

Abstract
Graft-versus-host disease (GvHD) is still the major non-relapse, life-limiting complication after hematopoietic stem cell transplantation. Modern pharmacologic immunosuppression is often insufficient and associated with significant side effects. Novel treatment strategies now include adoptive transfer of ex vivo expanded regulatory T cells (Tregs), but their efficacy in chronic GvHD is unknown. We treated three children suffering from severe, therapy-refractory GvHD with polyclonally expanded Tregs generated from the original stem cell donor. Third-line maintenance immunosuppression was tapered to cyclosporin A and low-dose steroids shortly before cell transfer. Regular follow-up included an assessment of the subjective and objective clinical development, safety parameters, and in-depth immune monitoring. All patients showed marked clinical improvement with substantially decreased GvHD activity. Laboratory follow-up showed a significant enhancement of the immunologic engraftment, including lymphocytes and dendritic cells. Monitoring the fate of Tregs by next-generation sequencing demonstrated clonal expansion. In summary, adoptive transfer of Tregs was well tolerated and able to modulate an established undesired T cell mediated allo-response. Although no signs of overimmunosuppression were detectable, the treatment of patients with invasive opportunistic infections should be undertaken with caution. Further controlled studies are necessary to confirm these encouraging effects and eventually pave the way for adoptive Treg therapy in chronic GvHD.
Keywords: hematopoietic stem cell transplantation, chronic graft-versus-host disease, regulatory T cells, adoptive T cell therapy, clinical immunology
Graphical abstract
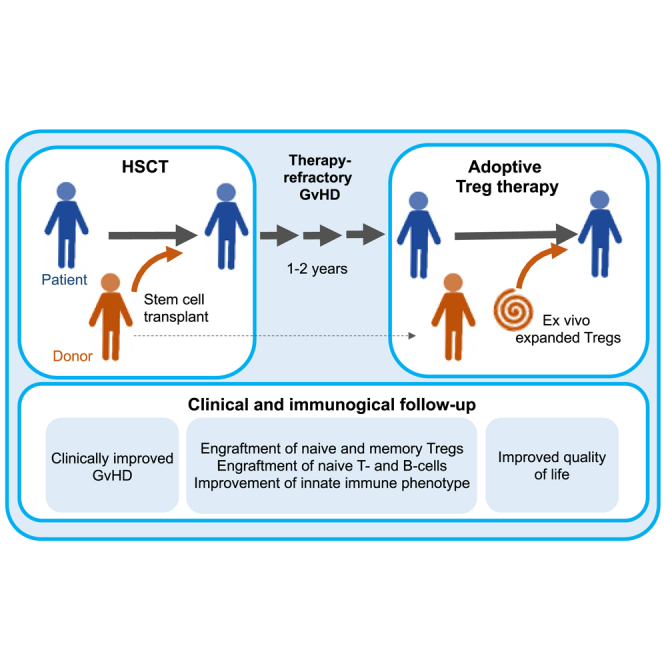
Landwehr-Kenzel et al. used ex vivo expanded Tregs to treat three children with therapy-refractory cGvHD. To support Treg function and survival, maintenance immunosuppression was tapered to cyclosporin A and low-dose steroids. All patients showed a substantial clinical benefit and improved immune functions with engraftment of naive T cells, B cells, and dendritic cells.
Introduction
Chronic graft-versus-host disease (cGvHD) remains the most significant post-transplant non-relapse cause of morbidity and mortality after hematopoietic stem cell transplantation (HSCT), the standard treatment for multiple malignant and benign hematologic diseases.1, 2, 3, 4 Despite being increasingly target-specific, modern pharmacologic immunosuppression has only insufficiently decreased the burden of cGvHD and is associated with significant side effects, including toxicities or an increased risk of infections and malignancies.5 During early phases, GvHD is usually triggered by chemotherapy or infection-induced tissue damage, which leads to a release of pro-inflammatory cytokines. This results in activation of antigen-presenting cells, which then present recipient-specific tissue antigens to alloreactive donor T cells.5,6 In case of cGvHD, donor lymphocytes remain continuously activated and fail to establish a host-specific tolerance.5 Therefore, concurrent with increasing indications for HSCT and driven by the medical need to combat cGvHD, the identification of thymic derived regulatory T cells (Tregs) has paved the way for the development of novel immunotherapeutic cell-based strategies.
Tregs are characterized as CD4+CD25+ cells expressing high levels of FoxP3,7 but very low levels of the IL-7 receptor α chain8 and have been demonstrated to play a critical role in the maintenance of immunological tolerance.9 As physiologic counterplayers of conventional T cells, Tregs can control overactivation and expansion of conventional T cells10 in various preclinical disease models.11, 12, 13, 14, 15, 16, 17 Recently, clinical grade Tregs products became available and first data on safety and efficacy has been published for the preventive/pre-emptive use of Tregs for GvHD in adults.18, 19, 20, 21, 22, 23 Reports on the therapeutic use of Tregs to treat GvHD are limited to individual case reports, and to the best of our knowledge, their use has not yet been reported in children.
We now report on three children who suffered from therapy-refractory, life-threatening GvHD and therefore received adoptive transfer of ex vivo expanded Tregs that were generated from the original stem cell donor on an individual treatment basis. Concomitant pharmacologic immunosuppression was tapered to cyclosporin A (CSA) and low-dose prednisolone shortly before cell transfer. We further demonstrate by clinical and immunological follow-up data that adoptive transfer of ex vivo expanded Treg products is well tolerated and can modulate established T cell-mediated allo-responses, supporting immune cell engraftment and ameliorating GvHD activity.
Results
Patient characteristics and clinical course
Patient 1, 8 years old, underwent peripheral blood stem cell transplantation from a 10/10 HLA related donor owing to transfusion dependent beta-thalassemia major (details listed in Table 1). Despite guideline-compliant GvHD prophylaxis, the patient developed acute GvHD 12 days after transplantation, which progressed to severe cGvHD. Skin and skin appendages were the main site of manifestation, but the gastrointestinal mucous membranes were also severely affected. Chronic pain, difficulties associated with food intake, contractures of the finger joints, dryness and burning of the eyes, and not least the disease-related stigma led to a significant impairment of the patient's quality of life (Figures 1A–1C and 1I–1O). The patient received first, second and third-line therapy (Table 1), including subcutaneous IL-2 applications as previously described by Koreth et al.24 None of these approaches significantly improved the clinical symptoms. On the contrary, the patient developed major complications, including arterial hypertension and two episodes of severe sepsis. Owing to the lack of therapeutic alternatives, adoptive Treg therapy was performed day +639 after HSCT at a dose of 3.0 × 106 Treg/kg i.v. During the 93-week follow-up period, we observed a significant clinical improvement. The Treg product was well tolerated. No signs of acute or chronic toxicity were observed; no inflammatory cytokine storm (Figure S1) and no signs of embolism or other acute adverse reactions occurred. Sixteen weeks after Treg transfer the patient presented with abdominal pain; the clinical course was highly suggestive for a beginning bacterial infection. Upon initiation of i.v. antibiotic therapy, the acute symptoms promptly resolved. No relevant additional infectious complications were recorded, in particular no critical virus reactivation or infection, no fungal infection, no additional bacterial infection and no other adverse events occurred during the follow-up period. With respect to cutaneous GvHD manifestation, continuous improvement with substantial benefits in the skin and joints was documented as depicted in Figures 1D–1F. The general status of the patient substantially improved; the parents reported less pain, improved food intake and a significant increase in vitality and recreational activity. The daily prednisolone dose was reduced from 10 mg (0.45 mg/kg) before Treg therapy to 5 mg (0.22 mg/kg). Quantification of the clinical improvement showed amelioration of ocular symptoms including xerophthalmia, photosensitivity, foreign body sensation, and epiphora conjunctival injection (Figure 1K). Additionally, decreased scleroderma and inflammation of the mucosa were observed (Figures 1L–1O).
Table 1.
