCC cells.
LINC01234 expression is associated with a poor prognosis for patients with HCC
We further examined the expression of LINC01234 in HCC tissues. LINC01234 was more highly expressed in HCC tissues than in paired adjacent normal tissues from TCGA database (Figure 1F) (n = 50, p = 0.0001). Consistently, we also found that LINC01234 was significantly overexpressed in HCC tissues from PKUCancer cohort (Figure 1G) (n = 52, p < 0.0001). Meanwhile, ASS1 was downregulated in HCC tissues from PKUCancer cohort (Figure 1G) (n = 52, p < 0.0001). We next determined the relationship between the LINC01234 expression and prognosis of the patients with HCC. A high LINC01234 level was correlated with a shorter overall survival and disease-free survival in patients with HCC in our cohort (Figure 1H). The TCGA dataset also showed that high LINC01234 expression predicted a shorter overall survival and disease-free survival in 345 cases of patients with HCC (Figure 1I). These findings indicated that LINC01234 is upregulated in HCC tissues and predicts a poor prognosis for patients with HCC.
Similar to many other lncRNAs, LINC01234 has tissue-specific expression patterns in different cancers. To determine the subcellular localization of LINC01234 in HCC cells, we detected LINC01234 expression in cytoplasmic and nuclear fractions by real-time quantitative PCR (RT-qPCR) analysis. The results revealed that LINC01234 was localized predominantly in the nucleus, with some localization in the cytoplasm (Figure 1J). This result was confirmed by RNA fluorescence in situ hybridization (FISH) assays in HepG2 and Huh7 cells (Figure 1K). Next, the expression levels of LINC01234 and ASS1 were evaluated in multiple HCC cell lines. As shown in Figure 1L, we observed a negative correlation between LINC01234 and ASS1 expression in HCC cell lines.
LINC01234 promotes HCC cell malignant behaviors and aspartate metabolic reprogramming
Since LINC01234 is potentially involved in HCC progression and aspartate metabolism, we further investigated the functional roles of LINC01234 in cellular behaviors. The overexpression of LINC01234 significantly promoted cell proliferation and increased colony formation (Figures 2A–2D). Moreover, the overexpression of LINC01234 enhanced cell mobility and increased their invasive capacity (Figures 2E and 2F). These results suggested that LINC01234 promotes HCC cell malignant behaviors, including proliferation, migration, and invasion. To explore the role of LINC01234 in metabolic reprogramming, we measured the aspartate in cells that overexpressed LINC01234. As shown in Figure 2G, the overexpression of LINC01234 increased the aspartate level in HCC cells.
Figure 2.

Overexpression of LINC01234 promotes HCC cell malignant behaviors and aspartate metabolic reprogramming
(A) RT-qPCR detection of LINC01234 overexpression in HepG2 and Huh7 cells.
(B) Cell proliferation was assessed by MTT assay in the indicated cells.
(C–D) Colony formation assay (C) and statistical analysis (D) were performed in cells transduced with LINC01234 vector or control (mean ± standard deviation).
(E-F) Transwell migration and invasion assays were performed in the control and LINC01234-overexpressed HepG2 and Huh7 cells.
(G) Cellular aspartate level was determined in cells transfected with the indicated vectors. Data are shown as mean ± standard deviation.
To verify the functional roles of LINC01234, we generated LINC01234-depleted cells (Figure 3A). Consistently, LINC01234 depletion inhibited cell proliferation and colony formation (Figures 3B–3D). The inhibition of LINC01234 also attenuated the migration and invasion of HCC cells (Figures 3E and 3F). Moreover, the depletion of LINC01234 significantly decreased the aspartate level (Figure 3G), indicating that LINC01234 is required for maintaining the aspartate level in HCC cells. Taken together, our data indicated that LINC01234 is functionally important in regulating cancer metabolic reprogramming and tumor cell malignant behavior.
Figure 3.

Depletion of LINC01234 attenuates HCC cell malignant behaviors and aspartate metabolic reprogramming
(A) RT-qPCR detection of LINC01234 depletion in HepG2 and Huh7 cells.
B) Cell proliferation was assessed by MTT assay in the indicated cells.
(C–D) Colony formation assay (C) and statistical analysis (D) were performed in cells transduced with LINC01234 siRNA1, siRNA2, or control siRNA.
(E–F) Transwell migration and invasion assays were performed in the control and LINC01234-depleted HepG2 or Huh7 cells.
(G) Cellular aspartate level was determined in cells transfected with the indicated shRNAs. Data are shown as mean ± standard deviation.
LINC01234 inhibits the expression of ASS1
To dissect the molecular mechanisms underlying LINC01234-mediated metabolic remodeling, we tried to identify LINC01234-involved pathways through a bioinformatics analysis. Based on the LINC01234 expression level, TCGA HCC samples were divided into two groups, including a LINC01234-low group and a LINC01234-high group. We identified the differentially expressed genes (DEGs) between the two groups. A KEGG pathway enrichment analysis revealed that the DEGs were enriched in ribosome, carbon metabolism, biosynthesis of amino acids, and aspartate metabolism (Figure 4A). These results further indicated that LINC01234 is involved in amino acids metabolism. To further explore the critical target of LINC01234, we examined the expression of six DEGs after LINC01234 was ablated, including GLUL, IDH3B, G6PD, ASNS, GOT1, and ASS1, all of which play important roles in cancer cell amino acids metabolism.34, 35, 36 Notably, LINC01234 depletion only regulated the expression of ASS1 (Figure 4B). Together, our results suggested that LINC01234 participates in cancer cell amino acids metabolism through regulating ASS1 expression.
Figure 4.

LINC01234 inhibits the expression of ASS1
(A) KEGG pathway enrichment analysis for the DEGs between LINC01234-upregulated and -downregulated HCC.
(B) Huh7 cells were stably transfected with LINC01234 shRNAs. RT-qPCR was performed for analyzing the mRNA levels of the indicated genes.
(C) HepG2 or Huh7 cells were stably transfected with LINC01234 shRNAs. Then, total RNAs were extracted from the cells and RT-qPCR was performed as indicated.
(D) Cells stably expressing LINC01234 were harvested. Then, total RNAs were extracted from the cells and RT-qPCR was performed as indicated.
(E) Cellular aspartate level was determined in the indicated cells.
(F) Total proteins were extracted from LINC01234-depleted cells and western blot was performed for the indicated proteins.
(G) LINC01234-overexpressed cells were harvested and lysed. The total proteins were extracted and subjected to western blot using the indicated antibodies. See also Figure S1.
