

Abstract
Clinical applications of hematopoietic stem cell (HSC) gene editing are limited due to their complex and expensive logistics. HSC editing is commonly performed ex vivo using electroporation and requires good manufacturing practice (GMP) facilities, similar to bone marrow transplant centers. In vivo gene editing could overcome this limitation; however, electroporation is unsuitable for systemic in vivo applications to HSCs. Here we evaluated polymer-based nanoparticles (NPs), which could also be used for in vivo administration, for the delivery of mRNA and nucleases to human granulocyte colony-stimulating factor (GCSF)-mobilized CD34+ cells. NP-mediated ex vivo delivery showed no toxicity, and the efficiency was directly correlated with the charge of the NPs. In a side-by-side comparison with electroporation, NP-mediated gene editing allowed for a 3-fold reduction in the amount of reagents, with similar efficiency. Furthermore, we observed enhanced engraftment potential of human HSCs in the NSG mouse xenograft model using NPs. Finally, mRNA- and nuclease-loaded NPs were successfully lyophilized for storage, maintaining their transfection potential after rehydration. In conclusion, we show that polymer-based NP delivery of mRNA and nucleases has the potential to overcome current limitations of HSC gene editing. The predictable transfection efficiency, low toxicity, and ability to lyophilize NPs will greatly enhance the portability and provide a highly promising platform for HSC gene therapy.
Keywords: polymeric nanoparticles, hematopoietic stem cells, gene editing nucleases, gene therapy, lyophilization, quality control standards
Graphical abstract
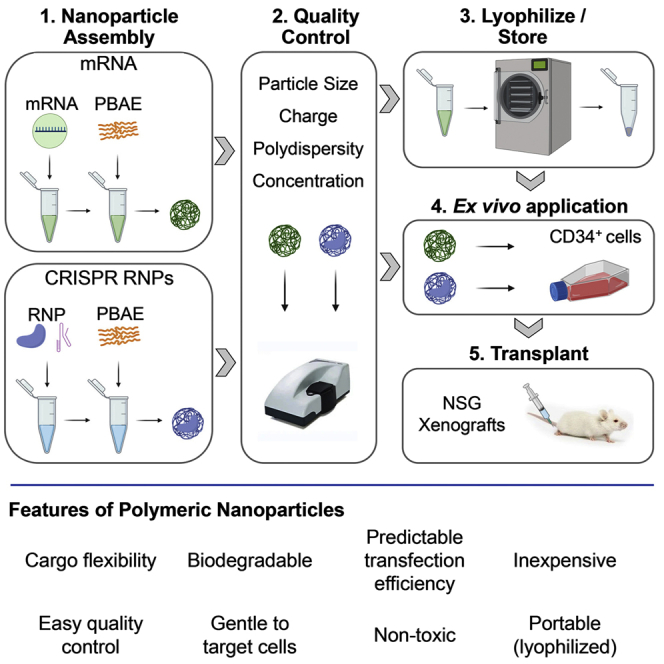
We show that polymeric nanoparticles (NPs) provide a gentle, efficient, and predictable method of mRNA and nuclease delivery for hematopoietic stem cell (HSC) gene therapy. NPs are inexpensive, can be used for targeted ex vivo and in vivo delivery, and can be lyophilized for enhanced portability and accessibility for HSC gene therapy.
Introduction
The current gold standard treatment for many hematological diseases and disorders is an allogeneic hematopoietic stem cell transplant (HSCT). While this approach can lead to a therapeutic benefit, most patients will not have a related histocompatibility leukocyte antigen (HLA)-matched donor, and transplants from alternative donors with inferior HLA matching can lead to significant graft-versus-host disease (GVHD) as well as infectious disease complications. Autologous transplantation of gene-corrected patient cells has the potential to overcome some of these hurdles.1,2 Hematopoietic stem cell (HSC) gene therapy trials using CRISPR-Cas9 nucleases to treat β-hemoglobinopathies3,4 have shown promising results, paving the way toward a more widespread application of gene editing for a multitude of diseases and disorders.
Despite these encouraging breakthroughs, therapeutic HSC gene editing is currently limited to only a few countries due to the logistical requirements. Clinical gene editing approaches rely on the modification of CD34+ hematopoietic stem and progenitor cells (HSPCs) outside the patient's body (ex vivo) and in their current routine application require highly specialized medical facilities equipped with specialized good manufacturing practice (GMP)-grade rooms. Administration of HSPC gene therapy agents directly into the patient (in vivo) using targeted agents could mitigate the myeloablation-related morbidity of current ex vivo applications as well as solve logistical issues, avoid the need for highly specialized GMP facilities, and enhance accessibility in countries with less-well-established infrastructure. However, the current delivery of gene editing reagents into HSPCs is electroporation, a technology that is not applicable for in vivo HSC gene therapy, and alternative modalities are needed for the delivery of gene editing reagents.
We have previously shown that polymeric nanoparticles (NPs) can be used to deliver mRNA into T cells and human umbilical cord blood-derived HSPCs.5 Here, we optimized our previously reported NP formulation to deliver mRNA into human granulocyte colony-stimulating factor (GCSF)-mobilized HSPCs, the most frequently used cell source for HSC gene therapy and editing, and specifically focused on the NP-mediated delivery of CRISPR-Cas9 gene editing reagents into these cells. Most importantly for a routine clinical application of this technology, we defined cargo-specific quality control parameters for NPs and established protocols to lyophilize and store formulated NPs for enhanced portability and future in vivo applications.
Results
Particle charge is the most important parameter for successful uptake of mRNA-loaded NPs
Poly-β-amino esters (PBAEs) have been successfully used to transfect a wide variety of cell types,5, 6, 7 but little is known about the optimal PBAE formulation for the transfection of human CD34+ cells. Here, we tested PBAE-based-polymer variations from our previously established protocol5 to minimize toxicity and optimize efficiency for the delivery of mRNA specifically into human GCSF-mobilized CD34+ cells.
In contrast to our previously published ratio of 1.1:1 (diacrylate to amine monomer ratio), two additional molar ratios of 1.05:1 and 1.2:1 were produced (Table S1). NPs were formed through electrostatic self-assembly by mixing cationic PBAE polymers and anionic mRNA at a weight-to-weight ratio of 60:1 in aqueous conditions.7 NPs were co-incubated with human CD34+ cells for 2 h at 37°C, washed, and cultured for 48 h. Transfected cells were analyzed for viability and transfection efficiency using an automated cell counter and flow cytometry, respectively.
The viability of cells was greater than 90% for all PBAE batches without any difference compared with untreated cells (Figure 1A). A wide range of GFP expression (from 1.6% to 92%) was observed in human CD34+ cells (Figures 1B and 1C). On average, the highest transfection efficiency was observed with PBAEs at a molar ratio of 1.2:1 (B7–B9), with polymer B7 demonstrating the overall greatest efficiency (Figure 1B).
Figure 1.

