

Abstract
Nonalcoholic steatohepatitis (NASH), characterized by hepatic steatosis, inflammation, and liver injury, has become a leading cause of end‐stage liver diseases and liver transplantation. Krüppel‐like factors 10 (KLF10) is a Cys2/His2 zinc finger transcription factor that regulates cell growth, apoptosis, and differentiation. However, whether it plays a role in the development and progression of NASH remains poorly understood. In the present study, we found that KLF10 expression was selectively upregulated in the mouse models and human patients with NASH, compared with simple steatosis (NAFL). Gain‐ and loss‐of function studies demonstrated that hepatocyte‐specific overexpression of KLF10 aggravated, whereas its depletion alleviated diet‐induced NASH pathogenesis in mice. Mechanistically, transcriptomic analysis and subsequent functional experiments showed that KLF10 promotes hepatic lipid accumulation and inflammation through the palmitoylation and plasma membrane localization of fatty acid translocase CD36 via transcriptionally activation of zDHHC7. Indeed, both expression of zDHHC7 and palmitoylation of CD36 are required for the pathogenic roles of KLF10 in NASH development. Thus, our results identify an important role for KLF10 in NAFL‐to‐NASH progression through zDHHC7‐mediated CD36 palmitoylation.
Keywords: CD36, KLF10, nonalcoholic steatohepatitis, palmitoylation, zDHHC7
Subject Categories: Membranes & Trafficking, Metabolism, Post-translational Modifications & Proteolysis
KLF10 enhances hepatic lipid accumulation, inflammation, and fibrosis by transcriptionally upregulating zDHHC7, which promotes palmitoylation and plasma membrane localization of CD36 in hepatocytes.
Introduction
Due to rapid changes in lifestyles, the global prevalence of nonalcoholic fatty liver disease (NAFLD) has reached more than 25% (Younossi et al, 2018; Zhou et al, 2019). While patients with simple steatosis (NAFL) have a low risk of poor prognosis (Loomba et al, 2021), about 25% of patients will develop nonalcoholic steatohepatitis (NASH), which is the main cause of end‐stage liver diseases and liver transplantation (Lindenmeyer & McCullough, 2018; Schwabe et al, 2020). During the past years, a number of studies have uncovered the pathogenesis in NAFL, such as insulin resistance, dysregulation of adipokines, and changes in gut microbiome (Friedman et al, 2018). However, the molecular mechanism of NAFL‐to‐NASH progression remains poorly understood.
Hepatocytes play a pivotal role in the hepatic and systemic lipid homeostasis through regulation of lipogenesis, fatty acid β‐oxidation, lipid uptake, and secretion. Dysregulation of these processes can lead to the development of NAFL and subsequent progression to NASH. Fatty acid translocase CD36 is a widely expressed membrane glycoprotein, which facilitates the uptake and intracellular trafficking of long‐chain fatty acids (Coburn et al, 2000; Hames et al, 2014). The substantial contribution of CD36 to the pathogenesis of NAFL and NASH has been well acknowledged by multiple animal and human studies (Rada et al, 2020). Besides, growing evidence indicates that the role played by CD36 extends far beyond the transport of fatty acids (FAs). CD36 also impacts FAs oxidation by influencing the activation of monophosphate‐activated protein kinase (AMPK; Samovski et al, 2015). Depletion of CD36 markedly reduced inflammatory response in high‐fat‐diet (HFD)‐induced obese mice by inhibiting the c‐JUN N‐terminal kinase (JNK) pathway (Kennedy et al, 2011). Interestingly, it has been demonstrated that CD36 protein is palmitoylated at cysteine residues (cys) 3, 7, 464, and 466 on both N‐ and C‐terminal cytoplasmic tails (Tao et al, 1996). Inhibition of CD36 palmitoylation results in the endoplasmic reticulum (ER) accumulation of CD36 and decreases its incorporation into plasma membrane rafts, thus reducing the efficiency of FAs uptake (Thorne et al, 2010). These findings suggest that targeting CD36 palmitoylation or its upstream regulators might be a promising therapeutic approach to treat NAFLD and NASH.
Multiple lines of evidence demonstrated that Krüppel‐like factors (KLFs), a family of Cys2/His2 zinc finger transcription factors, are involved in hepatic and systemic glucose and lipid homeostasis (Hsieh et al, 2019). For instance, KLF11 modulates hepatic lipid metabolism by inducing the expression of peroxisome proliferator‐activated receptor α and its target genes (Zhang et al, 2013). KLF15 can upregulate hepatic gluconeogenesis, and is an important therapeutic target for anti‐diabetic drug—metformin (Gray et al, 2007; Takashima et al, 2010). Besides, in response to hormonal signals, KLF9 is induced in hepatocytes and promotes hepatic glucose production and fatty acid oxidation through stimulating PGC‐1a gene expression (Cui et al, 2019). As such, understanding the roles of KLFs may provide new insight into the mechanisms of metabolic liver diseases (Li et al, 2020).
In the present study, through unbiased transcriptome analysis, we demonstrate that KLF10 is selectively increased in the livers of mouse models and human patients with nonalcoholic steatohepatitis. We further investigated the role and molecular mechanism of KLF10 in NAFL‐to‐NASH progression. Through in vivo gain‐ and loss‐of‐function studies, our findings identify hepatocyte KLF10 as a novel regulator in NASH progression.