Clinical and transplantation related characteristics of patient 1, patient 2 and patient 3
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Sex | Male | Male | Male |
| Indication for HSCT | β-Thalassemia major | Myelodysplastic syndrome | Septic granulomatous disease |
| Age at HSCT | 8 years | 11 years | 11 years |
| Conditioning | Busulfan Cyclophosphamide ATG-Fresenius |
Fludarabin Thiothepa ATG-Fresenius |
Fludarabin Busulfan∗∗ Alemtuzumab |
| Graft | PBSC | Bone marrow | Bone marrow |
| HLA match | 10/10 MRD | 10/10 MUD | 9/10 MUD |
| Cell dose (/kg) | 2.9 × 108 TNC 5.3 × 106 CD34+ cells 1.2 × 108 CD3+ T cells |
4.1 × 108 TNC 3.4 × 106 CD34+ cells 4.5 × 107 CD3+ T cells |
4.1 × 108 TNC 1.9 × 106 CD34+ cells 5.1 × 106 CD3+ T cells |
| GvHD prophylaxis | CSA, MTX, ATG | CSA, MTX | CSA, MMF, Prednisone |
| Retransplantation | – | d +61 | d +531 |
| 2nd conditioning | – | Thymoglobulin | CD34+ PBSC |
| Graft | – | PBSC | |
| GvHD manifestation sites | Skin Mukosa GI tract |
Skin Mukosa GI tract |
GI tract Skin Liver |
| GvHD therapy | CSA Prednisolone MMF MTX Azathioprine Budenoside Low-dose IL-2 s.c. |
CSA Prednisolone Methylprednisolone Tacrolimus Photopheresis Infliximab Methotrexate Ruxolitinib |
CSA Prednisolone MMF MTX Budenoside ECP Imatinib MSC d+341, d+348, d+373, d+378 |
| tTreg therapy post HSCT | d +639 | d +736/+675 | d +603 |
| tTreg product characteristics | |||
| Fold expansion rate | 3 193 | 20,751 | 1 291 |
| Viability | 98% | 98% | 97% |
| CD4+CD25+FoxP3+ | 95% | 97% | 94% |
| % IFNg producer | 1.5% | 1.3% | 7.61% |
| tTreg dose | 3 × 106/kg | 3 × 106/kg | 3 × 106/kg |
| Co-administered immunosuppression | CSA Low-dose prednisolone |
CSA Low-dose prednisolone |
CSA MMF |
GI tract, gastrointestinal tract; MMF, mycophenolate mofetil. Conditioning: Busulfan∗∗, only 75% of regular myeloablative busulfan i.v. dose (3 days instead of 4) with therapeutic drug monitoring for a targeted dose of 65,000 ng/h/mL; ATG-Fresenius+, antithymocyte globulin (Grafalon). Grafts: BM, bone marrow; MRD, matched related donor; MUD, matched unrelated donor; PBSC, peripheral blood stem cells. Cell dose: TNC, dose of total nucleated cells per kilogram of body weight infused; MSC, mesenchymal stem cells.
Figure 1.

Adoptive Treg transfer ameliorates clinical symptoms of cGvHD in individual patients
(A–H) document the clinical extent of cutaneous and mucosal GvHD in patient 1 (A–F, green lines) and patient 2 (G and H, blue lines) at time of Treg transfer (A–C) and 1 year later (D–F). As depicted in (A–F), patient 1 suffered from severe signs of inflammation, hyper- and hypopigmentation and skin lesions as well as finger contractures (A–C). Patient 2 showed severe sings of mucositis with leukoplakia of the oral mucosa (G). One year after Treg therapy not only leukoplakia and mucositis was substantially reduced, but their reduced clinical burden also substantially increased quality of life (H). (I–O) The overall patient status (I) and GvHD symptoms of the eyes such as xerophthalmia, photosensitivity foreign body sensation, epiphora conjunctival injection (K), the skin (L and M) and the oral mucosa (N and O) were further quantified by a clinical follow-up score, which evaluated the clinical situation as compared with the previous visit (scoring depicted in Table 2). As documented by the clinical scoring, the general condition, ocular disorders and symptoms of both the skin and the mucosa continuously improved over time (I, K, M, and O). In line with these observations, the percentage of skin affected from scleroderma and oral erythroderma substantially decreased (L and N). Most impressively, skin softening, which cannot be depicted in the photographs, was also observed.
Patient 2, 11 years old, underwent stem cell transplantation and retransplantation owing to myelodysplastic syndrome from a 10/10-ident unrelated donor (Table 1). The patient suffered from severe acute GvHD of the intestine starting 60 days after transplantation, which had progressed to cGvHD of the skin, the liver, the lung, and the intestine by day 103 after transplant. Signs of active cGvHD included facial erythema, multiple nummular skin lesions, severe oral (Figures 1G and 1H) and ocular, hepatic, and pulmonary manifestations. GvHD activity barely responded to first-, second- and third-line immunosuppressive regimens (Table 1), which induced various side effects, including severe osteonecrosis, which required regular pain medication. At the time when the patient was first presented to our team, he received quadruple immunosuppressive therapy with prednisolone, methotrexate, ruxolitinib, and weekly extracorporeal photopheresis. Owing to the lack of alternative treatment options, Treg product was generated from the original stem cell donor and the patient received 3.0 × 106 expanded Tregs/kg on days +736 and +675 after stem cell transplantation. The Treg product was well tolerated. No signs of acute or chronic toxicity were observed, nor was there an inflammatory cytokine storm, signs of embolism, or other acute adverse reactions. During the entire observation period, no infectious complications were recorded; in particular, no critical virus reactivation or infection, no fungal infection, no additional bacterial infections and no other adverse events occurred during this follow-up period. The patient's general condition substantially improved over time (Figure 1I). At time of Treg transfer, the patient required nightly high-flow oxygen supplementation; however, within 6 months after adoptive Treg transfer all oxygen supplementation could be terminated. Photophobia as well as foreign body sensation in the eyes and xerophthalmia (Figure 1K) and signs of cutaneous and mucosal inflammation decreased (Figures 1L–1O). Prednisolone could be tapered to 2.5 mg/d and the CSA trough levels were targeted at 80–100 μg/L. Radiologically, osteonecrosis did not progress further and partially improved; the patient did not require pain medication as of 6 months after Treg therapy. The overall clinical improvement continued for 15 months after Treg therapy until the patient suffered from primarily non-GvHD complications, including a urethral polyp and gastroesophagitis. Endoscopy of both sites was performed, but during microbiological, virological, and histological workup, no evidence of infectious triggers, GvHD, or malignancy was found. Nineteen months after Treg transfer, the patient suffered from sudden onset of hemoptysis. During bronchoscopy, the bronchial and pulmonary mucosa showed diffuse signs of bleeding without larger bleedings sources. Biopsies as well as microbiological and virological specimens were taken and thoroughly analyzed, but no signs of GvHD or infections were found. Owing to respiratory failure on mechanical ventilation the patient died 10 days after bleeding onset. Retrospectively, cGvHD-related coagulopathy seems possible.25
Patient 3, 11 years old, was stem cell transplanted owing to chronic granulomatous disease (a de novo mutation of the CYBB gene) from a 9/10 HLA-ident unrelated donor (Table 1). The patient developed mild aGvHD of the skin (day +10). Timely correlating with CSA trough levels below the target range aGvHD later progressed to severe cGvHD affecting the skin (moderately) and the gastrointestinal tract including the intestine and the liver (both severely). Multimodal immunosuppressive therapy promoted infectious complications with microbiological evidence of Candida glabrata, Staphylococcus epidermidis, Lactobacillus rhamnosus, Aspergillus fumigatus, BK virus, and adenovirus. Pulmonary computed tomography scans showed atypical infiltrates of the lung, as typically seen in pulmonary aspergillosis. Anti-infective therapy was escalated but options were limited by chronic kidney and liver insufficiency. Chronic GvHD treatment included first-, second-, and third-line treatment approaches including CSA, mycophenolate mofetil, prednisolone (max 5 mg/kg/day), methotrexate, budenoside, and extracorporeal photopheresis (Table 1). Owing to continuously uncontrolled intestinal GvHD the patient received four doses of mesenchymal stem cells between day +341 and day +378 (individual treatment approach). Subsequently, transplant failure occurred and a stem cell boost with CD34-selected stem cells of the initial donor became necessary on day +531. The clinical condition of the patient remained critical and cGvHD activity was persistently high. Finally, the patient received 3.0 × 106 expanded Tregs/kg as an individual treatment approach on day +603 after HSCT. Tregs were well tolerated without any signs of undesired cytokine release (Figure S1). The patient showed a remarkable clinical stabilization and improvement of GvHD symptoms. Steroids were reduced from a maximum of 20 mg (0.77 mg/kg) daily to a minimum of 10 mg (0.38 mg/kg) and the patient was mobilized. A computed tomography scan of the lung performed on day +19 after Treg transfer (day +622 after HSCT) showed declining, bilateral milk glass changes and infiltrates. Four weeks after Treg transfer, infectious parameters, fever, and GvHD activity increased again. The option of anti-infectious therapy escalation was limited by the pre-existing chronic liver and kidney disease. The clinical condition rapidly deteriorated and during an acute event, most likely a pulmonary hemorrhage owing to the underlying pulmonary aspergillosis, the patient died on day +35 after Treg therapy (day +638 after HSCT).
