

Abstract
As mediators of intercellular communication, extracellular vesicles containing molecular cargo, such as microRNAs, are secreted by cells and taken up by recipient cells to influence their cellular phenotype and function. Here we report that cardiac stress-induced differential microRNA content, with miR-200c-3p being one of the most enriched, in cardiomyocyte-derived extracellular vesicles mediates functional cross-talk with endothelial cells. Silencing of miR-200c-3p in mice subjected to chronic increased cardiac pressure overload resulted in attenuated hypertrophy, smaller fibrotic areas, higher capillary density, and preserved cardiac ejection fraction. We were able to maximally rescue microvascular and cardiac function with very low doses of antagomir, which specifically silences miR-200c-3p expression in non-myocyte cells. Our results reveal vesicle transfer of miR-200c-3p from cardiomyocytes to cardiac endothelial cells, underlining the importance of cardiac intercellular communication in the pathophysiology of heart failure.
Keywords: cardiac angiogenesis, microRNAs, extracellular vesicles, intercellular communication, cardiac remodeling
Graphical abstract
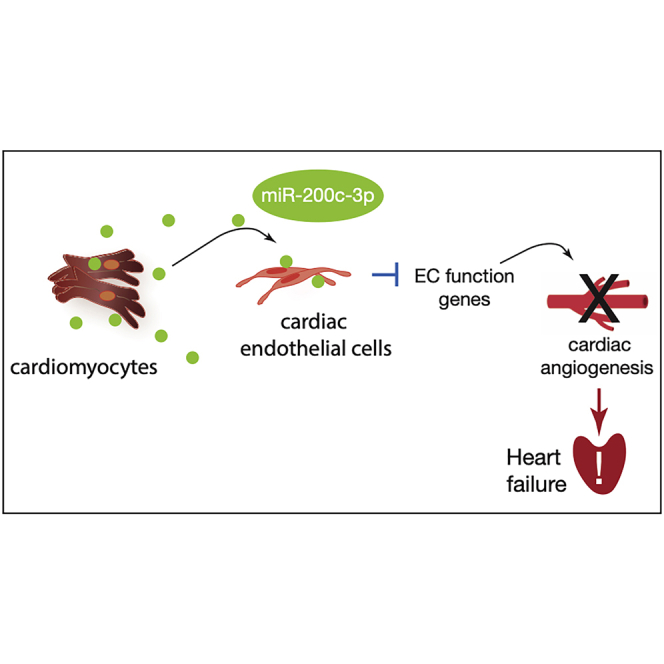
Ottaviani et al. report a new mechanism used by cardiac muscle cells under stress to influence neighboring cell function by releasing small vesicles containing miR-200c-3p. When taken up by endothelial cells, this miRNA impairs angiogenic function, worsening cardiac function. Inhibition of this process is a potential therapeutic target for heart failure.
Introduction
During postnatal development or physiological hypertrophy, the myocardium increases in size along with a matching response of the coronary vasculature, with consequent capillary growth, allowing a sufficient supply of oxygen and nutrients to reach the growing organ.1, 2, 3 On the contrary, in pathological cardiac hypertrophy, there is inadequate microvasculature growth that leads to hypoxia, cell death, autophagy, and fibrosis.4 Disparity between growth of cardiomyocytes (CMs) and capillaries is an essential factor in progression from hypertrophy to heart failure (HF).5,6 HF is one of the leading causes of mortality and morbidity worldwide, and, despite the different etiologies, the myocardium response is similar, with major changes at the molecular, cellular, and biochemical levels.7
The heart is a multicellular organ, and cell-cell communication is fundamental to maintain organ homeostasis and integrity and in response to injury. Short-range cellular communication is often not sufficient for the cell to adequately react to external stimuli. In the past years, extracellular vesicles (EVs) have been implicated as important players in the cross-talk between cardiac cells,8 carrying biological information and delivering cargos in a more or less targeted manner and, thus, modulating the response of recipient cells.9 EVs are membranous vesicles secreted into the extracellular space and can transport lipids, small proteins, messenger RNA (mRNA), and microRNA (miRNA). miRNAs are a class of endogenous, small (∼22 nt), single-stranded and highly conserved noncoding RNAs that exert their function by suppressing gene expression at the post-transcriptional level. Several miRNAs have been found to affect molecular mechanisms leading to pathological cardiac remodeling10 and HF, making these non-coding RNAs strong effective targets for cardiovascular disease treatment. Some miRNAs are not randomly sorted/loaded into vesicles, such as exosomes, but are selectively exported based on specific sequences that can change under specific pathophysiological conditions.11 After delivery, miRNAs derived from EVs can exert functional roles in recipient cells by modulating gene expression and, thus, influencing their phenotype. Here we show that, by perturbing the transfer of the anti-angiogenic miR-200c-3p from CMs to epithelial cells (ECs) under stress conditions, we can rescue microvascular and cardiac function. These results underlie the importance of cardiac intercellular communication in the response of the heart to chronic stress.
Results
miR-200c-3p is enriched in hypertrophic CM-derived EVs
The α- and β-adrenoreceptor agonists phenylephrine (PE) and isoproterenol (Iso), respectively, were used to induce cellular hypertrophic growth phenotype in primary neonatal rat CMs (NRCMs).12,13 After 48 h of stimulation, the cell culture supernatant was collected and used for isolation of EVs using an ultracentrifugation protocol adapted from Halkein et al.,14 followed by morphological EV characterization (Figure 1A). Assessment of the morphological characteristics of EVs isolated from untreated as well as PE-Iso-treated CMs revealed a cup-shaped structure observed by transmission electron microscopy (TEM) (Figure 1B) and an average diameter between 100 and 200 nm, as confirmed by nanoparticle tracking analysis (NTA) and in accordance with small EVs, as reported in previous studies (Figure 1C).15,16 Treatment of CMs with GW4869, a neutral sphingomyelinase antagonist and established inhibitor of EV biogenesis/release,17,18 inhibited EV release in comparison with untreated or PE-Iso-treated CMs (Figure 1C). By using flow cytometry as a second method of characterization, we were able to confirm expression of the tetraspanin CD63, an established ubiquitous EV marker15,16 (Figure 1D). To ensure that our preparations were enriched in small EVs, we performed western blot analyses to detect common EV markers and potential contaminants. Our results showed that the isolated EVs expressed the markers CD81, Alix, Hsp70, and GAPDH, whereas calnexin and ApoA-1, markers of cytoplasmic proteins and high-density lipoproteins, respectively, were absent (Figure 1E). Hence, we could confirm isolation of a purified EV population from the supernatant of cultured control or PE-Iso-treated CMs.
Figure 1.

miR-200c-3p is abundant in EVs derived from CMs under stress conditions
(A) Experimental setup of EV isolation from untreated and PE-Iso-treated CMs. (B) Representative electron microscopy images of EVs derived from the supernatant of cultured untreated or PE-Iso-treated CMs. (C) Analysis of EV size and concentration performed by NTA. (D) Dot plot of forward scatter (FS) versus CD63 or IgG1, showing a clear population of CD63 events for untreated or PE-Iso-treated CMs. (E) Western blot analysis for characterization of EVs derived from untreated or PE-Iso-treated CMs after additional purification steps. Expression levels of EV markers such as Alix, Hsp70, and CD81 as well as of the potential contaminants calnexin and ApoA-1 were assessed. (F) Profile of the miRNA content of EVs derived from PE-Iso-stimulated CMs. Light blue bars represent the most enriched and dark blue bars the most depleted miRNAs. (G) Real-time PCR validation of results obtained in (D) for miR-200c-3p after RNase treatment with or without Triton X-100. ∗p < 0.05 versus untreated CMs, #p < 0.05 versus untreated and RNase-treated EVs. (H) Real-time PCR assessment of miR-200c-3p expression in different cardiac cell types under sham and TAC conditions. ∗p < 0.05 versus the corresponding sham group (error bars indicate SEM), #p < 0.05 versus CMs.
EVs released by donor cells will exert key functions in recipient cells by transferring their cargos, depending on the status of the EV-producing (donor) cell. Interestingly, this bioactive cargo is enriched in miRNAs, ubiquitous regulators of physiological and pathophysiological processes. To determine the miRNA content of CM-derived EVs, we performed a qRT-PCR-based array and, compared with CMs under resting conditions (serum free [SF]), we identified more than 200 miRNAs differentially expressed in EVs derived from hypertrophic CMs (Table S1). Among those, miR-200c-3p was the most significantly enriched miRNA (5-fold change; Figure 1F). To ascertain that the results from our array are not influenced by the presence of protein-associated extracellular miRNAs and validate the presence of the identified miRNAs in the isolated EVs, RNase A digestion with or without Triton X-100 was performed before validating the array results in a larger number of samples. Our results showed profiles similar to the ones observed with our initial EV preparations (Table S2), with miR-200c-3p remaining highly enriched in EVs from hypertrophic CMs (Figure 1G), indicating that the identified miRNAs are encased within EVs.
miR-200c-3p is located in an intronic region of a long non-coding RNA (lncRNA; MIR200CHG-201) in human chromosome 12 (mouse chromosome 6), clustered together with miR-141, and it is highly conserved among species (Figure S1B). In the myocardium, miR-200c-3p is expressed at very low levels and mostly in non-CM cells. Cardiac stress induced by increased pressure overload resulted in increased levels of miR-200c-3p with specific upregulation in non-CM cells, including ECs and other cell types (Figure 1H).
Increased miR-200c-3p expression levels impair endothelial cell function
Next we investigated the intercellular uptake of CM-derived EVs by ECs and their functional effect on ECs. NRCMs treated with the hypertrophic stimuli PE and Iso or left untreated were seeded on a porous membrane to allow passage of EVs toward the lower compartment, in which human umbilical vein endothelial cells (HUVECs) were cultured (Figure 2A). To delve into the biological role of EV uptake by ECs, we performed endothelial functional assays on HUVECs after 72 h of co-culture with CMs. A clear decrease in the capacity of ECs to migrate (Figure 2B) and form vascular networks was observed when ECs were co-cultured with PE-Iso-treated CMs (Figures 2C and 2D). Treatment of hypertrophic NRCMs with GW4869 prior to co-culture with ECs restored the capacity of ECs to migrate and form tubes to control levels (Figures 2B–2D). To determine whether the observed effects on the endothelium could be driven by CM-derived miR-200c-3p, we transfected NRCMs with a specific precursor molecule (pre-200c) to increase its expression. Although we could overexpress the miRNA in CMs, the levels of overexpression were increased when we also treated the cells with GW4869, suggesting that miR-200c-3p could no longer be secreted through EVs but, instead, accumulated in CMs (Figure 2E). Overexpression in CMs resulted in upregulation of miR-200c-3p in ECs in the lower compartment (Figure 2F), which, in turn, impaired endothelial function, as reflected by a lower EC capacity to migrate (Figure 2G) and form tubes (Figures 2H and 2I). Treatment of NRCMs with GW4869 prior to co-culture confirmed that EVs mediate the transfer of miR-200c-3p between the two cell types because inhibition of their formation did not impair EC function, as revealed by increased endothelial angiogenic capacity (Figures 2G–2I).
