

Abstract
CXCL5 is overexpressed in colorectal cancer (CRC) and promotes distant metastasis and angiogenesis of tumors; however, the underlying mechanism that mediates CXCL5 overexpression in CRC remains unclear. Here, we successfully extracted and identified primary mesenchymal stromal cells (MSCs) and verified the promoting effects of tumor-associated MSCs on CRC proliferation and metastasis in vivo and in vitro. We found that MSCs not only promoted the expression of CXCL5 by secreting CCL7 but also secreted TGF-β to inhibit this process. After secretion, CCL7/CCR1 activated downstream CBP/P300 to acetylate KLF5 to promote CXCL5 transcription, while TGF-β reversed the effect of KLF5 on transcription activation by regulating SMAD4. Taken together, our results indicate that MSCs in the tumor microenvironment promoted the progression and metastasis of CRC and regulated the expression of CXCL5 in CRC cells by secreting CCL7 and TGF-β. KLF5 is the key site of these processes and plays a dual role in CXCL5 regulation. MSCs and their secreted factors may serve as potential therapeutic targets in the tumor environment.
Key words: mesenchymal stromal cells, CCL7, TGF-β, KLF5, acetylation
Graphical abstract
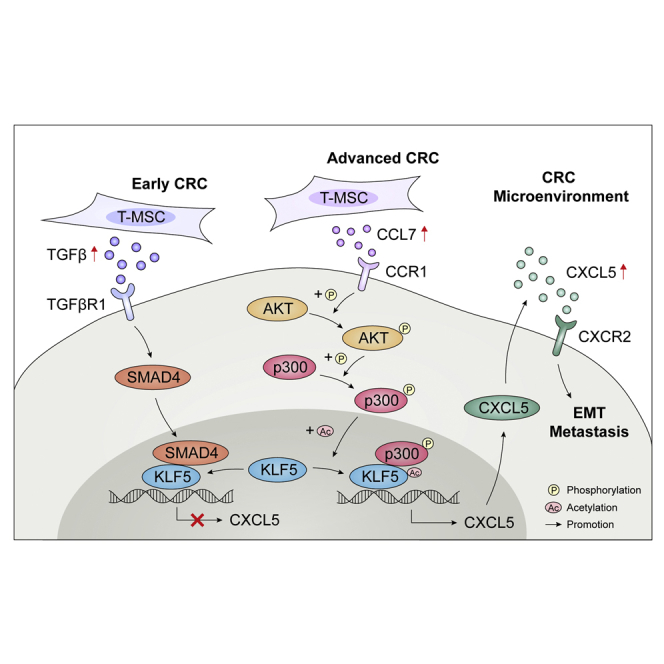
The role of mesenchymal stromal cells (MSCs) in the tumor microenvironment is a cutting-edge topic at present. Here, authors reported the specific markers to characterize MSCs and fibroblasts. They verified the promoting effects of MSCs on colorectal cancer proliferation and metastasis by secreting cytokines CCL7 and TGF-β.
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors of the digestive system, with morbidity and mortality ranking third in the world.1 Tumorigenesis, tumor development, and metastasis are complex and multistep processes between tumor cells and the tumor microenvironment (TME).2 A variety of cells and molecules participate in the regulation of this process, where chemokines play an important role.3 Our previous study found that C-X-C motif chemokine ligand 5 (CXCL5) is overexpressed in CRC tissues and promotes distant metastasis of tumors.4 CXCL5, also known as epithelial cell-derived neutrophil-activating peptide, is expressed in a variety of malignant tumors. By binding to the specific receptor CXCR2, CXCL5 mediates neutrophil migration, promotes tumor cell metastasis and invasion, and is related to tumor staging and poor prognosis.5,6 However, the underlying mechanism that mediates CXCL5 overexpression in CRC remains unclear.
Mesenchymal stromal cells (MSCs), also known as mesenchymal stem cells, are a type of heterogeneous mesenchymal progenitor cells that participate in tissue maintenance under normal homeostasis and are associated with pathological stromal responses to tissue damage and tumor formation.7,8 In the research on CRC, tumor-associated MSCs (T-MSCs) have been considered an activator for cancer progression and strongly promote CRC cell invasion and metastasis.9 Similarly, MSCs also showed analogous effects in other cancers, including lung cancer,10 prostate cancer,11 and breast cancer.12 However, currently, the molecular mechanism by which MSCs contribute to tumor pathogenesis and the mutual communication between MSCs and tumor cells in the TME are still unclear.
In this study, we explored the effects of MSCs and their secreted factors on CRC. We first extracted and identified primary MSCs from CRC tissues and normal tissues and verified the promoting effects of T-MSCs on CRC proliferation and metastasis in vivo and in vitro. We found that MSCs have a two-way pattern of regulation of CXCL5 expression in CRC cells. It not only promotes the expression of CXCL5 by secreting C-C motif chemokine ligand 7 (CCL7) but also secretes transforming growth factor β (TGF-β) to inhibit this process. CCL7/C-C motif chemokine receptor 1 (CCR1) activated downstream CREB binding protein (CBP)/P300 to acetylate KLF5 to promote CXCL5 transcription, while TGF-β reversed the effect of KLF5 on transcription activation by regulating SMAD4. Our research conclusively demonstrates that MSCs present in the CRC microenvironment up-regulated the expression of CXCL5 by secreting CCL7 to promote tumor progression and metastasis. However, at an early stage, MSCs also inhibited the expression of CXCL5 by secreting TGF-β, thereby inhibiting CRC progression. MSCs may become potential targets for tumor therapy owing to their ability to remodel the TME and regulate tumor cells.
Results
Isolation of MSCs from CRC tissue and normal tissue
To study the effect of MSCs in the TME on CRC cells, we collected matched CRC tissues and adjacent normal mucosa tissues from 20 patients and isolated MSCs from them (Figure 1A). According to different sources, these cells were named T-MSCs and normal MSCs (N-MSCs). We obtained 16 pairs of T-MSCs and N-MSCs and used them in subsequent experiments (Table S1). T-MSCs and N-MSCs isolated from patients have similar morphological characteristics, manifested as an adherent growth to plastic in minimum essential medium α (α-MEM) culture conditions, with elongated spindle (fibroblast-like) shape (Figure 1A). Next, we tested the ability of T-MSCs and N-MSCs to differentiate into adipocytes, osteoblasts, and chondrocytes in vitro (Figure 1B). There was no significant difference between the differentiation efficiency of T-MSCs and N-MSCs.
Figure 1.

Extraction and identification of MSCs
(A) Schematic diagram of extracting MSCs. H&E staining showed the adherent growth of MSCs. The MSCs showed in the figure were derived from patient P2. Scale, 100 μm. (B) Differentiation of MSCs. Alizarin red staining showed the osteogenic differentiation, oil red O staining showed adipogenic differentiation, and alcian blue staining showed chondrogenic differentiation. The MSCs showed in the figure were derived from patient P2. Scale, 100 μm. (C) The results of flow cytometry. Gray means blank control, red means T-MSCs, and blue means N-MSCs. The MSCs showed in the figure were derived from patient P8. (D) The results of immunofluorescence assay. The MSCs showed in the figure were derived from patient P8. Scale, 50 μm.