Improved T cell engraftment after Treg transfer
An analysis of the total T cell compartment (Figures 2A–2P, S2, and S3) by multicolor flow cytometry revealed distinct changes that together indicated substantial progress in T cell engraftment and normalization of T cell subset distributions. At the time of Treg transfer, all patients showed mild (patient 1) to severe (patient 2 and patient 3) CD3+ lymphopenia (Figures 2A–2C), with particularly reduced CD4+ T cell numbers (Figures 2D–2F) and reduced CD8+ T cells (Figures 2K–2M). While total the CD3+ and CD8+ T cells remained largely stable throughout the observation periods (Figures 2A–2C), we observed a remarkable and long-term increase of CD4+ T cells (Figures 2D–2F). To assess T cell differentiation states the expression of CD45RA and CCR7 were employed to identify naive (CD45RA+ CCR7+, TN), central memory (CD45RA-CCR7+, TCM), effector memory (CD45RA–CCR7–, TEM) and terminally differentiated CD45RA+ (CD45RA+ CCR7–, TEMRA) T cells. In addition to the increase in absolute CD4+ T cell counts, we found, for the first time since transplantation, peripherally detectable numbers of TN, which is a sign of intrinsic expansion and improved T cell engraftment (Figures 2G–2I). This was first detectable at 4–8 weeks after Treg transfer and continued throughout the observation periods reaching levels of approximately 50% of all Tregs in patient 1. Interestingly, in contrast to the robust induction of CD4+ T cells, absolute numbers of CD8+ T cells remained largely stable (Figures 2K–2M). However, addressing the proportional distribution of T cell maturation states in more detail we observed a substantial shift toward CD8+ TN while the frequency of TEM and TCM was reduced in patient 1 > patient 2 (Figures 2N–2P).
Figure 2.

Engraftment of naive T cells after Treg therapy
(A–P) Peripherally collected blood was analyzed by multicolor flow cytometry. Cells were gated as singlets, CD45+ leukocytes, lymphocytes, and the expression of CD3+ (A–C), CD3+CD4+ (D–I) and CD8+ (K–P). CD3+CD4+and CD3+CD8+ T cells were further analyzed for the expression of CD45RA and CCR7 (G–I and N–P). Subsequently, T cell maturation of the CD4+ and the CD8+ compartment was separately analyzed by the identification of naive as CD45RA+CCR7+ (TN), central memory as CD45RA−CCR7+ (TCM), effector memory as CD45RA−CCR7– (TEM), and terminally differentiated CD45RA+ T cells as CD45RA+CCR7– (TEMRA). Depicted are total leukocytes numbers per microliter as a continuous line in (A–C). All cells subsets are shown before Treg administration and in the weeks after Treg transfer (x axis). (A–F and K–M) Results are depicted as black solid lines in absolute cell numbers per microliter and assigned to the left y axis. Relative cell frequencies of the parental gate are depicted as gray lines in percent and assigned to the right y axis. (G, H, and N–P) CD4+ and CD8+ compartments were analyzed for their composition of TN, TCM, TEM, and TEMRA cells are depicted in relative frequencies.
T cell expansion occurs within HLA-DR– T cells
HLA-DR is expressed on activated T cells in vivo and in vitro and is associated with a significantly higher risk of acute and cGvHD. To monitor for T cell activation in vivo after Treg transfer, expression of HLA-DR was assessed on CD4+ and CD8+ T cells (Figures 3A–3M and S4). In line with the observation of naive T cell engraftment, we found an impressive increase of non-activated HLA-DR–CD4+ T cells in patient 1 and patient 2 (Figures 3A–3F). This effect was particularly strong within the CD4+ T cell compartment but paralleled by an increase in HLA-DR–CD8+ T cells. At the same time, the absolute number of activated, HLA-DR+ CD4+ and CD8+ T cells remained largely stable (Figures 3A–3C and 3G–3I). The expansion of HLA-DR– T cells was further confirmed by the concurrent proportional decrease in the relative counts of activated HLA-DR+ T cells in both the CD4+ and CD8+ T cell compartment (Figures 3D–3F and 3K–3M).
Figure 3.

T cell engraftment is not accompanied by increased HLA-DR+ cells
(A–M) CD4 and CD8 T cell compartments were identified from singlets, CD45+ leukocytes, lymphocytes, CD3+ cells and analyzed for the expression if HLA-DR+ by multicolor flow cytometry. (A–C and G–I): Depicted are absolute cell numbers of HLA-DR expressing CD4+ (A–C) and CD8+ (G–I) T cells. (D–F and K–M): Relative distribution of HLA-DR+ (yellow) and HLA-DR- (blue) cells within the CD4+ and CD8+ compartment are depicted in (D–F and K–M).
Engraftment of naive and expansion of memory Tregs
For monitoring the fate of Tregs, CD3+CD4+CD25highCD127low Treg counts were assessed whenever a blood collection was justified for the evaluation of the patients' status (Figures 4A–4I). Interestingly, a biphasic increase of CD3+CD4+CD25highCD127low Tregs was observed in all patients (Figures 4A–4C and S5). Owing to the short observation time, this increase is, however, difficult to interpret in patient 3 (Figure 4C). In patient 2, the absolute Treg numbers and Treg frequencies decreased again starting after week 40 post Treg transfusion (Figure 4B).
Figure 4.

Substantial engraftment of naive and memory Tregs
Numbers and frequencies of peripherally circulating Tregs were assessed by multicolor flow cytometry. Tregs were gated as singlets, CD45+ leukocytes, lymphocytes, CD3+CD4+ T cells and subsequently identified as CD25highCD127low expressing cells. (A–C) Total Tregs were further sub-analyzed as (D–F) naive CD45RA+ and (G–I) memory CD45RA− Tregs. Results are depicted as black solid lines in absolute cell numbers per microliter and assigned to the left y axis. Relative cell frequencies of the parental gate are depicted as gray lines in % and assigned to the right y axis. The s axis shows time from Treg transfer.
To better understand Treg biology in vivo, we further analyzed Treg maturation states according to their expression of CD45RA, the characteristic marker for naive T cells (Figures 4D–4I). At the time of Treg transfer all three patients showed very low numbers of Tregs; naive Tregs were—if at all—only barely detectable (Figures 4D–4F). Starting at about 3 months after adoptive Treg transfer, both patient 1 and patient 2 showed a remarkable increase of naive Treg, which occurred for the first time since HSCT and may indicate intrinsic Treg engraftment (Figures 4D and 4E). At 65 weeks after Treg transfer, patient 1 reached the lower limit of normal Treg numbers (Figure 4D). The biphasic increase of total Tregs was also observed in circulating memory Tregs and might be explained as adoptively transferred Tregs in the first phase and in vivo Treg expansion and/or maturation in the second phase (Figures 4G–4I). Further underlining and paralleled by the observation of relative and absolute Treg expansion, the relative frequency of memory Tregs decreased (Figures 4G and 4H). In line with the early peak of memory Tregs observed in patient 1 and patient 2, we found comparably high numbers of memory Tregs in patient 3 (Figure 4I).