Figure 2.

Increased miR-200c-3p expression impairs endothelial cell function
(A) Setup of the Transwell co-culture system. CMs were treated with PE-Iso or a precursor molecule for miR-200c-3p (pre-200c) in the absence or presence of GW4869 (GW) before being added to the upper compartment and co-cultured with HUVECs in the lower compartment. (B) Assessment of migration by quantification of wound closure within 24 h on HUVECs after 72 h in co-culture with untreated or PE-Iso-treated CMs and in the absence or presence of GW. (C) Representative images of tube formation assay on HUVECs under the same conditions as in (B). Color code: green for branches, yellow for master segments, light blue for meshes, red for nodes, pink for segments, and dark blue for junctions. (D) Quantification of relative tube length based on images in (C). (A–D) ∗p < 0.05 versus the control group, #p < 0.05 versus the PE-Iso group. (E and F) Real-time PCR analysis of miR-200c-3p expression in (E) CMs and (F) ECs after 72 h of co-culture in the presence or absence of GW. (G) Assessment of migration by quantification of wound closure within 24 h of HUVECs after 72 h in co-culture with CMs transfected with pre-200c or precursor control (scr). (H) Representative images of the tube formation assay on HUVECs under the same conditions as in (G). (I) Quantification of relative tube length based on images in (H). (E–I) ∗p < 0.05 versus the scrambled group, #p < 0.05 versus precursor-200c. (J) Experimental setup for adding purified EVs from untreated or PE-Iso-treated CMs directly to HUVEC cultures. (K) Assessment of migration by quantification of wound closure within 24 h of HUVECs after 48 h in culture with purified EVs from untreated or PE-Iso-treated CMs. (L) Representative images of the tube formation assay on HUVECs under the same conditions as in (K). (M) Quantification of relative tube length based on images in (L). (K–M) ∗p < 0.05 versus the control group. (N) Experimental setup for direct transfection of HUVECs with pre-200c or scr. (O) Quantification of the wound closure rate of HUVECs transfected with pre-200c or scr. (P) Representative images of the tube formation assay on HUVECs after transfection with pre-200c or scr. (Q) Quantification of the number of junctions as a measure of tube formation based on images in (P). (R) Quantification of EdU-positive cells as a readout of cell proliferation under the same conditions as in (P). (O–R) ∗p < 0.05 versus the scrambled group. Error bars indicate SEM.
CMs are also known to contribute to the inflammatory response by secreting cytokines such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-a)19 and to angiogenesis by releasing vascular endothelial growth factor (VEGF).20,21 To rule out a contribution of such CM-derived factors to the endothelial phenotypes observed by us, we determined the effect of purified EVs derived from untreated or PE-Iso-treated CMs (Figure 2J). As expected, hypertrophic CM-derived EVs significantly inhibited EC migration and tube formation (Figures 2K–2M).
Because CD45+ cells in the heart also express considerable amounts of miR-200c-3p, we performed functional assays where we co-cultured ECs with macrophage-like cells (MPs) to determine whether there was an effect on EC function (Figures S2A–S2C). Co-culture resulted in increased EC migration and tube formation (Figures S2B and S2C), suggesting stimulation of endothelial angiogenic capacity by CD45+ cells despite no increase in miR-200c-3p expression on ECs after co-culture with MPs (Figure S2D). To determine whether the pro-angiogenic effect observed was associated with EV release, we added conditioned medium derived from MPs that were left untreated or treated with GW4869 or transfected with a control scrambled anti-miRNA, an anti-miRNA-200c, to ECs (Figure S2E). We observed that treatment of MPs with GW4869 did not result in decreased EC migration or tube formation capacity, and neither did inhibition of miR-200c-3p on MPs (Figures S2F and S2G). Although treatment of MPs with GW4869 (GW) or control antimir did not change the baseline expression levels of miR-200c-3p in MPs, the levels of this miRNA were significantly reduced after transfection with an inhibitor of miR-200c-3p (Figure S2H). These results demonstrate that incubation of ECs with different conditioned media did not result in significant changes in miR-200c-3p expression on ECs, suggesting that the pro-angiogenic effect of MPs was not mediated by EVs or by miR-200c-3p (Figure S2I).
Because the content of CM-derived EVs is not restricted to this specific miRNA, we determined the direct effect of overexpressing only miR-200c-3p on EC function. Transfection of HUVECs with a specific miR-200c-3p precursor molecule (Figure 2N) significantly impaired endothelial angiogenic capacity by inhibiting EC migration capacity (Figure 2O) and by diminishing the ability to form a vascular network (Figures 2P and 2Q) and proliferate (Figure 2R). We demonstrate that impaired endothelial angiogenic capacity is induced by uptake of EVs derived from NRCMs and that their biological effect is mediated in part by transfer of miR-200c-3p.
Inhibition of miR-200c-3p in specific cardiac cell types
To explore the possibility of silencing miR-200c-3p in specific cardiac cell types with an antagomir, we used a CY3-labeled antisense oligonucleotide specifically targeting miR-200c-3p at two different doses: 5 mg/kg BW (body weight; low dose) to preferentially target non-myocyte cell types22 such as ECs, and 80 mg/kg BW (high dose) to target myocytes and non-myocyte cell populations in the myocardium (Figure 3A). The low dose was administered intraperitoneally on 3 consecutive days to assure maximal uptake by the first cells encountering the antagomir, whereas the high dose was administrated as a single intraperitoneal injection to achieve broader cellular uptake. The difference in regimens relates to the fact that, with one single injection of the low dose, we were not able to detect sufficient amounts of antagomir-positive cells for quantification, whereas in a previous study, we showed that one single injection of antagomir at the high dose was sufficient to inhibit a miRNA 7 days after administration.23 Isolation of different cell populations 1 week after delivery of the antagomirs by a Langendorff-free method,24 followed by separation of the main cardiac cell populations through fluorescence-activated cell sorting (FACS), revealed that low-dose CY3-labeled antagomir reached ∼15% of the total cardiac cell populations, whereas the high dose reached 80% or more of all cardiac cells (Figure 3B). We subsequently analyzed the antagomir distribution throughout the different cell populations in more detail. With the low dose, we found the antagomir mostly in ECs (CD31+CD45−), CD45+ hematopoietic cells, and, to a much lesser extent, fibroblasts (CD31−CD45−CD140a+) and CMs (gated by morphology) (Figure 3C). With the high dose, 80% or more of hematopoietic cells and ECs and 50% or more of CMs and fibroblasts were targeted (Figure 3D), indicating that, at the high dose, the antagomir reached all cell types rather than only hematopoietic cells and ECs. Confocal microscopy confirmed localization of the CY3-labeled antagomir mostly on ECs with the lower dose and more ubiquitous localization with the high dose (Figure 3E). These findings demonstrate the ability of antagomirs to target different cell populations in vivo in a dose-dependent manner.
Figure 3.

Different doses of antagomir for miR-200c-3p can target specific cardiac cell types
(A) Study design. Wild-type (WT) mice were injected with Cy3-labeled antagomir-200c-3p on 3 consecutive days with 5 mg/kg BW (red arrows) or once with 80 mg/kg BW (blue arrow). After 1 week, hearts were harvested for cardiac tissue and cell isolation. (B) Percentage of Cy3 labeling in isolated cardiac cell analysis. (C and D) Percentage of cells positive for Cy3 in different cardiac cell populations after administration of a (C) low or (D) high dose of antagomir. (E) Representative images of heart sections from untreated or antagomir-treated mice: staining of cell nuclei with Hoechst (first column), detection of Cy3-labeled antagomir (second column), detection of CMs with α-actinin antibody (third column), Isolectin B4 for endothelial cell detection (fourth column), and a merge of all different staining (fifth column). ∗p < 0.05 versus CMs (error bars indicate SEM).
Suppression of miR-200c-3p under cardiac stress promotes angiogenesis and improves cardiac function
To address the contribution of miR-200c-3p to cardiac remodeling induced by increased cardiac pressure overload, we subjected wild-type mice to sham or transverse aortic constriction (TAC) surgery, followed by treatment with antagomir against miR-200c-3p for 3 consecutive days at 5 mg/kg BW or 80 mg/kg BW, and assessed cardiac geometry and function 4 weeks after surgery (Figure 4A). Although cardiac miR-200c-3p expression levels in mice treated with 5 mg/kg BW was reduced by 70%, its expression was entirely abrogated with the high dose under sham and TAC conditions (Figure 4B). Curiously, mice that underwent TAC surgery displayed lower basal levels of miR-200c-3p (Figure 4B), which suggests that, under stress conditions, CMs, which compose most of the heart volume,25 could release this specific miRNA into the extracellular space through enriched vesicles.
Figure 4.

Suppression of miR-200c-3p stimulates angiogenesis and improves cardiac function in pressure-overloaded hearts
(A) Study design. WT mice were subjected to TAC or sham surgery and subsequently injected with a high or low dose of antagomir-200c-3p for 3 consecutive days. Cardiac geometry and function were determined by echocardiography 4 weeks after surgery. (B) Real-time PCR analysis of miR-200c-3p expression in animals subjected to TAC or sham surgery and left untreated or treated with low/high doses of antagomir. ∗p < 0.05 versus the respective control group. (C) Gravimetric analysis of corrected heart weights in sham and TAC animals treated with antagomir-200c-3p. (D) Representative images of whole hearts (top panel), Sirius red-stained sections (second panel), and GSI-stained sections (third panel) from sham and TAC animals treated with antagomir-200c-3p. (E) Quantification of the CM surface area in heart sections from (G) by wheat germ agglutinin (WGA) staining; n = 3 (60 microscopic fields for each animal/condition). (F) Quantification of the fibrotic area in hearts from conditions in (D) by Sirius red staining; n = 3 (60 microscopic fields for each animal/condition). (G) Representative images of heart sections from sham and TAC animals treated with antagomir-200c-3p: WGA-stained sections (first column), Isolectin B4-stained sections (second column), Hoechst-stained sections (third column), and a merge of all staining (fourth column). (H) Quantification of the total number of capillaries by Isolectin B4 staining; n = 3 (60 microscopic fields for each animal/condition). (I) Real-time PCR analysis of CD31 expression in animals subjected to TAC or sham surgery and left untreated or treated with low/high doses of antagomir. (J) Quantification of ejection fraction (EF) from conditions in (D). (C–J) ∗p < 0.05 versus the corresponding control group, #p < 0.05 versus the experimental group (error bars indicate SEM).