We then verified the mesenchymal phenotype of T-MSCs and N-MSCs by detecting surface marker expression (Figure 1C) and found that they expressed mesenchymal markers, including CD73, CD90, CD105, and CD29, but lacked expression of CD45 (bone marrow cells), CD31 (endothelial cells), or epithelial cell adhesion molecule (EpCAM; epithelial cells) (Figure S17A). In addition, we detected the expression of fibroblast markers in T-MSCs and N-MSCs and found that they expressed fibroblast specific protein-1 (FSP1), α smooth muscle actin (α-SMA), and vimentin (Figure 1D). Altogether, these data indicate that T-MSCs and N-MSCs isolated from patients have phenotypic characteristics of mesenchymal stem cells and fibroblasts.13
To further distinguish whether MSCs isolated from patients are mesenchymal stem cells or fibroblasts, we simultaneously extracted MSCs and fibroblasts, namely T-MSCs, N-MSCs, cancer-associated fibroblasts (CAFs), and normal fibroblasts (NFs), from tumors and normal tissues of the same CRC patients. We next determined their potential to differentiate into osteogenic, adipogenic, and chondrogenic cell types. The results showed that MSCs had a high differentiation potential, whereas NFs had an extremely low differentiation potential, and the differentiation potential of CAFs was between that of MSCs and NFs (Figures S1A and S2), indicating that T-MSCs and CAFs might represent a transitional state in the TME. In addition, we tested the expression of a series of mesenchymal-related markers in MSCs and fibroblasts, including fibroblast markers (CD106, CD26, CD10, CD29, α-SMA, FAP (fibroblast activation protein alpha), FSP1, PDGFRB (platelet derived growth factor receptor beta), and vimentin),14 mesenchymal cell markers (CD90, CD73, and CD105),15 and stem cell markers (CD44, CD34, CD133, and CD166).16 Among the mesenchymal cell markers, only CD90 was slightly down-regulated in fibroblasts, especially in NFs, and there was no significant difference in the expression of CD73 and CD105 among the different types of cells. Of the fibroblast markers, CD10 was found to be significantly less expressed in MSCs, especially in N-MSCs. In addition, CD106 expression was down-regulated in fibroblasts, but there was no significant difference in CD26 and CD29 expression among the four cell types. Among the stem cell markers, a pronounced up-regulation of CD34 was observed in fibroblasts, and there was no significant difference in the expression of CD44, CD133, and CD166 among the different types of cells (Figures S1B, S3, S4, S5, and S7). The results of the immunofluorescence assays showed that the fibroblast markers SMA, FAP, and PDGFRB were significantly up-regulated in fibroblasts, whereas FSP1 and vimentin levels were similar among the different types of cells. In particular, high expression of fibroblast activation markers, such as SMA, FAP, and CD10, was observed not only in fibroblasts but also in T-MSCs, representing an important feature that distinguishes them from N-MSCs (Figures S1C and S6). Thus, in this study, we designated these cells as MSCs according to published research.17
Effect of MSCs on CRC
To evaluate the effect of T-MSCs and N-MSCs on CRC cells, we designed two in vitro treatment methods (the MSC cell medium [MSC-CM]-treated and co-cultivation models) (Figure 2A). Cell counting kit-8 (CCK-8) and colony formation assays were performed to examine the effect of MSCs on CRC cell proliferation. In the MSC-CM-treated model, T-MSC-CM promoted the proliferation of HT29 and SW620 cells compared with N-MSC-CM. In contrast, both T-MSCs and N-MSCs promoted the proliferation of HT29 and SW620 cells in the co-cultivation model (Figures 2B–2D). In addition, the transwell assay revealed that T-MSC-CM promoted migration of HT29 and SW620 cells compared with N-MSC-CM in the MSC-CM treatment model, but there was no difference between T-MSCs and N-MSCs in the co-cultivation model (Figures 2E and 2F). We then used nude mice to assess the function of T-MSCs and N-MSCs in the CRC TME (Figures 2I and 2J) and found that, compared with the control, both T-MSC and N-MSC co-injection with CRC cells can significantly increase the volume and weight of xenografts, as well as the number of metastatic nodules, indicating that MSCs can promote tumor growth and metastasis in vivo.
Figure 2.

T-MSCs promoted the proliferation and migration of CRC cells
(A) Schematic diagram of MSC-CM treatment model and co-cultivation model. (B) The results of CCK-8 proliferation assay. The MSCs showed in the figure were derived from patient P8. (C and D) The results of colony formation assay. T-MSCs significantly promote the proliferation of HT29 and SW620 in MSC-CM treatment model, and both T-MSCs and N-MSCs promoted proliferation of HT29 and SW620 in the co-cultivation model. The MSCs showed in the figure were derived from patient P8. (E and F) The results of transwell assay. (G) The results of flow cytometry. The MSCs showed in the figure were derived from patient P8. Scale, 200 μm. (H) The results of immunofluorescence assay. The expression of α-SMA in T-MSCs was significantly higher than that in N-MSCs, and co-cultivation treatment significantly increased α-SMA expression in N-MSCs. The MSCs showed in the figure were derived from patient P8. Scale, 50 μm. (I) Diagram of xenograft tumors in nude mice. HT29 and SW620 were injected into nude mice alone or co-injected with MSCs. Both T-MSCs and N-MSCs significantly promoted CRC proliferation in vivo. The MSCs showed in the figure were derived from patient P2. (J) Diagram of liver metastasis in nude mice. HT29 and SW620 were injected into nude mice alone or co-injected with MSCs. Both T-MSCs and N-MSCs significantly promoted CRC metastasis in vivo. The MSCs showed in the figure were derived from patient P2. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Based on these results, we suspected that in a co-culture environment and in vivo, tumor cells might change the phenotype and function of N-MSCs. To verify this hypothesis, we tested α-SMA expression in N-MSCs, T-MSCs, and co-cultivation-treated N-MSCs. The results showed that the expression of α-SMA in T-MSCs was significantly higher than that in N-MSCs, and co-cultivation treatment significantly increased α-SMA expression in N-MSCs (Figures 2G and 2H), suggesting that tumor cells can activate N-MSCs in a co-culture environment and transform their phenotype similar to T-MSCs. Thus, we chose the MSC-CM-treated method to investigate the effect of T-MSCs on CRC cells in our follow-up research, and N-MSCs were used as a control.
T-MSCs promoted the expression of CXCL5 in CRC cells by secreting CCL7
Our previous study found that CXCL5 was overexpressed in CRC tissues and promoted distant metastasis, but the underlying mechanism of CXCL5 overexpression in CRC remains unclear. We have confirmed the effect of T-MSCs on the proliferation and metastasis of CRC in Figure 2. In addition, we also verified that T-MSC-CM treatment and co-cultivation with MSCs significantly promoted the epithelial-mesenchymal transition (EMT) phenotype of CRC cells, which resulted in tumor cells acquiring a mesenchymal, spindle-like morphology, decreased expression of the epithelial marker E-cadherin, and increased expression of the mesenchymal markers N-cadherin and vimentin (Figures S8 and S9). Thus, we explored whether T-MSCs can regulate the expression of CXCL5 in CRC cells and found that compared with N-MSC-CM, T-MSC-CM significantly promoted the expression of CXCL5 in both HT29 and SW620 cells (Figure S10A). We then collected the xenografts formed by CRC cells in the previous section (Figure 2I) and found that, compared with the control group, the MSC co-injection treatment significantly increased CXCL5 expression in HT29 and SW620 cells (Figures S10B and S10C). These results suggest that T-MSCs in the TME could promote the expression of CXCL5 in CRC cells. Next, 16 pairs of T-MSC-CM and N-MSC-CM were used to treat SW620 cells, and CXCL5 expression in SW620 cells was detected using western blotting and PCR analyses (Figures S10D and S10E). We selected the most effective T-MSCs (T2, T8, T12, T15, and T16) to promote CXCL5 expression and used them in subsequent experiments (Figures 3A and 3B).
Figure 3.