Characterization and testing of GFP mRNA-loaded NPs on human CD34+ cells
(A) Cell viability and (B) GFP expression after NP-mediated GFP mRNA delivery. Molar ratios of diacrylate to amine to synthesize polymer batches are indicated at the bottom. Untreated cells were used as a control. (C) GFP expression in NP-treated human CD34+ cells (scale bar, 20 μm). (D) Correlation between the transfection efficiency and average size of NP-mRNA. (E) Polydispersity index (PDI) of NP-mRNA. (F) Representative TEM image of NP-mRNA using polymer batch B7 (scale bar, 2 μm; insert, 50 nm). (G) Correlation between the transfection efficiency and the zeta potential of NP-mRNA. Size, PDI, and zeta potential were measured with a Zetasizer. Statistics: (A and B) mean ± SD.
We noted inconsistent transfection efficiency comparing identical formulations, suggesting batch-to-batch variability.8 We therefore aimed to identify chemical and physicochemical properties that correlate with the transfection efficiency, which can be used as quality control parameters for PBAE-mediated delivery of mRNA into human CD34+ cells.
First, the chemical structure of polymers was analyzed using 1H NMR. Formation and purity of PBAEs were highly similar without any obvious differences (Figure S1). Second, we determined the physicochemical properties of NPs by measuring the size, polydispersity index (PDI), morphology, and zeta potential of the NPs formed using transmission electron microscopy (TEM) and a Zetasizer. The average size of NPs ranged from 137 to 347 nm (Figure 1D) and showed only a weak correlation (R2 = 0.2222) with the transfection efficiency. The effective NP size achieving >50% transfection efficiency ranged from 178 to 347 nm. Low PDIs (0.10–0.26) were observed across all batches, confirming the homogeneity of the NPs (Figure 1E). Monodispersity and size of the NPs were confirmed by TEM for the most effective polymer batch, B7. NPs had a spherical shape with an average size of 203 nm as well as a uniform size distribution (Figure 1F). Last, we determined the surface charge of the NPs. NPs were positively charged (1.5–32 mV), and a strong correlation (R2 = 0.9094) of NP surface charge with the transfection efficiency of human CD34+ cells was found (Figure 1G).
These data suggest that the surface charge of NPs is the main factor mediating effective uptake of mRNA-loaded NPs without compromising the viability of human CD34+ cells.
Effective and predictable NP-mediated delivery of Cas9 RNPs
PBAE-based NPs are an efficient carrier system for delivering CRISPR-Cas9 complexes into cell lines.9,10 Here, we evaluated our PBAE batches for the delivery of Cas9 ribonucleoprotein (RNP) complexes into human CD34+ cells. We disrupted the CD33 gene using two guide RNAs (gRNAs) to excise exon 2 encoding the V-set extracellular immunoglobulin-like domain,11,12 a strategy to protect HSCs from anti-CD33 treatments in acute myeloid leukemia (AML) patients. NP-RNP formulations were tested in nine independent experiments on human CD34+ cells to evaluate the reproducibility of NP physicochemical properties and their cytotoxicity as well as gene editing efficiency.
High cell viability (>86%) was noted across all NPs loaded with RNPs (NP-RNPs) we tested. However, great variability was observed in the downregulation of CD33 surface expression, ranging from 13% to 85% (Figure 2A). Stable genomic excision of CD33 exon 2 was confirmed by PCR compared with untreated cells (Figure S2A). NP-RNPs were smaller than NPs loaded with mRNA (NP-mRNA), ranging from 98 to 214 nm. Like NP-mRNAs, NP-RNPs showed no correlation (R2 = 0.06045) between the size and CD33 loss (Figure 2B). A wide range of PDI (0.12–0.79) was observed in NP-RNP formulations, and high PDIs were associated with little CD33 downregulation (Figure 2C). However, even with low PDI, some formulations did show poor CD33 downregulation, indicating that the PDI is not the main factor for successful editing. NP-RNPs showed a positive surface charge (12–30 mV) that was strongly correlated (R2 = 0.8652) with CD33 downregulation, confirming also for NP-RNPs that the charge is the most important parameter for successful cargo delivery into human CD34+ cells (Figure 2D). The effective size range reaching greater than 50% knockout of CD33 was seen for 133 to 200 nm sized NP-RNPs with 20–30 mV surface charge (Figures 2B and 2D).
Figure 2.

Characterization and testing of RNP-loaded NPs on human CD34+ cells
(A) Cell viability and knockout of CD33 exon 2 on the cell surface (CD33ΔE2) determined 72 h after NP-mediated editing of human CD34+ cells. The editing efficiency was calculated relative to the background noise in untreated cells. (B) Correlation between the CD33 editing efficiency and size of NP-RNPs. (C) Polydispersity index (PDI) of NP-RNPs. (D) Correlation between the CD33 editing efficiency and zeta potential of NP-RNPs. Statistics: (C) mean ± SD.
In contrast to NP-mRNAs, NP-RNPs have multiple components that need to be assembled before NPs can be formed. Furthermore, gRNAs have been shown to be highly susceptible to degradation. Here, we tested whether the assessment of physicochemical properties can be used to determine the integrity of components and successful assembly of RNPs, as well as the formation of NP-RNPs. RNPs are small and negatively charged due to the gRNA incorporated into the positively charged Cas9 (Figure S2B). Positively charged Cas9 proteins, as well as single-guide RNAs (sgRNAs) alone with polymer, form 40 to 60 nm sized particles, have a slight positive charge, and are distinctly different from NP-RNPs in their physicochemical properties (Figure S2B). Only intact RNP complexes can generate particles that have the polymer in the correct size and charge range shown to be effective and predictive for gene editing.
We show that our modified PBAE formulation can efficiently encapsulate and deliver RNPs into human CD34+ cells without signs of cytotoxicity. Most importantly, the editing efficiency of NP-RNPs correlates with the surface charge, providing an important quality control to predict the gene editing efficiency in human CD34+ cells. In addition, the combined assessment of size and charge can ensure the integrity of gene editing reagents and successful formation of NPs containing RNP complexes.
NP-RNP-edited HSPCs retain long-term multilineage engraftment and CD33 downregulation in vivo
To confirm that NP-mediated gene editing has no adverse effects on the engraftment and multilineage differentiation potential, ex vivo NP-edited CD34+ cells were transplanted into sublethally irradiated adult NSG mice. Human chimerism and, specifically, the knockdown of CD33 surface expression were tracked longitudinally in the peripheral blood (PB). In addition, multilineage differentiation in tissues, as well as engraftment of gene-edited human CD34+ cells in the bone marrow (BM) stem cell compartment, was determined at necropsy (Figure 3A).
Figure 3.

Long-term engraftment of NP-RNP-edited human CD34+ cells
(A–C) Schematic of the experimental design. Longitudinal flow cytometric assessment of (B) human chimerism and (C) CD14+ monocytes in the PB. (D) PCR validation of CD33 editing in human cells in the PB at weeks (W) 8 and 10 post-transplant. (E) Quantification of PCR bands in (D). (F) Longitudinal tracking of human CD14+ monocytes in the PB lacking CD33 exon 2 on the cell surface (CD33ΔE2) via flow cytometry. (G) Frequency of human chimerism in the bone marrow (BM), spleen, and thymus. (H) Frequency of engrafted human CD34+ cells (left y axis) and the HSC-enriched CD34+CD90+ subset (right y axis) in the BM. (I) Genomic analysis of the CD33 genotype (+/+, wild type; +/−, heterozygous knockout; −/−, homozygous knockout) of individual CD34+- and CD34+CD90+-derived colonies (representative gel pictures shown in Figure S3I). Statistics: (G and H) mean ± SEM, Wilcoxon signed-rank test.