Wild-type mice subjected to TAC surgery and left untreated showed severe adverse cardiac remodeling at 4 weeks, as reflected by increased heart weight, interstitial fibrosis, severe myocyte disarray, left ventricular (LV) dilation, systolic and diastolic dysfunction, and a decreased number of capillaries, with subsequent lower expression of CD31 as a result of capillary rarefaction (Figures 4C–4J; Table 1). Depletion of miR-200c-3p resulted in normalized heart size and morphology with subsequent improved cardiac function, as reflected by reduced LV dimensions and increased ejection fraction (EF) (Figures 4C–4E and 4J; Table 1). Although there was an increase in CD45 expression levels in myocardial tissue 4 weeks after TAC, these levels were reduced to control levels upon antagomir treatment, suggesting attenuation of the inflammatory response (Figure S2G). Remarkably, no differences in phenotypes and cardiac function were observed between mice that received the low or high dose of antagomir. In fact, the number of cardiac capillaries was increased in TAC mice already at a low dose of antagomir (Figures 4D and 4G–4I), suggesting that the biological role of miR-200c-3p is mainly exerted in non-CM cells.
Table 1.
Morphometric and echocardiographic characteristics
| Sham |
TAC |
|||||
|---|---|---|---|---|---|---|
| Vehicle | Antagomir (5 mg/kg) | Antagomir (80 mg/kg) | Vehicle | Antagomir (5 mg/kg) | Antagomir (80 mg/kg) | |
| n | 7 | 8 | 6 | 9 | 11 | 12 |
| HW/BW | 0.0057 ± 0.00012 | 0.0059 ± 0.00026 | 0.0058 ± 0.00031 | 0.0097 ± 0.00024a | 0.0085 ± 0.00026a,b | 0.0084 ± 0.00034a,b |
| IVSd (mm) | 0.7410 ± 0.04573 | 0.7625 ± 0.02301 | 0.6806 ± 0.02610 | 1.189 ± 0.04951a | 0.9521 ± 0.04848a,b | 0.9333 ± 0.04201a,b |
| IVSs (mm) | 1.318 ± 0.07709 | 1.116 ± 0.03662 | 1.189 ± 0.05166 | 1.454 ± 0.04483 | 1.350 ± 0.05318 | 1.436 ± 0.05857 |
| LVIDd (mm) | 4.365 ± 0.1047 | 4.146 ± 0.1421 | 4.483 ± 0.06956 | 3.900 ± 0.1442 | 4.225 ± 0.1189 | 4.388 ± 0.1164 |
| LVIDs (mm) | 2.906 ± 0.1069 | 2.941 ± 0.1255 | 3.123 ± 0.08436 | 3.184 ± 0.1246 | 3.194 ± 0.1412 | 3.233 ± 0.1275 |
| LVPWd (mm) | 0.7519 ± 0.04728 | 0.7058 ± 0.03970 | 0.6678 ± 0.05265 | 1.325 ± 0.06442a | 1.068 ± 0.06496a,b | 1.068 ± 0.06564a,b |
| LVPWs (mm) | 1.124 ± 0.04511 | 1.124 ± 0.07504 | 0.9872 ± 0.05838 | 1.529 ± 0.07893a | 1.373 ± 0.06596 | 1.395 ± 0.07262 |
| FS (%) | 33.39 ± 1.966 | 29.19 ± 0.9220 | 30.38 ± 1.244 | 18.37 ± 1.101a | 24.75 ± 1.473a,b | 26.52 ± 1.440a,b |
| EF (%) | 61.96 ± 2.641 | 56.35 ± 1.469 | 57.79 ± 1.890 | 38.48 ± 2.020a | 49.10 ± 2.432a,b | 51.78 ± 2.269a,b |
Data are expressed as means ± SEM. Sham, sham-operated control group; TAC, transverse aortic constriction; HW, heart weight; BW, body weight; LV, left ventricular; IVSd, interventricular septal thickness at end-diastole; IVSs, interventricular septal thickness at end-systole; LVIDd, left ventricular internal dimension at end-diastole; LVIDs, left ventricular internal dimension at end-systole; LVPwd, left ventricular posterior wall thickness at end-diastole; LVPws, left ventricular posterior wall thickness at end-systole; EF, ejection fraction; FS, fractional shortening.
p < 0.05 versus the sham vehicle group.
p < 0.05 versus the TAC vehicle group.
miR-200c-3p is a negative regulator of cell communication
To explore the molecular mechanism underlying the anti-angiogenic effects of miR-200c-3p, we performed RNA sequencing to assess the transcriptomic changes in ECs after overexpression of this miRNA. Data analysis revealed 194 genes differentially regulated after miR-200c-3p overexpression, of which 80 were downregulated (at 1.0 reads per kilobase of exon model per million mapped reads [RPKM] cutoff and 1.3-fold-change cutoff) (Figure 5A; Table S2). Based on these results, we performed a protein-protein interaction (PPI) network and GO analysis, which revealed enrichment in genes involved in regulation of cellular component movement, such as BMP4 (bone morphogenetic protein 4), SMAD7 (SMAD family member 7), MET (MET proto-oncogene, receptor tyrosine kinase), and NEDD4L (neural precursor cell expressed, developmentally down-regulated 4-like, E3 ubiquitin protein ligase); in cell communication, such as VAMP7 (vesicle-associated membrane protein 7), TRIB2 (Tribbles pseudokinase 2), LRRK2 (leucine-rich repeat kinase 2), and TLR4 (Toll-like receptor 4); and in cell migration, such as CXCL2 (C-X-C motif chemokine ligand 2), CXCL1, HMOX2 (heme oxygenase 2), and BCL2 (BCL2 apoptosis regulator) (Figure 5B). Next, we performed a bioinformatics prediction of the miR-200c-3p seed sequence interaction with the 3′ UTR of the downregulated genes (Figures S1B and S3A) revealed by RNA sequencing and validated the transcript levels of IL-8 (CXCL8), KDELC1 (protein O-glucosyltransferase 2), CFL2 (Cofilin 2), and NDN (Necdin; a MAGE family member) in HUVECs transfected with precursor molecules for miR-200c-3p (Figure 5C). To address whether any of these potential target genes of miR-200c-3p could make a prominent contribution to the angiogenic properties of ECs, we performed high-throughput screening to determine the proliferative capacity of ECs after transfection with a library consisting of individually selected specific siRNAs against the 31 most downregulated genes upon mir-200c-3p overexpression (Figure 5D; Table S3). Using this approach, CFL2, KDELC1, CXCL8, RECK (reversion-inducing cysteine-rich protein with Kazal motifs), and NDN were experimentally validated as inhibitors of EC proliferation (Figure 5E). To determine whether expression of these genes was also affected in MPs upon increased expression of miR-200c-3p, we transfected MPs with precursor molecules for miR-200c-3p and showed that only CFL2 and KDELC1 were inhibited in MPs (Figure S3B), suggesting different regulatory mechanisms triggered by this miRNA in different types of cells.
Figure 5.

miR-200c-3p regulates cell communication, migration, and proliferation
(A) Heatmap representation of the RNA-seq data derived from HUVECs transfected with a control precursor (scr) or a precursor for miR-200c-3p (pre-200c). Shown in bright blue are the most downregulated and in bright yellow the most upregulated genes. (B) Network analysis of the differentially expressed genes, where each node represents a protein (blue or yellow, depending on fold change), and the colored outer ring refers to the corresponding GO term. (C) Real-time PCR analysis of the expression of four representative downregulated genes (CXCL8, KDELC1, CFL2, and NDN). ∗p < 0.05 versus the scrambled group. (D) Distribution of standardized fold change, calculated based on the percentage of EdU-positive ECs for each specific siRNA and relative to the mock siRNA. Each dot corresponds to one single siRNA, and the siRNAs corresponding to the blue dots are depicted in (E) representative images of merged DAPI and EdU (top row), and EdU-positive ECs (bottom row) for the most effective siRNAs. (F) Western blot analysis to asses protein levels of CFL2, KDELC1, CXCL8, and NDN in myocardial samples derived from mice subjected to sham or TAC surgery, left untreated or treated with low-dose antagomir-200c. (G) Quantification of results obtained in (F). ∗p < 0.05 versus the respective sham or TAC group. Error bars indicate SEM.
To determine whether expression of these genes is altered in cardiac pressure overload, we assessed their protein levels by western blot analyses. We observed that, under cardiac stress induced by pressure overload, and when miR-200c-3p was enriched in CM-derived EVs, the protein levels of CFL2 and KDELC1 were downregulated (Figures 5F and 5G). No clear alterations were observed for CXCL8 or NDN. Treatment with a low-dose antagomir was sufficient to restore CFL2 and KDELC1 protein levels above baseline (Figures 5F and 5G).
These data demonstrate that miR-200c-3p affects endothelial function by targeting genes that directly affect cell proliferation and migration, as well as cell communication, and explaining how this miRNA exerts detrimental anti-angiogenic functions in vitro and in vivo.
Discussion
Pathological cardiac hypertrophy is often associated with cardiac structural remodeling, interstitial fibrosis, and contractile dysfunction.26 Progression of hypertrophic growth to HF occurs without additional new blood vessel formation, leading to capillary rarefaction and inadequate growth of the cardiac microvasculature.27,28 To date, perturbed cross-talk between CMs and ECs defining the pathogenesis of several heart conditions29 has been mainly associated with paracrine growth factors from CMs, mostly including secreted proteins and peptides such as growth factors, hormones, cytokines, and extracellular matrix proteins.1 Here we identified a new mechanism by which CMs are able to influence the function of the surrounding endothelium through release of EVs enriched with miR-200c-3p. We found that CM-derived miR-200c-3p is transferred to cardiac ECs in a vesicle-dependent manner. In ECs, this miRNA acts as an anti-angiogenic factor because its increased expression results in impaired endothelial migration and tube formation as well as a lower proliferation capacity. The overall result is a decrease in capillary density that worsens cardiac outcome under stress conditions.