T-MSCs promoted the expression of CXCL5 in CRC cells by secreting CCL7
(A and B) The results of western blot. T-MSC-CM promoted the expression of CXCL5 in HT29 and SW620. The MSCs showed in the figure were derived from patients P2, P8, P12, P15, and P16. (C) HT29 and SW620 were treated with product of centrifugal filtration, and 3 kDa concentrate and 30 kDa filtrate enhanced the expression of CXCL5. The MSCs showed in the figure were derived from patient P15. (D) The results of qPCR showed the RNA expression of related cytokine in paired MSCs. The MSCs showed in the figure were derived from patients P2, P8, P12, P15, and P16. (E) The results of ELISA. T-MSCs secreted high levels of CCL7 and CCL8. The MSCs showed in the figure were derived from patients P2, P8, P12, P15, and P16. (F) The results of western blot. Expression of CXCL5 was increased in rhCCL7-treated HT29 and SW620 cell lines. (G) The results of western blot. Anti-CCL7 antibody neutralized the CXCL5 promotion effect of T-MSC-CM. The MSCs showed in the figure were derived from patient P15. (H) The results of western blot. Knocking down CCR1 decreased the expression of CXCL5 induced by rhCCL7. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We then attempted to analyze the contents of T-MSC-CM to explore the potential mechanism of its effect on regulating CXCL5 expression. T-MSC-CM was divided into six subsets, namely 3 kDa concentrate, 3 kDa filtrate, 10 kDa concentrate, 10 kDa filtrate, 30 kDa concentrate, and 30 kDa filtrate, using centrifugal filtration. The expression of CXCL5 in CRC cells increased after treatment with the 3 kDa concentrate and 30 kDa filtrate (Figure 3C), indicating that the molecular weight of the target protein was approximately 10 kDa. Secretory cytokines related to MSCs or CAFs were supported according to the reports and screened on the basis of their molecular mass (Table S9)18,19; 21 cytokines were finally selected. qPCR was used to screen the related RNA expression and revealed that only CCL7 and CCL8 were significantly overexpressed in all the T-MSCs compared with N-MSCs (Figure 3D). ELISA was used to verify the qPCR findings (Figure 3E). In addition, Oncomine and Gene Expression Profiling Interactive Analysis (GEPIA) software showed that the expression of CCL7, CCL8, and CXCL5 was highly correlated (Figure S10F).
Recombinant human CCL7 (rhCCL7), rhCCL8, anti-CCL7, and anti-CCL8 were used to validate the effects of CCL7 and CCL8. The results showed that the expression of CXCL5 in HT29 and SW620 cells increased significantly after rhCCL7 stimulation but remained unchanged after rhCCL8 stimulation (Figure 3F). Moreover, anti-CCL7 neutralized the CXCL5 promotion effect of T-MSC-CM, while anti-CCL8 had no significant effect on T-MSC-CM function (Figure 3G). In addition, when the concentration of rhCCL7 reached 1 ng/mL, the expression of CXCL5 increased significantly (Figures S10G and S10H), which was consistent with the concentration of CCL7 in T-MSC-CM (Figures 3E and S10I). Therefore, we concluded that T-MSC-CM promoted the expression of CXCL5 in CRC cells by secreting CCL7.
KLF5 induced by CCL7/CCR1 promoted the transcription of CXCL5 in CRC cells
The chemokine ligands in the microenvironment must bind to chemokine receptors on the cell membrane to function, and the specific receptors of the chemokine CCL7 include CCR1, CCR2, and CCR5.20 Western blotting analysis showed that CCR1 knockdown significantly decreased the expression of CXCL5 induced by CCL7, while knockdown of CCR2 or CCR5 did not affect the expression of CXCL5 in HT29 and SW620 cells (Figures 3H and S11A).
Studies have shown that transcription factors regulate gene transcription and protein levels by binding to specific sequences in the gene promoter regions.21 Because both T-MSC-CM and rhCCL7 could increase the expression of CXCL5 at the RNA level (Figures S10E and S10H), we speculated that this process was mediated by transcription factors. The JASPAR database was used to analyze the transcription factor binding sites in the promoter region of CXCL5, and the top 50 binding sites were selected (Table S10), which corresponded to 40 transcription factors. Next, the qPCR assay was used to detect the expression of the above 40 transcription factors, and the results showed that the expression of KLF5 and MEF2C was significantly up-regulated after rhCCL7 treatment (Figure 4A). Western blotting was used to verify the qPCR results (Figure 4B). Then, small interfering RNA (siRNA) was used to confirm the function of KLF5 and MEF2C, and the results showed that only when KLF5 was knocked down that the effect of rhCCL7 in promoting CXCL5 expression was blocked, while knocking down MEF2C did not significantly change CXCL5 expression (Figure 4C). These results suggest that CCL7/CCR1 promotes the expression of CXCL5 by inducing the expression of the downstream transcription factor KLF5.
Figure 4.

KLF5 induced by CCL7/CCR1 promoted the transcription of CXCL5 in CRC cells
(A) The results of qPCR showed the RNA expression of related transcription factors in HT29 and SW620. The expression of KLF5 and MEF2C were up-regulated by rhCCL7. (B) The results of western blot. rhCCL7 promoted the expression of KLF5 and MEF2C in HT29 and SW620. (C) The results of western blot. Knocking down KLF5 decreased the expression of CXCL5 induced by rhCCL7. (D) KLF5-binding elements (KBE1–5) on CXCL5 promoter region. (E) The results of luciferase reporter assay. KLF5 promoted the luciferase activity of pGL3-CXCL5-FL and truncated 1 plasmids. (F–H) ChIP assay showed the direct binding of KLF5 to the predicted site (KBE2) of the CXCL5 promoter. (I) The results of luciferase reporter assay. KLF5 could not promoted the luciferase activity of CXCL5-KBE2-Mut. (J and K) The results of ChIP assay. rhCCL7 promoted the binding efficiency of KLF5 to KBE2, which can be reversed by ML264(a KLF5 inhibitor). Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further verify whether KLF5 can regulate CXCL5 transcriptional expression by directly binding to its promoter region, we predicted the potential KLF5-binding elements (KBE1–5) on the CXCL5 promoter region using the JASPAR database (Table S11). Then, the plasmids of the full-length CXCL5 promoter region (pGL3-CXCL5-FL) and four truncated CXCL5 promoter fragments (truncated 1: −1200 to +100 nt; truncated 2: −600 to +100 nt; truncated 3: −100 to +100 nt; and truncated 4: −20 to +100 nt) were constructed (Figure 4D). The results showed that overexpression of KLF5 promoted the luciferase activity of pGL3-CXCL5-FL and truncated 1 plasmid (−1200 to +100 nt). In contrast, in the truncated 2 plasmid (−600 to +100 nt), which did not contain the KBE2 and KBE3 sequences, KLF5 overexpression had no significant effect on luciferase activity (Figure 4E). In truncated 3 and 4 plasmids, the luciferase activity was too weak and was not significantly different from that of pGL3-basic, suggesting that they were not effective promoter regions of CXCL5. Then, chromatin immunoprecipitation (ChIP) was used to detect the binding ability of KLF5 to these two binding sites, and the direct binding of KLF5 to KBE2 (−1112 bp) of the CXCL5 promoter (Figures 4F–4H). In addition, the sequence of KBE2 was deleted from pGL3-CXCL5-FL to construct the pGL3-CXCL5-KBE2-Mut plasmid. As expected, KBE2 mutation significantly reduced the CXCL5 promoter induced by KLF5 (Figure 4I). Subsequently, ChIP showed that rhCCL7 treatment promoted the binding efficiency of KLF5 to KBE2 in HT29 and SW620 cells, whereas ML264 (a KLF5 inhibitor) reversed this effect (Figures 4J and 4K). Therefore, we confirmed that KLF5 can promote the transcriptional expression of CXCL5 in CRC cells by specifically binding to the KBE2 (−1112 bp) sequence in the CXCL5 promoter region, and this effect was regulated by CCL7 and could be reversed by KLF5 inhibition.
The acetylation of KLF5 by CCL7/P300 promoted CXCL5 transcription by increasing KLF5 binding to the CXCL5 promoter region
The activation of transcription factors is a key driving factor in cancer development. KLF5 has been reported to form a transcription complex with other transcription factors or acetyltransferases, thereby elevating KLF5 acetylation or histone acetylation to regulate transcription.21,22 The immunoprecipitation (IP) assay was used to examine the acetylation level of KLF5 and showed that CCL7 stimulation significantly increased KLF5 acetylation (Figure 5A). In addition, the protein-protein interaction (PPI) network of KLF5 showed a strong interaction between CREBBP (CBP), EP300 (P300), and KLF5 (Figure 5B). Therefore, we next validated the interaction between KLF5 and CBP/P300 and found that CBP/P300 was significantly enriched by the KLF5 antibody (Figure 5C). Notably, CBP/P300 contains histone acetyltransferase (HAT) activity,23 and CBP/P300 can be activated by phosphorylation and acetylation.24 Therefore, we examined the phosphorylation and acetylation levels of CBP/P300 after CCL7 treatment and found that rhCCL7 significantly promoted the phosphorylation of CBP/P300, which can be blocked by si-CCR1 [small interfering-CCR1]) treatment (Figure 5D). In contrast, the acetylation of CBP/P300 was not affected by rhCCL7 or si-CCR1. A485, a CBP/P300-specific inhibitor25 was used to detect the effect of p300/CBP on CCL7-mediated CXCL5 overexpression. The results showed that the effect of rhCCL7 in promoting CXCL5 expression was reversed by A485 (Figure 5E), indicating that CCL7 promotes CXCL5 expression by increasing the phosphorylation of the KLF5 co-activator CBP/P300. The results of western blotting showed that rhCCL7 treatment increased the combination of CBP/P300 and KLF5 (Figures 5A and S11B). These results indicate that rhCCL7 stimulation promoted the CBP/P300-KLF5 combination and acetylation of KLF5 by CBP/P300.