Polymer formulation B7 was used to edit human CD34+ cells ex vivo. NP-RNPs were 200 nm in diameter with a PDI of 0.2, and the surface charge was highly positive (30 mV). High viability (85%) of human CD34+ cells was observed after NP-mediated editing. Approximately 85% downregulation of CD33 surface expression was seen ex vivo compared with untreated control cells. For the mouse xenograft studies, 2.0 × 105 NP-edited and 5.0 × 105 unedited cells were transplanted per mouse and the animals were followed longitudinally.
Overall human engraftment in the PB was comparable (Figure 3B), and multilineage differentiation was seen in both groups (Figures 3C and S3A). Genomic analysis confirmed excision of the CD33 exon 2 in bulk PB cells with on average 60%–70% editing (Figures 3D and 3E). Consistent downregulation of CD33 surface expression greater than 90% was seen by flow cytometry on PB monocytes across all time points (Figure 3F).
Analysis of the BM, spleen, and thymus confirmed human engraftment in mice receiving untreated and NP-RNP-treated cells (Figure 3G) with multilineage contribution in BM, spleen, and thymus in both experimental groups (Figures S3B–S3D). Immunophenotypic characterization of the BM stem cell compartment demonstrated successful engraftment of CD34+ HSPCs and the HSC-enriched CD34+CD90+ subset (Figure 3H). Phenotypical human CD34+ and CD34+CD90+ cells were introduced into colony-forming cell (CFC) assays by fluorescence-activated cell sorting (FACS) to confirm their multilineage differentiation potential. Both phenotypes were capable of realizing erythroid, myeloid, and erythromyeloid colonies (Figures S3E–S3H).
Genomic analysis of individual CD34+- and CD34+CD90+-derived colonies confirmed CD33 editing exclusively in the NP-RNP-treated group (Figure S3I). Approximately 56% of CD34+-derived colonies and 26% of CD34+CD90+-derived colonies demonstrated editing of the CD33 exon 2 (Figure 3I). Interestingly, the majority of gene-modified CD34+-derived colonies demonstrated monoallelic editing (62%), whereas more biallelic knockout of CD33 exon 2 (61%) was observed in colonies derived from the HSC-enriched CD34+CD90+ subset.
In summary, NP-RNP-edited CD34+ cells demonstrate long-term multilineage engraftment in the mouse xenograft model. Most importantly, gene-edited CD34+ as well as primitive CD34+CD90+ cells efficiently engrafted in the BM stem cell compartment, persisted long term, and maintained their multilineage differentiation potential.
Enhanced feasibility of NP-RNPs in comparison with electroporation-mediated gene editing
Electroporation (EP) is the state-of-the-art technology to deliver gene-editing reagents into human CD34+ cells for experimental, pre-clinical, and clinical applications. Here, we compared NP-RNPs side by side with EP for the delivery of RNPs into human GCSF-mobilized CD34+ cells. Similar to the previous experiment, we used our polymer formulation B7 to deliver RNPs for the knockout CD33 exon 2. In parallel, CD34+ cells were edited with our established EP protocol.11 Twenty-four hours after editing, NP-edited, EP-edited, and unedited CD34+ cells were evaluated for viability; downregulation of CD33 was assessed by flow cytometry; and edited cells were transplanted into sublethally irradiated NSG mice to evaluate the long-term multilineage engraftment potential.
NP-RNPs were 200 nm in diameter with a PDI of 0.2, and the surface charge was highly positive (30 mV). Cell viability was slightly lower after NP (85%) and EP (72%) treatment in comparison with untreated CD34+ cells (93%) (Figure 4A). CD33 expression was successfully downregulated in NP-edited (88%) as well as EP-edited (76%) CD34+ cells (Figure 4B). Of note, downregulation of CD33 with NPs required 3-fold smaller amounts of CD33-RNP reagents compared with EP.
Figure 4.

Long-term peripheral blood chimerism in mice transplanted with NP- versus EP-edited human CD34+ cells
(A) Cell viability and (B) reduction of CD33 cell-surface expression determined 72 h after NP- and EP-mediated editing of human CD34+ cells. Untreated cells were used as a control. (C and D) Longitudinal flow cytometric assessment of (C) human chimerism and (D) CD14+ monocytes in the PB in mice transplanted with untreated, EP-RNP-, and NP-RNP-edited human CD34+ cells. (E) Human multilineage engraftment in the PB at week 20 post-transplant. (F) PCR validation of CD33 editing in human cells in the PB. (G) Longitudinal monitoring of human CD14+ monocytes in the PB lacking CD33 exon 2 on the cell surface (CD33ΔE2) via flow cytometry. (H) Quantification of PCR bands in (F). Statistics: (A and B) mean ± SD, one-way ANOVA.
For the mouse xenograft studies, 4.0 × 105 NP-edited, 3.2 × 105 EP-edited, and 5.0 × 105 unedited cells were transplanted per mouse. Longitudinal flow cytometric assessment of the PB showed human multilineage engraftment in all three groups (Figures 4C, 4D, and 4E). Genomic analysis and flow cytometry confirmed the successful deletion of CD33 exon 2 (Figure 4F) and downregulation of CD33 cell-surface expression on monocytes (Figure 4G) in mice engrafted with edited CD34+ cells, respectively. Semiquantitative assessment of the genomic analysis (NP-RNP, 30%–55%; EP, 25%–45%) was slightly lower than the observed downregulation of CD33 by flow cytometry on monocytes (Figure 4H).
Overall, NP-mediated delivery of gene-editing reagents is similar in efficiency to the current gold standard EP approach on human GCSF-mobilized CD34+ cells. However, 3-fold smaller amounts of gene-editing reagents are required in comparison with EP to reach similar efficiencies of CD33 knockout and downregulation.
Improved reconstitution of the BM stem cell compartment with NP-RNP-edited human CD34+ cells
We next evaluated the engraftment and multilineage differentiation potential of NP- and EP-edited human CD34+ cells in tissues. Mice were euthanized 20 weeks post-transplant, and multilineage engraftment was determined flow cytometrically in the BM, spleen, and thymus. In addition, recovery of the BM stem cell compartment was determined by flow cytometry, and engrafted human CD34+ cells as well as the HSC-enriched CD34+CD90+ subset were isolated and introduced into CFC assays.
No significant differences in the frequency of human chimerism and multilineage engraftment were seen in the BM of mice transplanted with NP-edited, EP-edited, or unedited CD34+ cells (Figures 5A and S4A). However, human chimerism in mice transplanted with EP-edited CD34+ cells was three to five times lower than in the control group and in animals receiving NP-edited CD34+ cells. Similar trends were seen in the spleen and thymus (Figures 5A, S4B, and S4C).
Figure 5.