Results
KLF10 is significantly increased in the livers of NASH mice and patients
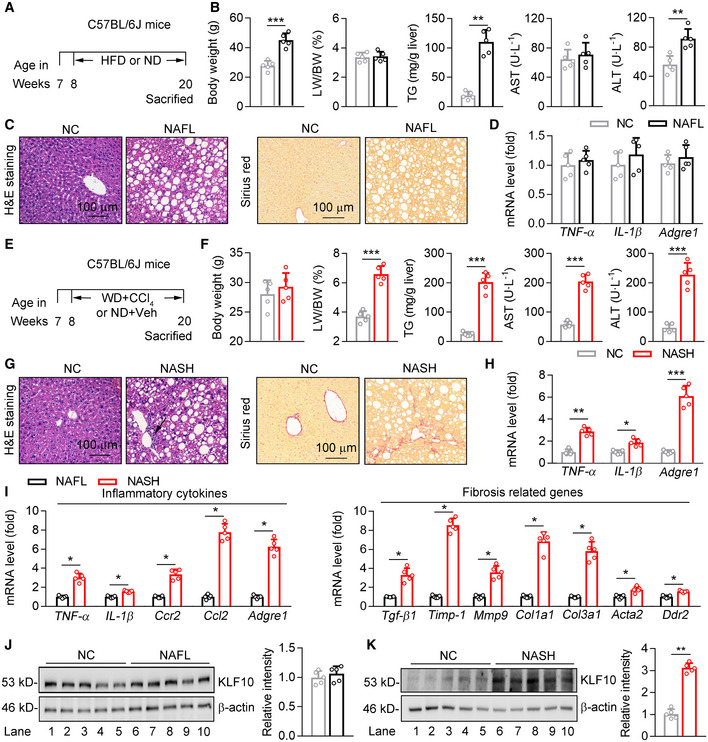
High‐fat diet (HFD)‐fed mice usually developed obesity, insulin resistance, and hepatic steatosis (NAFL) without dramatic inflammatory response and obvious liver injury and fibrosis (Charlton et al, 2011; Asgharpour et al, 2016). In contrast, western diet (WD)‐feeding plus CCl4 treatment (WD/CCl4) can induce histological, immunological, and transcriptomic features resembling the pathogenesis of human NASH (Tsuchida et al, 2018). Therefore, to identify the key factors in the progression of NAFL to NASH, 8‐week‐old C57BL/6 male mice were divided into two groups. In the first group, mice were fed with HFD or normal diet (ND) for 12 weeks (Fig EV1A). As expected, compared with mice fed an ND, HFD‐feeding increased body weights, hepatic triglyceride (TG) contents and serum alanine transaminase (ALT) levels without significant changes in serum aspartate transaminase (AST) levels, liver fibrosis, and expression levels of inflammatory genes (TNF‐α, IL‐1β, and Adgre1; Fig EV1B–D). In the second group, mice were treated with WD/CCl4 or ND/vehicle control for 12 weeks (Fig EV1E). Body weights were comparable between WD/CCl4‐ and ND/vehicle control‐treated mice (Fig EV1F), which is consistent with previous observations (Tsuchida et al, 2018). However, compared with ND/vehicle control‐treated mice, mice kept on the WD/CCl4 had a dramatic increase of liver/body weight ratio, hepatic TG contents, and serum AST and ALT levels (Fig EV1F). In addition, lobular inflammation and fibrosis were detected by H&E and Sirius red staining in the livers of WD/CCl4‐treated mice (Fig EV1G). The mRNA levels of hepatic inflammatory genes were also significantly upregulated in WD/CCl4‐treated mice (Fig EV1H). Furthermore, when compared with HFD mice, expression of inflammation‐ and fibrosis‐related genes was significantly increased in the livers of WD/CCl4 mice (Fig EV1I). Collectively, these findings suggest that HFD‐feeding may induce NAFL while WD/CCl4 treatment can induce NASH.
Figure EV1. KLF10 expression was selectively induced in the livers from NASH mice.
-
A–D8‐week‐old male C57BL/6J mice were kept on high‐fat‐diet‐feeding (HFD) or normal diet (ND) for 12 weeks to induce NAFL model. n = 5 per group. (A) Scheme for the experimental strategy. (B) Body weights, liver/body weight ratio, hepatic TG contents, and serum AST and ALT levels. (C) Representative H&E staining and Sirius red staining of liver sections. (D) Relative mRNA levels of hepatic inflammatory genes.
-
E–H8‐week‐old male C57BL/6J mice were kept on WD/CCl4 or ND/Vehicle control for 12 weeks to induce NASH model. n = 5 per group. (E) Scheme for the experimental strategy. (F) Body weights, liver/body weight ratio, hepatic TG contents, and serum AST and ALT levels. (G) Representative H&E staining and Sirius red staining of liver sections. (H) Relative mRNA levels of hepatic inflammatory genes.
-
I–KComparison of WD/CCl4‐treated mice (NASH) and HFD‐fed mice (NAFL). n = 5 per group. (I) Relative mRNA level of genes involved in the hepatic inflammation and fibrosis. (J) Protein levels of KLF10 in the liver, and its normalization to β‐actin. (K) Representative KLF10 immunofluorescence staining of liver sections.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (B, D, F, H, I–K).
Source data are available online for this figure.