Next, we examined the effect of LINC01234 on ASS1 expression. As shown in Figure 4C, ASS1 was significantly upregulated in LINC01234-depleted HepG2 and Huh7 cells. Consistently, LINC01234 overexpression downregulated mRNA level of ASS1 (Figure 4D). As the rate-limiting enzyme in the urea cycle, ASS1 promotes the conversion from aspartate to urea and ASS1 deficiency in cancer cells leads to upregulated cellular aspartate level, which further activates the mTORC1 pathway to support cancer progression. Indeed, it has been observed that LINC01234 regulated the cellular aspartate level in an ASS1-dependent manner (Figure 4E). We then evaluated mTOR activity after LINC01234 interfered. The protein level of ASS1 was increased in LINC01234-depleted cells (Figure 4F). Importantly, decreased mTORC1 activity was detected by evaluating the phosphorylation of the mTORC1 downstream substrate p70 S6K1 (ribosomal S6 protein kinase 1) (Figure 4F). Similarly, the overexpression of LINC01234 decreased the ASS1 protein level and increased mTORC1 activity (Figure 4G). Moreover, we found that LINC01234 failed to activate mTORC1 activity in ASS1-overexpressed cells (Figure S1). Together, these results uncovered that LINC01234 inhibits the expression of ASS1, leading to accumulated cellular aspartate levels and increased mTORC1 activity in cancer cells.
LINC01234 impairs the transcriptional activation of ASS1
We further determined how LINC01234 affects the expression of ASS1. Luciferase assays revealed that LINC01234 overexpression inhibited ASS1 transactivation (Figure 5A). Similarly, LINC01234 depletion upregulated ASS1 transactivation (Figure S2A). Through in silico analysis, we found that LINC01234 interacted with tumor suppressor p53 (Figure S2B). Previous studies have demonstrated that ASS1 is transcriptionally activated by p53.15 Therefore, we hypothesized that LINC01234 impairs p53-mediated transcriptional activation of ASS1. RNA immunoprecipitation (RIP) assays suggested that LINC01234 interacted with p53 (Figure 5B). To further characterize the interaction between LINC01234 and p53 in cells, we performed MS2-tagged RNA affinity purification and western blotting. Compared with the expression of the negative control, the co-expression of the MS2-LINC01234 and MS2-GFP plasmids led to a significant enrichment of p53, demonstrating that p53 binds to LINC01234 in cells (Figure 5C). An immunofluorescence colocalization analysis showed that LINC01234 and p53 colocalized mainly in the nucleus (Figure 5D), suggesting that the LINC01234 binds to p53 in the nucleus. Similar results were obtained by RIP assay (Figure 5E).
Figure 5.

LINC01234 suppresses the transcriptional activation of ASS1
(A) Luciferase activity in HEK293T cells transfected with indicated vectors was evaluated. Data are presented as the relative ratio luciferase activity to renilla activity.
(B) An RIP analysis was performed to detect p53-bound LINC01234 using anti-p53 antibody.
(C) HCC cells were transfected with the indicated vectors. Then, the whole cell lysates were subjected to co-immunoprecipitation using anti-p53 antibody.
(D) The colocalization of LINC01234 and p53 was determined. The nuclei were stained with DAPI. Scale bar, 10 μm.
(E) The subcellular colocalization of LINC01234 and p53 was determined by RIP assay.
(F) HepG2 and Huh7 cells were transfected as indicated. Western blot was performed to determine the protein level of p53.
(G) Chromatin immunoprecipitation (ChIP)-qPCR analyses were employed to analyze the occupancy on the promoter of ASS1, P21, and Puma in HepG2 cells.
(H) An RT-qPCR analysis was used to detect the p21 and puma levels in HepG2 cells.
(I) Cells transfected with the indicated vectors were harvested. The cell lysates were subjected to western blot using the indicated antibodies. Data are shown as mean ± standard deviation. See also Figure S2.
We next analyzed the effect of LINC01234 on p53 activity. LINC01234 had no effect on the p53 protein level (Figure 5F), indicating that LINC01234 may regulate p53-mediated transcriptional activity. The enrichment of p53 on ASS1 promoter was evaluated by chromatin immunoprecipitation experiments. As shown in Figure 5G, LINC01234 decreased the enrichment of p53 on the ASS1 promoter. However, LINC01234 had little effect on the enrichment of p53 on p21 or Puma promoters, the two typical downstream genes of p5337 (Figure 5G). Moreover, LINC01234 depletion increased the enrichment of p53 on ASS1 promoter, but had no effect on the enrichment of p53 on p21 or Puma promoter (Figure S2C). Consistently, the overexpression or knockdown of LINC01234 had no influence on mRNA levels of p21 and puma (Figure 5H). Reintroducing p53 impaired the effect of LINC01234 overexpression on ASS1 expression (Figure 5I). Furthermore, depletion of p53 ablated the effect of LINC01234 knockdown on the expression of ASS1 in HepG2 cells (Figure S2D). These results suggest that LINC01234 inhibits the expression of ASS1 induced by p53, which is an important transcriptional activator for ASS1 expression.
It is of interest to note why LINC01234 specifically modulates the expression of ASS1 other than p21 or puma. Recent studies have shown that lncRNAs modulate the transcription of target genes by directly forming DNA-RNA complexes with the promoter of downstream targets and subsequently affecting the activities of TFs.38,39 It is possible that LINC01234 interacts with the promoter of ASS1 and impairs the activity of p53. Therefore, we used sequence alignment to identify a sequence that potentially binds to ASS1 promoter as previously described.39 The results revealed that LINC01234 has three potential binding sites on the ASS1 promoter (Figure S2E). To further verify the results, the primers were designed according to the predicted binding sites and chromatin isolation by RNA purification assays were conducted to explore whether LINC01234 has the ability to bind to these sites. Among the three promoter fragments, the results showed that LINC01234 capture ASS1-promoter 1 (−807/–797) and promoter 2 (−614/–603) (Figure S2F). To clarify whether LINC01234 impairs the transcriptional activation of ASS1 by binding to these two sites, we generated the ASS1 reporter lacking binding sites (Figure S2G). The results revealed that the mutants significantly increased luciferase expression compared to the wild-type ASS1-pGL3 reporter vector after co-transfection with the LINC01234 expression plasmid (Figure S2H). Thus, these data revealed that LINC01234 directly forms RNA-DNA complex with the promoter of ASS1 and impairs transcriptional activation of ASS1.
HepG2 cells carry wild-type p53 while Huh7 cells have mutant p53. It is important to note that mutant p53 often loses the functions as tumor suppressor and fails to activate of ASS1.15 Thus, we explored whether LINC01234 regulates the expression of ASS1 after depleting mutant p53 in Huh7 cells. As shown in Figure S2I, LINC01234 still regulated the expression level of ASS1 in p53-depleted Huh7 cells. Moreover, the luciferase assay revealed that the binding sites are critical for the LINC01234-mediated activation of ASS1 in Huh7 cells (Figure S2J). These data suggested that LINC01234 regulates ASS1 expression by directly forming an RNA-DNA complex with the promoter of ASS1 and impairing transcriptional modulation of ASS1 by TFs, such as p53.