Most studies so far have attributed to EVs a potential role as (cardiac) disease biomarkers because they contain certain miRNAs, including miR-1, miR-133a/b, miR-208a, and miR-499, that are detectable even before traditional cardiac disease markers such as troponin or creatine kinase.30,31 Despite the importance of EVs in paracrine signaling, less is known about the function of secreted EVs and their paracrine actions among the different cell types of the heart. We identified miR-200c-3p as the most enriched miRNA in EVs derived from stressed CMs, not only in comparison with other miRNAs derived from EVs but also with the cellular endogenous expression levels. In line with our findings, several studies have shown that the miRNA content of CM-derived EVs does not always reflect expression levels in the parental cells, as described for vesicles derived from certain cancer cells,32,33 inflammatory cells,34,35 and CMs.36 Curiously, most of the miRNAs found to be enriched or depleted in CM-derived EVs are derived from passenger strands (3p), generally believed to be degraded within a cell.37 This observation, apart from supporting regulatory roles for miRNA passenger strands,38,39 also strengthens the theory of a specific transport mechanism of passenger miRNAs.36,40
Although endogenous miR-200c-3p expression in CMs is not upregulated upon increased cardiac pressure overload, increased levels of this miRNA were observed in CMs from ischemic41 and diabetic42 hearts, suggesting that regulation of this miRNA in the heart is dependent on the type of cardiac stress and that increased pressure overload induces selective release of CM-derived miR-200c-3p through EVs. The underlying mechanisms for triage of specific miRNAs into EVs remains unknown.43 Recent studies have revealed that recognition of particular motifs in the 3′ sequence of the miRNA by specific RNA-binding proteins causes those miRNAs to be packed into EVs even though no binding motifs have been identified yet.29,44 Others have shown, mainly for circulating cells, that post-transcriptional modifications of the 3′ end of miRNAs, such as uridylation or adenylation, influence whether a specific miRNA stays in the cell or will be mainly shuttled to EVs released by that same cell.45,46 The types of EVs that are responsible for transfer of miRNA-200c from CMs to cardiac ECs remain to be determined.
Increased levels of miR-200c-3p in EVs released by hypertrophic CMs impairs the angiogenic capacity of the endothelium, suggesting a prominent role of CM-derived EVs in the process of cardiac capillary rarefaction and endothelial dysfunction. Because cardiac perfusion and blood supply are largely determined at the level of the microvasculature,47 any reduction in capillary density results in cardiac subperfusion, tissue hypoxia, contractile dysfunction, and, ultimately, HF. Although numerous endogenous noncoding RNAs have been associated previously with EC function,48 this study is the first report of functional EV-mediated transfer of a CM-derived miRNA to the surrounding endothelium in heart disease. EV-mediated delivery of miR-200c-3p to ECs induces capillary rarefaction and reduces angiogenic capacity by perturbing endogenous expression levels of numerous genes that were associated previously with maintenance of proper cell growth and mobility (CFL2,49 IL-8,50,51 RECK,52 and NDN53,54), cell apoptosis (PMAIP155,56), regulation of notch signaling (KDELC157), or cell-cell communication (VAMP758,59 and LRRK260), all determinant processes for proper EC function. In fact, inhibition of miR-200c in non-myocyte cells in the heart is sufficient to prevent capillary rarefaction and cardiac remodeling, precluding development of HF. This is in line with previous preclinical strategies to enhance myocardial angiogenesis by delivery of angiogenic genes or growth factors.61 However, such potential treatment strategies are currently limited to the preclinical stage because, for some of them, apart from demonstration of increased vascularity, very few results with clinical significance have been obtained. This lack of promising preclinical results highlights the need for a better understanding of the pathways that drive blood vessel dysfunction and cardiac angiogenesis to more accurately identify new therapeutic targets and/or strategies.
CM-derived EVs have been shown to be preferentially taken up by ECs,62 but how and why this is the case remains to be investigated. Why CMs release a miRNA that negatively affects the surrounding environment is unknown, as is the molecular mechanism underlying the increased release of miR-200c-3p from CMs through EVs. The mechanism may rely on the type of stress to which the heart is exposed because it has been shown that CM-derived EVs can induce eNOS activation in cardiac microvascular ECs and in this way protect against ischemia/reperfusion injury.63 In atherosclerosis, upregulation of miR-200c family members has been shown to correlate with inflammatory markers.64 The contribution of miR-200c-3p to the cardiac inflammatory response induced by pressure overload has not been studied, even though our data indicate considerable expression levels of this miRNA in CD45+ cells. Although we observed stimulation of angiogenesis by CD45+ cells, this was independent of EV release and transfer of miR-200c-3p by these cells toward ECs. This may have to do with the role of monocyte-like cells and heart-resident macrophages in stimulating angiogenesis and inhibiting fibrosis, with a potential role in cardiac neovascularization under conditions of stress,65,66 but more research is needed to establish the contribution of these immune cells to cardiac pathologic remodeling and cardiac disease. Interestingly, miR-200c can be induced by oxidative stress, and its overexpression induces cell senescence in vitro,67 suggesting a role in (cardiac) aging. In fact, among the CM-derived enriched miRNAs detected by us, miR-30d has been associated with cardiac fibrosis and inflammation68 and shown to promote tumor angiogenesis (miR-30d).69 Increased cardiac endogenous expression of miR-212 has been shown to induce hypertrophic growth70 and affect EC function,71 but whether this was the 3p strand was not reported. Although the role of the remaining identified miRNAs in (cardiac) pathologies has not yet been studied, the miRNA cocktail present in hypertrophic CM-derived EVs seems to instigate remodeling responses in the stressed heart by triggering an orchestrated response involving different cardiac cell types, where ECs seem to have a directing role.
Here we demonstrate the existence of CM-EC communication via EVs as an essential feature of normal cardiac function and reinforce the concept that cardiac heterocellularity needs to be carefully considered in the pathogenesis of HF, particularly when implementing new platforms to screen drug candidates or lead optimization. Exploitation of EV-mediated CM-EC communication could lead to identification of additional therapeutic targets such as miR-200c-3p to enhance cardiac angiogenesis in the stressed heart.
Materials and methods
Primary CM cultures
CMs were isolated from hearts collected from 1- to 2-day-old neonatal rats. Atrial tissue was removed, and the ventricles were cut into 1 mm3 pieces before enzymatic digestion.72 Tissue was subjected to multiple rounds of enzymatic digestion using 0.05% pancreatin (Sigma, P3292) and 290 units/mg of collagenase (Gibco, 17100-017). Cells were collected by centrifugation at 1,500 rpm for 6 min and resuspended in DMEM (Gibco, 21885-025) supplemented with 15% fetal bovine serum (Gibco, 10270-106) and gentamicin (10 mg/mL, Gibco, 15710). Subsequently, cells were differentially plated for 1 h on uncoated cell culture dishes to remove non-myocytes. The CMs were then plated on 0.2% gelatin cell culture dishes and cultured the next day in SF DMEM containing 20% M199 (Gibco, 31153-026), 15% fetal bovine serum (FBS), 100 units/mL penicillin/streptomycin (Gibco, 15140-122), and gentamicin (10 mg/mL). After 24 h, CMs were maintained in SF DMEM and treated with 10 μM PE (Sigma, P6126) and 10μM Iso (Sigma, I5627) for 48 h.
EV isolation
Five to six million NRCMs were seeded on 10-cm dishes and incubated for 48 h with PE (10 μM) and Iso (10 μM). The medium was collected and centrifuged at 2,000 × g for 15 min at 4°C. EVs were purified from the obtained supernatant by differential centrifugation as described previously.73 Briefly, samples were ultracentrifuged twice at 10,000 × g for 30 min at 4°C, the pellet was discarded, and the supernatant was submitted to ultracentrifugation at 100,000 × g for 2 h at 4°C to pellet EVs. Finally, the pellet from the last step was washed with cold PBS, ultracentrifuged again at 100,000 × g for 2 h at 4°C, resuspended in 150 μL of cold PBS, and stored at −80°C for later use. Ultracentrifugation steps were performed using a swinging bucket rotor SW 32 Ti in an Optima XPN 100K ultracentrifuge (Beckman Coulter, CA, USA) and 28.7-mL polyallomer conical tubes (Beckman Coulter).
EV characterization by dynamic light scattering (DLS)
DLS measurements were done on a Zetasizer Nano ZS ZEN3600 (Malvern Instruments, Malvern, UK), equipped with a solid-state He-Ne laser at 633-nm wavelength. The sample was pre-equilibrated at 37°C for at least 60 s, and each measurement was the average of 11 runs. Three consecutive measurements were performed for each sample to evaluate its stability. The results were analyzed by the equipment software, considering the viscosity and refractive index of water at the measurement temperature and a refractive index of 1.59 for the scattering particles. The average size was taken from the analysis in volume distribution of particles.
EV characterization by flow cytometry analysis
EVs were analyzed for the presence of the exosomal marker CD63 by flow cytometry. Isolated EVs were resuspended in PBS and bound to magnetic CD63-coated Dynabeads (Life Technologies) during overnight incubation at 4°C. The next day, the Dynabead-bound EVs were stained with CD63-fluorescence in situ hybridization (FITC) (Abcam, ab108949) or the corresponding isotype control (FITC mouse immunoglobulin G1 [IgG1], Abcam, ab91356) and analyzed by flow cytometry (BD Accuri C6 Plus cytometer).
EV characterization by TEM
EV samples were diluted 1:1 in 4% (v/v) paraformaldehyde (PFA) and placed on formvar carbon-coated grids (TAAB Technologies) for 20 min at room temperature (RT). After washing 4 times with PBS, grids were placed on a drop of 1% (v/v) glutaraldehyde for 5 min, followed by 5 washes with distilled water, 1 min each. In a dark environment, grids were incubated with uranyl-oxalate solution (pH 7) for 5 min and then placed on ice in contact with a solution of methyl cellulose (9:1) for 10 min. EV imaging was performed in a JEM-1400 (JEOL) electron microscope operated at 80 kV.
EV characterization by NTA
Size and concentration of sEVs were determined by NTA using the NanoSight NS300 (Malvern Instruments). The system used an O-ring top plate, and the sample was injected manually at an approximate flow of 1 mL every 20 s. sEVs were diluted in PBS until a concentration between 15 and 45 particles/frame was reached. For each sample, 5 videos of 30 s were recorded with the camera level set at 16. All videos were processed with NTA 3.2 analytical software, using a software threshold between 2 and 4 depending on the quality of the videos.