To explore the potential contributing factors in the progression from NAFL to NASH, transcriptome RNA sequencing was performed using the livers of HFD feeding‐induced NAFL mice and WD/CCl4‐induced NASH mice (Fig 1A). Differentially expressed genes were identified using an adjusted P‐value < 0.05 and fold change > 2.0 (Dataset EV1). As a result, compared with NAFL mice, 514 genes were significantly and differentially expressed in the livers of NASH mice. Among these genes, 328 genes showed an increased expression, while 186 genes showed a decreased expression. The most highly upregulated genes included a range of novel genes but also those known to regulate NASH progression, such as Lcn2 (Ye et al, 2016), Mmp12 (Weng et al, 2018), Ccl2 (Baeck et al, 2012). Of note, our screen revealed a markedly upregulation of KLF10 in the livers of NASH mice (Fig 1A). The upregulation of KLF10 was further confirmed by quantitative real‐time PCR, western blots, and immunohistochemistry (Fig 1B–D). Indeed, KLF10 expression was comparable between HFD mice and their controls (Fig EV1J), but substantially increased in the WD/CCl4‐treated mice when compared to ND/vehicle control‐treated mice (Fig EV1K). We also found that hepatic KLF10 expression is positively correlated with NAFLD activity score (NAS), inflammation score, fibrosis score, and `triglyceride contents in mouse livers (Fig 1E).
Figure 1. Hepatic KLF10 is induced in NASH mice and patients.
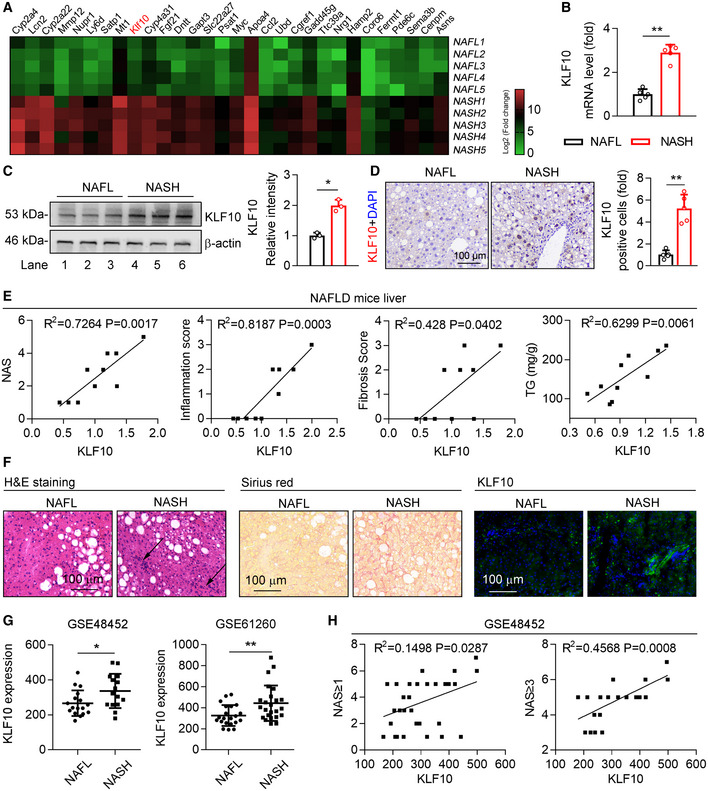
- Heatmap of genes in the livers of HFD‐feeding induced NAFL mice and WD/CCl4‐treatment induced NASH mice. n = 5. The color gradient represents log2 (Fold change).
- Relative mRNA levels of KLF10 in the livers from NAFL and NASH mice. n = 5.
- Protein levels of KLF10 examined by western blots (left) and its expression was normalized to the β‐actin (right). n = 3. Results represent three independent experiments.
- Representative immunohistochemistry of KLF10 protein expression in the liver sections from NAFL and NASH mice. The bar chart showed the fold change of KLF10 positive cells per field (NASH vs. NAFL). n = 5.
- Correlations between KLF10 and NAS, inflammation score, fibrosis scores, and hepatic triglyceride contents (TG) from NAFL mice (n = 7) and NASH mice (n = 8).
- H&E staining, Sirius red staining, and KLF10 immunofluorescence staining of liver sections from NAFL and NASH patients. n = 3.
- Spearman correlations between KLF10 expression and NAS in liver tissues from NAFL and NASH patients using GEO datasets (GSE48452). Left panel, n = 32 (including 17 NAFL patients and 15 NASH patients). Right panel, n = 21 (including 6 NAFL and 15 NASH patients).
Data information: *P < 0.05, **P < 0.01‐. Results are shown as mean ± SD. Student’s t‐test (B–D, G). Spearman’s correlation (E, H).
Source data are available online for this figure.
To assess the clinical relevance of our findings, liver biopsies from patients with NAFL and NASH were collected. Immunostaining showed that KLF10 protein abundance was increased in the livers of NASH patients compared with patients with NAFL (Fig 1F). We also performed gene expression analysis in NASH patients using Gene Expression Omnibus (GEO) datasets (GSE48452 and GSE61260) obtained from NCBI database. As shown in Fig 1G, KLF10 expression was increased in the liver tissues of NASH patients compared to NAFL patients in two databases (Fig 1G). Additionally, KLF10 expression was positively correlated with NAS score (GSE48452) (Fig 1H).