Figure 5.

The acetylation of KLF5 by CCL7/P300 promoted CXCL5 transcription by increasing KLF5 binding to CXCL5 promoter region
(A) The results of IP and western blot. rhCCL7 increased KLF5 acetylation and the combination of CBP/P300 and KLF5. (B) PPI network of KLF5 constructed by STRING database and inBio Discover software. (C) The results of co-IP showed the interaction between KLF5 and CBP/P300. (D) The results of western blot. rhCCL7 significantly promoted the phosphorylation of CBP/P300. (E) A485 reversed the effect of rhCCL7 in promoting CXCL5 expression by inhibited the CBP/P300 function. (F) The results of luciferase reporter assay. Co-transfection with EP300 and KLF5 significantly increased the luciferase activity of pGL3-CXCL5-FL. (G) The results of ChIP assay showed that P300 and KLF5 formed a complex and bind to the CXCL5 promoter. (H) The results of western blot showed that rhCCL7 enhanced the phosphorylation level of AKT. (I) LY294002 inhibited the effect of rhCCL7 on promoting P300 phosphorylation and CXCL5 expression. (J) The results of IP and western blot. LY294002 and A485 blocked the function of CCL7 on acetylating KLF5. (K) ChIP assay showed that LY294002 and A485 significantly reduced the KLF5 binding on CXCL5 promoter. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The dbPTM database (https://awi.cuhk.edu.cn/dbPTM/) showed that KLF5 contains four acetylation sites (Table S12): lysine 31 (K31), lysine 335 (K335), lysine 369 (K369), and glutamine 278 (F278). We selected two sites with high scores (K335 and K369), and mutated the lysine (K) at both sites to non-acetylated arginine (R) to construct a non-acetylated KLF5 plasmid (KLF5-K335/369R). The results of the luciferase reporter assay showed that compared with cells transfected with EP300 or KLF5, co-transfection with EP300 and KLF5 significantly increased the luciferase activity of pGL3-CXCL5-FL (Figure 5F). This effect can be eliminated by replacing KLF5 with KLF5-K335/369R or replacing pGL3-CXCL5-FL with pGL3-CXCL5-KBE2-Mut, indicating that P300 amplified the effect of KLF5 on promoting CXCL5 transcription by acetylating KLF5. ChIP and Re-ChIP assays showed that P300 and KLF5 could form a complex and bind to the KBE2 region (−1192 to −1080 nt) of the CXCL5 promoter (Figure 5G). The pull-down assay showed that both KLF5 and CBP/P300 were significantly enriched by the KBE biotin-labeled probe, and CCL7 treatment increased the content of KLF5 and phosphorylation level of P300 in the protein precipitation (Figure S11C).
Next, we explored the mechanism underlying CCL7 promoting the phosphorylation of P300. A variety of protein kinases downstream of chemokine receptors have been reported to regulate P300 phosphorylation, including PI3K (phosphoinositide 3-kinase), AKT (protein kinase B), ERK (extracellular signal-regulated kinase),26 and JNK (c-Jun N-terminal kinase).27 Thus, we analyzed the expression of related proteins in HT29 and SW620 cells treated with or without rhCCL7 and found that the phosphorylation level of AKT was significantly enhanced by rhCCL7 treatment (Figures 5H and S11D). Next, an inhibitor of PI3K (LY294002) was used to verify the effect of PI3K/AKT on CCL7-related P300 phosphorylation and CXCL5 overexpression. Western blotting showed that the function of rhCCL7 in promoting P300 phosphorylation and then increasing CXCL5 expression could be reversed by LY294002 (Figures 5I and S11E). In addition, we also examined the effect of the PI3K inhibitor (LY294002) and CBP/P300 inhibitor (A485) on the KLF5 acetylation of CCL7 and found that both LY294002 and A485 can block the function of CCL7 in acetylating KLF5 (Figure 5J). ChIP assay verified that both LY294002 and A485 significantly reduced KLF5 binding to the CXCL5 promoter (Figure 5K), indicating that CCL7 can induce the formation of the KLF5-P300 complex on the CXCL5 promoter, which was regulated by the phosphorylation of AKT and P300.
MSC inhibited the expression of CXCL5 in CRC cells by secreting TGF-β
As shown in Figure 3, we screened the expression of specific RNAs in paired T-MSCs and N-MSCs. In particular, we were concerned that some RNAs were expressed at low levels in T-MSCs compared with paired N-MSCs, such as TGF-β (Figure 3D). Next, ELISA analysis showed that the expression patterns of TGF-β in MSCs derived from different patients were significantly different (Figure 6A). Only 37.5% of T-MSCs (n = 6) had higher TGF-β secretion compared with their matched N-MSCs. It has been reported that, in the early stage of tumor progression, TGF-β can promote cell apoptosis by inducing EMT, thereby acting as a tumor suppressor.28,29 Our results showed that T-MSCs derived from tumors at earlier stages showed higher secretion of TGF-β (Figure 6B). Similarly, immunohistochemistry (IHC) staining on 78 CRC tissues4 showed that TGF-β expression in the earlier stages (I + II) was much higher than that in the advanced tumor stages (III + Ⅳ) (Figures S12A and S12B). In addition, our previous research reported that high expression of CXCL5 was significantly correlated with tumor stage. Consistently, CRC cells treated with T-MSC-CM from advanced tumors showed higher CXCL5 expression (Figure 6C). Combined with the previous results on the effect of these 16 pairs of MSC-CM on regulating CXCL5 expression in CRC cells, we concluded that the content of TGF-β in T-MSC-CM was negatively correlated with CXCL5 expression in the CRC cells they treated (Figure 6D), which strongly suggested that TGF-β secreted by T-MSCs may have an inhibitory effect on CXCL5 expression.
Figure 6.

MSC inhibited the expression of CXCL5 in CRC cells by secreting TGF-β
(A) ELISA analysis showed the secretion of TGF-β in 16 pairs of MSCs. (B) T-MSCs derived from tumors with earlier TNM stage had higher secretion of TGF-β. (C) The relative RNA expression of CXCL5 of SW620 after treated with MSC-CM. Expression of CXCL5 was increased in SW620 after treated with T-MSC-CM from advanced tumor. (D) The content of TGF-β in T-MSC-CM was detected by ELISA, and the expression of CXCL5 of SW620 was detected by qPCR. The content of TGF-β in T-MSC-CM was negatively correlated with CXCL5 expression in SW620 they treated. (E) Western blot showed the expression of SMAD2/3 and SMAD4 in CRC cell lines. (F) The results of western blot. TGF-β treatment significantly reduced the CXCL5 expression in SW620 and HCT116. (G) Western blot showed the expression of KLF5 and CXCL5 in SW620 and HCT116 after indicated treatment. (H and I) Western blot showed the expression of CXCL5 in SW620 and HCT116 after indicated treatment. Knocking down SMAD4 and KLF5 recovered the inhibitory effect of TGF-β on the expression of CXCL5, and knocking down both SMAD4 and KLF5 increased the expression of CXCL5. (J) The results of luciferase reporter assay. Co-transfection with KLF5 and SMAD4 inhibited the luciferase activity of pGL3-CXCL5-FL. (K) The results of co-IP showed the interaction between KLF5 and SMAD4. (L and M) The results of co-IP and western blot. TGF-β promoted the combination of SMAD4 and KLF5. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
In CRC, the TGF-β downstream molecule SMAD4 is commonly mutated or inactivated, thereby escaping the inhibitory effect of TGF-β.30 Therefore, we screened the expression of SMAD2/3 and SMAD4 in CRC cell lines. Western blotting indicated that only SW620 and HCT116 cells expressed high levels of SMAD2/3 and SMAD4, and TGF-β treatment significantly reduced their CXCL5 expression (Figures 6E and 6F). Moreover, western blotting showed that the expression of CXCL5 decreased significantly when the concentration of TGF-β reached 2 ng/mL (Figure S12C), which was consistent with the concentration of TGF-β in the supernatant from high TGF-β-secreted T-MSCs (Figures 6A and S12D). Thus, we concluded that MSCs could inhibit the expression of CXCL5 in CRC cells by secreting TGF-β.