T cell receptor repertoire analyses
In line with our previously published reports,26 our Treg products expressed a polyclonal T cell receptor (TCR) repertoire comparable with those of freshly isolated Tregs but distinct from non-Tregs (Figure 5). To track the fate of adoptively transferred Tregs, we compared the TCR repertoire of the respective Treg product with those of PBMC collected during follow-up of our patients. As shown in Figure 5, the donor-derived Treg product contains overlapping T cell clones with PBMC of the respective HSCT recipient, representing the few Treg cells within the PBMC. The number of distinct Treg TCRs overlapping PBMC samples rose from 256 (patient 1) and 281 (patient 3) before Treg infusion up to more than 1,000 in both patients within the first weeks. Treg TCR clones were detectable at high levels throughout the observation periods of both patients. Interestingly, in patient 1 there was a temporary decrease in the overlapping clone counts to 484 in association with a bacterial infection at 16 weeks after Treg therapy, which recovered fast after clearance of the infection. Of major importance, we found that the number of distinct Treg TCRs (Figures 5C and 5D) paralleled the frequency of total Tregs in the peripheral blood detected by flow cytometric analysis (Figures 4A and 4C). The biphasic increase of Treg engraftment described in Figure 4 was, however, not only represented by the dynamic of unique TCRs, but also detected when the total amount of overlapping Treg TCRs (Figures 5E and 5F) and frequencies of Treg TCRs within all T cells (Figures 5G and 5H) were assessed. In summary, these data support the hypothesis of long-term engraftment of adoptively transferred Tregs and their survival for the full follow-up time of more than 9 months.
Figure 5.

Adoptively transferred Tregs expand in vivo and show long-term survival
(A–H) A clonotype analysis was performed from an aliquot of the adoptively transferred Treg product. The clonotype analysis was further conducted in peripherally collected PBMC before Treg therapy and serially thereafter. Overlapping clonotypes were assessed and allowed cellular tracking of the Treg product in the peripheral patient blood. (A and B) TCR clones detected in the Treg product are depicted on the y axis, TCR clones detected in the peripheral PBMC preparations are depicted on the x axes. Overlapping clones, present in the Treg product and the peripheral blood, are depicted between both axes. As a control, Treg product clones of patient 1 were analyzed in PBMC preparations of patient 3 and vice versa. The number of distinct TCRs present in both the Treg product and PBMCs are indicated above each panel. (C and D) Number of distinct TCRs present in Treg products and patient samples. (E and F) Depicted are total numbers of Treg product TCRs detected in PBMC samples. (G and H) Depicted are frequencies of different Treg TCRs relative to all TCRs detected in each PBMC sample.
B cell engraftment after Treg transfer
Long lasting B cell deficiency is a common phenomenon in HSCT patients suffering from cGvHD.27,28 Deficient B cell development at early maturation states has previously been reported to be both a cause and consequence of active GvHD. Therapeutic approaches to combat acute and cGvHD frequently cause further delays to B cell engraftment. To assess the effects of adoptive Treg therapy on B cell development, the maturation state of B cells was analyzed, ranging from naive B cells to plasmablasts, as well as their engraftment (Figures 6A–6S and S6). Before Treg infusion, CD19+ B cells were barely detectable in patient 1 and patient 3 and substantially decreased in patient 2 (Figures 6A–6C); these data were in line with very low numbers of naive B cell (Figures 6D–6F). Approximately 20 weeks after Treg transfer, we observed for the first time since transplantation a remarkable expansion within the naive B cell compartment (Figures 6D–6F), which also resulted in an absolute increase of in the total number of B cells (Figures 6A–6C). Transitional (Figures 6G–6I) and non-class-switched (Figures 6K–6M) B cell initially followed the same trend in expansion as naive B cells; however, this expansion effect was lost over time as depicted here for transitional (Figures 6G–6H), non-class-switched (Figures 6K–6M), class-switched (Figures 6N–6P) B cells and plasmablasts (Figures 6Q–6S).
Figure 6.

Treg transfer induces engraftment of circulating B cells in early maturation states
(A–C) Peripherally circulating B cells were identified in freshly collected blood as singlets, CD45+ leukocytes, lymphocytes, and the expression of CD19. (D–F) Naive B cells were detected as CD27−IgD+ within CD19+ cells. (G–I) Cells expressing CD38highCD24high of CD19+IgM+CD27− B cells were defined as transitional B cells. (K–P) CD19+IgD−IgM+ cells were further analyzed for the expression of CD27 and CD38 to define non-class-switched B cells as CD27+CD38high and class-switched B cells as CD27+CD38low. (Q–S) Plasmablasts were identified as CD19+IgD−IgM− and CD27intCD38high cells. All results are depicted as black solid lines in absolute cell numbers per microliter and assigned to the left y axis. Relative cell frequencies of the parental gate are depicted as gray lines in percent and assigned to the right y axis. The x axis shows time from Treg transfer. A more detailed description of the gating protocol was previously described by Streitz et al.24
Adoptive Treg transfer improves dendritic cell engraftment
The effect of adoptively transferred Tregs on innate immune cells critically involved in early infection control is largely unknown. To elucidate if Tregs can support the engraftment of innate immune cells as well as lymphocytes, we analyzed the number of granulocytes, monocytes, and dendritic cells (DC) (Figures 7A–7P and S7). After Treg transfer, granulocyte (Figures 7A–7C) and monocyte (Figures 7D–7F) numbers were largely stable, although we found both cell compartments to be transiently increased after Treg transfer with repetitive peaks at later stages. Impaired reconstitution of DC in patients suffering from GvHD has previously been described29, 30, 31 and was also observed in our patients with cGvHD before Treg therapy (Figure 7). Serial follow-up, however, showed that adoptive Treg therapy seems to support DC engraftment (Figures 7G–7P). After Treg transfer, and in parallel with the engraftment of naive CD4+ T cells, we observed an increase in LIN−HLA-DR+CD11c+ DC (Figures 7G–7I), CD16+ DCs (Figures 7K–7M), and mDC1 (Figures 7N–7P).
Figure 7.

Engraftment after Treg therapy is not restricted to the lymphocyte compartment but similarly observed in DC
Possible alterations within the innate immune system were monitored using the DURAClone IM Phenotyping Basic (A–F) and the DURAClone IM Dendritic Cells (G–P) kits using peripherally collected blood. (A–C) Granulocytes were defined as a distinct population in the side scatter from singlet CD45+ leukocytes. (D–F) Within the non-granulocytic population, monocytes were defined as CD14+ cells. (G–P) Lin−HLA-DR+CD11c+ myeloid DC (mDC) were gated within the HLA-DR+ DC population of CD45+ leukocytes. CD16+ monocytic DC were identified as CD16+Clec9–, while mDC1 were defined as CD1c+CD16–. All results are depicted as black solid lines in absolute cell numbers per microliter and assigned to the left y axis. Relative cell frequencies of the parental gate are depicted as gray lines in percent and assigned to the right y axis. The x axis shows time from Treg transfer. (A–C) In addition to absolute and relative granulocyte numbers, total leukocyte numbers are depicted in black without symbols.
Discussion
Our data suggest that the adoptive transfer of polyclonal ex vivo expanded Tregs from the original stem cell donor at a single dose of 3 × 106/kg body weight is well tolerated and can improve the clinical condition in children suffering from therapy-refractory cGvHD. We did not observe adverse effects directly related to Treg transfer, such as allergic reactions, cytokine storm, transfusion-related lung injury, or pulmonary embolism. All three severely ill patients showed fast improvement of cGVHD symptoms and engraftment of naive lymphocytes and DC. In two patients this effect lasted more than 1 year. One patient died of an unexpected pulmonary hemorrhage 19 months after Treg transfer; however, no infectious cause or active pulmonary GvHD could be detected. The third patient could not be followed long term because he died of a pre-existing invasive aspergillus infection at day 35 after Treg treatment.
The encouraging results on the course of cGvHD are even more pronounced considering the tapering of maintenance third-line immunosuppression in all three patients to CSA and a low-dose steroid treatment just before Treg transfer to allow Treg survival and functionality. The improvement of cGvHD symptoms that allowed further tapering of immunosuppression, underlines the efficacy of Treg transfer. Furthermore, the reduction in immunosuppression had the knock-on effect of decreasing drug-related side effects such as organ toxicity, osteonecrosis and the incidence of clinically relevant infection episodes. There are several reports that calcineurin inhibitors may decrease Treg survival and function in vivo;32, 33, 34, 35, 36 however, our recent data in a GvHD mouse model rather suggests that low-dose CSA is beneficial for Treg functionality, whereas steroid treatment should be avoided.37 The long-lasting increased levels of Tregs and the sustainable amelioration of GvHD in our low-dose CSA-treated patients supports our previous findings.