KLF10 aggravates hepatic lipid accumulation and inflammatory response in NASH mice
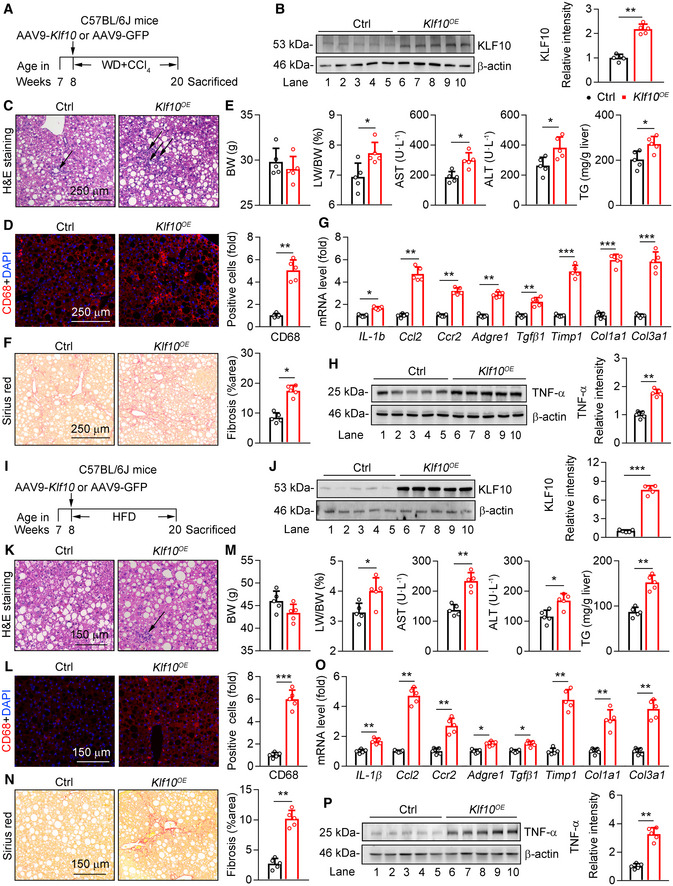
To explore the role of KLF10 in NASH progression, we performed mouse experiments by two different ways. Firstly, thyroxine binding globulin (TBG) promoter‐driven adeno‐associated virus (AAV9) containing Klf10 or GFP was administered into C57BL/6J mice via tail vein injection. Two groups of mice were then treated with WD/CCl4 for 12 weeks (Fig 2A). This approach would explore whether liver‐specific KLF10 overexpression (KLF10OE) could aggravate NASH progression. Our findings showed that protein levels of KLF10 were significantly increased in the livers of mice with AAV‐KLF10 injection (Fig 2B). As a result, KLF10OE promoted inflammatory cell infiltration in mice as shown by H&E staining and CD68 immunofluorescence, respectively (Fig 2C and D). Besides, although body weights were comparable (Fig 2E), liver/body weight ratio, serum AST and ALT levels, and hepatic TG contents were significantly increased in mice with KLF10 overexpression (Fig 2E). Hepatic KLF10OE also aggravated liver fibrosis in mice as shown by Sirius red staining (Fig 2F). In agreement, mRNA levels of genes involved in hepatic inflammation and fibrosis were upregulated in mice overexpressing KLF10 (Fig 2G). Protein contents of TNF‐α in the livers were also increased (Fig 2H).
Figure 2. Hepatic KLF10 overexpression promotes NASH pathogenesis in mice.
-
A–H8‐week‐old male C57BL/6J mice were administered with AAV‐GFP or AAV ‐Klf10 through tail vein injection, and then kept on WD/CCl4 for 12 weeks. n = 5 per group. (A) Scheme for the experimental strategy. (B) Protein levels of KLF10 in the livers (left) and its expression was normalized to the β‐actin (right). (C) H&E staining of liver sections. (D) CD68 immunofluorescence staining of liver sections (left) and its quantitation (right). (E) Body weight (BW), liver/body weight ratio, serum AST and ALT levels, and hepatic TG contents of two groups of mice. (F) Sirius red staining of liver sections (left) and its quantitation (right). (G) Relative mRNA levels of genes involved in the hepatic inflammation and fibrosis. (H) Protein levels of TNF‐α in the livers (left) and its expression was normalized to the β‐actin (right).
-
I–P8‐week‐old male C57BL/6J mice were administered with AAV‐GFP or AAV ‐Klf10 through tail vein injection, and then kept on HFD‐feeding for 12 weeks. n = 5 per group. (I) Scheme for the experimental strategy. (J) Protein levels of KLF10 in the livers (left) and its expression was normalized to β‐actin (right). (K) H&E staining of liver sections. (L) CD68 immunofluorescence staining of liver sections (left) and its quantitation (right). (M) Body weight (BW), liver/body weight ratio, serum AST and ALT levels, and hepatic TG contents. (N) Sirius red staining of liver sections (left) and its quantitation (right). (O) Relative mRNA levels of genes involved in the hepatic inflammation and fibrosis. (P) Protein levels of TNF‐α in the livers (left) and its expression was normalized to the β‐actin (right).
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (B, D–G, H, J, L–P).
Source data are available online for this figure.
Secondly, C57BL/6J mice were administered with AAV‐Klf10 or ‐GFP and then fed with HFD for 12 weeks (Fig 2I and J). This approach would explore whether KLF10 overexpression could promote NAFL‐to‐NASH development. As expected, KLF10 overexpression induced obvious liver damage, as shown by an increase of inflammatory cells infiltration (Fig 2K and L). While KLF10 overexpression did not affect body weight (Fig 2M), it led to a higher liver/body weight ratio, serum ALT and AST levels, and hepatic TG contents (Fig 2M). Liver fibrosis was observed in HFD mice expressing AAV‐KLF10 (Fig 2N). Consistently, the mRNA levels of inflammatory and fibrotic genes were upregulated (Fig 2O). Protein levels of TNF‐α were also increased (Fig 2P). Collectively, both experiments indicated that hepatic KLF10OE can promote diet‐induced NASH pathogenesis in mice.