Tissue engraftment and reconstitution of the BM stem cell compartment in mice transplanted with NP- versus EP-edited human CD34+ cells
(A) Frequency of human chimerism in the BM, spleen, and thymus. (B) Frequency of engrafted human CD34+ cells (left y axis) and the HSC-enriched CD34+CD90+ subset (right y axis) engrafted in the BM. (C and D) Total CFC potential (left graphs) and quantification of erythroid, myeloid, and erythromyeloid colonies (right graphs) of human (C) CD34+ and (D) CD34+CD90+ cells. CFU, colony-forming unit; CFU-M, CFU monocyte/macrophage; CFU-G, CFU granulocyte; CFU-GM, CFU granulocyte/monocyte/macrophage; BFU-E, burst-forming unit erythrocyte; CFU-Mix, CFU containing a mix of erythroid and myeloid cells. (E) Quantification of the PCR-based assessment of CD33 exon 2 knockout (CD33ΔE2) in PB and BM at week 20 shown in Figure S4D. (F) Genomic analysis of the CD33 genotype (+/+, wild type; +/−, heterozygous knockout; −/−, homozygous knockout) of individual CD34+- and CD34+CD90+-derived colonies (representative gel pictures shown in Figures S4E and S4F). Statistics: (A and B) mean ± SEM, one-way ANOVA; (C and D) mean ± SD, one-way ANOVA.
Nearly identical frequencies of CD34+ and CD34+CD90+ cells were found in mice receiving untreated or NP-edited cells (Figure 5B). In comparison, mice transplanted with EP-edited CD34+ cells contained on average two to three times less human CD34+ as well as CD34+CD90+ cells in the BM, suggesting impaired engraftment potential of EP-edited CD34+ cells. Qualitatively, engrafted human CD34+ cells from all three groups were highly similar in their overall colony-forming potential, with almost identical proportions of erythroid, myeloid, and erythromyeloid differentiation potentials (Figure 5C). The greatest overall CFU potential was found for NP-edited CD34+CD90+ cells in comparison with untreated and EP-edited cells without any obvious change in the composition of colony types (Figure 5D). Of note, phenotypic CD34+CD90+ cells in two of four EP-edited mice did not form any colonies.
Finally, we determined the knockout of CD33 exon 2 in PB and BM white blood cells (WBCs) at necropsy (week 20) as well as from individual CD34+- and CD34+CD90+-derived colonies. Similar to the cell-surface downregulation of CD33 on PB monocytes (Figure 4G), 21%/49% (EP/NP) and 63%/47% (EP/NP) knockout of CD33 exon 2 was seen in PB and BM WBCs, respectively (Figures 5E and S4D). Up to 68%/72% of CD34+ colonies showed excision of CD33 exon 2 in the NP- as well as the EP-edited group, respectively (Figures 5F and S4E). Enhanced biallelic editing was seen within NP-edited CD34-derived colonies (77%) in comparison with EP-edited colonies (30%). Less overall editing (NP, 36%; EP, 49%) was seen in CD34+CD90+-derived colonies, with similar trends in the biallelic editing between the NP- and the EP-edited groups (Figures 5F and S4F).
These findings demonstrate that NP-edited CD34+ cells show potential for engraftment and BM reconstitution, like unedited cells. Most importantly, enhanced homing of primitive CD34+CD90+ cells into the BM was found for cells edited with NPs in comparison with EP.
Lyophilized NPs retain gene-editing potential
To allow for an off-the-shelf availability of gene-editing agents, we evaluated the lyophilization of mRNA- as well as RNP-loaded NPs to enable an upfront production, quality control, and storage of NPs for experimental, pre-clinical, and clinical applications. NPs were cryoprotected in sucrose,13 stored at −20°C for 4–6 weeks, and rehydrated with water. Rehydrated particles were analyzed for size, charge, and PDI and applied to human CD34+ cells for functional testing.
Rehydrated NP-mRNAs showed an insignificant reduction in size and charge compared with freshly produced NP-mRNAs (Figure 6A). Similarly, a slight reduction of transfection efficiency was observed for lyophilized mRNA-loaded RNPs (Figure 6B). To determine whether the observed loss in efficiency is connected to the reduced charge, we overlaid the values with our previous correlation (Figure 6C). Charges measured for lyophilized and rehydrated NPs matched our previous correlation, confirming that the same quality control (QC) and predictive parameters can be applied to lyophilized NP-mRNAs after rehydration.
Figure 6.

Physicochemical properties and functional testing of lyophilized GFP mRNA- and CD33 RNP-loaded NPs
(A) Size and zeta potential of lyophilized NP-mRNAs. (B) Cell viability and transfection efficiency of human CD34+ cells treated with either fresh or lyophilized and rehydrated NP-mRNAs (n = 2). (C) Overlay of the previously determined correlation of NP-mRNA charge and transfection efficiency shown in Figure 1F (black dots and line) with the charge of freshly prepared (F) and lyophilized (L) NP-mRNAs. (D) Size and zeta potential of lyophilized NP-RNPs. (E) Cell viability and CD33 exon 2 knockout (CD33ΔE2) efficiency of human CD34+ cells treated with either fresh or lyophilized and rehydrated NP-RNPs (n = 2). (F) Overlay of the previously determined correlation of NP-RNP charge and transfection efficiency shown in Figure 2D (black dots and line) with the charge of freshly prepared (F) and lyophilized (L) NP-RNPs. Statistics: (A, B, D, and E) mean ± SD; (A and D) Wilcoxon signed-rank test.
Finally, we tested lyophilized NP-RNPs on human CD34+ cells. No change in size and minimal reduction in charge was observed in lyophilized and rehydrated NPs in comparison with freshly generated NPs (Figure 6D). Again, a slight reduction in activity was observed for lyophilized RNP-loaded NPs when applied to CD34+ cells (Figure 6E). Similar to mRNA-loaded NPs, the observed change in charge and efficiency for rehydrated NP-RNPs fit our previously determined correlation for freshly prepared NP-RNPs (Figure 6F).
These data demonstrate that mRNA- and RNP-loaded NPs can be lyophilized, stored, and rehydrated with minimal loss of activity. Furthermore, previously determined QC parameters with freshly prepared NPs for the prediction of activity on CD34+ cells do also apply to lyophilized and rehydrated NPs.
Discussion
Here, we show that our PBAE-based NP formulation can efficiently deliver mRNA as well as nucleases into GCSF-mobilized human CD34+ cells. Cargo-dependent QC parameters for NPs enabled us to determine the integrity of the gene-editing reagents (gRNA, Cas9) and reliably predict the efficiency of CD34+ cell transfection. NP-mediated delivery required 3-fold smaller amounts of reagents in comparison with EP, and enhanced reconstitution potential of NP-edited cells in the BM stem cell compartment of the NSG mice was observed. Finally, mRNA- as well as nuclease-loaded NPs can be lyophilized and stored at −20°C, maintaining their transfection potential after rehydrating.