The only other case reports describing a therapeutic approach of adoptive Treg transfer in cGvHD were published by Theil et al.23 (five patients) and Trzonkowski et al.19 (one patient) with cell doses ranging from 1 × 105/kg to 4.5 × 106/kg. In line with our approach, Theil et al. administered Treg products that were produced from the original stem cell donor after Treg isolation by magnetic bead separation (CliniMACS technology). In contrast, however, unstimulated leukapheresis products were used as the starting material. Further, while our products were expanded for 3 weeks and reached a purity of greater than 94% at an average fold expansion rate of 8,411, Theil et al.23 conducted Treg expansion over 12 days only and reached an average of 4.5-fold expansion with 78% purity. The patient with cGvHD described by Trzonkowski et al.19 received Tregs that were produced from a buffy coat of the original family stem cell donor. CD4+ cells were isolated by negative immunomagnetic sorting (StemCell Technologies) and subsequently sorted using the FACSARIA sorter (BD Biosciences) for CD3+CD4+CD25highCD127−doublet–lineage–dead–. Tregs were cultured for 2 weeks to reach sufficient cell numbers at a purity for FoxP3 of 90%.19 In both studies, co-administered GvHD immunosuppressive treatments were also tapered before Treg infusion.19,23 In the cohort published by Theil et al., patients received between 0.97 and 4.45 × 106 Tregs/kg body weight; one patient received a second Treg dose, and three of five patients received concomitant low-dose IL-2 applications, which complicates the differentiation between Treg transfusion related and direct IL-2 related impacts on the clinical course.23 The patient treated by Trzonkowski et al.19 received 1 × 105 Treg/kg body weight. In line with our data, both groups reported partial response or stable disease within the limited follow-up time of 5–23 weeks after Treg infusion. Similar to one patient with cGvHD described by Trzonkowski et al.,19 patient 2's symptoms of pulmonary fibrosis unexpectedly but substantially improved over time. The underlying pathophysiology for the diffuse mucosal hemorrhage in patient 2 remains unexplained as there were no direct or indirect indicators of GvHD or infections found.
From clinical approaches in humans, we know that it is very difficult to describe correlations between the dose and the effect of adoptively transferred Tregs in the transplant setting. Multiple clinical trials for various clinical indications are currently ongoing and registered at www.clinicaltrials.gov. Our group conducted a phase I/IIa study (NCT02371434 (ONEnTreg13) and EudraCT:2011-004,301-24 (ONErgt11) treating living donor kidney transplant patients with escalating doses of Tregs (3 + 3 design, n = 11, 0.5, 1.0, or 2.5–3.0×106 cells/kg body weight) while tapering standard immunosuppressive comedication. This study was conducted as part of the ONE study consortium, which aimed to investigate the feasibility and safety of different cell therapeutic approaches. Study patients were compared with a reference group, in which patients received standard of care. Although it seems counterintuitive that higher Treg doses are not superior to lower Treg doses, we did not find any dose-response association for any of the parameters investigated.38 These observations were in line with a parallel study conducted in two centers in the UK.39 Harden et al.39 found that a dose-dependent effect can be observed on an immunological but not on a clinical level, even if Tregs were administered at a dose as high as 10 × 106 per kg. Reports on Treg therapy in human patients suffering from GvHD are heterogeneous with respect to cell dose, Treg source, and patient characteristics. The administered Treg doses varied between 1 × 105 and 5 × 106, but no clear dose-response correlation can be derived from these data.19, 20, 21, 22, 23 Last but not least, one should bear in mind that the production process of Tregs varies from center to center and may lead to substantially different product characteristics; comparability is, therefore, limited and should be treated with caution. In summary, we chose the dose of 3.0×106 cells/kg body weight based on our clinical experience from the ONE study, in which we learnt that 3.0×106 cells/kg are safe, at least in adult patients, and after intensive discussions in a multidisciplinary team. The life-threatening condition of all patients made the treatment approach necessary, and the dose was chosen that was most likely to be safe and, more important, hopefully effective.
Concerns that the immunosuppressive properties of polyclonal Treg infusion could increase susceptibility to infections may have hampered the progressive development of broad Treg applications. In autoimmune and solid organ transplant recipients, however, there is no evidence of an increased risk, and immune monitoring has not revealed signs of over-immunosuppression.25 In 23 HSCT patients treated with Tregs as a GvHD prophylactic approach, Brunstein et al.40 showed a higher cumulative density (accounting for multiple infections in one individual patient per 1,000 patient-days) of opportunistic viral infections caused by human herpesvirus 6, CMV and parainfluenza compared with historical controls. However, the cumulative incidence remained comparable. We closely monitored all patients for viral infections or reactivation and found viral loads continuously at or below the lowest limit of detection, indicating sufficient control. Only patient 2 showed a short-term, low-level Epstein-Barr virus reactivation, which was promptly controlled by CSA tapering.
Di Ianni et al.20 reported in 2011 on 4 of 28 patients who underwent HLA-haploidentical HSCT and were prophylactically treated with Tregs but died from acute exacerbation of an pre-existing invasive aspergillosis. After Treg therapy, the authors, however, observed an improved immune reconstitution of protective T cells toward different pathogens, including opportunistic infections, most likely mediated by early T cell reconstitution after.20 Accordingly, if the pre-transplant conditioning regimen caused the loss of infection control and how Treg infusion may have additionally contributed to the exacerbation remains unresolved. Our results, in addition to previously published data, suggest that the overall low incidence of opportunistic infections, including aspergillosis, after Treg therapy in immunocompromised patients demonstrates a favorable safety profile. However, in the case of preexisting, active invasive fungal disease, Treg therapy might not be beneficial.
GvHD and the associated therapeutic interventions are known to substantially hamper T cell41, 42, 43, 44, 45, 46 and B cell47, 48, 49 engraftment and maturation. Accordingly, patients with cGvHD suffer from combined immunodeficiency. In addition, the cellular phenotype in cGvHD is not only characterized by low total lymphocyte counts, particularly of naive lymphocytes, but also by an imbalance between regulatory and effector T cells with persistently decreased frequencies of Tregs, and a shift toward a pro-inflammatory environment.42,50 A positive impact of Treg therapy on bone marrow functions, but limited to erythrocyte formation only, was first reported by Trzonkowski et al.,19 showing stabilized hemoglobin levels after Treg infusion in cGvHD. In our patients, we found a remarkable and lasting increase in circulating Tregs after cell infusion. The expansion of naive T and B cells is of particular importance and, thus, a good marker for the reconstitution of primary immune organs and related immunocompetence after transplantation. The TCR clonotypic analyses during the follow-up confirmed the long-lasting expansion of Tregs in the patients. We observed a very low frequency of clonal overlap between the patients' blood and the products before Treg therapy, despite long-lasting contact with the recipients' alloantigen and active GvHD releasing inflammatory triggers. Clearly, intrinsic HSCT-derived donor Tregs were not sufficiently activated in the host. Further, an increased expansion of donor Treg clonotypes with shared clones with the Treg product is paralleled by the increase of absolute Treg counts and relative Treg frequencies, suggesting the in vivo expansion of the Treg preactivated during manufacturing process. Additionally, the biphasic increase in circulating Treg numbers most likely reflected (i) adoptively transferred memory Tregs early after cell infusion, while (ii) at later stages the increase in circulating Tregs seems to be an additive effect of both (a) expansion of adoptively transferred Tregs and (b) the detection of naive Tregs, indicating intrinsic engraftment and expansion of this compartment. Indeed, naive Tregs and the increase of conventional naive CD4+ T cell, CD8+ T cell, and B cell subsets were detected after adoptive Treg transfer, even in the absence of IL-2 administration.
This observation is in contrast with finding from Theil et al.,23 who demonstrated slightly increased frequencies of naive Tregs only in patients who received combined IL-2/Treg therapy, suggesting that naive Treg induction might be IL-2 mediated.24 Another interesting observation was the increased number of DC after Treg infusion, underlining the improved competence of the adaptive immune system. Granulocytic and monocytic functions were not hampered after Treg therapy. These findings are of particular importance, since none of the previously investigated immunosuppressive drugs have been demonstrated to improve engraftment of naive B and T cells or DC to this extent, indicating true de novo synthesis from the bone marrow.
When interpreting these results, the following limiting factors should be considered: (i) we report on the individual case treatments of just three patients, and (ii) the patients suffered from cGvHD for a mean time of more than 2 years and received first-, second-, and third-line immunosuppressive therapies without satisfactory efficacy before Treg infusion, thus there may already have been irreversible tissue destruction in affected organs. As there is no standard for second and third-line or therapy-refractory GvHD therapy, our patients had received a heterogeneous treatment regimen before adoptive Treg therapy. Thus, the inter-individual comparability between the three treated patients is limited. As all patients with this severity of cGvHD have a very poor prognosis with a high short-term mortality risk, historical controls do not exist. For ethical reasons, we did not routinely perform comparable immunomonitoring in patients who did not receive Treg therapy on an individual treatment basis. However, since time since transplantation exceeded 600 days we consider the likelihood of improvement by spontaneous events or owing to alternative third-line treatment options very unlikely. Therefore, we consider the clinical and immunological of course each patient themselves as their matched historical control.