KLF10 upregulates zDHHC7 expression through binding to its promoter
To explore the molecular mechanism(s) of KLF10 in promoting NASH pathogenesis, we performed RNA sequencing analysis in mouse primary hepatocytes (MPHs) transfected with lentivirus expressing KLF10 or empty vector as a negative control (Fig 3A and B). The top 10 upregulated genes and 10 downregulated genes were shown in Fig 3A (P < 0.001). Among the dysregulated genes induced by KLF10 overexpression, zDHHC7 and Gadd45g were also upregulated in the livers of WD/CCl4‐treated mice compared with HFD‐fed mice (highlighted in red in Dataset EV1). zDHHC7 belongs to a large family of DHHC‐containing palmitoyl acyltransferases. Among them, zDHHC4 and zDHHC5 have been shown to regulate palmitoylation and plasma membrane localization of CD36, thereby modulating lipid metabolism in white adipocytes (Wang et al, 2019). Therefore, zDHHC7 was chosen as a potential downstream target gene of KLF10 for further analysis. We confirmed that mRNA and protein levels of zDHHC7 were upregulated in MPHs after KLF10 overexpression (Fig 3B). Furthermore, expression levels of KLF10 and zDHHC7 were positively correlated in the liver tissues from GEO database (GSE61260 and GSE48452) (Fig 3C and D). KLF10 overexpression also significantly upregulated zDHHC7 expression in HepG2 cells (Fig EV2A and B). However, mRNA expression of other zDHHCs were not affected (Fig 3E), suggesting a specific regulation of zDHHC7 by KLF10. Consistently, protein levels of zDHHC7 were also dramatically increased in the livers of two mouse models with KLF10 overexpression as described in Fig 2 (Fig 3F).
Figure 3. KLF10 transcriptionally activates zDHHC7 expression.
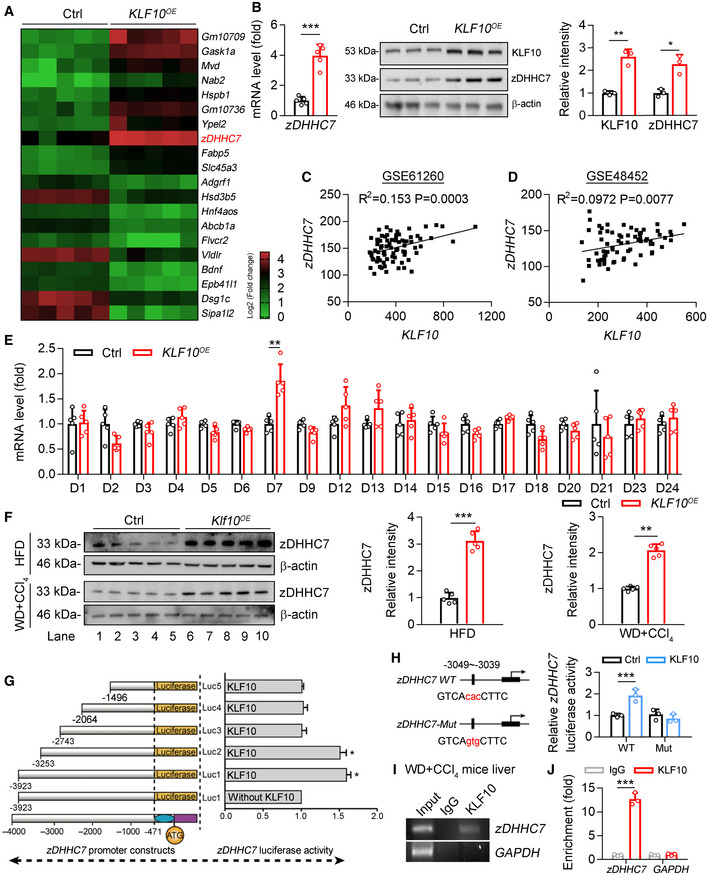
-
AHeatmap of genes in mouse primary hepatocytes transfected with lentivirus vectors expressing KLF10 (KLF10 OE ) or empty vector (Ctrl), P < 0.001. n = 5 per group. The color gradient represents log2 (Fold change).
-
BRelative mRNA (n = 5) and protein (n = 3) levels of zDHHC7 in MPHs expressing KLF10 or empty vector (Ctrl). The protein level of KLF10 or zDHHC7 was normalized to β‐actin. Results represent three independent experiments.
-
C, DSpearman correlations between KLF10 and zDHHC7 in liver tissues from NASH and NAFL patients using GEO datasets (GSE48452 and GSE61260). In the dataset GSE61260 (C), n = 83 (including 38 healthy controls, 21 NAFL patients, and 24 NASH patients). In the dataset GSE48452 (D), n = 73 (including 41 healthy controls, 17 NAFL patients, and 15 NASH patients).
-
ERelative mRNA levels of DHHCs family in HepG2 cells overexpressing KLF10 or empty vector (ctrl). n = 5. Results represent three independent experiments.
-
FProtein levels of zDHHC7 in the livers of HFD mice or WD/CCl4 mice as described in Fig 2. The protein level of zDHHC7 was normalized to the β‐actin. n = 5.
-
GLuciferase reporter assays showing the activity of different truncation of zDHHC7 promoter fragments in HepG2 cells. n = 3. Results represent three independent experiments.