To clarify whether MSCs are the main cell type that secrete CCL7 and TGF-β, the public database the Human Protein Atlas (https://www.proteinatlas.org/) and a single-cell sequencing dataset (Gene Expression Series [GSE]: GSE178318) were used to analyze the expression of CCL7 and TGF-β in different cell types. The results showed that CCL7 was mainly secreted by immune cells, mesenchymal cells (CAFs), and epithelial cells (cancer cells), while TGF-β was mainly secreted by immune cells, mesenchymal cells, epithelial cells, and endothelial cells (Figures S13A–S13C). In addition, IHC was used to stain and locate CCL7 and TGF-β in tumor tissues, revealing that CCL7 was expressed in tumor cells and stromal cells, while TGF-β was mainly expressed in stromal cells (Figure S13D). Because tumor cells and mesenchymal cells were the main cell populations present in the experimental environment in vitro, we analyzed the secretion of CCL7 and TGF-β in CRC cells and MSCs using ELISA. Results showed that the secretion of CCL7 and TGF-β by CRC cells was significantly lower than that by T-MSCs and was below the optimal concentrations reported for CCL7 and TGF-β (Figure S13E).
The inhibitory effect of TGF-β on the expression of CXCL5 in CRC cells was Smad4/KLF5 dependent
LY2109761, a dual inhibitor of TGF-β receptor type I/II (TGF-βRI/II),31 was used to verify the effect of TGF-β and its receptor TGF-βR on CCL7-induced CXCL5 overexpression in CRC cells. Overexpression of KLF5 and CXCL5 induced by CCL7 was abolished by TGF-β, while LY2109761 recovered the expression of KLF5 and CXCL5 in SW620 and HCT116 cells after TGF-β treatment (Figure 6G). In addition, TGF-β significantly increased the phosphorylation level of SMAD2, which was abolished by LY2109761 (Figure S12E). In particular, although western blotting showed that the total expression of SMAD4 had no obvious difference in CRC cells after different treatments, the content of SMAD4 in nuclear protein was significantly increased after TGF-β treatment, and this effect could be reversed by LY2109761(Figures S12F and S12G).
siRNA technology was used to knock down KLF5 and TGF-β downstream molecules SMAD2, SMAD3, and SMAD4 in SW620 and HCT116 cells under TGF-β stimulation, and the results showed that knockdown of SMAD4 could recover the inhibitory effect of TGF-β on the expression of CXCL5 in CRC cells, whereas knockdown of SMAD2 or SMAD3 had no significant effect on CXCL5 expression compared with small interfering negative control (si-NC). Interestingly, as shown in Figure 4, we confirmed that KLF5 promoted CXCL5 expression, but when we knocked down KLF5 under TGF-β treatment, the expression of CXCL5 increased (Figure 6H). However, compared with knockdown of SMAD4 or KLF5 alone, knockdown of both SMAD4 and KLF5 increased the expression of CXCL5 (Figure 6I), suggesting that SMAD4 and KLF5 may play a dual regulatory role in TGF-β inhibition of CXCL5 expression in CRC cells. Next, luciferase reporter assay showed that transfection with SMAD2, SMAD3, or SMAD4 plasmids showed no significant effect on the luciferase activity of pGL3-CXCL5-FL when KLF5 was not present. In contrast, when KLF5 was present, co-transfection with SMAD4 significantly inhibited the function of KLF5 in promoting the luciferase activity of pGL3-CXCL5-FL (Figure 6J).
The luciferase reporter assay confirmed that SMAD4 and KLF5 were the main elements in the TGF-β-inhibiting CXCL5 expression process, so we investigated whether they have direct interaction. In the PPI network of KLF5, SMAD4 was located in the center and gained a high score, strongly suggesting that SMAD4 and KLF5 interact directly (Figure 5B). Co-IP assay confirmed the direct interaction between KLF5 and SMAD4 (Figure 6K). In addition, TGF-β stimulation promoted the combination of KLF5 and SMAD4 (Figures 6L and 6M).
SMAD4 activated by TGF-β reversed the effect of KLF5 on transcription activation of CXCL5 in CRC cells
The JASPAR database was used to predict the potential SMAD4-binding elements (SBE1 and SBE2) in the CXCL5 promoter region (Table S13) (Figure 7A). SMAD4 also binds to the consensus TGF-β inhibitory element (TIE).32 Based on the multiple sequence alignment of TIEs from genes for human urokinase, collagenase, peroxisome proliferator-activated receptor γ (PPARγ), and c-Myc,33 we predicted a new TIE sequence of CXCL5, which was 100% matched to the consensus TIE sequence (Figure S12H). The results showed that overexpression of SMAD4 reversed the effect of the KLF5 plasmid on the luciferase activity of pGL3-CXCL5-FL, indicating that only pGL3-CXCL5-FL contained the most effective inhibitory element (TIE) of SMAD4 on the CXCL5 promoter (Figure 7B). ChIP showed that SMAD4 bound both to the regions of the CXCL5 promoter that contained TIE (−1508 to −1360 nt) and SBE1 (−1192 to −1081 nt) (Figures 7C and 7D). In addition, TGF-β treatment only promoted the binding efficiency of SMAD4 to TIE and LY2109761 inhibited this effect, whereas the binding efficiency of SMAD4 to SBE1 was not influenced by TGF-β (Figures 7E and 7F).
Figure 7.

SMAD4 activated by TGF-β reversed the effect of KLF5 on transcription activation of CXCL5 in CRC cells
(A) SMAD4-binding elements (SBE1 and SBE2), TGF-β inhibitory element (TIE), and KBE on CXCL5 promoter region. (B) The results of luciferase reporter assay. SMAD4 reversed the effect of KLF5 plasmid on promoting luciferase activity of pGL3-CXCL5-FL. (C and D) ChIP assay showed that SMAD4 both bound to the regions of CXCL5 promoter that contained TIE (−1508 to −1360 nt) and SBE1 (−1192 to −1081 nt). (E and F) The results of ChIP assay. TGF-β promoted the binding efficiency of SMAD4 to TIE. (G) The results of luciferase reporter assay. SMAD4 could not reversed the effect of KLF5 on promoting luciferase activity of CXCL5-TIE-Mut. (H) ChIP assay showed that ML264 (the KLF5 inhibitor) inhibited the effect of TGF-β on promoting the binding efficiency of SMAD4 to TIE. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we deleted the TIE sequence from the pGL3-CXCL5-FL plasmid to construct pGL3-CXCL5-TIE-Mut and found that the mutation of TIE significantly abolished the inhibitory effect of SMAD4 on KLF5 and induced CXCL5 transcription activation (Figure 7G). The results of ChIP showed that TGF-β promoted the binding efficiency of SMAD4 to TIE, whereas ML264 (the KLF5 inhibitor) inhibited this effect (Figure 7H). In addition, pull-down assay also verified that both KLF5 and SMAD4 were significantly enriched by the TIE biotin-labeled probe (Figure 7I), which was increased by TGF-β treatment (Figure S12I). Therefore, we confirmed that the inhibitory effect of TGF-β on the expression of CXCL5 was completed by SMAD4, which can form a transcription complex with KLF5 on the CXCL5 promoter and reverse the transcription activation effect of KLF5.