Based on this experience, a clinical trial is currently being planned with a standardized clinical protocol for the administration of adoptive Treg therapy in patients with cGvHD. Whether complete remission of cGvHD or, alternatively, control of chronically established GvHD and prevention of further progress is a realistic goal is debatable. Adoptive Treg therapy, will have to be compared to alternative immunomodulatory therapy approaches, e.g., the administration of the JAK1/2 inhibitor ruxolitinib51, 52, 53 and adoptive transfer of mesenchymal stem cells (MSC),54 for which promising results in the prevention and treatment of GvHD recently became available. Although beneficial effects were demonstrated for ruxolitinib, considerable side effects seem to limit the long-term administration in both adults and children.51,55 In contrast, despite the clinical efficacy reported for MSCs by some authors, data are contradictory and based on a recent Cochrane analysis further data are necessary to prove MSCs to be effective in preventing and/or treating GvHD.54 Based on our experience, we believe that, in addition to its good tolerability, the adoptive transfer of Tregs may be effective even in long-lasting cGVHD, but will most likely improve the overall outcome more effectively at a stage when the patient is (i) not yet critically ill, (ii) has suffered from less organ toxicity, and (iii) is at a lower risk for acute infection exacerbation.
Materials and methods
Study design
Three children with therapy-refractory or steroid-dependent GvHD were treated by adoptive transfer of ex vivo expanded Tregs on an individual treatment basis. All patients and their parents provided written informed consent after receiving detailed oral and written information. Before adoptive Treg therapy, maintenance immunosuppression, initiated before making the decision to undergo Treg therapy, was tapered to CSA (target trough level 80–100 μg/L) and the lowest clinically possible dose of prednisolone before cell therapy. A single dose of 3 × 106 HSCT donor-derived Treg/kg was administered by intravenous infusion. Single dose antipyretic and antihistamine was given 30 min before Treg application. Clinical and laboratory follow-up was performed using standardized clinical evaluation sheets as well as a laboratory work-up, including biochemistry, CSA trough levels, and hematologic, immunologic, and microbiologic parameters. Clinical follow-up was performed in accordance with the recommendations summarized by the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. The 2014 Response Criteria Working Group Report.56
To ensure a high validity of our data we used a combination of consistently having the same physician visiting all patients at every study visit and a systematic, validated documentation system. To quantify clinical changes the clinical follow-up score depicted in Table 2 was used to evaluate the patient's current status as compared the previous visit (Table 2).
Table 2.
Clinical follow-up score
| None | CR | Significantly |
Moderately |
Slightly |
Equal | Slightly |
Moderately |
Significantly |
|
|---|---|---|---|---|---|---|---|---|---|
| Improved | Worse | ||||||||
| General | +3 | +2 | +1 | 0 | −1 | −2 | −3 | ||
| Skin | +3 | +2 | +1 | 0 | −1 | −2 | −3 | ||
| Oral mucosa | +3 | +2 | +1 | 0 | −1 | −2 | −3 | ||
| GI tract | +3 | +2 | +1 | 0 | −1 | −2 | −3 | ||
| Eyes SCORE (sum) | +3 | +2 | +1 | 0 | −1 | −2 | −3 | ||
To monitor and quantify the clinical course of GvHD symptoms, the general condition as well as the status of the skin, mucosa, eyes and the GI-Tract were documented. Symptoms were scored, as compared to the previous visit, by +1 to +3 points for improvement of symptoms, while worsening of disease activity was scored by −1 to −3. Scores were cumulatively added up throughout the entire follow-up period. CR, complete remission; GI, gastrointestinal.
Treg product manufacturing
The clinical grade Treg products were manufactured at our GMP facility, Berlin Center for Advanced Therapies, as recently described in detail.38 The manufacturing procedure has been authorized by the regional and national regulatory authorities (LAGeSo-Berlin and Paul-Ehrlich Institute, respectively). Briefly, 50 mL peripheral blood was collected from the original hematopoietic stem cell donors. Tregs were isolated by depletion of CD8+ and enrichment of CD25+ cells using CliniMACs technology (Miltenyi Biotech). Subsequently, Tregs were stimulated by Treg-expansion beads (Miltenyi Biotech) and cultured for 23 days in the presence of IL-2 and a mammalian target of rapamycin inhibitor. The robust manufacturing process demonstrated high expansion rates, high purity, and high viability (Table 2). All products met our release criteria and complied with the safety-relevant parameters (mycoplasmas, endotoxins, sterility). The end-product was resuspended in 50 mL 0.9% saline and administered within 8 h after filling at a rate of 2 mL/min.
Immune monitoring
Safety and hematologic data were monitored at each clinical follow-up visit by routine measures. On a voluntary basis, additional clinical and laboratory parameters were assessed to monitor safety and pharmacokinetics/pharmacodynamics in more detail. Specifically, peripherally collected blood counts and standardized multi-color flow cytometry were used to assess more than 60 immune cell subsets as described by Streitz et al.57 Briefly, freshly collected blood (EDTA Vacutainers, BD) was stained using the DURAClone IM Phenotyping Basic, IM T cell Subsets IM Treg, IM TCRs, IM B Cell, and IM DC kits according to the manufacturer's protocol (Beckman Coulters). All samples were analyzed on CE-labeled 10-color Navios flow cytometers (Beckman Coulter). Exemplary plots of all gating strategies of subsets described in this manuscript are depicted in Figures S2–S7. The T cell repertoire was assessed in all Treg products by using next-generation sequencing, as well as in peripheral whole blood samples collected during follow-up of Treg-treated patients as described elsewhere.58
Data and materials availability
Most of the data are presented in the text, figures, and legends. Any additional data will be made available upon request by the corresponding author (Landwehr-kenzel.sybille@mh-hannover.de).
Acknowledgments
We are grateful to M. Streitz and Katrin Vogt (Institute of Medical Immunology, Charité University Medicine Berlin) for expert technical assistance. We thank Nicola Brindle for careful proofreading. Last but not least we thank all technicians at the BCRT, the Berlin Center for Advanced Therapies GMP Team, patients, parents, physicians, and nurses for their individual and collaborative support. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 825392 (RESHAPE) and from the European Union, Seventh Framework Programme [FP7/2007-2013], under grant agreement no HEALTH-F5-2010-260687 The ONE Study. The project was further funded by the German Federal Ministry of Education and Research (BMBF) under grant agreement with the Berlin Institute of Health and BIH Center for Regenerative Therapies (BCRT). S.L-K. was supported by a personal “Clinical Scientist” grant (BSRT/BCRT) and the Rahel-Hirsch-Habilitation Grant (Charité). The funders had no role in considering the study design or in the collection, analysis, interpretation of data, writing of the report, or decision to submit the article for publication.
Author contributions
Conceptualization: S.L.K., J.S.K., H.D.V., P.R.
Methodology: S.L.K., L.M.J., H.H., S.M., D.K., A.R., M.S.H., U.S., N.B., H.D.V., P.R.
Investigation: S.L.K., L.M.J., J.S.K., H.H., S.M., D.K., A.R., H.v.B., M.V., B.G., U.S., N.B.
Visualization: S.L.K., H.H., S.M., M.V., U.S.
Funding acquisition: S.L.K., M.A., N.B., H.D.V., P.R.
Project administration: S.L.K., M.A., D.K., P.R.
Supervision: S.L.K., H.D.V., P.R.
Writing – original draft: S.L.K., H.D.V., P.R.
Writing – review and editing: S.L.K., L.M.J., J.S.K., M.A., H.H., S.M., D.K., A.R., H.v.B., M.V., M.S.H., B.G., U.S., N.B., H.D.V., P.R.
Declaration of interests
Authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.02.025.