EV characterization by western blot analysis
Western blot analysis for detection of EV markers and contaminants was performed. Briefly, up to 15 μL of concentrated EV preparations in PBS (0.5–4 μg) were mixed with 5 μL 4× Laemmli buffer (0.25 M Tris base, 8% SDS, 40% glycerol, 200 mg bromophenol blue, and 10% 2-mercaptoethanol) and boiled at 96°C for 10 min. For analysis of tetraspanins, Laemmli buffer was prepared without reducing agents. Samples were loaded in 30-μL wells of Any kD Mini-PROTEAN TGX Stain-Free Protein Gel (Bio-Rad, 4568123) and gel electrophoresis was performed in 1× Tris/glycine/SDS buffer prepared from a commercial 10× concentrated stock (10× Tris/glycine/SDS electrophoresis buffer, Bio-Rad, 1610772), at a constant voltage of 120 V for 75 min. Then gels were placed in blotting buffer (25 mM Tris, 192 mM glycine, and 20% methanol in water) for 10 min to equilibrate. Then the gel was stacked on top of a nitrocellulose membrane (GE Healthcare, 10600016), and both were assembled in a transfer system. Transfer was performed under wet conditions at 200 mA for 90 min. Then the membrane was removed and blocked in 1:1 PBS-Tween 20 (0.2% [v/v]) with Intercept blocking buffer (LI-COR Biosciences, 927-70001) solution for 1 h at RT. Membranes were then washed with PBS-Tween 20 and left to incubate overnight at 4°C with the appropriate primary antibodies and according to the manufacturer’s recommendations (see antibody details below). Then membranes were washed 3 times with PBS-Tween and incubated for 1 h at RT with secondary antibodies. Membranes were then washed 3 times and viewed in the Odyssey CLx system (LI-COR Biosciences) at the 700-nm and 800-nm wavelengths. Antibodies were diluted 1:1,000 for primary incubation or 1:10,000 for secondary incubation. The antibodies used were as follows: GAPDH (Millipore, MAB374), Calnexin (Santa Cruz #sc-23954), ApoA-1 (Santa Cruz Biotechnology, sc-376818), Alix (Cell Signaling Technology, 2171S), CD81 (Santa Cruz, sc-166029), HSP70 (Enzo Life Sciences, ADI-SPA-820), and IRDye 800CW goat anti-mouse IgG secondary antibody (LI-COR Biosciences, 926-32210).
Culture of HUVECs, functional assays, and Transwell co-culture
Pooled HUVECs were purchased from Lonza (CC-2519). Cells were cultured in EGM-2 medium consisting of EBM-2 basal medium (Lonza, CC-3156) supplemented with the Endothelial Cell Growth Medium-2 Bullet Kit (CC-4176) and cultured at 37°C with 5% CO2. Cells in passages 6 and 7 were used. 15 × 104 HUVECs were cultured in 6-well plates. Upon reaching 70% confluency, they were transfected with 10 nM of a precursor molecule for hsa-miR-200c-3p or a scramble precursor (AM17100 and AM17110, respectively; Life Technologies) as a control for 24 h. Next, HUVECs were trypsinized (trypsin 0.05%, Lonza, cc-5012) before plating according to the different functional assays to be performed. For the migration assays, a confluent monolayer of HUVECs in a 6-well plate was scratched using a 200-μL pipette tip, and pictures were taken immediately after scratching and at 8 h and 24 h with a Canon EO600D camera linked to an inverted microscope (Eclipse TS100, Nikon). Quantification of the closure rate was performed by measuring the area covered by HUVECs with ImageJ software. For tube formation, Matrigel (Thermo Fisher Scientific, 356230) was plated and left to set for 1 h at 37°C with 5% CO2. 1 × 104 HUVECs were cultured on Matrigel, and 16 h later, pictures were taken with a Canon EO600D camera linked to an inverted microscope (Eclipse TS100, Nikon). To characterize the tube structures, an ImageJ macro (Angiogenesis Analyser) was used. To evaluate proliferation, 1 μM 5-ethynyl-2′-deoxyuridine (EdU; Life Technologies) was added for 16 h to 5 × 103 HUVECs. To assess EdU incorporation, cells were fixed with 4% PFA for 15 min and processed using the Click-IT EdU 555 Imaging Kit (Thermo Fisher Scientific, C10339) according to the manufacturer’s instructions. The cell nuclei were stained with Hoechst 33342 (Life Technologies). The fluorescence signals were visualized using a DMI3000B microscope (Leica). For the co-culture experiments, 35 × 104 NRCMs were cultured in Thinserts with 0.4-μm pores (Greiner, 65641). One day later, cells were transfected with 25 nM of a precursor molecule of hsa-miR-200c-3p or a scramble precursor as a control (AM17100 and AM17110, respectively; Life Technologies). In some instances, 4 h after transfection, the culture medium was replaced with fresh medium with 10 μM GW (Millipore, 567715) for 24 h. Alternatively, NRCMs were incubated with 10 μM PE and 10 μM Iso for 48 h. Subsequently, the Thinserts were transferred onto 6-well plates previously seeded with HUVECs and cultured for another 72 h. After this, HUVECs were used for the different functional assays as described above.
THP-1 cells
THP-1 cells (ATTC) were cultured in 25-cm2 sterile culture flasks (Corning) in complete medium consisting of RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS (Sigma) and 1% penicillin-streptomycin (Life Technologies). The cells were maintained at a density of 106 cells/mL, and flasks with cell suspension were kept at 37°C and 5% CO2. The culture medium was exchanged every 2 days. THP-1 cells were plated in 6-well culture plates at a concentration of 5 × 105 cells/mL in complete medium, treated with 100 ng/mL of 4α-phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich), and incubated for 48 h at 37°C and 5% CO2. The PMA-induced THP-1 cells transdifferentiated into MPs that were able to adhere to plastic. Adherent cells were washed to remove PMA and cultured with 1 mL SF RPMI medium for 48 h. Cells, or the resulting conditioned medium, were then used for (co)culture experiments with HUVECs. In the co-culture experiments with HUVECs, 2 × 105 MPs were cultured in Thinserts with 0.4-μm pores (Greiner, 65641) for 24 h. Subsequently, the Thinserts were transferred onto 6-well plates previously seeded with HUVECs and cultured for another 72 h. After this, HUVECs were used for the different functional assays as described above. Conditioned medium was clarified by centrifugation at 13,000 rpm and 4°C for 5 min before adding it at 25% to HUVEC culture medium for 24 h, followed by different functional assays as described above. In some instances, MPs were treated with 10 μM GW (Millipore 567715) for 24 h or transfected with 25 nM of a control or a hsa-miR-200c-3p inhibitor (QIAGEN) for 8 h.
Aortic banding and transthoracic echocardiography
Mice used for this study were male and female B6CBAF1/JRJ (Charles River Laboratories), wild type, 2–3 months old. All protocols were performed according to guidelines approved by the local Animal Care and Use Committee. Mice were housed in a climate-controlled environment under a 12-h light/dark cycle with ad libitum access to chow and water. TAC or sham surgery was performed on mice anesthetized with ketamine (75 mg kg−1 BW)/medetomidine (1 mg kg−1 BW) and maintained during surgery with isoflurane (3% in oxygen). TAC was performed as described previously.74 In brief, the aorta was subjected to a 27G constriction between the first and second truncus of the aortic arch. Four weeks after surgery, mice were shaved and underwent light anesthesia with isoflurane (1% in oxygen), and Doppler echocardiography was performed. Echocardiographic parameters were measured using an RMV707B (15–45 MHz) scan head interfaced with a 770 high-frequency ultrasound system (VisualSonics). The pressure gradient between the proximal and distal sites of the TAC was calculated with the Doppler, and only mice with a pressure gradient greater than 50 mm Hg were included.
Antagomir delivery in vivo
Chemically modified antisense oligonucleotides, antagomirs, (Fidelity Oligos) were designed to target mmu-miR-200c-3p. To distinguish the types of cells that take up the antagomir, we used 3′-conjugated cholesterol and 5′-conjugated fluorescent Cy3 antagomirs. Female and male mice were injected intraperitoneally once with 80 mg/kg BW or for 3 consecutive days with 5 mg/kg BW of labeled antagomir-200c-3p. Control mice were injected for 3 consecutive days with vehicle (sterile PBS, 0.1 mL). Mice were sacrificed 1 week after the first antagomir injection. For the functional studies, we injected unlabeled antagomirs into female and male mice intraperitoneally for 3 consecutive days with 5 mg/kg BW or 80 mg/kg BW, and control animals were injected for 3 consecutive days with vehicle (sterile PBS, 0.1 mL).
Single-cell suspension and flow cytometry analysis
Isolation of different cell populations from murine hearts was performed using a Langendorff-free method as described previously.19 In brief, mouse hearts were perfused and digested with collagenase II (Worthington, LS004176), IV (Worthington Biochemical, LS004188) and protease (Sigma-Aldrich, P5147), cut into small pieces, and completely dissociated by pipetting. Cell suspensions were passed through a strainer, and myocytes settled for 20 min through gravity. Cells from the supernatant, which contained the non-myocyte cell population, were incubated on ice for 20 min with conjugated antibodies: CD140a-APC (BioLegend, 135907), CD31-APC (BioLegend, 102516), TER119-FITC (BioLegend, 116206), and CD45 FITC (BioLegend, 103107). Cells were analyzed with a FACSCanto II cytometer (BD Biosciences). To isolate cardiac ECs, the non-CMs cell suspension was incubated for 15 min at 4°C with magnetic CD146-labeled MicroBeads (Miltenyi Biotec, 130-092-007), followed by magnetic separation and 3 washing steps with PBS. FACS was also used for EV characterization. EVs were incubated for 30 min with latex beads (Invitrogen, A37304), followed by overnight incubation (RT) of the EV-coated beads with purified anti-CD63 antibody (FC-5.01, Zymed).
Histological analysis and (immunofluorescence) microscopy
Hearts were fixed in 4% PFA in PBS, embedded in paraffin, and sectioned at 4 μm. Sections were deparaffinized, rehydrated, and used for Sirius red or Isolectin B4 (GSI-biotin, Vector Laboratories, 1:1,000). Imaging was performed using a DM200 microscope (Leica). Fibrotic areas and capillary density were quantified using ImageJ imaging software.