A great variety of NP formulations have been used for the delivery of mRNA, DNA, proteins, peptides, etc., into various healthy or malignant cell types both ex vivo and in vivo.6,14,15 However, only very a few NP-based nonviral delivery strategies have been reported so far specifically for the delivery of cargo into human CD34+ HSPCs. The most frequently used particle types for CD34+ HSPCs are lipid NPs (LNPs),16, 17, 18 gold NPs (GNPs),19,20 and polymer-based NPs (PNPs).5,21, 22, 23 Successful NP-mediated RNP delivery into human HSPCs has been previously shown by Shahbazi et al. using GNPs19 and Cruz et al. with PNPs.21 Similar to our data but focusing on the knockout of the γ-globin promoter to reactive fetal hemoglobin for the treatment of sickle cell disease (SCD), Cruz et al. reached greater editing efficiencies in human GCSF-mobilized CD34+ cells with PNPs than with an EP-based delivery in the ex vivo setting.21 Here, we demonstrate in the NSG xenograft model that PNP-edited human CD34+ cells also maintain their multilineage long-term engraftment potential and show enhanced potential to reconstitute the BM stem cell compartment, in contrast to EP-edited cells. Similarly, enhanced efficiency in Cas9 RNP delivery has been shown for PNPs in comparison with LNPs when tested on human cell lines ex vivo or in mice in vivo.24,25 Collective data from these publications illustrate that PNP-mediated delivery specifically of RNPs has great potential for ex vivo as well as in vivo gene-editing protocols.
A major bottleneck of HSC gene editing is the fragility of the individual RNP components. Despite chemical modification, gRNAs can be subject to degradation due to environmental RNases, and interruptions of low-temperature (−80°C) cooling chains hold the risk of diminished nuclease activity. Functionality assessment before EP-mediated delivery of these components is commonly not done or technically feasible. Therefore, the efficiency of gene editing after EP-mediated delivery remains oftentimes uncertain until cells can be assessed either by flow cytometry, with next-generation sequencing, or in functional transplantation assays days, weeks, or months later. Lyophilization to stabilize NPs with various cargoes has been shown to be feasible in many applications.21,26,27 Furthermore, polymers were shown to protect degradation-sensitive RNA from RNases.27,28 Here, we show that polymeric NPs loaded with mRNA as well as RNP complexes can be easily lyophilized, stored at −20°C, and rehydrated, maintaining transfection efficiency for human CD34+ cells. Furthermore, we demonstrate that successful formation of RNP complexes incorporated into the PNPs can be easily assessed for measuring the size and charge of particles, providing an instant QC for the gene-editing reagents before use. These QC parameters remained the same for freshly prepared as well as lyophilized and rehydrated PNPs, providing valuable guidelines for the assessment of particles for experimental, pre-clinical, and clinical applications.
The ability to easily assess the batch-to-batch variability of polymers and individual NP formulations without long-lasting experiments, as well as the opportunity to preserve quality-controlled mRNA- or RNP-loaded PNPs will be of great value to increase the standardization, reproducibility, portability, and accessibility of HSC gene therapy. Most importantly, it allows the use of HSC gene therapy in facilities without the availability of equipment to assess the physicochemical properties of NPs or availability of low-temperature storage capabilities. In addition, GMP facilities are very limited in their daily throughput for ex vivo HSC gene therapy due to the risk of contamination and cross-contamination. Even large GMP facilities are not able to handle more than a few or up to a dozen patients simultaneously, making it impossible to treat the millions of patients who could benefit from this promising technology. Consequently, a goal that many groups are working toward is the in vivo delivery of gene-editing reagents to be fully GMP facility independent. However, in vivo administration of gene therapy vectors comes with new challenges. Target specificity, especially when delivering nucleases, will be of greatest importance to avoid unwanted off-target effects in cells other than HSCs to prevent secondary diseases. Modifications in the polymer26 or lipid,29 targeting with antigen-specific antibody fragments,5,30, 31, 32, 33, 34, 35 or exploiting/modifying the natural tropism of the vector36 are promising strategies to enable HSC specificity and provide enhanced safety of in vivo HSC gene therapy.
The need for standardization of formulations and the definition of QC parameters for the use of NPs in clinical applications is governed by the Nanotechnology Characterization Laboratory (NCL) founded by the NIH. In addition to their defined criteria for the characterization of NP formulations, we here provide a direct correlation of QC parameters with the efficiency of delivery and gene editing on human CD34+ cells. The ability to easily predict the outcome based on physicochemical properties will be of great value, especially due to the batch-to-batch variability seen in the reagents that are not possible to capture otherwise. Interestingly, the QC parameters defined here were specific for human GCSF-mobilized CD34+ cells and did not apply to cell lines (data not shown). This cell-type specificity may help to enhance on-target delivery of gene therapy agents using PNPs for in vivo HSC gene therapy.
This proof-of-concept study focuses on the CRISPR-based knockout of CD33. However, sgRNAs can be easily exchanged for alternative HSC gene therapy approaches aiming for the treatment of other hematological diseases and disorders such as HIV/AIDS37,38 and sickle cell disease,39 opening up the availability of this treatment option to many patients. While our PNP-based CRISPR approach relies on DNA double-strand breaks and bears the risk of unwanted genomic alterations,40 treatment approaches relying on a single nucleotide exchange may focus on NP-mediated delivery using base/prime editors.41 Other approaches likely benefiting from NP-mediated delivery systems include the homology-directed repair (HDR)-mediated integration of genomic information into specific loci. While current HDR-mediated approaches rely on a two-step protocol delivering the nuclease with EP and the homology template for integration by viral transduction, both components can be delivered within the same or in two independent NPs simultaneously reducing the toxicity and increasing the efficiency to reach the same cell.
In conclusion, polymeric PBAE-based PNPs are a feasible, chemically defined, and highly portable delivery platform with the potential to overcome currently existing limitations and bottlenecks toward a routine clinical application of HSC gene therapy. Due to their low toxicity, biocompatibility, and engineerable surface, PNPs are a promising technology for the HSC-targeted in vivo delivery of gene-editing materials. PNP formulations are highly versatile, their cargoes interchangeable for disease-specific applications; their quality is easy to control; and they allow predictable gene editing of human HSCs.
Materials and methods
Polymer synthesis
PBAE polymers were synthesized using a method similar to that described by Mangraviti et al. and Stephan et al.5,7 Briefly, the base monomer, 1,4-butanediol diacrylate, was combined with a side-chain monomer, 4-amino-1-butanol, in a 1.05:1, 1.1:1, or 1.2:1 molar ratio. The mixture was heated to 90°C and stirred for 24 h to produce diacrylate-terminated polymers. The polymers were dissolved in 2 mL anhydrous tetrahydrofuran (THF) and added to 5 mmol of 1-(3-aminopropyl)-4-methylpiperazine to form piperazine-capped polymers. The reactions were conducted for 2 h at room temperature while stirring. Polymers were purified to remove unreacted monomers and oligomers via precipitation in diethyl ether. Polymers were dried in a vacuum desiccant for 24 h to remove traces of ether. Finally, PBAEs were dissolved in anhydrous dimethyl sulfoxide (DMSO) at 100 mg/mL and stored at −20°C with desiccant in small aliquots to limit freeze-thaw cycles.