-
HThe transcriptional activity of wild‐type or mutant zDHHC7 promoter in HepG2 cells. n = 3. Results represent three independent experiments.
-
IChromatin immunoprecipitation analysis of KLF10 binding activity at the zDHHC7 promoter. Soluble chromatin was prepared from the livers of WD/CCl4 treatment‐induced NASH mice and immunoprecipitated with antibodies against KLF10 or against IgG. The final DNA extractions were amplified using pairs of primers that cover the regions of zDHHC7 and GAPDH gene promoters.
-
JQuantitative real‐time RCR results of ChIP assays. IgG, immunoglobulin G. n = 3. Results represent three independent experiments.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (B, E, F, H, J). Spearman’s correlation (C, D).
Source data are available online for this figure.
Figure EV2. The roles of KLF10 overexpression and zDHHC7 knockdown in cultured cells.
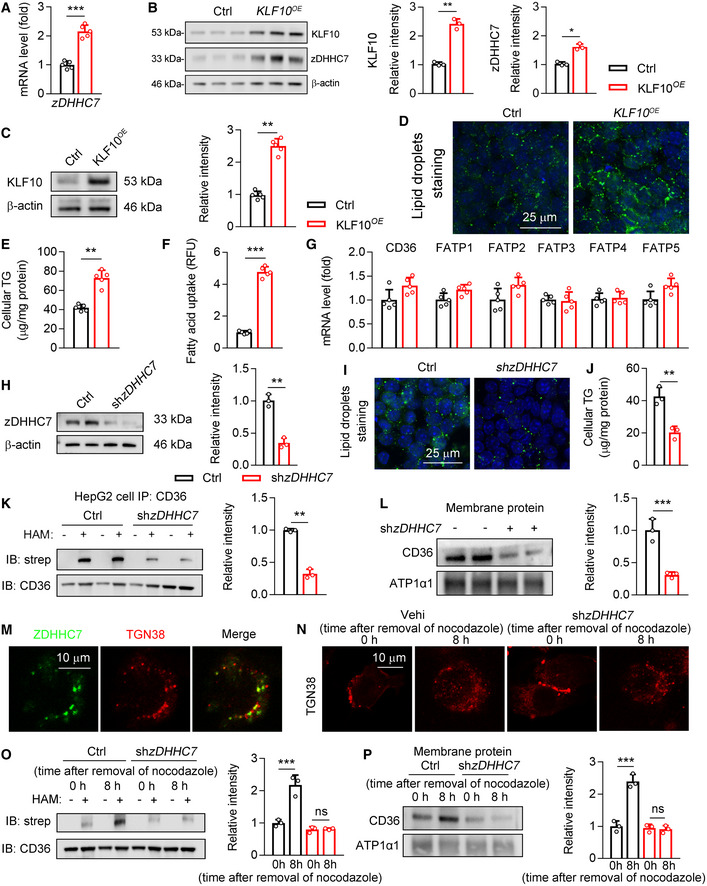
-
A, BHepG2 cells were transfected with lentivirus expressing KLF10 or negative control (Ctrl). n = 5 independent experiments. (A) Relative mRNA of zDHHC7. (B) Protein levels of KLF10 and zDHHC7, and their normalization to β‐actin.
-
C–GHepG2 cells were transfected with lentivirus expressing KLF10 or negative control (Ctrl). n = 5 independent experiments. (C) Protein levels of KLF10, and its normalization to β‐actin. (D) BODIPY staining for lipid droplets. (E) Cellular TG contents. (F) Fatty acids uptake assays. (G) Relative mRNA level of fatty acid transporters.
-
H–LHepG2 cells were treated with lentivirus expressing shRNA targeting zDHHC7 or negative control (Ctrl). n = 3 independent experiments. (H) Protein levels of zDHHC7, and its normalization to β‐actin. (I) BODIPY staining for lipid droplets in HepG2 cells. (J) Cellular TG contents. (K) Protein levels of palmitoylated CD36. (L) CD36 protein expression in the plasma membrane fractions, and its normalization to ATP1a1.
-
MRepresentative immunofluorescence staining of zDHHC7 and TGN38 in HepG2 cells.
-
N, OOn day 0, HepG2 cells were transduced with lentivirus expressing negative control (Ctrl) or shzDHHC7. On day 2, cells were treated with 1 mM nocodazole for 12 h. On day 3, cells were fixed or collected at 0 and 8 h after removal of nocodazole. n = 3. Results represent three independent experiments. (N) Immunofluorescence staining of TGN38 (red). (O) Protein levels of palmitoylated CD36 in HepG2 cells.
-
PCD36 protein expression in the plasma membrane fractions, and its normalization to ATP1a1. n = 3. Results represent three independent experiments.
Data information: **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (A–C, E–H, J–L). One‐way ANOVA (O, P).
Source data are available online for this figure.