ASS1 restoration diminishes LINC01234-associated oncogenic properties in HCC cells
To determine whether ASS1 is critical for the LINC01234-mediated oncogenic effect, we depleted LINC01234 in ASS1-ablated cells and performed cell proliferation assays. As shown in Figures 6A and 6B, the knockdown of ASS1 abrogated LINC01234-related cell proliferation. LINC01234 had slight effect on colony formation rate when ASS1 was silenced (Figures 6C and 6D). Moreover, reintroduced ASS1 abolished the cell colony formation ability induced by LINC01234 (Figures 6E–6G). Thus, these results demonstrated that LINC01234 induced HCC cells proliferation owing to ASS1 inhibition.
Figure 6.

ASS1 restoration attenuates the effects of LINC01234 in HCC cells
(A) ASS1 was silenced in LINC01234-knockdown HepG2 and Huh7 cell lines. Western blot was used to analyze the indicated proteins.
(B) HepG2 and Huh7 cells were transfected as indicated and proliferation was assessed by MTT assays.
(C) A colony formation assay was performed as indicated.
(D) The number of colony formation in (C) was summarized from three independent experiments (mean ± standard deviation).
(E) LINC01234-overexpressed cells were transfected as indicated. Western blot was used to detect the indicated proteins.
(F) Colony formation assay was performed as indicated.
(G) The number of colony formation in (F) was summarized from three independent experiments (mean ± standard deviation).
Depletion of LINC01234 attenuates HCC progression
Then, we investigated the role of LINC01234 in tumorigenesis in vivo. LINC01234 knockdown significantly repressed cell-based xenograft tumor growth (Figures 7A–7C). As indicated by the hematoxylin and eosin and immunohistochemistry results, LINC01234 decrease the levels of the cell proliferation marker Ki67 (Figure 7D). We also examined the expression of ASS1 in mice xenografts. Elevated ASS1 level was observed in the LINC01234-depleted group (Figure 7E). Consistently, LINC01234 overexpression significantly promoted xenograft tumor growth (Figures S3A–S3C). These results are consistent with in vitro experimental results.
Figure 7.

Depletion of LINC01234 suppresses HCC growth in vivo and enhances the antitumor efficacy of sorafenib
(A) Tumor formation was performed in nude mice injected with LINC01234 shRNA1, LINC01234 shRNA2, or control cells (n = 6).
(B) The volume of xenograft obtained in (A) was measured and summarized.
(C) The weight of xenograft obtained in (A) was measured and summarized.
(D) The Ki67 expression was evaluated and summarized by immunohistochemistry in the mouse xenografts. Scale bar, 20 μm.
(E) The ASS1 level of xenografts were detected by western blot.
(F) The HCC cells were treated with sorafenib as indicated. Then, the cell number was evaluated by MTT assay.
(G) Colony number quantification of the HepG2 cells was determined after treatment with sorafenib.
(H) Colony number quantification of the Huh7 cells was determined after treatment with sorafenib. See also Figure S3.
Increased activation of mTORC1 activities is associated with resistance of sorafenib treatment in HCC.40 Since LINC01234 enhances mTORC1 activities in HCC cells, we asked if depletion of LINC01234 sensitized HCC cells to sorafenib treatment. Indeed, we found that LINC01234 depletion increased the sensitivity of HCC cells to the drug treatment (Figures 7F–7H). Together, our data suggested that the depletion of LINC01234 attenuates HCC progression and increases chemosensitivity of HCC cells to sorafenib.
Discussion
In cancer cells, the expression of some enzymes in metabolism is often dysregulated, which is beneficial to reprogram cancer cells metabolism pathways to increase their biomass and sustain their malignancies.41,42 Many tumors, including HCC, malignant melanoma, malignant pleural mesothelioma, and renal cancer, lack ASS1 to remodel their amino acid metabolism.11,12,43,44 The overexpression of ASS1 or activation of ASS1 by small molecule inhibits cancer progression.14 Here, we also observed a loss of ASS1 in HCC through bioinformatics analyses, further identifying ASS1 as a tumor suppressor in HCC. Some cancers lose ASS1 expression because of epigenetic silencing of the ASS1 promoter.43,45 The mechanisms by which ASS1 is downregulated in HCC remain unclear. Here, we identified that the lncRNA LINC01234 inhibits the expression of ASS1. We also demonstrated that LINC01234 interacts with the promoter of ASS1 and impairs the transcriptional activation of ASS1. Importantly, LINC01234 level is negatively correlated with the ASS1 mRNA level using the TCGA HCC cohort and our cohort. We also explored the relationship of LINC01234 and ASS1 expression in other cancers using the TCGA database. LINC01234 is also negatively associated with ASS1 expression in some cancers including colon adenocarcinoma and testicular germ cell tumors (Figure S4). These findings suggest that LINC01234 is one of critical regulators in ASS1 expression and amino acid metabolism. Recent studies have reported other mechanisms for ASS1 downregulation in human cancers. It has been observed that aberrant CpG methylation is associated with the decreased ASS1 expression.46 The histone acetyltransferase p300 and TFs, including p53 and HIF1α, also participate in regulating ASS1 expression.47,48 Together with our results, these studies suggest that the expression of ASS1 is controlled by multiple factors. It is important to further explore the relationships among these factors, especially in tumorigenesis.
As the rate-limiting enzyme in ureagenesis, ASS1 is responsible for catalyzing argininosuccinate formation from citrulline and aspartate. Elevated ASS1 activities accelerate the conversion of nitrogen from aspartate to urea.13 Consistently, we found that LINC01234 increases cellular aspartate in HCC cells. Aspartate is indispensable for cell survival and growth owing to its critical role in protein synthesis and nucleotide biosynthesis.49 Cells rely on their intracellular aspartate biosynthesis because circulating levels of aspartate are low compared with other amino acids. For cancer cells, they must program aspartate metabolism to increase aspartate production and decrease aspartate degradation for maintaining their malignancies.50 LINC01234 regulates cellular aspartate in an ASS1-dependent manner, suggesting that LINC01234 is mainly involved in remodeling aspartate catabolism in cancer cells. We also observed that LINC01234 had no effect on the expression of glutamic-oxaloacetic transaminase 1, the critical enzyme for aspartate synthesis.