Polymer characterization
To confirm the formation and purity of the produced polymers, NMR spectroscopy via 1H NMR (Bruker 499.956 MHz) was used to characterize the polymer structure.
Nanoparticle preparation
NP-mRNA
PBAE polymer and GFP mRNA (TriLink Biotechnologies, San Diego, CA, USA) were separately dissolved in 25 mM nuclease-free sodium acetate buffer (pH 5.2) and combined at equal volumes, with a 60:1 mass ratio of polymer to mRNA. NPs were allowed to assemble for 5 min at room temperature.
NP-RNP
To prepare NPs encapsulating CRISPR RNPs, two synthetic sgRNAs flanking exon 2 of CD33 (Table S2; Synthego, Redwood City, CA, USA) were mixed together at a 1:1 volume ratio. Cas9 protein (TrueCut Cas9 v.2; Thermo Fisher Scientific, Waltham, MA, USA) and sgRNAs were then mixed at a 5:1 molar ratio at room temperature for 10 min to allow RNP assembly. Next, RNPs were mixed with PBAE polymers at a 1:1 volume ratio to form NPs.
Nanoparticle lyophilization
NPs loaded with GFP mRNA or CD33-RNPs were lyophilized by adding 60 mg/mL D-sucrose as a cryoprotectant before flash-freezing on dry ice. NPs were processed in a FreeZone 2.5 L freeze-dry system (Labconco, Fort Scott, KS, USA). Lyophilized NPs were stored at −20°C until use. For application, lyophilized NPs were reconstituted in sterile water to restore their original concentration.
NP characterization
Zetasizer
The hydrodynamic diameter, PDI, and zeta potential of NPs were measured using a Zetasizer Nano S device (Malvern Analytical, Malvern, UK). Measurements were carried out in triplicate and data were analyzed using the Zetasizer software (version 7.13). For measurements, NPs were diluted 1:50 in 25 mM nuclease-free sodium acetate buffer.
TEM
The size and shape of NPs were determined using TEM. A volume of 25 μL of sample was applied to a glow-discharge-activated 200 mesh carbon/formvar-coated copper grid. After 30 s, a drop of ½ Karnovsky's fixative, a drop of 0.1 M cacodylate buffer, 8 drops of deionized water, and a drop of 1% (w/v) filtered uranyl acetate were added to the grids sequentially. Grids were dried inside a desiccator and imaged by TEM (JEOL JEM 1400, Peabody, MA, USA).
Enrichment and culture of human CD34+ cells
Primary human CD34+ cells were purchased from the Co-operative Center for Excellence in Hematology (CCEH) at the Fred Hutchinson Cancer Research Center. Collections were performed according to the Declaration of Helsinki and were approved by a local ethics committee/institutional review board of the Fred Hutchinson Cancer Research Center. All healthy adult donors were mobilized with GCSF. Human CD34+ cells were enriched as previously described on a CliniMACS Prodigy according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany).42
Human CD34+ cells were cultured in StemSpan (STEMCELL Technologies, Vancouver, BC, Canada) medium supplemented with penicillin-streptomycin (PS) (100 U/mL) (Gibco by Life Technologies, Waltham, MA, USA) and 100 ng/mL of each stem cell factor (PeproTech, Cranbury, NJ, USA), thrombopoietin (TPO; PeproTech), and Fms-related tyrosine kinase 3 ligand (FLT3-L; Miltenyi Biotec). Cells were cultured at 37°C, 85% relative humidity, and 5% CO2.
In vitro transfection of human CD34+ cells with nanoparticles
Human CD34+ cells were thawed and cultured overnight in StemSpan medium with PS and cytokines. Next day, cells were harvested, counted, and resuspended in StemSpan without PS and cytokines at a density of 150,000 cells per 150 μL per well of a 96 well plate (Corning, Corning, NY, USA). Freshly prepared or lyophilized NPs were incubated with human CD34+ cells for 2 h at 37°C. For 150,000 cells per 96-well-plate well, a total volume of 15 μL NPs (60:1 mass ratio of polymer to mRNA) was added. After 2 h, transfected cells were centrifuged and recultured in fresh StemSpan medium with PS and cytokines for 48 to 72 h at 37°C until analysis.
Electroporation of human CD34+ cells
EP of nuclease components was carried out as employed previously.43 Briefly, a volume of 0.69 μL RNPs was formed as described above in an Eppendorf tube. For small-scale ex vivo experiments, 150,000 human CD34+ cells were resuspended in 49.3 μL BTX buffer and added to the same tube, and the mixture was then transferred into a 1 mm EP cuvette and electroporated with 125 V and 5 ms pulse duration using the ECM 380 Square Wave Electroporation System (Harvard Apparatus, Cambridge, MA, USA). For large-scale mouse transplants, cell numbers and volumes were scaled to 500,000 cells per mouse and EP was performed in 2 mm cuvettes with 250 V and 5 ms pulse duration. Electroporated cells were cultured in StemSpan medium containing PS and cytokines at a density of 1 × 106/mL for 24 to 72 h at 37°C until analysis or transplant.
Viability assessment of human CD34+ cells
The cell viability was analyzed using the Countess II FL automated cell counter (Thermo Fisher Scientific). A 10 μL volume of trypan blue stain (0.4%) (Invitrogen, Waltham, MA, USA) was mixed with 10 μL of cell suspension, and 10 μL of the mixture was applied to a disposable cell counting chamber slide and inserted into the device. The percentage of cell viability of each sample was recorded in duplicate and reported as the mean ± SEM. To confirm the cell viability, 5 μL 4′,6-diamidino-2-phenylindole (DAPI; cat. no. 564907, lot no. 9294998; BD Biosciences, Franklin Lakes, NJ, USA) was used during the flow cytometry.
Mouse xenograft transplantation
For the in vivo assessment of engineered human CD34+ cells, adult (8 to 12 weeks old) NSG (non-obese diabetic [NOD].Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice were irradiated at 275 cGy. Four hours later, the mice were intravenously injected with 200 μL of either untreated, NP-RNP, or EP gene-edited human CD34+ cells. Beginning at 8 weeks post-injection, blood samples were collected every 2 to 4 weeks and analyzed by flow cytometry. After 19 to 20 weeks, the animals were sacrificed and tissues harvested and analyzed. All animal studies were carried out at the Fred Hutchinson Cancer Research Center in compliance with the approved Institutional Animal Care and Use Committee (IACUC) protocol 1483.
Flow cytometry analysis and cell sorting
Flow cytometric analysis and sorting of human CD34+ cells were performed using the fluorochrome-conjugated antibodies listed in Table S3. Dead cells and debris were excluded via forward light scatter (FSC)/side light scatter (SSC) gating and DAPI staining. Flow cytometric analysis and cell sorting were performed on a FACSymphony A5, FACSCelesta, FACSAria IIu, and Symphony S6 (BD Biosciences). Data were acquired using FACSDiva version 6.1.3 and newer (BD Biosciences). Data analysis was performed using FlowJo version 8 and higher (BD Biosciences).