For immunofluorescence, sections were deparaffinized and rehydrated, followed by antigen retrieval. Slides were submerged in sodium citrate buffer and boiled for 15 min. After cooling down, slides underwent blocking and permeabilization with 1% BSA (Millipore, 82-100-6) and 0.5% Triton X-100 (Sigma, T8787) for 1 h at RT. CM staining was performed with anti-α-sarcomeric actinin (Abcam, ab68167 1:100), followed by Alexa Fluor 647 goat anti-rabbit IgG (H+L) (Invitrogen, A-21245, 1:1,000). For EC staining, Isolectin Griffonia simplicifolia conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, I21411, 1:50) was used. For membrane staining, wheat germ agglutinin (WGA), Alexa Fluor 647 conjugate (Thermo Fisher Scientific, W32466, 50μg/mL) was used. Imaging was performed with a confocal laser-scanning microscope (Leica TCSNT) equipped with argon-krypton and helium-neon lasers.
qPCR and qPCR-based array
Total RNA was isolated from mouse heart tissue or cultured cells using Direct-zol RNA MiniPrep (Base Clear, R2053) according to manufacturer’s instructions. 1 μg RNA was reverse-transcribed with M-MLV reverse transcriptase (Promega, Madison, WI, USA) for mRNAs or 5 ng/μL RNA with Universal cDNA Synthesis Kit II (Exiqon, 203301) for miRNAs. For mRNAs, real-time PCR was performed on an iCycler (Bio-Rad) using SYBR Green Supermix (Bio-Rad). For miRNAs, ExiLENT SYBR Green Master Mix (QIAGEN) was used. Transcript quantities were compared using the relative Ct method, where the amount of target was normalized to the amount of endogenous control (L7 for mRNAs and 5s for miRNAs) fold changes were determined using the 2−ΔΔCT method. Primers for human sequences used for this study were as follows: human L7, Fw-ACCTGCAGAACCCAAATTGG and Rv-TTGACGAAGGCGAAGAAGCT-5′; human CXCL-8, Fw-GAAGAAACCACCGGAAGGAA and Rv-CTTGGCAAAACTGCACCTTC; human Kdelc1, Fw-CCGTCATACTTCTCTTGCTCCTCT and Rv-CACATCTGGCATCTTCACCAGTCT; human Cfl2, Fw-GAAGGATGAATGGGCAGGATGAGT and Rv-CCAGAAGCCATGTAAGTCGTCCAA; human Ndn, Fw-AGACTGCTCCTGCAGAGTTTGGA and Rv-TGTTTTCTGTGCCCAGCAAGGT. Primers for mouse sequences used for this study were as follows: mouse L7, Fw-GAAGCTCATCTATGAGAAGGC and Rv-AAGACGAAGGAGCTGCAGAAC; mouse CD31, Fw-CCAAAGCCAGTAGCATCATGGTC and Rv-GGATGGTGAAGTTGGCTACAGG; mouse CD45, Fw-CTTCAGTGGTCCCATTGTGGTG and Rv-TCAGACACCTCTGTCGCCTTAG; mouse CXCL8, Fw-CGTGGCTCTCTTGGCAGCCTTC and Rv-TCCACAACCCTCTGCACCCAGTT; mouse Kdelc1, Fw-TACATTCGGGCGGTGGATACCT and Rv-AACCTGGACACCAACTCTCGTG; mouse Cfl2, Fw-GCTCCTGAAAGTGCACCGTTAAA and Rv-GCGGTCCTTAATATCGTCCAAGC; mouse NDN, Fw-CCTCTGGTTTCCAGACATGGTG and Rv-ATGGTGTGGAGATTGGTCAGCC.
Primers for miRNA were as follows: 5S (QIAGEN, YP00203906) and miR-200c-3p (QIAGEN, YP00204482).
For the qPCR-based array, cDNA synthesis and real-time qPCR was performed using the miRCURY LNA Universal RT microRNA PCR System (Exiqon) according to the manufacturer’s instructions. In brief, the RNA was tailed with a poly(A) sequence at the 3′ end and then reverse transcribed into cDNA using a universal poly(T) primer with a 3′-end degenerate anchor and a 5′-end universal tag. The cDNA products were subsequently diluted 125-fold, transferred to the Ready-to-Use microRNA PCR Mouse & Rat Panel, and quantified using SYBR Green-based real-time PCR and LNA-enhanced miRNA-specific primers. Real-time PCRs were run on an iCycler (Bio-Rad) using the thermal cycling parameters recommended by Exiqon. Raw Ct values were analyzed using the relative Ct method, where the amount of target was normalized to the amount of endogenous control (L7 for mRNAs and 5s for miRNAs) fold changes were determined using the 2−ΔΔCT method. Validation of the qPCR-based array was performed on EV samples after performing RNase A digestion with or without Triton X-100. Prior to subjecting all samples to 0.4 μg/mL RNase A (Thermo Fisher Scientific) treatment for 10 min at 37°C, some samples were suspended in 2 mL of PBS and treated with 100 μL of Triton X-100 (Sigma Aldrich) to achieve 5% Triton concentration. Samples were then used for RNA isolation and further experiments.
RNA sequencing
Deep sequencing of total RNA, isolated from HUVECs was performed 24 h after transfection with 10 nM of hsa-miR-200c-3p or scrambled (control) precursor molecules (AM17100 and AM17110, respectively; Life Technologies). RNA purity and integrity were determined with an RNA Nano 6000 Kit for the 2100 Bioanalyzer (Agilent Technologies). rRNA deletion and subsequent NGS library preparation was performed according to the manufacturer’s instructions using the NEBNext rRNA depletion and RNA Ultra II Library Prep Kit (New England Biolabs). Quality control of the resulting DNA library was carried out using the DNA 1000 Kit (2100 Bioanalyzer, Agilent Technologies). Library molarity was determined by qPCR (NEBNext Library Quant Kit, NEB) for subsequent equimolar pooling and sequenced on the NextSeq500 instrument using v2 chemistry in a single read mode for 75 cycles. The Bcl2fastq tool (Illumina, v.2.20.0.422) was applied to compile raw fastq data per sample. Raw sequence files were quality checked using FASTQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and trimmed to remove Illumina adaptor using Cutadapt software. The raw sequencing reads were then mapped to the Ensembl Homo sapiens reference genome (Ensembl GRCh37 PAR-masked) using STAR software. Rounded gene counts were normalized to RPKM using the rpkm function in the Bioconductor package edgeR. Genes with RPKM values greater than 2.00 in miRNA- and scramble-transfected HUVECs were considered expressed genes. Fold changes were taken with respect to the expression upon scramble transfection. Genes whose expression fold change was greater than 1.3 were considered differentially expressed.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE192586 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192586).
Bioinformatics analyses: PPI and GO network
Based on the results of the miRNA deep sequencing, a PPI network analysis was conducted and generated using Cytoscape software (v.3.7.1) with the built-in STRING plug-in (v.1.4.1), a software package for biological network visualization and interactive network generation. The filter applied was the limitation to the species source of Homo sapiens. The network interactions from the STRING software and genes involved in the GO and pathway analysis results were obtained synchronously. GO terms were selected manually to be those of most interest. In the PPI network, each node represents a protein, and each edge between two nodes represents an interaction between these two proteins. The color of the nodes corresponds to the value of the log2 fold change calculated in the RNA sequencing (RNA-seq). The outer ring colors refer to the corresponding GO term.
Small interfering RNA (siRNA)-based high-throughput screening
HUVECs were reverse transfected with a specific library of siRNAs (SMARTpool siRNA, Dharmacon). The library of siRNAs was robotically (Hamilton StarLab) cherrypicked from stock siRNA whole-genome library plates (SMARTpool siRNA, Dharmacon) to 384-well black optical cell culture plates (PerkinElmer, Cellcarrier 384) coated with gelatin (20 μg/mL) and fibronectin (5 μg/mL). 5 μL of each siRNA (500 nM) was spotted into each well, leaving empty columns 1, 2, 11, 12, 23, and 24 for addition of controls, such as siRNA resuspension buffer and siRNA negative control 4 (Dharmacon, B-002000-UB-100 and D-001210-04, respectively). siRNAs were transfected at a final concentration of 50 nM. Briefly, the transfection reagent (Lipofectamine RNAiMAX, Life Technologies) was diluted in OPTI-MEM (Life Technologies) and incubated for 5 min (the final volume was calculated considering 0.06 μL of RNAiMAX and 15 μL of OPTI-MEM for each well). The mixture of transfection reagent and OPTI-MEM was robotically added to the siRNAs arrayed on 384-well plates using a Multidrop reagent dispenser (Thermo Fisher Scientific). The transfection mix and siRNA were incubated for 30 min, and immediately thereafter, cells were robotically dispensed at a final concentration of 1.4 × 103 cells/well on top of the transfection mix. 66 h after transfection, EdU was added to the cells at a final concentration of 10 μM. The proliferation assay was run for 6 h. Cells were fixed in 4% buffered PFA for 12 min, blocked with 1% BSA in PBS for 30 min at RT, and then permeabilized with 0.5% Triton X-100 in PBS. EdU incorporation was detected using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Thermo Scientific). Nucleus staining was performed using Hoechst dye. Plates were imaged using an automated high-content screening microscope (Operetta, PerkinElmer). 15 images/wavelength/well were taken with a 20× magnification objective (NA, 0.45). Images were analyzed by Harmony software (PerkinElmer), and the total number of cells (nuclear count by segmentation based on nuclear size and fluorescence intensity) and the total number of EdU-positive nuclei (count by 488 intensity in the nuclear compartment) was calculated. The number of EdU-positive cells is expressed as log2 fold over control-treated cells (siRNA negative control 4). The screening was performed at the International Center for Genetic Engineering and Biotechnology (ICGEB) High-Throughput Screening Facility (http://www.icgeb.org/high-throughput-screening.html).