Increased cellular aspartate also promotes activation of mTORC1 pathway, which is the crucial sensor of nutrients.51 Dysregulated mTOR signaling promotes tumor cell proliferation, metastasis and chemoresistance.52 Inhibitors of mTORC1 have been applied clinical trials for treating cancers, including HCC.53 Our results suggest that LINC01234 increases mTORC1 activity through inhibiting ASS1 expression. Moreover, LINC01234 facilitates cancer cell proliferation and migration. Thus, mTOR signaling is involved in LINC01234-associated cancer progression. Recently, it has been observed that endoplasmic reticulum (ER) stress induces the translocation of ASS1 from the cytoplasm to the ER and translocated ASS1 enhances ER stress-related cell death via the induction of CHOP expression, revealing that ASS1 induces cancer cell apoptosis in an amino acid metabolism-independent way.12 It needs to be investigated whether LINC01234 regulates ASS1-induced apoptosis.
At present, the list of lncRNAs involved in tumor progression is rapidly expanding. Many lncRNAs have been implicated in cancer metabolism regulation, but the underlying mechanisms remain poorly understood. We found that LINC01234 promotes HCC progression and uncovered the mechanism by which LINC01234 regulates aspartate metabolism. Recent studies have reported that LINC01234 exerts oncogenic effects in human cancers, including colorectal cancer, clear cell renal cancer, gastric cancer, non-small-cell lung cancer and esophageal cancer.22,23,25,27,29,31,54,55 LINC01234 was mainly identified as a ceRNA to competitively bind miRNAs, such as miR-642a-5p, miR-124-3p, miR-1284, and miR-433.21,29,56 Here, we uncovered that LINC01234 decreases ASS1 expression by inhibiting transcription of ASS1. Instead of working as a ceRNA, LINC01234 serves to affect activities of TFs by directly forming DNA-RNA complexes with an ASS1 promoter. In cells carrying wild-type p53, LINC01234 regulates p53-mediated ASS1 transcription. Moreover, LINC01234 still modulates the transcription of ASS1 in cells harboring mutant p53 although mutant p53 fails to activate transcription of ASS1,15 suggesting that LINC01234 may affect activities of other TFs in these cells. These findings expand our understanding about the roles of LINC01234 in cancer progression. Our study showed that LINC01234 is highly expressed in HCC and that high expression of LINC01234 is closely linked to unfavorable outcomes. Together with previous studies, these findings suggest that LINC01234 is highly expressed in multiple types of cancer and LINC01234 might be a promising pan-cancer therapeutic target.
Despite high-profile failures of multiple vascular endothelial growth factor receptor (VEGFR)-targeting agents against HCC, substantial efforts continue in this area of the endoplasmic reticulum.57 Sorafenib, a multi-kinase inhibitor, is commonly used in HCC therapy after being approved for treating advanced HCC in 2007.58 Mechanistically, sorafenib shows the ability to repress the Raf/MEK/ERK pathway, as well as regulating (platelet-derived growth factor receptor β, Fms-like tyrosine kinase, and VEGFR.59 However, patients are vulnerable to develop resistance to sorafenib during treatment.60 The mechanisms of sorafenib resistance are complicated.59 As an important downstream target of PI3K/AKT, the mTORC1 pathway is reported to be associated with sorafenib resistance. Activated mTORC1 phosphorylates its two downstream targets p70 S6K1 and eukaryotic initiation factor 4E binding protein 1 to support protein synthesis and drive cancer cell growth.42 Recent studies have reported that aspirin, an inhibitor of the mTOR pathway, promotes the anti-cancer activities of sorafenib.61 Results from clinical trials also showed that mTORC1 inhibitors, such as rapamycin and everolimus, seem to be an effective approach to increase sensitivity of sorafenib.62,63 In our study, LINC01234 upregulated mTORC1 activity by inhibiting ASS1 expression, indicating that LINC01234 may relate to sorafenib sensitivity. Indeed, our further results support the notion that downregulation of LINC01234 promotes sensitivity of HCC cells to sorafenib. In a future study, it will be more meaningful to validate the roles of LINC01234 in vivo. Since sorafenib is a multi-kinase inhibitor, it also needs to be further analyzed the latent alternative pathway by which LINC01234 relates to sorafenib sensitivity.
Conclusion
Our study uncovered that LINC01234 has a pivotal role in reprogramming aspartate metabolism in HCC. LINC01234 inhibits the transcription of ASS1 and decreases ASS1 activity, resulting in enhanced mTORC1 activation to promote HCC progression (Figure 8). Moreover, the depletion of LINC01234 attenuates the growth, migration, and drug resistance of HCC cells. Our findings provide a potential basis for RNA interference-based strategies that target lncRNAs and cancer metabolism for cancer treatment. It is unknown whether targeting LINC01234-mediated metabolic reprogramming based on RNA interference can be used to treat human cancers, including HCC. This needs to be further explored in future study.
Figure 8.

A working model explaining how LINC01234 promotes HCC progression
Materials and methods
Patient samples
Frozen human HCC tissues and adjacent non-tumoral tissues were randomly obtained from the patients who underwent resections in the Peking University Cancer Hospital from 2009 to 2012. The clinical pathologic characteristics of patients including gender, age, maximum tumor diameter, number of tumor nodules, hepatitis B virus, hepatitis C virus, alpha fetoprotein level, total bilirubin level, cirrhosis, Edmondson-Steiner grading, TNM grading, venous infiltration, and lymph node infiltration are summarized in Table S1. This study was reviewed and approved by the Ethics Committee of Peking University Cancer Hospital & Institute, and written informed consent was obtained from all patients.
Animal study
Female athymic BALB/c nude mice (4–6 weeks old, Beijing Vital River Laboratory Animal Technology Co., Ltd) were randomly divided into five groups. The indicated Huh7 cells in 200 μL PBS with 10% Matrigel were subcutaneously injected in right axilla of mice. One month later, the mice were euthanized and tumors were dissected. The tumor volume is the length × width2/2. The animal study was reviewed and approved by the Ethics Review Committee of Peking University Health Science Center. The approval number of present investigation assigned by review ethics committee is LA2021208.
Cell culture
HepG2, Huh7, SMMC-7721, MHCC-97H, PLC, or HEK293T cell lines were purchased from Cell Resource Center, IBMS, CAMS/PUMC. HepG2, PLC, and HEK293T cells were cultured in the DMEM medium supplemented with 10% fetal bovine serum. Huh7 and SMMC-7721 cells were cultured in the RPMI1640 medium containing 10% fetal bovine serum. Cells were maintained in 5% CO2 at 37°C in incubators with 100% humidity.
RNA extraction and RT-qPCR
Total RNA was extracted from the HCC tissues or cultured cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized from 5 μg of RNA using the superscript first-strand synthesis system (Roche). Then, the RT-qPCR was performed using the 7500 Real-Time PCR System. The sequences of the qPCR primers are shown in Table S2.