CFC assay
For CFC assays, 400 FACS-purified CD34+ and CD34+CD90+ cells were seeded on 30 mm plates in 1.1 mL of methylcellulose (MethoCult H4435, STEMCELL Technologies). Colonies were counted and scored after 12–14 days according to morphology into colony-forming unit (CFU)-granulocyte (CFU-G), CFU-macrophage (CFU-M), granulocyte-macrophage (CFU-GM), and burst-forming unit-erythrocyte (BFU-E). Colonies consisting of erythroid and myeloid cells were scored as CFU-Mix.
PCR analysis of CD33 exon 2
White blood cells
Genomic DNA was extracted from samples using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Excision of exon 2 was detected by PCR amplification on 40–60 ng genomic DNA using 20 μM forward and reverse primers, listed in Table S2. The target sequence was amplified running 35 cycles (assumed to be within the window of linearity), and the resulting amplicon was run on a 2% agarose gel (UltraPure agarose, Thermo Fisher Scientific) with a 100 bp ladder (Thermo Fisher Scientific). Bands corresponding to 200 bp indicated the deletion of exon 2, while bands corresponding to 600 bp indicated the wild-type exon. To allow semiquantitative assessment of gel bands, only 5%–10% of the PCR product was loaded to not exceed the detection limit during exposure and data acquisition on the G:Box Chemi-XRQ system (Genesys, Daly City, CA, USA). Exposure times were adjusted to not exceed the detection limit. Semiquantitative analysis of gel bands was performed with ImageJ (see also https://imagej.nih.gov/ij/docs/menus/analyze.html#gels).
CFC colonies
Individual colonies were picked and stored in QuickExtract DNA extraction solution (Lucigen; QE09050). Colonies were heat lysed at 65°C for 20 min, 100°C for 10 min, and then cooled down. Five microliters of the lysed colony was used as a template for PCR amplification using the forward and reverse primers listed above. Resulting amplicons were run on a 2% agarose gel with 100 bp ladder. Bands corresponding to 200 bp indicated the deletion of exon 2, while bands corresponding to 600 bp indicate the wild-type exon.
Statistical analysis
All data are reported as means ± SEM. Statistical analyses were performed using the Wilcoxon signed-rank test or one-way ANOVA where applicable, using GraphPad Prism software, version 6 (GraphPad Software, USA).
Acknowledgments
We thank Helen Crawford for her help in preparing the manuscript and figures. We thank Ted A. Gooley for his support in determining the statistical tests for the data analysis. This work was supported in part by grants to H.P.K. from the National Institutes of Health (R01AI135953 and HL136135). H.P.K. also received support as a Markey Molecular Medicine Investigator, as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research, and as the Stephanus Family Endowed Chair for Cell and Gene Therapy.
Author contributions
R.E.K., S.R., and H.P.K. designed the study. R.E.K., K.B., S.R., R.M., M.C., G.C., H.M., C.W., and A.M.P. performed experiments. O.H. provided assistance in some experiments. R.E.K. and S.R. generated the figures. H.P.K. funded the study. S.R., R.E.K., and H.P.K. wrote the manuscript. All authors reviewed and edited the final manuscript.
Declaration of interests
S.R. is a consultant to Forty-Seven, Inc. (Gilead Sciences), and Ensoma, Inc. H.P.K. is or was a consultant to and has or had ownership interests with Rocket Pharmaceuticals, Homology Medicines, VOR Biopharma, and Ensoma, Inc. H.P.K. has also been a consultant to CSL Behring and Magenta Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.02.026.
Contributor Information
Hans-Peter Kiem, Email: hkiem@fredhutch.org.
Stefan Radtke, Email: sradtke@fredhutch.org.
Supplemental information
References
- 1.Morgan R.A., Gray D., Lomova A., Kohn D.B. Hematopoietic stem cell gene therapy: progress and lessons learned. Cell Stem Cell. 2017;21:574–590. doi: 10.1016/j.stem.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naldini L. Gene therapy returns to centre stage (Review) Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 3.Staal F.J.T., Aiuti A., Cavazzana M. Autologous stem-cell-based gene therapy for inherited disorders: state of the art and perspectives. Front Pediatr. 2019;7:443. doi: 10.3389/fped.2019.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 cene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 5.Moffett H.F., Coon M.E., Radtke S., Stephan S.B., McKnight L., Lambert A., Stoddard B.L., Kiem H.P., Stephan M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017;8:389. doi: 10.1038/s41467-017-00505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sunshine J.C., Sunshine S.B., Bhutto I., Handa J.T., Green J.J. Poly(beta-amino ester)-nanoparticle mediated transfection of retinal pigment epithelial cells in vitro and in vivo. PLoS ONE. 2012;7:e37543. doi: 10.1371/journal.pone.0037543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mangraviti A., Tzeng S.Y., Kozielski K.L., Wang Y., Jin Y., Gullotti D., Pedone M., Buaron N., Liu A., Wilson D.R., et al. Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano. 2015;9:1236–1249. doi: 10.1021/nn504905q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keeney M., Ong S.G., Padilla A., Yao Z., Goodman S., Wu J.C., Yang F. Development of poly(beta-amino ester)-based biodegradable nanoparticles for nonviral delivery of minicircle DNA. ACS nano. 2013;7:7241–7250. doi: 10.1021/nn402657d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rui Y., Varanasi M., Mendes S., Yamagata H.M., Wilson D.R., Green J.J. Poly(beta-amino ester) nanoparticles enable nonviral delivery of CRISPR-Cas9 plasmids for gene knockout and gene deletion. Mol. Ther. Nucleic Acids. 2020;20:661–672. doi: 10.1016/j.omtn.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu D., Shen H., Tan S., Hu Z., Wang L., Yu L., Tian X., Ding W., Ren C., Gao C., et al. Nanoparticles based on poly (beta-amino ester) and HPV16-targeting CRISPR/shRNA as potential drugs for HPV16-related cervical malignancy. Mol. Ther. 2018;26:2443–2455. doi: 10.1016/j.ymthe.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humbert O., Laszlo G.S., Sichel S., Ironside C., Haworth K.G., Bates O.M., Beddoe M.E., Carrillo R.R., Kiem H.P., Walter R.B. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. 2019;33:762–808. doi: 10.1038/s41375-018-0277-8. [DOI] [PubMed] [Google Scholar]
- 12.Shaw B.C., Estus S. Pseudogene-mediated gene conversion after CRISPR-Cas9 editing demonstrated by partial CD33 conversion with SIGLEC22P. CRISPR J. 2021;4:699–709. doi: 10.1089/crispr.2021.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tzeng S.Y., Guerrero-Cazares H., Martinez E.E., Sunshine J.C., Quinones-Hinojosa A., Green J.J. Non-viral gene delivery nanoparticles based on poly(beta-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32:5402–5410. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L., Hu S., Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials. 2018;171:207–218. doi: 10.1016/j.biomaterials.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durymanov M., Reineke J. Non-viral delivery of nucleic acids: insight into mechanisms of overcoming intracellular barriers. Front Pharmacol. 2018;9:971. doi: 10.3389/fphar.2018.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison G.S., Wang Y., Tomczak J., Hogan C., Shpall E.J., Curiel T.J., Felgner P.L. Optimization of gene transfer using cationic lipids in cell lines and primary human CD4+ and CD34+ hematopoietic cells. BioTechniques. 1995;19:816–823. [PubMed] [Google Scholar]