Discussion
The study of the interactions between tumor cells and the TME has been the subject of current tumor progression and metastasis research. Many types of cells, including MSCs, endothelial progenitor cells, and immune cells, are recruited into the microenvironment of developing tumors.34 Besides cellular components, the tumor stroma also contains acellular components, including extracellular matrix and soluble proteins (cytokines, chemokines, and growth factors).35,36 Therefore, the metastatic phenotype of tumor cells depends not only on reprogramming in tumor cells but also on other cellular and acellular components in the TME. Recent studies have shown that the residence of MSCs in the TME and their interaction with inflammatory factors play an important role in tumor progression.37
MSCs exist in the stroma of normal tissues and tumor tissues, and they can differentiate into adipocytes, osteoblasts, and chondrocytes. MSCs express specific markers, such as CD73, CD90, and CD105, but lack CD34, CD45, and CD19 expression.15 However, in our study, fibroblasts, especially CAFs, also possessed differentiation ability and expressed specific markers similar to MSCs. It has been reported that fibroblasts can be divided into four subsets based on the fibroblast markers FAP, CD29, α-SMA, PDGFRB, and PDPN (podoplanin).14 It has also been proposed that there are high proportions of CAF-S1 and CAF-S4 subsets in the TME, while there are high proportions of CAF-S2 and CAF-S2 subsets in the non-tumor environment. According to this classification method, combined with our results, the expression patterns of the fibroblast markers expressed by T-MSCs, CAFs, N-MSCs, and NFs were consistent with those of CAF-S4, CAF-S1, CAF-S3, and S1 subsets, respectively. Therefore, we speculated that in previous classification methods, MSCs were classified as fibroblasts and were thus defined as one or more subsets. For CAFs (S1 subset) and T-MSCs (S4 subset) in the TME, we concluded that they could be distinguished based on the expression of FAP and PDGFRB. However, we still believe that MSCs and fibroblasts are in a transitional state; more specifically, T-MSCs differentiate into CAFs in the TME and show half of the physiological characteristics of MSCs and half those of fibroblasts.
MSCs secrete a variety of secretory cytokines, which can modify extracellular matrix components to regulate tumor cell growth and metastasis.37 However, the role of MSCs in tumor stroma is controversial. Previous studies have reported that MSCs can act as activators of tumorigenesis and metastasis.38, 39, 40, 41, 42 In contrast, others have proposed that MSCs may play an inhibitory role in tumor cells.43,44 The controversy in these studies may be, at least in part, due to the different sources of MSCs involved in the experiment and the differences in the culture system. Most of these studies were performed with MSC lines or healthy donor-derived MSCs,43,44 and studies on T-MSCs in cancer patients are limited. In addition, in most studies, a co-culture system of MSCs and cancer cells was used to detect the effect of MSCs on the TME.40 Because MSCs within the TME might have an altered phenotype, this model cannot truly reflect the function of healthy donor-derived MSCs. In our study, we found that tumor cells can activate N-MSCs in a co-culture environment and transform their phenotype to T-MSCs. We believe that the tumor-promoting phenotype of MSCs in the TME (T-MSC) was conferred by tumor cells, and N-MSCs did not have a tumor-promoting phenotype.
CXCL5 is overexpressed in CRC and promotes tumor metastasis and invasion and has been reported that CXCL5 secretion can be regulated by a variety of cytokines, including interleukin-17A (IL-17A), IL-1β, and tumor necrosis factor α (TNF-α).5 In this study, we explored the upstream mechanism of CXCL5 overexpression in CRC, which is highly related to CCL7 and TGF-β secreted by MSCs. CCL7 is overexpressed in many cancers and is associated with disparities in overall survival.45,46 Importantly, CCL7 can be secreted by CAFs and promotes tumor invasion and migration.47, 48, 49 In our study, we reported that CCL7 was highly expressed in T-MSCs, which increased the expression of CXCL5 in CRC cells. KLF5 is highly expressed in rapidly dividing intestinal epithelial cells and multiple epithelial cancer types.50 KLF5 is generally considered protumorigenic51,52 and promotes the proliferation and progression of cancer cells. Previous studies have reported that KLF5 can form a transcription complex with acetyltransferases, thereby elevating KLF5 acetylation21 or histone acetylation23 to regulate transcription. Our study confirmed that CCL7 treatment increased KLF5 acetylation and significantly promoted the transcription of CXCL5.
In contrast, according to previous reports, TGF-β is closely related to MSCs, which are not only secreted by MSCs as the main source but also regulate the differentiation state and function of MSCs.53 However, the role of MSC-derived TGF-β in the TME remains controversial. MSC-derived TGF-β1 promotes tumor progression.38,54 In contrast to the above conclusion, some studies have reported the tumor-suppressive effect of TGF-β.55,56 In our study, we found that the expression of TGF-β in T-MSCs was related to the TNM staging of the primary tumor. This finding might help us to better understand the dual role of TGF-β in the TME.
Classically, TGF-β binds to TGFBR2 at the cell surface, causing the recruitment and phosphorylation of TGFBR1, which subsequently propagates the signal through Smad activation.57 By recruiting other cofactors to achieve high affinity and selectivity interaction with DNA, R-Smad–Smad4 complexes regulate gene activation or inhibition responses.56 To date, many transcription factors have been reported to act as cofactors, including AP1, forkhead, bHLH, and zinc finger transcription factors.58 We confirmed that SMAD4 and KLF5 were the main components in the TGF-β-inhibiting CXCL5 expression process through the formation of SMAD4-KLF5 complexes on the CXCL5 promoter.
Increasingly, studies have shown that most transcription factors play a dual role in transcription regulation and are often affected by internal and external environment stimulation.59 In this article, we report that T-MSCs derived from early-stage CRC patients secreted higher levels of TGF-β, and high-dose TGF-β reversed the promotion effect of CCL7 on CXCL5 expression in vitro. The key to triggering the switch of the two-way pattern was the transcription factor KLF5. Many studies have shown that KLF5 acts as a transcriptional activator or repressor, depending on the environment. Our study also reported that exogenous CCL7 stimulation triggered transcriptional activation by KLF5, whereas exogenous TGF-β triggered the repressive functions of KLF5. We proposed that the difference in TGF-β secretion by T-MSCs from different patients was related to the clinical CRC stage. We found that TGF-β expression in the earlier CRC stages (I + II) was much higher than that in the advanced CRC stages (III + Ⅳ). In addition, CCL7 expression in advanced stages was slightly higher than that in early stages, but the difference between the two groups was not statistically significant. Therefore, we believe that different levels of TGF-β and CCL7 secretion by MSCs in the TME at different stages of CRC progression are important factors in triggering of the two-way pattern switch.
Conclusion
In conclusion, our studies revealed that T-MSCs had a tumor-promoting phenotype, while N-MSCs did not have a tumor-promoting phenotype. In addition, tumor cells can activate N-MSCs in a co-culture environment and transform their phenotype similar to that of T-MSCs, indicating that the tumor-promoting phenotype of T-MSCs was conferred by tumor cells. In CRC cells, T-MSCs enhanced CXCL5 expression by increasing CCL7/CCR1 secretion and downstream regulation of CBP/P300 and KLF5 pathway. At the same time, MSC-derived TGF-β negatively regulated the expression of CXCL5 in CRC cells in a Smad4/KLF5-dependent pattern, and SMAD4 reversed the effect of KLF5 on transcription activation of CXCL5.
Methods and materials
Patients and specimens
Tissue samples from patients with CRC were collected after obtaining informed consent from the Biomedical Ethics Committee of Ruijin Hospital. Between 2017 and 2021, we collected tumor tissues and adjacent normal tissues from 23 patients (Table S1) at the Shanghai Minimally Invasive Surgery Medical Center of Ruijin Hospital. All patients were pathologically diagnosed with CRC and underwent laparoscopic surgery at our center. Patients who received preoperative treatment (such as radiotherapy or chemotherapy) were excluded from the study. Based on the 2015 National Comprehensive Cancer Network (NCCN) guidelines, TNM staging of the slice of each patient was performed. All patients were fully informed and signed an informed consent form.