Supplemental information
References
- 1.Atkinson K. Chronic graft-versus-host disease. Bone Marrow Transplant. 1990;5:69–82. [PubMed] [Google Scholar]
- 2.Deeg H.J., Leisenring W., Storb R., Nims J., Flowers M.E., Witherspoon R.P., Sanders J., Sullivan K.M. Long-term outcome after marrow transplantation for severe aplastic anemia. Blood. 1998;91:3637–3645. [PubMed] [Google Scholar]
- 3.Lee S.J., Klein J.P., Barrett A.J., Ringden O., Antin J.H., Cahn J.-Y., Carabasi M.H., Gale R.P., Giralt S., Hale G.A., et al. Severity of chronic graft-versus-host disease: association with treatment-related mortality and relapse. Blood. 2002;100:406–414. doi: 10.1182/blood.v100.2.406. [DOI] [PubMed] [Google Scholar]
- 4.Goerner M., Gooley T., Flowers M.E.D., Sullivan K.M., Kiem H.-P., Sanders J.E., Martin P.J., Storb R. Morbidity and mortality of chronic GVHD after hematopoietic stem cell transplantation from HLA-identical siblings for patients with aplastic or refractory anemias. Biol. Blood Marrow Transplant. 2002;8:47–56. doi: 10.1053/bbmt.2002.v8.pm11858190. [DOI] [PubMed] [Google Scholar]
- 5.Zeiser R., Blazar B.R. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N. Engl. J. Med. 2017;377:2565–2579. doi: 10.1056/NEJMra1703472. [DOI] [PubMed] [Google Scholar]
- 6.Ferrara J.L., Levine J.E., Reddy P., Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–1561. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakaguchi S., Sakaguchi N., Asano M., Itoh M., Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 8.Seddiki N., Santner-Nanan B., Martinson J., Zaunders J., Sasson S., Landay A., Solomon M., Selby W., Alexander S.I., Nanan R., et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 2006;203:1693–1700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edinger M. Regulatory T cells for the prevention of graft-versus-host disease: professionals defeat amateurs. Eur. J. Immunol. 2009;39:2966–2968. doi: 10.1002/eji.200940030. [DOI] [PubMed] [Google Scholar]
- 10.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 11.Taylor P.A., Lees C.J., Blazar B.R. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493–3499. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 12.Siepert A., Ahrlich S., Vogt K., Appelt C., Stanko K., Kühl A., van den Brandt J., Reichardt H.M., Nizze H., Lehmann M., et al. Permanent CNI treatment for prevention of renal allograft rejection in sensitized hosts can Be replaced by regulatory T cells. Am. J. Transplant. 2012;12:2384–2394. doi: 10.1111/j.1600-6143.2012.04143.x. [DOI] [PubMed] [Google Scholar]
- 13.Orlando G., Hematti P., Stratta R.J., Burke G.W., Di Cocco P., Pisani F., Soker S., Wood K. Clinical operational tolerance after renal transplantation: current status and future challenges. Ann. Surg. 2010;252:915–928. doi: 10.1097/SLA.0b013e3181f3efb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews J.B., Ramos E., Bluestone J.A. Clinical trials of transplant tolerance: slow but steady progress. Am. J. Transplant. 2003;3:794–803. doi: 10.1046/j.1600-6135.2003.0154.x. [DOI] [PubMed] [Google Scholar]
- 15.Wood K.J., Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat. Rev. Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 16.Wood K.J., Bushell A., Hester J. Regulatory immune cells in transplantation. Nat. Rev. Immunol. 2012;12:417–430. doi: 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- 17.Landwehr-Kenzel S., Issa F., Luu S.-H., Schmück M., Lei H., Zobel A., Thiel A., Babel N., Wood K., Volk H.-D., et al. Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am. J. Transplant. 2014;14:594–606. doi: 10.1111/ajt.12629. [DOI] [PubMed] [Google Scholar]
- 18.Valujskikh A., Baldwin W.M., Fairchild R.L. Recent progress and new perspectives in studying T cell responses to allografts. Am. J. Transplant. 2010;10:1117–1125. doi: 10.1111/j.1600-6143.2010.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trzonkowski P., Bieniaszewska M., Juścińska J., Dobyszuk A., Krzystyniak A., Marek N., Myśliwska J., Hellmann A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol. 2009;133:22–26. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Ianni M.D., Falzetti F., Carotti A., Terenzi A., Castellino F., Papa B.D., Zei T., Ostini R.I., Cecchini D., Aloisi T., et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 21.Edinger M., Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr. Opin. Immunol. 2011;23:679–684. doi: 10.1016/j.coi.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Brunstein C.G., Miller J.S., Cao Q., McKenna D.H., Hippen K.L., Curtsinger J., Defor T., Levine B.L., June C.H., Rubinstein P., et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theil A., Tuve S., Oelschlägel U., Maiwald A., Döhler D., Oßmann D., Zenkel A., Wilhelm C., Middeke J.M., Shayegi N., et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy. 2015;17:473–486. doi: 10.1016/j.jcyt.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Koreth J., Matsuoka K., Kim H., McDonough S.M., Bindra B., Alyea E.P., III, Armand P., Cutler D., Ho V., Treister N., et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2014;365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulanic D., Lozier J.N., Pavletic S.Z. Thrombocytopenia and hemostatic disorders in chronic graft versus host disease. Bone Marrow Transplant. 2009;44:393–403. doi: 10.1038/bmt.2009.196. [DOI] [PubMed] [Google Scholar]
- 26.Lei H., Reinke P., Volk H.D., Lv Y., Wu R. Mechanisms of immune tolerance in liver transplantation-crosstalk between alloreactive T cells and liver cells with therapeutic prospects. Front. Immunol. 2019;10:1–12. doi: 10.3389/fimmu.2019.02667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Der Maas N.G., Berghuis D., Van Der Burg M., Lankester A.C. B cell reconstitution and influencing factors after hematopoietic stem cell transplantation in children. Front. Immunol. 2019;10:782. doi: 10.3389/fimmu.2019.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanco E., Pérez-Andrés M., Arriba-Méndez S., Contreras-Sanfeliciano T., Criado I., Pelak O., Serra-Caetano A., Romero A., Puig N., Remesal A., et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 2018;141:2208–2219.e16. doi: 10.1016/j.jaci.2018.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Stenger E.O., Turnquist H.R., Mapara M.Y., Thomson A.W. Dendritic cells and regulation of graft-versus-host disease and graft-versus-leukemia activity. Blood. 2012;119:5088–5103. doi: 10.1182/blood-2011-11-364091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinze A., Elze M.C., Kloess S., Ciocarlie O., Königs C., Betz S., Bremm M., Esser R., Klingebiel T., Serban M., et al. Age-matched dendritic cell subpopulations reference values in childhood. Scand. J. Immunol. 2013;77:213–220. doi: 10.1111/sji.12024. [DOI] [PubMed] [Google Scholar]
- 31.Li X., Gao Q., Feng Y., Zhang X. Developing role of B cells in the pathogenesis and treatment of chronic GVHD. Br. J. Haematol. 2019;184:323–336. doi: 10.1111/bjh.15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Serres S., Sayegh M., Najafian N. Immunosuppressive drugs and Tregs: a critical evaluation! Clin. J. Am. Soc. Nephrol. 2009;4:1661–1669. doi: 10.2215/CJN.03180509. [DOI] [PubMed] [Google Scholar]
- 33.Segundo D.S., Ruiz J.C., Fernández-Fresnedo G., Izquierdo M., Gómez-Alamillo C., Cacho E., Benito M.J., Rodrigo E., Palomar R., López-Hoyos M., et al. Calcineurin inhibitors affect circulating regulatory T cells in stable renal transplant recipients. Transplant. Proc. 2006;38:2391–2393. doi: 10.1016/j.transproceed.2006.08.081. [DOI] [PubMed] [Google Scholar]
- 34.Demirkiran A., Sewgobind V.D., Van Der Weijde J., Kok A., Baan C.C., Kwekkeboom J., Tilanus H.W., Metselaar H.J., Van Der Laan L.J.W. Conversion from calcineurin inhibitor to mycophenolate mofetil-based immunosuppression changes the frequency and phenotype of CD4+FOXP3+ regulatory T cells. Transplantation. 2009;87:1062–1068. doi: 10.1097/TP.0b013e31819d2032. [DOI] [PubMed] [Google Scholar]