Transfection
Cells were transfected with plasmid DNA using Polyethylenimine (Polyplus, France) according to the manufacturer’ protocol. Cells were transfected with siRNA duplexes using lipofectamine 2000 (Invitrogen) according to the manufacturer’ protocol. In transient transfection experiments, plasmid DNA concentrations were kept at a constant level with an empty vector. The sequences of short hairpin RNAs (shRNAs) and siRNAs are listed in the Table S3.
Antibodies and plasmids
Anti-ASS1 antibody (A9084) was purchased from Abclonal Technology. Anti-p53 antibody (DO-1) and anti-β-actin antibody were purchased from Santa Cruz Biotechnology. Anti-p70 S6 Kinase (49D7) and anti-phospho-p70 S6 Kinase (Thr389) (108D2) antibodies were purchased from Cell Signaling Technology. LINC01234 was cloned into the pcDNA3.1 or PCDH-CMV-MCS-EF1-GFP vector. Flag-tagged p53 was cloned into pCI-neo vector. LINC01234 shRNA1 and shRNA2 were cloned into pLKO.1 vector. All plasmids cloned with PCR inserts were confirmed by DNA sequencing (RioBiotech). pLKO.1-shRNA-lentivirus was packaged using a three-plasmids system, including pMD2.G, psPAX2 as helper plasmids, and pLKO.1 shRNA plasmid. PCDH-lentivirus was packaged using a four-plasmids system, including pMDLg/pRRE, pRSV-Rev, pMD2.G as helper plasmids, and PCDH plasmid.
Cell proliferation assay
Cell proliferation assay was performed using MTT (3-(4,5)-dimethylthiahiazo (-z-y1)-2,5-di- phenytetrazoliumromide) kit (Promega). Cells treated as indicated were seeded into 96-well plate (500 cells/well). Cell number was evaluated by MTT assay according to the manufacturer’ protocol for each day.
Cell migration and invasion assays
Cell migration and invasion assays were carried out in 24-well transwell chambers with 8-μm-pore polycarbonate filter inserts (Corning Inc.). Briefly, HepG2 and Huh7 cells were seeded in uncoated or matrigel-coated inserts (BD Biosciences). The lower chambers were added with 10% fetal bovine serum-supplemented DMEM as chemoattractant. Then, cells on the upper side of the filter were removed and those on the lower surface of the insert were fixed in 4% paraformaldehyde and stained with crystal violet (Beyotime Biotechnology). Finally, the migrated cells were quantified and summarized.
Colony formation assay
Cells were treated as indicated and subsequently seeded into 6-well plate (500 cells per well). After cultured for 14 days, the colonies were fixed with 4% paraformaldehyde (VETEC) and stained with 0.1% crystal violet (Sigma-Aldrich). The visible colonies were counted and summarized.
Western blotting
Cells were collected and total proteins were extracted. Protein concentration was determined using Coomassie brilliant blue method (Beyotime, Shanghai). After denaturation, the total proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Then, the proteins were transferred onto nitrocellulose filter membrane. The membrane was incubated with the indicated primary antibodies after blocked with 5% milk. Next, the membrane was treated with anti-rabbit or anti-mouse secondary antibodies for 1 h. Finally, all bands were detected by an ECL western blot kit ( Gene-Protein Link). A β-actin was used as the control.
Immunohistochemistry
The xenograft tumorous tissues were performed on paraffin-embedded sections. The slices were treated for dewaxing and rehydrating. After antigen retrieval, the endogenous peroxidase activity was blocked with 3% hydrogen peroxide. Then, the sections were treated with 10% goat serum for 30 min at room temperature. Sections were incubated with the primary antibody at 4°C overnight and then with HRP-conjugated goat anti-rabbit IgG (Zhongshan Golden Bridge Biotechnology) at 37°C for 30 min. The slides were stained with 3,3-diaminobezidine (DAKO) for visualization. Finally, the slides were counterstained with light hematoxylin.
Drug treatment
Sorafenib (SC0236, Beyotime Biotechnology) was resolved in DMSO to 10 μM for storage. The drug was diluted in cell culture medium for usage.
Fluorescence in situ hybridization
FISH experiment was performed using the Ribo Fluorescent In Situ Hybridization Kit (RiboBio) according to the manufacturer’ protocol. Briefly, cells were plated on the slides. After fixed with 4% PFA for 15 min in room temperature, the cells were permeabilized with 0.5% Triton X-100. Then, the Cy3-labeled probes were added for detecting the indicated RNAs, following by DAPI staining to visualize the nuclei.
RNA subcellular fraction
Cytoplasmic and nuclear RNA purification kit (Norgen Biotek) was used for RNA subcellular fractionation. RNA was extracted from the cytoplasmic and nuclear fractions according to the manufacturer's instructions. Then, RT-qPCR was employed to detect LINC01234 level. β-Actin was used as the cytoplasmic marker, and U6 was used as the nuclear marker.
Luciferase activity assay
Reporter assays were performed using the Dual-Luciferase Assay System (Promega). Briefly, the cells were co-transfected with ASS1 promoter luciferase reporter constructs or pGL3-basic reporter control plasmid and a Renilla luciferase control reporter vector (Promega). Forty-eight hours later, the luciferase assays were carried out according to the manufacturer's instruction.
RIP
A RIP assay was performed as previously described.64 In brief, cells were irradiated with UV light and lysed in high salt lysis buffer (25 mM Tris-Cl pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5% NP-40, 1 mM NaF, 1 mM DTT, 100 U/mL Rinonuclease inhibitors [RNasin), an EDTA-free protease inhibitor). Then, magnetic beads coated with 5 μg of anti-p53 antibodies were incubated with the prepared cell lysates overnight at 4°C. The RNA-protein complexes were washed six times and incubated with proteinase K digestion buffer. RNA was extracted by Trizol-chloroform RNA extraction methods. The relative expression of RNA was evaluated by RT-qPCR and normalized to the input.
Chromatin immunoprecipitation
Briefly, HCC cells transfected as indicated were cross-linked with 1% formaldehyde for 10 min, followed by glycine treatment to stop cross-linking. Then, cells were lysed in nuclear lysis buffer and the lysate was sonicated to shear DNA to approximately 200–1000 bp. Supernatant from 1 × 106 cells was used for each immunoprecipitation with the indicated antibodies. After immunoprecipitation, the DNA fragments were purified using TIANamp Genomic DNA Kit (TIANGEN BIOTECH) and were quantified by RT-qPCR using the indicated primers. Human ASS1 binding site was amplified by using the following primers: (forward) 5′-AGAGTCCACTCCCGAGCAG-3′ and (reverse) 5′-ATCAAAGCCCAAGTCCCCTA-3′.