Specimens processing and cell culture
Paired CRC tissues and adjacent normal tissues were collected and washed with PBS containing penicillin-streptomycin solution. Tweezers and disposable surgical blades were then used to mince the tissues into meat emulsions. The tissue fragments were transferred to a 50 mL centrifuge tube and digested with 3 mg/mL collagenase IV (Sigma) and 5 MU/mL DNase I (Calbiochem) in α-MEM at 37°C for 30 min until fully digested. The digested tissue suspension was passed through a 100 μm mesh filter to remove residual tissue. The filtrate was collected and washed three times with PBS. The collected cells were then inoculated in α-MEM (MSCs) or DMEM/F12 (fibroblasts) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and cultured at 37°C, 5% CO2, and 20% O2. On the second day after culture, the supernatant and suspended cells were discarded. After 10 days of culture, the adherent cells were harvested and stored as frozen stock (first generation), and 2–10 passages of cells were used for subsequent experiments.
Statistical analysis
All statistical analyses were performed using SPSS version 20.0. Quantitative variables were analyzed using the Student’s t test. The data are shown as the mean ± SD. All the experiments were performed in triplicate. Statistical significance was set at p < 0.05.
Abbreviations
CXCL5 C-X-C motif chemokine ligand 5
CRC colorectal cancer
TME tumor microenvironment
MSC mesenchymal stromal cell
T-MSC tumor-associated mesenchymal stromal cell
CM cell medium
α-SMA α-smooth muscle actin
CAF cancer-associated fibroblast
KBE KLF5-binding element
PPI protein-protein interaction
HAT histone acetyltransferase
EMT epithelial-mesenchymal transition
TGF-βRI/II TGF-β receptor type I/II
SBE SMAD4-binding element
TIE TGF-β inhibitory element
IHC immunohistochemical
ChIP chromatin immunoprecipitation
IP immunoprecipitation.
Acknowledgments
This study was supported by National Natural Science Foundation of China (81871933, 81802326) and Foundation of Shanghai Municipal Health Committee (202040144) funding. The funders of this project had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author contributions
All authors meet the authorship requirements. Conception and design: Yaping Z., J.Z., and A.L. development of methodology: Zhuoqing X., H.G., and Yuchen Z. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.: Zhuoqing X., H.G., J.H., and Zifeng X. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Zhuoqing X., Wenchang L., and W.F. Writing, review, and/or revision of the manuscript: Zhuoqing X., J.Z., and A.L. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Z.L., F.C., and X.S. Study supervision: Wangyi L., Y.M., and X.S. Revision director: J.Z. and A.L. All authors read and approved the final manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.005.
Contributor Information
Yaping Zong, Email: angela_zyp@126.com.
Jingkun Zhao, Email: zhaojk8891@126.com.
Aiguo Lu, Email: luaiguo1965@163.com.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Bussard K.M., Mutkus L., Stumpf K., Gomez-Manzano C., Marini F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84. doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J., Knaut H. Chemokine signaling in development and disease. Development. 2014;141:4199–4205. doi: 10.1242/dev.101071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao J., Ou B., Han D., Wang P., Zong Y., Zhu C., Liu D., Zheng M., Sun J., Feng H., Lu A. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3beta/beta-catenin pathways. Mol. Cancer. 2017;16:70. doi: 10.1186/s12943-017-0629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia J., Xu X., Huang P., He M., Wang X. The potential of CXCL5 as a target for liver cancer - what do we know so far? Expert Opin. Ther. Targets. 2015;19:141–146. doi: 10.1517/14728222.2014.993317. [DOI] [PubMed] [Google Scholar]
- 6.Chen C., Xu Z.Q., Zong Y.P., Ou B.C., Shen X.H., Feng H., Zheng M.H., Zhao J.K., Lu A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-kappaB pathway in colorectal cancer. Cell Death Dis. 2019;10:178. doi: 10.1038/s41419-019-1431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuiffo B.G., Karnoub A.E. Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh. Migr. 2012;6:220–230. doi: 10.4161/cam.20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prockop D.J., Brenner M., Fibbe W.E., Horwitz E., Le Blanc K., Phinney D.G., Simmons P.J., Sensebe L., Keating A. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy. 2010;12:576–578. doi: 10.3109/14653249.2010.507330. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X., Hu F., Li G., Li G., Yang X., Liu L., Zhang R., Zhang B., Feng Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018;9:25. doi: 10.1038/s41419-017-0176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo D., Hu S., Tang C., Liu G. Mesenchymal stem cells promote cell invasion and migration and autophagy-induced epithelial-mesenchymal transition in A549 lung adenocarcinoma cells. Cell Biochem. Funct. 2018;36:88–94. doi: 10.1002/cbf.3320. [DOI] [PubMed] [Google Scholar]
- 11.Prantl L., Muehlberg F., Navone N.M., Song Y.H., Vykoukal J., Logothetis C.J., Alt E.U. Adipose tissue-derived stem cells promote prostate tumor growth. Prostate. 2010;70:1709–1715. doi: 10.1002/pros.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaturvedi P., Gilkes D.M., Wong C.C., Kshitiz, Luo W., Zhang H., Wei H., Takano N., Schito L., Levchenko A., Semenza G.L. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J. Clin. Invest. 2013;123:189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borriello L., Nakata R., Sheard M.A., Fernandez G.E., Sposto R., Malvar J., Blavier L., Shimada H., Asgharzadeh S., Seeger R.C., DeClerck Y.A. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 2017;77:5142–5157. doi: 10.1158/0008-5472.CAN-16-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelon F., Bourachot B., Kieffer Y., Magagna I., Mermet-Meillon F., Bonnet I., Costa A., Givel A.M., Attieh Y., Barbazan J., et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020;11:404. doi: 10.1038/s41467-019-14134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poggi A., Varesano S., Zocchi M.R. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front. Immunol. 2018;9:262. doi: 10.3389/fimmu.2018.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calloni R., Cordero E.A., Henriques J.A., Bonatto D. Reviewing and updating the major molecular markers for stem cells. Stem Cells Dev. 2013;22:1455–1476. doi: 10.1089/scd.2012.0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y., Du L., Lin L., Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat. Rev. Drug Discov. 2017;16:35–52. doi: 10.1038/nrd.2016.193. [DOI] [PubMed] [Google Scholar]
- 18.Harrell C.R., Jovicic N., Djonov V., Volarevic V. Therapeutic use of mesenchymal stem cell-derived exosomes: from basic science to clinics. Pharmaceutics. 2020;12 doi: 10.3390/pharmaceutics12050474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra P., Banerjee D., Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J. Leukoc. Biol. 2011;89:31–39. doi: 10.1189/jlb.0310182. [DOI] [PubMed] [Google Scholar]
- 20.Zlotnik A., Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao C., Li Y., Qiu W., He F., Zhang W., Zhao D., Zhang Z., Zhang E., Ma P., Liu Y., et al. C5a induces A549 cell proliferation of non-small cell lung cancer via GDF15 gene activation mediated by GCN5-dependent KLF5 acetylation. Oncogene. 2018;37:4821–4837. doi: 10.1038/s41388-018-0298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B., Li Y., Wu Q., Xie L., Barwick B., Fu C., Li X., Wu D., Xia S., Chen J., et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat. Commun. 2021;12:1714. doi: 10.1038/s41467-021-21976-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Guo B., Aguilera-Jimenez E., Chu V.S., Zhou J., Wu Z., Francis J.M., Yang X., Choi P.S., Bailey S.D., Zhang X. Chromatin looping shapes KLF5-dependent transcriptional programs in human epithelial cancers. Cancer Res. 2020;80:5464–5477. doi: 10.1158/0008-5472.CAN-20-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang F., Marshall C.B., Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol. Life Sci. 2013;70:3989–4008. doi: 10.1007/s00018-012-1254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang R., He Y., Robinson V., Yang Z., Hessler P., Lasko L.M., Lu X., Bhathena A., Lai A., Uziel T., Lam L.T. Targeting lineage-specific MITF pathway in human melanoma cell lines by A-485, the selective small-molecule inhibitor of p300/CBP. Mol. Cancer Ther. 2018;17:2543–2550. doi: 10.1158/1535-7163.MCT-18-0511. [DOI] [PubMed] [Google Scholar]