- 35.Korczak-Kowalska G., Wierzbicki P., Bocian K., Klosowska D., Niemczyk M., Wyzgal J., Korecka A., Durlik M., Chmura A., Paczek L., et al. The influence of immuosuppressive therapy on the development of CD4+CD25+ T cells after renal transplantation. Transplant. Proc. 2007;39:2721–2723. doi: 10.1016/j.transproceed.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Segundo D.S., Ruiz J.C., Izquierdo M., Fernández-Fresnedo G., Gómez-Alamillo C., Merino R., Benito M.J., Cacho E., Rodrigo E., Palomar R., et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4 +CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation. 2006;82:550–557. doi: 10.1097/01.tp.0000229473.95202.50. [DOI] [PubMed] [Google Scholar]
- 37.Landwehr-Kenzel S., Zobel A., Schmitt-Knosalla I., Forke A., Hoffmann H., Schmueck-Henneresse M., Klopfleisch R., Volk H., Reinke P. Cyclosporine A but not corticosteroids support efficacy of ex vivo expanded, adoptively transferred human Tregs in GvHD. Front. Immunol. 2021;12:716629. doi: 10.3389/fimmu.2021.716629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roemhild A., Otto N.M., Moll G., Abou-El-Enein M., Kaiser D., Bold G., Schachtner T., Choi M., Oellinger R., Landwehr-Kenzel S., et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. BMJ. 2020;371:m3734. doi: 10.1136/bmj.m3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harden P.N., Game D.S., Sawitzki B., Van der Net J.B., Hester J., Bushell A., Issa F., Brook M.O., Alzhrani A., Schlickeiser S., et al. Feasibility, long-term safety, and immune monitoring of regulatory T cell therapy in living donor kidney transplant recipients. Am. J. Transplant. 2021;21:1603–1611. doi: 10.1111/ajt.16395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunstein C.G., Blazar B.R., Miller J.S., Cao Q., Hippen K.L., McKenna D.H., Curtsinger J., McGlave P.B., Wagner J.E. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol. Blood Marrow Transplant. 2013;19:1271–1273. doi: 10.1016/j.bbmt.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kook H., Goldman F., Padley D., Giller R., Rumelhart S., Holida M., Lee N., Peters C., Comito M., Huling D., et al. Reconstruction of the immune system after unrelated or partially matched T-cell-depleted bone marrow transplantation in children: immunophenotypic analysis and factors affecting the speed of recovery. Blood. 1996;88:1089–1097. [PubMed] [Google Scholar]
- 42.Alho A.C., Kim H.T., Chammas M.J., Reynolds C.G., Matos T.R., Forcade E., Whangbo J., Nikiforow S., Cutler C.S., Koreth J., et al. Unbalanced recovery of regulatory and effector T cells after allogeneic stem cell transplantation contributes to chronic GVHD. Blood. 2016;127:646–657. doi: 10.1182/blood-2015-10-672345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson D., Defor T., Burns L., Mcglave P., Miller J., Wagner J., Weisdorf D. A comparison of related donor peripheral blood and bone marrow transplants: importance of late-onset chronic graft-versus-host disease and infections. Biol. Blood Marrow Transplant. 2003;9:52–59. doi: 10.1053/bbmt.2003.50000. [DOI] [PubMed] [Google Scholar]
- 44.Törlén J., Gaballa A., Remberger M., Mörk L.M., Sundberg B., Mattsson J., Uhlin M. Effect of graft-versus-host disease prophylaxis regimens on T and B cell reconstitution after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2019;25:1260–1268. doi: 10.1016/j.bbmt.2019.01.029. [DOI] [PubMed] [Google Scholar]
- 45.Van Roessel I., Prockop S.E., Klein E., Boulad F., Scaradavou A., Spitzer B., Kung A., Curran K., Cancio M., O’Reilly R.J., et al. Early CD4+ T cell reconstruction as predictor for outcomes after allogeneic hematopoietic cell transplantation in pediatric and young adult patients: a validation cohort analyses. Biol. Blood Marrow Transplant. 2020;26:S302–S303. [Google Scholar]
- 46.MacDonald K.P.A., Hill G.R., Blazar B.R. Chronic graft-versus-host disease: biological insights from preclinical and clinical studies. Blood. 2017;129:13–21. doi: 10.1182/blood-2016-06-686618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scarselli A., Di Cesare S., Capponi C., Cascioli S., Romiti M.L., Di Matteo G., Simonetti A., Palma P., Finocchi A., Lucarelli B., et al. Longitudinal evaluation of immune reconstitution and B-cell function after hematopoietic cell transplantation for primary immunodeficiency. J. Clin. Immunol. 2015;35:373–383. doi: 10.1007/s10875-015-0154-4. [DOI] [PubMed] [Google Scholar]
- 48.Hilgendorf I., Mueller-Hilke B., Kundt G., Holler E., Hoffmann P., Edinger M., Freund M., Wolff D. The lack of memory B cells including T cell independent IgM + IgD + memory B cells in chronic graft-versus host disease is associated with susceptibility to infection. Transpl. Int. 2012;25:87–96. doi: 10.1111/j.1432-2277.2011.01388.x. [DOI] [PubMed] [Google Scholar]
- 49.Abdel-Azim H., Elshoury A., Mahadeo K.M., Parkman R., Kapoor N. Humoral immune reconstitution kinetics after allogeneic hematopoietic stem cell transplantation in children: a maturation block of IgM memory B cells may lead to impaired antibody immune reconstitution. Biol. Blood Marrow Transplant. 2017;23:1437–1446. doi: 10.1016/j.bbmt.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Beres A.J., Drobyski W.R. The role of regulatory T cells in the biology of graft versus host disease. Front. Immunol. 2013;4:163. doi: 10.3389/fimmu.2013.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mozo Y., Bueno D., Sisinni L., Fernández-Arroyo A., Rosich B., Martínez A.P., Benítez-Carabante M.I., Alonso L., Uría M.L., Heredia C.D.D., et al. Ruxolitinib for steroid-refractory graft versus host disease in pediatric HSCT: high response rate and manageable toxicity. Pediatr. Hematol. Oncol. 2021;38:331–345. doi: 10.1080/08880018.2020.1868637. [DOI] [PubMed] [Google Scholar]
- 52.Jagasia M., Perales M.A., Schroeder M.A., Ali H., Shah N.N., Chen Y.B., Fazal S., Dawkins F.W., Arbushites M.C., Tian C., et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood. 2020;135:1739–1749. doi: 10.1182/blood.2020004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hechinger A.K., Smith B.A.H., Flynn R., Hanke K., McDonald-Hyman C., Taylor P.A., Pfeifer D., Hackanson B., Leonhardt F., Prinz G., et al. Therapeutic activity of multiple common γ-chain cytokine inhibition in acute and chronic GVHD. Blood. 2015;125:570–580. doi: 10.1182/blood-2014-06-581793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fisher S.A., Cutler A., Doree C., Brunskill S.J., Stanworth S.J., Navarrete C., Girdlestone J. Mesenchymal stromal cells as treatment or prophylaxis for acute or chronic graft-versus-host disease in haematopoietic stem cell transplant (HSCT) recipients with a haematological condition. Cochrane Database Syst. Rev. 2019;1:CD009768. doi: 10.1002/14651858.CD009768.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeiser R., von Bubnoff N., Butler J., Mohty M., Niederwieser D., Or R., Szer J., Wagner E.M., Zuckerman T., Mahuzier B., et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 2020;382:1800–1810. doi: 10.1056/NEJMoa1917635. [DOI] [PubMed] [Google Scholar]
- 56.Lee S.J., Wolff D., Kitko C., Koreth J., Inamoto Y., Jagasia M., Pidala J., Olivieri A., Martin P.J., Przepiorka D., et al. Measuring therapeutic response in chronic graft-versus-host disease. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: IV. The 2014 Response Criteria Working Group report. Biol. Blood Marrow Transplant. 2015;21:984–999. doi: 10.1016/j.bbmt.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Streitz M., Miloud T., Kapinsky M., Reed M.R., Magari R., Geissler E.K., Hutchinson J.A., Vogt K., Schlickeiser S., Kverneland A.H., et al. Standardization of whole blood immune phenotype monitoring for clinical trials: panels and methods from the ONE study. Transplant. Res. 2013;2:17. doi: 10.1186/2047-1440-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dziubianau M., Hecht J., Kuchenbecker L., Sattler A., Stervbo U., Rödelsperger C., Nickel P., Neumann A.U., Robinson P.N., Mundlos S., et al. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology. Am. J. Transplant. 2013;13:2842–2854. doi: 10.1111/ajt.12431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.