Aspartate assay
An aspartate assay kit (MAK095, Sigma) was used to detect intracellular aspartate level. According to the manufacturer’ protocol, cells were washed with PBS twice. Then, cells were homogenized in 200 μL of the aspartate assay buffer. The samples were centrifuged at 13,000 × g for 10 min to remove insoluble material. The sample was added with reaction buffer and the mixture was incubated for 30 min at room temperature. Finally, the absorbance was measured at 570 nm.
The contents of aspartate were normalized to the protein concentration.
Statistical analysis
The significance of two groups was assessed by Student t test. The Kaplan-Meier method test was used for disease-free survival and overall survival analyses. Spearman correlation analysis was used to calculate the correlation between LINC01234 and ASS1 level. All statistical analyses were performed using GraphPad 8.0 software. A p value of less than 0.05 was determined statistical significance. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Data are shown as mean ± standard deviation.
Acknowledgments
The authors thank Dr Qihua He (Peking University Health Science Center) for assistance with confocal microscopy. This work was supported by the grants from the National Natural Science Foundation of China [Grant No. 81874143], the National Natural Science Foundation of China [Grant No. 81802305], the Beijing Natural Science Foundation [Grant No. 7212061], and the Science Foundation of Peking University Cancer Hospital 2020-1.
Author contributions
M.H.C., X.F.L., and B.C.X. designed the project. M.H.C., C.F.Z., and X.F.L. performed the experiments and collected data. M.H.C., X.F.L., and X.J.D. analyzed the data. C.F.Z. and W.L. collected tissue samples. X.F.L. and B.C.X. supervised the project. M.H.C., X.J.D., X.F.L., and B.C.X. prepared the manuscript with input from all authors. The authors read and approved the final manuscript.
Declaration of interests
The authors declare no conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.02.020.
Contributor Information
Xiaofeng Liu, Email: liuxiaofeng100@bjmu.edu.cn.
Baocai Xing, Email: xingbaocai88@sina.com.
Supplemental information
References
- 1.Forner A., Reig M., Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 2.Xu D., Liu X., Wang L., Xing B. Hepatectomy plus adjuvant transcatheter arterial chemoembolization improves the survival rate of patients with multicentric occurrence of hepatocellular carcinoma. Oncol. Lett. 2018;16:5882–5890. doi: 10.3892/ol.2018.9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pranzini E., Pardella E., Paoli P., Fendt S.M., Taddei M.L. Metabolic reprogramming in anticancer drug resistance: a focus on amino acids. Trends Cancer. 2021;7:682–699. doi: 10.1016/j.trecan.2021.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Xiao F., Wang C., Yin H., Yu J., Chen S., Fang J., Guo F. Leucine deprivation inhibits proliferation and induces apoptosis of human breast cancer cells via fatty acid synthase. Oncotarget. 2016;7:63679–63689. doi: 10.18632/oncotarget.11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maddocks O.D., Berkers C.R., Mason S.M., Zheng L., Blyth K., Gottlieb E., Vousden K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly M.P., Jungbluth A.A., Wu B.W., Bomalaski J., Old L.J., Ritter G. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br. J. Cancer. 2012;106:324–332. doi: 10.1038/bjc.2011.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane A.N., Fan T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015;43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayers J.R., Vander Heiden M.G. Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 2015;40:130–140. doi: 10.1016/j.tibs.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimmock D., Maranda B., Dionisi-Vici C., Wang J., Kleppe S., Fiermonte G., Bai R., Hainline B., Hamosh A., O'Brien W.E., et al. Citrin deficiency, a perplexing global disorder. Mol. Genet. Metab. 2009;96:44–49. doi: 10.1016/j.ymgme.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Phillips M.M., Sheaff M.T., Szlosarek P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res. Treat. 2013;45:251–262. doi: 10.4143/crt.2013.45.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S., Lee M., Song Y., Lee S.Y., Choi I., Park I.S., Kim J., Kim J.S., Kim K.M., Seo H.R. Argininosuccinate synthase 1 suppresses tumor progression through activation of PERK/eIF2alpha/ATF4/CHOP axis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021;40:127. doi: 10.1186/s13046-021-01912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabinovich S., Adler L., Yizhak K., Sarver A., Silberman A., Agron S., Stettner N., Sun Q., Brandis A., Helbling D., et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou Z., Hu X., Luo T., Ming Z., Chen X., Xia L., Luo W., Li J., Xu N., Chen L., et al. Naturally-occurring spinosyn A and its derivatives function as argininosuccinate synthase activator and tumor inhibitor. Nat. Commun. 2021;12:2263. doi: 10.1038/s41467-021-22235-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto T., Lo P.H.Y., Saichi N., Ueda K., Hirata M., Tanikawa C., Matsuda K. Argininosuccinate synthase 1 is an intrinsic Akt repressor transactivated by p53. Sci. Adv. 2017;3:e1603204. doi: 10.1126/sciadv.1603204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao W., Liu J.N., Liu Z., Wang X., Han Z.G., Ji T., Chen W.T., Zou X. A three-lncRNA signature derived from the Atlas of ncRNA in cancer (TANRIC) database predicts the survival of patients with head and neck squamous cell carcinoma. Oral Oncol. 2017;65:94–101. doi: 10.1016/j.oraloncology.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 17.Yuan L., Xu Z.Y., Ruan S.M., Mo S., Qin J.J., Cheng X.D. Long non-coding RNAs towards precision medicine in gastric cancer: early diagnosis, treatment, and drug resistance. Mol. Cancer. 2020;19:96. doi: 10.1186/s12943-020-01219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu K., Jiang Y., Zhou W., Zhang B., Li Y., Xie F., Zhang J., Wang X., Yan M., Xu Q., et al. Long noncoding RNA RC3H2 facilitates cell proliferation and invasion by targeting MicroRNA-101-3p/EZH2 Axis in OSCC. Mol. Ther. Nucleic Acids. 2020;20:97–110. doi: 10.1016/j.omtn.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grillone K., Riillo C., Scionti F., Rocca R., Tradigo G., Guzzi P.H., Alcaro S., Di Martino M.T., Tagliaferri P., Tassone P. Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter”. J. Exp. Clin. Cancer Res. 2020;39:117. doi: 10.1186/s13046-020-01622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z., Chen X., Lu B., Gu Y., Chen Q., Lei T., Nie F., Gu J., Huang J., Wei C., et al. Up-regulated LINC01234 promotes non-small-cell lung cancer cell metastasis by activating VAV3 and repressing BTG2 expression. J. Hematol. Oncol. 2020;13:7. doi: 10.1186/s13045-019-0842-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu D., Jian X., Xu P., Zhu R., Wang Y. Linc01234 promotes cell proliferation and metastasis in oral squamous cell carcinoma via miR-433/PAK4 axis. BMC Cancer. 2020;20:107. doi: 10.1186/s12885-020-6541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu P., Zhu R., Wang Y., Yang F. Long non-coding RNA LINC01234 regulates proliferation, migration and invasion via HIF-2α pathways in clear cell renal cell carcinoma cells. BMC Cancer. 2020;8:e10149. doi: 10.7717/peerj.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X., Chen Z., Yu S., Nie F., Yan S., Ma P., Chen Q., Wei C., Fu H., Xu T., et al. Long noncoding RNA LINC01234 functions as a competing endogenous RNA to regulate CBFB expression by sponging miR-204-5p in gastric cancer. Clin. Cancer Res. 2018;24:2002–2014. doi: 10.1158/1078-0432.CCR-17-2376. [DOI] [PubMed] [Google Scholar]
- 24.Li H., Gao C., Liu L., Zhuang J., Yang J., Liu C., Zhou C., Feng F., Sun C. 7-lncRNA assessment model for monitoring and prognosis of breast cancer patients: based on cox regression and Co-expression analysis. Environ. Toxicol. 2019;9:1348. doi: 10.3389/fonc.2019.01348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghaffar M., Khodahemmati S., Li J., Shahzad M., Wang M., Wang Y., Li C., Chen S., Zeng Y. Long non-coding RNA LINC01234 regulates proliferation, invasion and apoptosis in esophageal cancer cells. J. Cell Biochem. 2018;9:4242–4249. doi: 10.7150/jca.26095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z., Liao X., Zhan W., Zhang J., Cheng Z., Li L., Tian T., Yu L., Li R. Long noncoding RNA LINC01234 promoted cell proliferation and invasion via miR-1284/TRAF6 axis in colorectal cancer. J. Hematol. Oncol. 2020;121:4295–4309. doi: 10.1002/jcb.29618. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z., Chen X., Lei T., Gu Y., Gu J., Huang J., Lu B., Yuan L., Sun M., Wang Z. Integrative analysis of NSCLC identifies LINC01234 as an oncogenic lncRNA that interacts with HNRNPA2B1 and regulates miR-106b biogenesis. Mol. Ther. 2020;28:1479–1493. doi: 10.1016/j.ymthe.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J., Han L.N., Song J.R., Bai X.M., Wang J.Z., Meng L.F., Li J., Zhou W., Feng Y., Feng W.R., et al. Long noncoding RNA LINC01234 silencing exerts an anti-oncogenic effect in esophageal cancer cells through microRNA-193a-5p-mediated CCNE1 downregulation. Cell Oncol. (Dordr) 2020;43:377–394. doi: 10.1007/s13402-019-00493-5. [DOI] [PubMed] [Google Scholar]
- 29.Liao X., Zhan W., Zhang J., Cheng Z., Li L., Tian T., Yu L., Li R. Long noncoding RNA LINC01234 promoted cell proliferation and invasion via miR-1284/TRAF6 axis in colorectal cancer. J. Cell Biochem. 2020;121:4295–4309. doi: 10.1002/jcb.29618. [DOI] [PubMed] [Google Scholar]
- 30.Chen X., Liu Y., Yang Z., Zhang J., Chen S., Cheng J. LINC01234 promotes multiple myeloma progression by regulating miR-124-3p/GRB2 axis. Am. J. Transl Res. 2019;11:6600–6618. [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y., Zhao H., Li H., Feng X., Tang H., Qiu C., Zhang J., Fu B. LINC01234/MicroRNA-31-5p/MAGEA3 Axis mediates the proliferation and chemoresistance of hepatocellular carcinoma cells. Mol. Ther. Nucleic Acids. 2020;19:168–178. doi: 10.1016/j.omtn.2019.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Rogers L.C., Zhou J., Baker A., Schutt C.R., Panda P.K., Van Tine B.A. Intracellular arginine-dependent translation sensor reveals the dynamics of arginine starvation response and resistance in ASS1-negative cells. Cancer Metab. 2021;9:4. doi: 10.1186/s40170-021-00238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji J.X., Cochrane D.R., Tessier-Cloutier B., Chen S.Y., Ho G., Pathak K.V., Alcazar I.N., Farnell D., Leung S., Cheng A., et al. Arginine depletion therapy with ADI-PEG20 limits tumor growth in argininosuccinate synthase-deficient ovarian cancer, including small-cell carcinoma of the ovary, hypercalcemic type. Clin. Cancer Res. 2020;26:4402–4413. doi: 10.1158/1078-0432.CCR-19-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams R.T., Guarecuco R., Gates L.A., Barrows D., Passarelli M.C., Carey B., Baudrier L., Jeewajee S., La K., Prizer B., et al. ZBTB1 regulates asparagine synthesis and leukemia cell response to L-asparaginase. Cell Metab. 2020;31:852–861e856. doi: 10.1016/j.cmet.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y., Pusch S., Innes J., Sidlauskas K., Ellis M., Lau J., El-Hassan T., Aley N., Launchbury F., Richard-Loendt A., et al. Mutant IDH sensitizes gliomas to endoplasmic reticulum stress and triggers apoptosis via miR-183-mediated inhibition of semaphorin 3E. Cancer Res. 2019;79:4994–5007. doi: 10.1158/0008-5472.CAN-19-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bott A.J., Shen J., Tonelli C., Zhan L., Sivaram N., Jiang Y.P., Yu X., Bhatt V., Chiles E., Zhong H., et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 2019;29:1287–1298e1286. doi: 10.1016/j.celrep.2019.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X., Tan Y., Zhang C., Zhang Y., Zhang L., Ren P., Deng H., Luo J., Ke Y., Du X. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016;17:349–366. doi: 10.15252/embr.201540505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Postepska-Igielska A., Giwojna A., Gasri-Plotnitsky L., Schmitt N., Dold A., Ginsberg D., Grummt I. LncRNA Khps1 regulates expression of the proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol. Cell. 2015;60:626–636. doi: 10.1016/j.molcel.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Su F., He W., Chen C., Liu M., Liu H., Xue F., Bi J., Xu D., Zhao Y., Huang J., et al. The long non-coding RNA FOXD2-AS1 promotes bladder cancer progression and recurrence through a positive feedback loop with Akt and E2F1. Cell Death Dis. 2018;9:233. doi: 10.1038/s41419-018-0275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim M.N., Lee S.M., Kim J.S., Hwang S.G. Preclinical efficacy of a novel dual PI3K/mTOR inhibitor, CMG002, alone and in combination with sorafenib in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2019;84:809–817. doi: 10.1007/s00280-019-03918-y. [DOI] [PubMed] [Google Scholar]