Next, to determine the molecular basis for the upregulation of zDHHC7 by KLF10, a series of luciferase reporter constructs containing the human zDHHC7 gene promoter were employed. Luciferase reporter assays showed that KLF10 overexpression activated the transcription of −3253Luc, but not −2743Luc, in HepG2 cells (Fig 3G), suggesting this region spanning from −3,253 bp to −2,743 bp could be required for the transcriptional action of zDHHC7 by KLF10. Bioinformatic analysis further identified a potential KLF binding site exits in the −3049 to −3039 region. Therefore, a mutant luciferase reporter plasmid was constructed containing a point mutation (GTCAcacCTTC → GTCAgtgCTTC) in the KLF10 binding site, which has been termed here as zDHHC7‐Mut (Fig 3H). The reporter system containing a wild‐type (WT) zDHHC7 promoter was used as control. KLF10 overexpression increased the luciferase activity when cells were transfected with the zDHHC7 WT reporter system; however, this effect was abolished when the KLF10 binding site was mutated (Fig 3H). Next, chromatin immunoprecipitation assays (ChIP) were performed to investigate whether endogenous KLF10 protein could directly bind to this region of zDHHC7 promoter in vivo. The zDHHC7 promoter fragment containing a KLF10 binding site could be amplified from the precipitates using anti‐KLF10 antibody in livers from WD/CCl4 treatment‐induced NASH mice (Fig 3I and J). Collectively, these results demonstrated that KLF10 transcriptional activates zDHHC7 expression through binding to its promoter region.
KLF10 promoted CD36 palmitoylation in the mouse livers of NASH mice
We then explored whether KLF10‐zDHHC7 regulatory axis can modulate CD36 palmitoylation and functions. Firstly, overexpression of KLF10 increased lipid droplets accumulation and cellular TG contents in HepG2 cells (Fig EV2C–E). Fatty acids uptake was also enhanced by KLF10 expression (Fig EV2F), while expression levels of fatty acid transporters remained unaffected (Fig EV2G). Next, lentivirus expressing shRNA‐targeting zDHHC7 or negative control (Ctrl) was transfected into HepG2 cells (Fig EV2H). As a result, lipid droplets accumulation and cellular TG contents were significantly reduced (Fig EV2I and J). Besides, CD36 was mainly trapped inside the cells and its palmitoylation was markedly inhibited (Fig EV2K and L). Immunofluorescence staining showed that zDHHC7 was colocalized with TGN38 in HepG2 cells, a marker of Golgi apparatus, suggesting that zDHHC7 protein is mainly localized in the Golgi apparatus (Fig EV2M). We therefore explored whether CD36 could be palmitoylated by zDHHC7 at Golgi. Nocodazole, which can reversibly inhibit microtubule polymerization, was used in HepG2 cells. As a result, the Golgi structures were disrupted after incubation of nocodazole for 12 h (Fig EV2N). Following nocodazole withdrawal for 8 h, Golgi returned to normal, which was accompanied by an increase in the palmitoylation and plasma membrane localization of CD36 (Fig EV2O and P). In contrast, in zDHHC7‐depleted cells, CD36 cannot localize to the plasma membrane after nocodazole withdrawal (Fig EV2O and P). Collectively, these findings demonstrated that zDHHC7‐mediated palmitoylation is required for the plasma membrane localization of CD36 in hepatocytes.
Next, the distributions of CD36 on plasma membranes were examined in livers of WD/CCl4 mice expressing AAV‐KLF10 or AAV‐GFP. Our results showed that the distributions of CD36 on hepatocyte plasma membranes were significantly increased in KLF10OE mice than control mice (Fig 4A and B). Consistently, while total CD36 protein levels remained unaffected, the abundance of CD36 in plasma membrane of hepatocytes was markedly increased (Fig 4C). The palmitoylation of hepatic CD36 was further investigated by immunoprecipitation and acyl‐biotin exchange (IP‐ABE) (Brigidi & Bamji, 2013). This assay showed that palmitoylated CD36 protein was significantly increased in the livers of mice overexpressing KLF10 (Fig 4D and E). In agreement, CD36 membrane localization and palmitoylation were also increased in the livers of HFD mice with AAV9‐Klf10 injection (Fig 4F–J). Taken together, our findings demonstrated that hepatic KLF10 overexpression enhanced CD36 palmitoylation, and promoted the distribution of CD36 on plasma membranes in hepatocytes.
Figure 4. KLF10 overexpression promotes CD36 palmitoylation in hepatocytes.
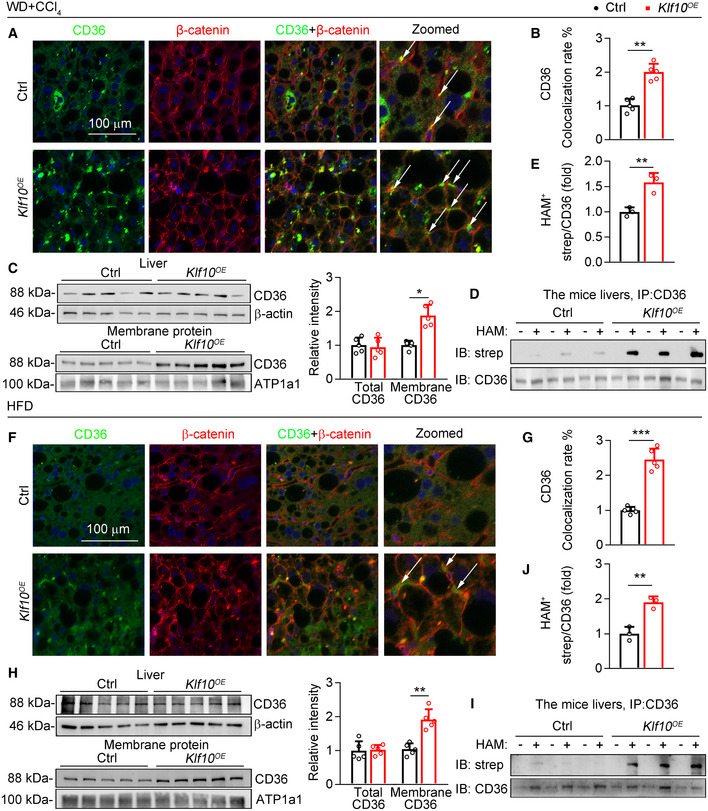
-
A–EThe liver tissues were collected from WD/CCl4‐treated mice expressing KLF10 or control as described in the Fig 2A. (A) Representative immunofluorescence staining of CD36 and β‐catenin. The arrows represent the colocalization of CD36 and β‐catenin. (B) Quantitative results of CD36 colocalization rate. n = 5. Results represent three independent experiments. (C) Expression of CD36 in whole livers and plasma membrane fractions. Protein levels were normalized to the β‐actin or ATP1a1, respectively. n = 5. Results represent three independent experiments. (D) Protein levels of palmitoylated CD36 in mouse livers. (E) Quantitative results of CD36 colocalization rate. n = 3. Results represent three independent experiments.