Western blot analysis
SDS-PAGE electrophoresis and blotting were performed as described previously.61 In short, whole tissue or cell lysates were produced in RIPA buffer supplemented with PhosSTOP (Roche) and Protease inhibitor cocktail (Roche). Then samples were boiled in 4× Laemmli buffer, including 2% β-mercaptoethanol, for 5 min at 95°C. SDS-PAGE and western blotting were performed using the Mini-PROTEAN 3 system (Bio-Rad). Blotted membranes were blocked in 5% BSA/Tris-buffered saline (TBS)-Tween. Primary antibody labeling was performed overnight at 4°C. Secondary IgG-horseradish peroxidase (HRP)-conjugated antibodies were applied for 2 h at RT. After each antibody incubation, blots were washed three times for 10 min in TBS-Tween. Images were generated using Supersignal West Dura extended duration enhanced chemiluminescence (ECL) substrate (Pierce) and the LAS-3000 documentation system (FujiFilm Life Science). Stripping was performed with Restore western blot stripping buffer (Pierce). Outputs were normalized for loading, and results are expressed as an n-fold increase over the values of the control group in densitometric arbitrary units. Primary antibodies used included rabbit polyclonal anti-CXCL8 (Novus Biologicals, 1:500, detection at 9 kDa), mouse monoclonal anti-KDELC1 (Novus Biologicals, 1:1,000, detection at 46 kDa), rabbit polyclonal anti-CFL2 (Proteintech, 1:1,500, detection at 19 kDa), monoclonal mouse anti-NDN (Novus Biologicals, 1:1,000, detection at 36 kDa), and mouse monoclonal anti-GAPDH (Millipore, 1: 10,000, detection at 36 kDa). Secondary antibodies included polyclonal rabbit anti-mouse IgG-HRP (Dako, 1:5,000) and polyclonal swine anti-rabbit IgG-HRP (Dako, 1:5,000).
Statistical analysis
The results are presented as mean ± standard error of the mean (SEM). Statistical approaches for bioinformatics analyses are described above. All other statistical analyses were performed using Prism software (GraphPad) and consisted of ANOVA followed by Newman-Keuls multiple comparisons test when group differences were detected at the 5% significance level or Student’s t test when comparing two experimental groups. Differences were considered significant when p < 0.05.
Acknowledgments
L.J.d.W. acknowledges support from the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centers, ZonMw, and the Royal Netherlands Academy of Sciences (CVON2017-ARENA PRIME). L.J.d.W. was also supported by ERC Consolidator Grant 311549 CALMIRS and VICI Award 918-156-47 from NWO. J.M.M.K. and P.A.d.C.M. were supported by the Dutch CardioVascular Alliance (DCVA) awarded to the Phaedra Consortium as well as the Impulse Grant 2018 awarded to the Phaedra IMPACT Consortium (2012-08, 2014-11). L.O. and P.d.C.M. were supported by a Dutch Heart Foundation grant (NHS2015T066). V.S.-P. and R.C.d.A. were supported by Foundation for Science and Technology of Portugal (FCT) grants (SFRH/BD/111799/2015 and SFRH/BD/129317/2017, respectively). D.N. was supported by a Foundation for Science and Technology of Portugal (FCT) grant (POCI-01-0145-FEDER-030985). This work was also partially supported by the H2020 Twinning project RESETageing (GA 952266). We are grateful to the Core Facility Genomics (CFG) of the Medical Faculty of the University of Münster, Germany, for performing RNA sequencing and helping with data analysis.
Author contributions
L.O., R.P.J., D.S.N., R.C.d.A., and P.A.d.C.M. conceived and planned the experiments. L.O., R.P.J., M.S., J.H., R.C.d.A., H.e.A., C.d.J., and D.S.N. carried out the experiments. V.S.-P., A.C.E., J.M.M.K., H.F., and S.O. contributed to sample preparation and analysis. L.O., R.P.J., V.S.-P., L.F., D.S.N., L.J.d.W., and P.A.d.C.M. contributed to the interpretation of the results. K.K. contributed to imaging acquisition and the respective data analysis. F.R., J.C.H., and M.S. contributed to the RNA sequencing data and respective analysis. J.C.H. contributed to RNA sequencing data analysis and visualization. L.O., R.P.J., and P.A.d.C.M. wrote the manuscript. P.A.d.C.M. supervised the execution and development of the project. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Declaration of interests
L.J.d.W. and P.A.d.C.M. are cofounders of Mirabilis Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.002.
Supplemental information
References
- 1.Gogiraju R., Bochenek M.L., Schafer K. Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front. Cardiovasc. Med. 2019;6:20. doi: 10.3389/fcvm.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutherland M.R., Ng K.W., Drenckhahn J.D., Wlodek M.E., Black M.J. Impact of intrauterine growth restriction on the capillarization of the early postnatal rat heart. Anat. Rec. 2019;302:1580–1586. doi: 10.1002/ar.24037. [DOI] [PubMed] [Google Scholar]
- 3.Tirziu D., Chorianopoulos E., Moodie K.L., Palac R.T., Zhuang Z.W., Tjwa M., Roncal C., Eriksson U., Fu Q., Elfenbein A., et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Invest. 2007;117:3188–3319. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hein S., Arnon E., Kostin S., Schönburg M., Elsässer A., Polyakova V., Bauer E.P., Klövekorn W.P., Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart:structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 5.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W.S., Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhee S., Chung J.I., King D.A., D'Amato G., Paik D.T., Duan A., Chang A., Nagelberg D., Sharma B., Jeong Y., et al. Endothelial deletion of Ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat. Commun. 2018;9:368. doi: 10.1038/s41467-017-02796-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kehat I., Molkentin J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sluijter J.P., Verhage V., Deddens J.C., van den Akker F., Doevendans P.A. Microvesicles and exosomes for intracardiac communication. Cardiovasc. Res. 2014;102:302–311. doi: 10.1093/cvr/cvu022. [DOI] [PubMed] [Google Scholar]
- 9.Paiva R.A., Martins-Marques T., Jesus K., Ribeiro-Rodrigues T., Zuzarte M., Silva A., Reis L., da Silva M., Pereira P., Vader P., et al. Ischaemia alters the effects of cardiomyocyte-derived extracellular vesicles on macrophage activation. J. Cell Mol. Med. 2019;23:1137–1151. doi: 10.1111/jcmm.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ottaviani L., da Costa Martins P.A. Non-coding RNAs in cardiac hypertrophy. J. Physiol. 2017;595:4037–4050. doi: 10.1113/JP273129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Li S., Li L., Li M., Guo C., Yao J., Mi S. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics. 2015;13:17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knowlton S.T., Michel M.C., Itani M., Shubeita H.E., Ishihara K., Brown J.H., Chien K.R. The alpha 1A-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy. J. Biol. Chem. 1993;268:15373–15380. [PubMed] [Google Scholar]
- 13.Zou M.Y., Yao A., Zhu W., Kudoh S., Hiroi Y., Shimoyama M., Uozumi H., Kohmoto O., Takahashi T., Shibasaki F., et al. Isoproterenol activates extracellular signal–regulated protein kinases in cardiomyocytes through calcineurin. Circulation. 2001;104:102–108. doi: 10.1161/hc2601.090987. [DOI] [PubMed] [Google Scholar]
- 14.Halkein J., Tabruyn S.P., Ricke-Hoch M., Haghikia A., Nguyen N.Q.N., Scherr M., Castermans K., Malvaux L., Lambert V., Thiry M., et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Invest. 2013;123:2143–2154. doi: 10.1172/JCI64365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lötvall J., Hill A.F., Hochberg F., Buzás E.I., Di Vizio D., Gardiner C., Gho Y.S., Kurochkin I.V., Mathivanan S., Quesenberry P., et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colombo M., Moita C., van Niel G., Kowal J., Vigneron J., Benaroch P., Manel N., Moita L.F., Théry C., Raposo G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013;126:5553–5565. doi: 10.1242/jcs.128868. [DOI] [PubMed] [Google Scholar]
- 17.Marchesini N., Luberto C., Hannun Y.A. Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J. Biol. Chem. 2003;278:13775–13783. doi: 10.1074/jbc.M212262200. [DOI] [PubMed] [Google Scholar]
- 18.Essandoh K., Yang L., Wang X., Huang W., Qin D., Hao J., Wang Y., Zingarelli B., Peng T., Fan G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta. 2015;1852:2362–2371. doi: 10.1016/j.bbadis.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song X., Kusakari Y., Xiao C.Y., Kinsella S.D., Rosenberg M.A., Scherrer-Crosbie M., Hara K., Rosenzweig A., Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am. J. Physiol. Cell Physiol. 2010;299:C1256–C1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueda K., Toko H., Komuro I. Endothelial cell–derived angiocrines elicit physiological cardiomyocyte hypertrophy. Circulation. 2019;139:2585–2587. doi: 10.1161/CIRCULATIONAHA.119.040632. [DOI] [PubMed] [Google Scholar]
- 21.Talman V., Kivelä R. Cardiomyocyte-endothelial cell interactions in cardiac remodeling and regeneration. Front. Cardiovasc. Med. 2018;26:101. doi: 10.3389/fcvm.2018.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiedler J., Jazbutyte V., Kirchmaier B.C., Gupta S.K., Lorenzen J., Hartmann D., Galuppo P., Kneitz S., Pena J.T., Sohn-Lee C., et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 23.Duygu B., Juni R., Ottaviani L., Bitsch N., Wit J.B.M., De Windt L.J., da Costa Martins P.A. Comparison of different chemically modified inhibitors of miR-199b in vivo. Biochem. Pharmacol. 2019;159:106–115. doi: 10.1016/j.bcp.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Ackers-Johnson M., Li P.Y., Holmes A.P., O'Brien S.M., Pavlovic D., Foo R.S. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 2016;119:909–920. doi: 10.1161/CIRCRESAHA.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou P., Pu W.T. Recounting cardiac cellular composition. Circ. Res. 2016;118:368–370. doi: 10.1161/CIRCRESAHA.116.308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maillet M., van Berlo J.H., Molkentin J.D. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sano M., Minamino T., Toko H., Miyauchi H., Orimo M., Qin Y., Akazawa H., Tateno K., Kayama Y., Harada M., et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 28.Izumiya Y., Shiojima I., Sawyer D.B., Colucci W.S., Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim S.L., Lam C.S., Segers V.F., Brutsaert D.L., De Keulenaer G.W. Cardiac endothelium-myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur. Heart J. 2015;36:2050–2060. doi: 10.1093/eurheartj/ehv132. [DOI] [PubMed] [Google Scholar]