- 41.Evers T.M.J., Holt L.J., Alberti S., Mashaghi A. Reciprocal regulation of cellular mechanics and metabolism. Nat. Metab. 2021;3:456–468. doi: 10.1038/s42255-021-00384-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X.F., Cai S.Y., Zhang C.F., Liu Z.Z., Luo J.Y., Xing B.C., Du X.J. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 2018;46:9601–9616. doi: 10.1093/nar/gky777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delage B., Luong P., Maharaj L., O'Riain C., Syed N., Crook T., Hatzimichael E., Papoudou-Bai A., Mitchell T.J., Whittaker S.J., et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. 2012;3:e342. doi: 10.1038/cddis.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai C.Y., Chi H.C., Chi L.M., Yang H.Y., Tsai M.M., Lee K.F., Huang H.W., Chou L.F., Cheng A.J., Yang C.W., et al. Argininosuccinate synthetase 1 contributes to gastric cancer invasion and progression by modulating autophagy. FASEB J. 2018;32:2601–2614. doi: 10.1096/fj.201700094R. [DOI] [PubMed] [Google Scholar]
- 45.Nicholson L.J., Smith P.R., Hiller L., Szlosarek P.W., Kimberley C., Sehouli J., Koensgen D., Mustea A., Schmid P., Crook T. Epigenetic silencing of argininosuccinate synthetase confers resistance to platinum-induced cell death but collateral sensitivity to arginine auxotrophy in ovarian cancer. Int. J. Cancer. 2009;125:1454–1463. doi: 10.1002/ijc.24546. [DOI] [PubMed] [Google Scholar]
- 46.Tao X., Zuo Q., Ruan H., Wang H., Jin H., Cheng Z., Lv Y., Qin W., Wang C. Argininosuccinate synthase 1 suppresses cancer cell invasion by inhibiting STAT3 pathway in hepatocellular carcinoma. Acta Biochim. Biophys. Sinica. 2019;51:263–276. doi: 10.1093/abbs/gmz005. [DOI] [PubMed] [Google Scholar]
- 47.Tsai W.B., Long Y., Chang J.T., Savaraj N., Feun L.G., Jung M., Chen H.H.W., Kuo M.T. Chromatin remodeling system p300-HDAC2-Sin3A is involved in Arginine Starvation-Induced HIF-1α Degradation at the ASS1 promoter for ASS1 Derepression. Scientific Rep. 2017;7:10814. doi: 10.1038/s41598-017-11445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silberman A., Goldman O., Boukobza Assayag O., Jacob A., Rabinovich S., Adler L., Lee J.S., Keshet R., Sarver A., Frug J., et al. Acid-induced downregulation of ASS1 contributes to the maintenance of intracellular pH in cancer. Cancer Res. 2019;79:518–533. doi: 10.1158/0008-5472.CAN-18-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi B.H., Coloff J.L. The diverse functions of non-essential amino acids in cancer. Cancers (Basel) 2019;11:675. doi: 10.3390/cancers11050675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melendez-Rodriguez F., Urrutia A.A., Lorendeau D., Rinaldi G., Roche O., Bogurcu-Seidel N., Ortega Muelas M., Mesa-Ciller C., Turiel G., Bouthelier A., et al. HIF1alpha suppresses tumor cell proliferation through inhibition of aspartate biosynthesis. Cell Rep. 2019;26:2257–2265e2254. doi: 10.1016/j.celrep.2019.01.106. [DOI] [PubMed] [Google Scholar]
- 51.Krall A.S., Mullen P.J., Surjono F., Momcilovic M., Schmid E.W., Halbrook C.J., Thambundit A., Mittelman S.D., Lyssiotis C.A., Shackelford D.B., et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021;33:1013–1026 e1016. doi: 10.1016/j.cmet.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 53.Sun S.Y. mTOR-targeted cancer therapy: great target but disappointing clinical outcomes, why? Front. Med. 2021;15:221–231. doi: 10.1007/s11684-020-0812-7. [DOI] [PubMed] [Google Scholar]
- 54.Xie J.J., Guo Q.Y., Jin J.Y., Jin D. SP1-mediated overexpression of lncRNA LINC01234 as a ceRNA facilitates non-small-cell lung cancer progression via regulating OTUB1. J. Cell Physiol. 2019;234:22845–22856. doi: 10.1002/jcp.28848. [DOI] [PubMed] [Google Scholar]
- 55.Zhang C., Huang D., Liu A., Xu Y., Na R., Xu D. Genome-wide screening and cohorts validation identifying novel lncRNAs as prognostic biomarkers for clear cell renal cell carcinoma. J. Cell Biochem. 2020;121:2559–2570. doi: 10.1002/jcb.29478. [DOI] [PubMed] [Google Scholar]
- 56.Lin C., Zhang Y., Chen Y., Bai Y., Zhang Y. Long noncoding RNA LINC01234 promotes serine hydroxymethyltransferase 2 expression and proliferation by competitively binding miR-642a-5p in colon cancer. Cell Death Dis. 2019;10:137. doi: 10.1038/s41419-019-1352-4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Zhu A.X., Duda D.G., Sahani D.V., Jain R.K. HCC and angiogenesis: possible targets and future directions. Nat. Rev. Clin. Oncol. 2011;8:292–301. doi: 10.1038/nrclinonc.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilhelm S.M., Adnane L., Newell P., Villanueva A., Llovet J.M., Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PubMed] [Google Scholar]
- 59.Hu X., Zhu H., Shen Y., Zhang X., He X., Xu X. The role of non-coding RNAs in the sorafenib resistance of hepatocellular carcinoma. Front. Oncol. 2021;11:696705. doi: 10.3389/fonc.2021.696705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia S., Pan Y., Liang Y., Xu J., Cai X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine. 2020;51:102610. doi: 10.1016/j.ebiom.2019.102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun D., Liu H., Dai X., Zheng X., Yan J., Wei R., Fu X., Huang M., Shen A., Huang X., et al. Aspirin disrupts the mTOR-Raptor complex and potentiates the anti-cancer activities of sorafenib via mTORC1 inhibition. Cancer Lett. 2017;406:105–115. doi: 10.1016/j.canlet.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 62.Chan J.A., Mayer R.J., Jackson N., Malinowski P., Regan E., Kulke M.H. Phase I study of sorafenib in combination with everolimus (RAD001) in patients with advanced neuroendocrine tumors. Cancer Chemother. Pharmacol. 2013;71:1241–1246. doi: 10.1007/s00280-013-2118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gangadhar T., Cohen E.E., Janisch L., House L.K., Ratain M.J. A drug interaction study of sorafenib (S) and rapamycin (R) in patients with advanced malignancies. J. Clin. Oncol. 2008;26:2545. [Google Scholar]
- 64.Liu X., Su K., Sun X., Jiang Y., Wang L., Hu C., Zhang C., Lu M., Du X., Xing B. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2021;40:132. doi: 10.1186/s13046-021-01934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.