- 17.Floch V., Le Bolc'h G., Audrezet M.P., Yaouanc J.J., Clement J.C., des Abbayes H., Mercier B., Abgrall J.F., Ferec C. Cationic phosphonolipids as non viral vectors for DNA transfection in hematopoietic cell lines and CD34+ cells. Blood Cells Mol Dis. 1997;23:69–87. doi: 10.1006/bcmd.1997.0123. [DOI] [PubMed] [Google Scholar]
- 18.Loukogeorgakis S.P., Fachin C.G., Dias A., Li H., Tang L., Kim A.G., Vrecenak J.D., Stratigis J.D., Ahn N.J., Nissim I., et al. Donor cell engineering with GSK3 inhibitor-loaded nanoparticles enhances engraftment after in utero transplantation. Blood. 2019;134:1983–1995. doi: 10.1182/blood.2019001037. [DOI] [PubMed] [Google Scholar]
- 19.Shahbazi R., Sghia-Hughes G., Reid J.L., Kubek S., Haworth K.G., Humbert O., Kiem H.P., Adair J.E. Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nat. Mater. 2019;18:1124–1132. doi: 10.1038/s41563-019-0385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma S., Woffendin C., Bahner I., Ranga U., Xu L., Yang Z.Y., King S.R., Kohn D.B., Nabel G.J. Gene transfer into human umbilical cord blood-derived CD34+ cells by particle-mediated gene transfer. Gene Ther. 1998;5:692–699. doi: 10.1038/sj.gt.3300656. [DOI] [PubMed] [Google Scholar]
- 21.Cruz L.J., van Dijk T., Vepris O., Li T., Schomann T., Baldazzi F., Kurita R., Nakamura Y., Grosveld F., Philipsen S., et al. PLGA-nanoparticles for intracellular delivery of the CRISPR-complex to elevate fetal globin expression in erythroid cells. Biomaterials. 2021;268:120580. doi: 10.1016/j.biomaterials.2020.120580. [DOI] [PubMed] [Google Scholar]
- 22.McNeer N.A., Chin J.Y., Schleifman E.B., Fields R.J., Glazer P.M., Saltzman W.M. Nanoparticles deliver triplex-forming PNAs for site-specific genomic recombination in CD34+ human hematopoietic progenitors. Mol. Ther. J. Am. Soc. Gene Ther. 2011;19:172–180. doi: 10.1038/mt.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duinhouwer L.E., van Rossum B.J., van Tiel S.T., van der Werf R.M., Doeswijk G.N., Haeck J.C., Rombouts E.W., Ter Borg M.N., Kotek G., Braakman E., et al. Magnetic resonance detection of CD34+ cells from umbilical cord blood using a 19F label. PLoS ONE. 2015;10:e0138572. doi: 10.1371/journal.pone.0138572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.Y., Liu D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M., Zuris J.A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U S A. 2016;113:2868–2873. doi: 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rui Y., Wilson D.R., Choi J., Varanasi M., Sanders K., Karlsson J., Lim M., Green J.J. Carboxylated branched poly(beta-amino ester) nanoparticles enable robust cytosolic protein delivery and CRISPR-Cas9 gene editing. Sci. Adv. 2019;5:eaay3255. doi: 10.1126/sciadv.aay3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen D.N., Roth T.L., Li P.J., Chen P.A., Apathy R., Mamedov M.R., Vo L.T., Tobin V.R., Goodman D., Shifrut E., et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020;38:44–49. doi: 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su X., Fricke J., Kavanagh D.G., Irvine D.J. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol. Pharmaceutics. 2011;8:774–787. doi: 10.1021/mp100390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei T., Cheng Q., Min Y.L., Olson E.N., Siegwart D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11:3232. doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veiga N., Goldsmith M., Granot Y., Rosenblum D., Dammes N., Kedmi R., Ramishetti S., Peer D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018;9:4493. doi: 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenblum D., Gutkin A., Kedmi R., Ramishetti S., Veiga N., Jacobi A.M., Schubert M.S., Friedmann-Morvinski D., Cohen Z.R., Behlke M.A., et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020;6:eabc9450. doi: 10.1126/sciadv.abc9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kedmi R., Veiga N., Ramishetti S., Goldsmith M., Rosenblum D., Dammes N., Hazan-Halevy I., Nahary L., Leviatan-Ben-Arye S., Harlev M., et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018;13:214–219. doi: 10.1038/s41565-017-0043-5. [DOI] [PubMed] [Google Scholar]
- 33.Niu F., Yan J., Ma B., Li S., Shao Y., He P., Zhang W., He W., Ma P.X., Lu W. Lanthanide-doped nanoparticles conjugated with an anti-CD33 antibody and a p53-activating peptide for acute myeloid leukemia therapy. Biomaterials. 2018;167:132–142. doi: 10.1016/j.biomaterials.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benedict C.A., Tun R.Y., Rubinstein D.B., Guillaume T., Cannon P.M., Anderson W.F. Targeting retroviral vectors to CD34-expressing cells: binding to CD34 does not catalyze virus-cell fusion. Hum. Gene Ther. 1999;10:545–557. doi: 10.1089/10430349950018625. [DOI] [PubMed] [Google Scholar]
- 35.Brendel C., Goebel B., Daniela A., Brugman M., Kneissl S., Schwable J., Kaufmann K.B., Muller-Kuller U., Kunkel H., Chen-Wichmann L., et al. CD133-targeted gene transfer into long-term repopulating hematopoietic stem cells. Mol. Ther. 2015;23:63–70. doi: 10.1038/mt.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C., Wang H., Georgakopoulou A., Gil S., Yannaki E., Lieber A. In vivo HSC gene therapy using a bi-modular HDAd5/35++ vector cures sckle cell disease in a mouse model. Mol. Ther. 2021;29:822–837. doi: 10.1016/j.ymthe.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maynard L.H., Humbert O., Peterson C.W., Kiem H.P. Genome editing in large animal models. Mol. Ther. J. Am. Soc. Gene Ther. 2021;29:3140–3152. doi: 10.1016/j.ymthe.2021.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson C.W., Kiem H.P. Lessons from London and Berlin: designing a scalable gene therapy approach for HIV cure. Cell Stem Cell. 2019;24:685–687. doi: 10.1016/j.stem.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Esrick E.B., Bauer D.E. Genetic therapies for sickle cell disease. Semin. Hematol. 2018;55:76–86. doi: 10.1053/j.seminhematol.2018.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radtke S., Pande D., Cui M., Perez A.M., Chan Y.Y., Enstrom M., Schmuck S., Berger A., Eunson T., Adair J.E., et al. Purification of human CD34(+)CD90(+) HSCs reduces target cell population and improves lentiviral transduction for gene therapy. Mol. Ther. Methods Clin. Dev. 2020;18:679–691. doi: 10.1016/j.omtm.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Humbert O., Peterson C.W., Norgaard Z.K., Radtke S., Kiem H.P. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta-hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018;8:75–86. doi: 10.1016/j.omtm.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.