- 26.Serra C., Palacios D., Mozzetta C., Forcales S.V., Morantte I., Ripani M., Jones D.R., Du K., Jhala U.S., Simone C., Puri P.L. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell. 2007;28:200–213. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su B., Luo T., Zhu J., Fu J., Zhao X., Chen L., Zhang H., Ren Y., Yu L., Yang X., et al. Interleukin-1beta/Iinterleukin-1 receptor-associated kinase 1 inflammatory signaling contributes to persistent Gankyrin activation during hepatocarcinogenesis. Hepatology. 2015;61:585–597. doi: 10.1002/hep.27551. [DOI] [PubMed] [Google Scholar]
- 28.Siraj A.K., Pratheeshkumar P., Divya S.P., Parvathareddy S.K., Bu R., Masoodi T., Kong Y., Thangavel S., Al-Sanea N., Ashari L.H., et al. zz in CRC. Mol. Cancer Ther. 2019;18:1312–1322. doi: 10.1158/1535-7163.MCT-18-1378. [DOI] [PubMed] [Google Scholar]
- 29.David C.J., Huang Y.H., Chen M., Su J., Zou Y., Bardeesy N., Iacobuzio-Donahue C.A., Massague J. TGF-beta tumor suppression through a lethal EMT. Cell. 2016;164:1015–1030. doi: 10.1016/j.cell.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B., Halder S.K., Kashikar N.D., Cho Y.J., Datta A., Gorden D.L., Datta P.K. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138:969–980.e1-3. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melisi D., Ishiyama S., Sclabas G.M., Fleming J.B., Xia Q., Tortora G., Abbruzzese J.L., Chiao P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itatani Y., Kawada K., Fujishita T., Kakizaki F., Hirai H., Matsumoto T., Iwamoto M., Inamoto S., Hatano E., Hasegawa S., et al. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology. 2013;145:1064–1075.e11. doi: 10.1053/j.gastro.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 33.Lakshmi S.P., Reddy A.T., Reddy R.C. Transforming growth factor beta suppresses peroxisome proliferator-activated receptor gamma expression via both SMAD binding and novel TGF-beta inhibitory elements. Biochem. J. 2017;474:1531–1546. doi: 10.1042/BCJ20160943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poggi A., Giuliani M. Mesenchymal stromal cells can regulate the immune response in the tumor microenvironment. Vaccines (Basel) 2016;4 doi: 10.3390/vaccines4040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosein A.N., Brekken R.A., Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020;17:487–505. doi: 10.1038/s41575-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouahoud S., Voorneveld P.W., van der Burg L.R.A., de Jonge-Muller E.S.M., Schoonderwoerd M.J.A., Paauwe M., de Vos T., de Wit S., van Pelt G.W., Mesker W.E., et al. Bidirectional tumor/stroma crosstalk promotes metastasis in mesenchymal colorectal cancer. Oncogene. 2020;39:2453–2466. doi: 10.1038/s41388-020-1157-z. [DOI] [PubMed] [Google Scholar]
- 37.Melzer C., Yang Y., Hass R. Interaction of MSC with tumor cells. Cell Commun. Signal. 2016;14:20. doi: 10.1186/s12964-016-0143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He W., Liang B., Wang C., Li S., Zhao Y., Huang Q., Liu Z., Yao Z., Wu Q., Liao W., et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene. 2019;38:4637–4654. doi: 10.1038/s41388-019-0747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuiffo B.G., Campagne A., Bell G.W., Lembo A., Orso F., Lien E.C., Bhasin M.K., Raimo M., Hanson S.E., Marusyk A., et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. 2014;15:762–774. doi: 10.1016/j.stem.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Karnoub A.E., Dash A.B., Vo A.P., Sullivan A., Brooks M.W., Bell G.W., Richardson A.L., Polyak K., Tubo R., Weinberg R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 41.Waghray M., Yalamanchili M., Dziubinski M., Zeinali M., Erkkinen M., Yang H., Schradle K.A., Urs S., Pasca Di Magliano M., Welling T.H., et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov. 2016;6:886–899. doi: 10.1158/2159-8290.CD-15-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin J.T., Wang J.Y., Chen M.K., Chen H.C., Chang T.H., Su B.W., Chang P.J. Colon cancer mesenchymal stem cells modulate the tumorigenicity of colon cancer through interleukin 6. Exp. Cell Res. 2013;319:2216–2229. doi: 10.1016/j.yexcr.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Qiao L., Xu Z.L., Zhao T.J., Ye L.H., Zhang X.D. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008;269:67–77. doi: 10.1016/j.canlet.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y.R., Yuan Y., Wang X.J., Wei L.L., Chen Y.N., Cong C., Li S.F., Long D., Tan W.D., Mao Y.Q., et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol. Ther. 2008;7:245–251. doi: 10.4161/cbt.7.2.5296. [DOI] [PubMed] [Google Scholar]
- 45.Thomas J.K., Mir H., Kapur N., Bae S., Singh S. CC chemokines are differentially expressed in Breast Cancer and are associated with disparity in overall survival. Sci. Rep. 2019;9:4014. doi: 10.1038/s41598-019-40514-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma I., Singh A., Sharma K., Saxena S. Gene expression profiling of chemokines and their receptors in low and high grade Astrocytoma. Asian Pac. J. Cancer Prev. 2017;18:1307–1313. doi: 10.22034/APJCP.2017.18.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korbecki J., Kojder K., Siminska D., Bohatyrewicz R., Gutowska I., Chlubek D., Baranowska-Bosiacka I. CC chemokines in a tumor: a review of pro-cancer and anti-cancer properties of the ligands of receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21218412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung D.W., Che Z.M., Kim J., Kim K., Kim K.Y., Williams D., Kim J. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int. J. Cancer. 2010;127:332–344. doi: 10.1002/ijc.25060. [DOI] [PubMed] [Google Scholar]
- 49.Liu J., Chen S., Wang W., Ning B.F., Chen F., Shen W., Ding J., Chen W., Xie W.F., Zhang X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-beta pathways. Cancer Lett. 2016;379:49–59. doi: 10.1016/j.canlet.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 50.McConnell B.B., Yang V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia L., Zhou Z., Liang H., Wu J., Shi P., Li F., Wang Z., Wang C., Chen W., Zhang H., et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene. 2016;35:2040–2051. doi: 10.1038/onc.2015.263. [DOI] [PubMed] [Google Scholar]
- 52.Wu Q., Fu C., Li M., Li J., Li Z., Qi L., Ci X., Ma G., Gao A., Fu X., et al. CINP is a novel cofactor of KLF5 required for its role in the promotion of cell proliferation, survival and tumor growth. Int. J. Cancer. 2019;144:582–594. doi: 10.1002/ijc.31908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caja L., Dituri F., Mancarella S., Caballero-Diaz D., Moustakas A., Giannelli G., Fabregat I. TGF-beta and the tissue microenvironment: relevance in fibrosis and cancer. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao X., Wu X., Qian M., Song Y., Wu D., Zhang W. Knockdown of TGF-beta1 expression in human umbilical cord mesenchymal stem cells reverts their exosome-mediated EMT promoting effect on lung cancer cells. Cancer Lett. 2018;428:34–44. doi: 10.1016/j.canlet.2018.04.026. [DOI] [PubMed] [Google Scholar]
- 55.Chen J., J A.G. Dysregulated PJA1-TGF-beta signaling in cancer stem cell-associated liver cancers. Oncoscience. 2020;7:88–95. doi: 10.18632/oncoscience.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seoane J., Gomis R.R. TGF-beta family signaling in tumor suppression and cancer progression. Cold Spring Harb. Perspect. Biol. 2017;9 doi: 10.1101/cshperspect.a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 58.Feng X.H., Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 59.Diakiw S.M., D'Andrea R.J., Brown A.L. The double life of KLF5: opposing roles in regulation of gene-expression, cellular function, and transformation. IUBMB Life. 2013;65:999–1011. doi: 10.1002/iub.1233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.