- 30.Schulte C., Zeller T. microRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc. Diagn. Ther. 2015;5:17–36. doi: 10.3978/j.issn.2223-3652.2014.12.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuwabara Y., Ono K., Horie T., Nishi H., Nagao K., Kinoshita M., Watanabe S., Baba O., Kojima Y., Shizuta S., et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011;4:446–454. doi: 10.1161/CIRCGENETICS.110.958975. [DOI] [PubMed] [Google Scholar]
- 32.Kogure T., Lin W.L., Yan I.K., Braconi C., Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237–1248. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Q., Yang J., Zheng J., Hsueh C., Guo Y., Zhou L. Characterization of selective exosomal microRNA expression profile derived from laryngeal squamous cell carcinoma detected by next generation sequencing. Oncol. Rep. 2018;40:2584–2594. doi: 10.3892/or.2018.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.L., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 35.Mittelbrunn M., Gutiérrez-Vásquez C., Villarroya-Beltri C., González S., Sánchez-Cabo F., González M.A., Bernad A., Sánchez-Madrid F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011;2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bang C., Batkai S., Dangwal S., Gupta S.K., Foinquinos A., Holzmann A., Just A., Remke J., Zimmer K., Zeug A. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest. 2014;124:2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okamura K., Balla S., Martin R., Liu N., Lai E.C. Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nat. Struct. Mol. Biol. 2008;15:581–590. doi: 10.1038/nsmb.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J.S., Phillips M.D., Betel D., Mu P., Ventura A., Siepel A.C., Chen K.C., Lai E.C. Widespread regulatory activity of vertebrate microRNA∗ species. RNA. 2011;17:312–326. doi: 10.1261/rna.2537911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiba M., Kimura M., Asari S. Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma HepG2 and lung cancer A549 cell lines. Oncol. Rep. 2012;28:1551–1558. doi: 10.3892/or.2012.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Z., Zhang S., Guo C., Li J., Sang W. Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. Int. J. Mol. Med. 2017;39:1589–1596. doi: 10.3892/ijmm.2017.2959. [DOI] [PubMed] [Google Scholar]
- 42.Reddy M.A., Natarajan R. Targeting miR-200c to ameliorate diabetes-induced endothelial dysfunction. Diabetes. 2016;65:1152–1154. doi: 10.2337/dbi16-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hessvik N.P., Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018;75:193–208. doi: 10.1007/s00018-017-2595-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villarroya-Beltri C., Gutiérrez-Vázquez C., Sánchez-Cabo F., Pérez-Hernández D., Vázquez J., Martin-Cofreces N., Martinez-Herrera D.J., Pascual-Montano A., Mittelbrunn M., Sánchez-Madrid F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koppers-Lalic D., Hackenberg M., Bijnsdorp I., van Eijndhoven M.A.J., Sadek P., Sie D., Zini N., Middeldorp J.M., Ylstra B., de Menezes R.X., et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014;8:1649–1658. doi: 10.1016/j.celrep.2014.08.027. [DOI] [PubMed] [Google Scholar]
- 46.Janas T., Janas M.M., Sapon K., Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015;589:1391–1398. doi: 10.1016/j.febslet.2015.04.036. [DOI] [PubMed] [Google Scholar]
- 47.Kaul S., Jayaweera A.R. Myocardial capillaries and coronary flow reserve. J. Am. Coll. Cardiol. 2008;52:1399–1401. doi: 10.1016/j.jacc.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 48.Juni R.P., Abreu R.C., da Costa Martins P.A. Regulation of microvascularization in heart failure - an endothelial cell, non-coding RNAs and exosome liaison. Noncoding RNA Res. 2017;2:45–55. doi: 10.1016/j.ncrna.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keezer S.M., Ivie S.E., Krutzsch H.C., Tandle A., Libutti S.K., Roberts D.D. Angiogenesis inhibitors target the endothelial cell cytoskeleton through altered regulation of heat shock protein 27 and cofilin. Cancer Res. 2003;63:6405–6412. [PubMed] [Google Scholar]
- 50.Apostolakis S., Vogiatzi K., Amanatidou V., Spandidos D.A. Interleukin 8 and cardiovascular disease. Cardiovasc. Res. 2009;84:353–360. doi: 10.1093/cvr/cvp241. [DOI] [PubMed] [Google Scholar]
- 51.Li A., Dubey S., Varney M.L., Dave B.J., Singh R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 52.Siddesha J.S., Valente A.J., Sakamuri S.S.V.P., Yoshida T., Gardner J.D., Somanna N., Takahashi C., Noda M., Chandrasekar B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell Cardiol. 2013;65:9–18. doi: 10.1016/j.yjmcc.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang Z., Fujiwara K., Minamide R., Hasegawa K., Yoshikawa K. Necdin controls proliferation and apoptosis of embryonic neural stem cells in an oxygen tension-dependent manner. J. Neurosci. 2013;33:10362–10373. doi: 10.1523/JNEUROSCI.5682-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Faveri L.E., Hurst C.D., Platt F.M., Taylor C.F., Roulson J.-A., Sanchez-Carbayo M., Knowles M.A., Chapman E.J. Putative tumour suppressor gene necdin is hypermethylated and mutated in human cancer. Br. J. Cancer. 2013;108:1368–1377. doi: 10.1038/bjc.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J.Y., An Y.M., Choi W.H., Kim J.M., Cho S., Yoo B.R., Kang J.W., Lee Y.S., Lee Y.J., Cho J. Pro-apoptotic Noxa is involved in ablative focal irradiation-induced lung injury. J. Cell Mol. Med. 2017;21:711–719. doi: 10.1111/jcmm.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wensveen F.M., Alves N.L., Derks I.A., Reedquist K.A., Eldering E. Apoptosis induced by overall metabolic stress converges on the Bcl-2 family proteins Noxa and Mcl-1. Apoptosis. 2011;16:708–721. doi: 10.1007/s10495-011-0599-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Potente M., Gerhardt H., Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 58.Vogel G.F., Klee K.M.C., Janecke A.R., Müller T., Hess M.W., Huber L.A. Cargo-selective apical exocytosis in epithelial cells is conducted by Myo5B, Slp4a, Vamp7, and Syntaxin 3. J. Cell Biol. 2015;211:587–604. doi: 10.1083/jcb.201506112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuster A., Nola S., Dingli F., Vacca B., Gauchy C., Beaujouan J.C., Nunez M., Moncion T., Loew D., Formstecher E., et al. The Q-soluble N-Ethylmaleimide-sensitive factor Attachment protein receptor (Q-SNARE) SNAP-47 regulates trafficking of selected vesicle-associated membrane proteins (VAMPs) J. Biol. Chem. 2015;290:28056–28069. doi: 10.1074/jbc.M115.666362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piccoli G., Onofri F., Cirnaru M.D., Kaiser C.J.O., Jagtap P., Kastenmüller A., Pischedda F., Marte A., von Zweydorf F., Vogt A., et al. Leucine-rich repeat kinase 2 binds to neuronal vesicles through protein interactions mediated by its C-terminal WD40 domain. Mol. Cell Biol. 2014;34:2147–2161. doi: 10.1128/MCB.00914-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oka T., Akazawa H., Naito A.T., Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ. Res. 2014;114:565–571. doi: 10.1161/CIRCRESAHA.114.300507. [DOI] [PubMed] [Google Scholar]
- 62.Zwi-Dantsis L., Winter C.W., Kauscher U., Ferrini A., Wang B., Whittaker T.E., Hood S.R., Terracciano C.M., Stevens M.M. Highly purified extracellular vesicles from human cardiomyocytes demonstrate preferential uptake by human endothelial cells. Nanoscale. 2020;12:19844–19854. doi: 10.1039/d0nr04278a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen G., Xu C., Gillette T.G., Huang T., Huang P., Li Q., Li X., Li Q., Ning Y., Tang R., et al. Cardiomyocyte-derived small extracellular vesicles can signal eNOS activation in cardiac microvascular endothelial cells to protect against Ischemia/Reperfusion injury. Theranostics. 2020;10:11754–11774. doi: 10.7150/thno.43163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magenta A., Sileno S., D'Agostino M., Persiani F., Beji S., Paolini A., Camilli D., Platone A., Capogrossi M.C., Furgiuele S. Atherosclerotic plaque instability in carotid arteries: miR-200c as a promising biomarker. Clin. Sci. 2018;132:2423–2436. doi: 10.1042/CS20180684. [DOI] [PubMed] [Google Scholar]
- 65.Revelo X.S., Parthiban P., Chen C., Barrow F., Fredrickson G., Wang H., Yücel D., Herman A., van Berlo J.H. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ. Res. 2021;129:1086–1101. doi: 10.1161/CIRCRESAHA.121.319737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Couto G. Macrophages in cardiac repair: environmental cues and therapeutic strategies. Exp. Mol. Med. 2019;51:1–10. doi: 10.1038/s12276-019-0269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magenta A., Cencioni C., Fasanaro P., Zaccagnini G., Greco S., Sarra-Ferraris G., Antonini A., Martelli F., Capogrossi M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18:1628–1639. doi: 10.1038/cdd.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J., Salvador A.M., Li G., Valkov N., Ziegler O., Yeri A., Yang Xiao C., Meechoovet B., Alsop E., Rodosthenous R.S., et al. Mir-30d regulates cardiac remodeling by intracellular and paracrine signaling. Circ. Res. 2011;128:e1–e23. doi: 10.1161/CIRCRESAHA.120.317244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin Z.Y., Chen G., Zhang Y.Q., He H.C., Liang Y.X., Ye J.H., Liang Y.K., Mo R.J., Lu J.M., Zhuo Y.J., et al. MicroRNA-30d promotes angiogenesis and tumor growth via MYPT1/c-JUN/VEGFA pathway and predicts aggressive outcome in prostate cancer. Mol. Cancer. 2017;16:48. doi: 10.1186/s12943-017-0615-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Ucar A., Gupta S., Fiedler J., Erikci E., Kardasinski M., Batkai S., Dangwal S., Kumarswamy R., Bang C., Holzmann A., et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumarswamy R., Volkmann I., Beermann J., Napp L.C., Jabs O., Bhayadia R., Melk A., Ucar A., Chowdhury K., Lorenzen J.M., et al. Vascular importance of the miR-212/132 cluster. Eur. Heart J. 2014;35:3224–3231. doi: 10.1093/eurheartj/ehu344. [DOI] [PubMed] [Google Scholar]
- 72.De Windt L.J., Lim H.W., Haq S., Force T., Molkentin J.D. Calcineurin promotes protein kinase C and c-Jun NH2-terminal kinase activation in the heart. Cross-talk between cardiac hypertrophic signaling pathways. J. Biol. Chem. 2000;275:13571–13579. doi: 10.1074/jbc.275.18.13571. [DOI] [PubMed] [Google Scholar]
- 73.Théry C., Amigorena S., Raposo G., Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006;3:3.22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 74.Bourajjaj M., Armand A.S., da Costa Martins P.A., Weijts B., van der Nagel R., Heeneman S., Wehrens X.H., De Windt L.J. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J. Biol. Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.