-
F–JThe liver tissues were collected from HFD‐fed mice expressing KLF10 or control as described in the Fig 2I. (F) Representative immunofluorescence staining of CD36 and β‐catenin. The arrows represent the colocalization of CD36 and β‐catenin. (G) Quantitative results of CD36 colocalization rate. Results represent three independent experiments. (H) Expression of CD36 in whole livers and plasma membrane fractions. Protein levels were normalized to the β‐actin or ATP1a1, respectively. n = 5. Results represent three independent experiments. (I) Protein levels of palmitoylated CD36 in mouse livers. (J) Quantitative results of CD36 colocalization rate. n = 3. Results represent three independent experiments.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (B, C, E, G, H, J).
Source data are available online for this figure.
DHHC7 is required for KLF10‐induced CD36 palmitoylation and plasma membrane localization
On the other hand, knockdown of KLF10 decreased CD36 palmitoylation and its localization on plasma membrane in HepG2 cells (Fig 5A–C). Then, 2‐bromopalmitate (2‐BP), an inhibitor of palmitoylation, were used to treat HepG2 cells in the absence or presence of KLF10 overexpression (Fig 5D). As a result, the palmitoylation of CD36 (Fig 5E), localization of CD36 on plasma membrane (Fig 5F), and accumulation of lipid droplets (Fig 5G) induced by KLF10 overexpression were attenuated by 2‐BP treatment in HepG2 cells. To rule out the nonspecific effects of 2‐BP, endogenous zDHHC7 expression in HepG2 cells was depleted by transfection of shRNA (Fig 5H). In agreement, zDHHC7 deficiency also suppressed the roles of KLF10 overexpression on CD36 palmitoylation, plasma membrane localization, cellular lipid droplets accumulation, and TG retention (Fig 5I–L).
Figure 5. zDHHC7 is required for KLF10‐induced CD36 palmitoylation and plasma membrane localization.
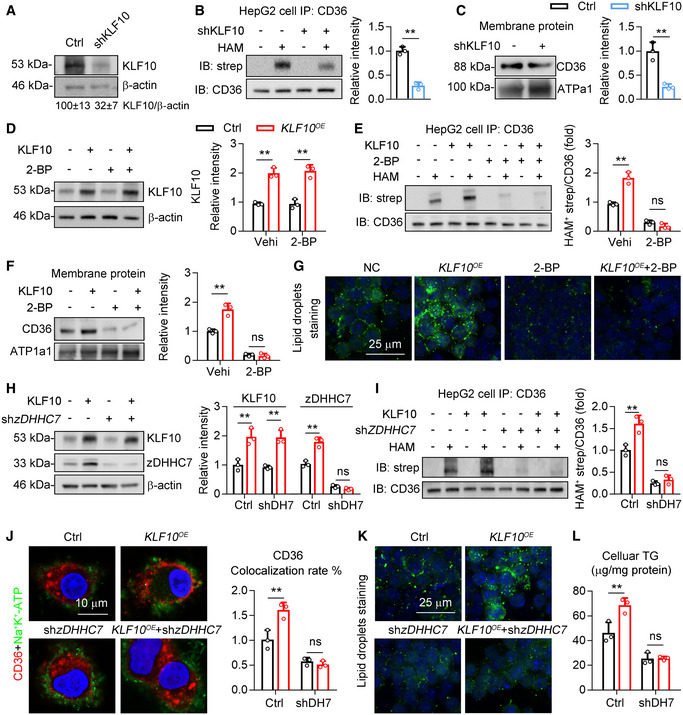
-
A–CHepG2 cells were transfected with lentivirus vectors expressing shRNA targeting KLF10 or negative control (Ctrl). n = 3. Results represent three independent experiments. (A) Protein level of KLF10, and its normalization to β‐actin. (B) Protein levels of palmitoylated CD36. (C) CD36 protein expression in the plasma membrane fractions, and its normalization to ATP1a1.
-
D–GHepG2 cells were treated with 2‐BP and/or lentivirus vectors expressing KLF10. n = 3. Results represent three independent experiments. (D) Protein levels of KLF10, and its normalization to β‐actin. (E) Protein levels of palmitoylated CD36. (F) CD36 protein expression in the plasma membrane fractions, and its normalization to ATP1a1. (G) BODIPY staining showing the lipid droplets.
-
H–LHepG2 cells were transfected with lentiviruses expressing KLF10 and/or shRNA targeting zDHHC7. n = 3 independent experiments. (H) Protein levels of KLF10 and zDHHC7, and its normalization to β‐actin. (I) Protein levels of palmitoylated CD36. (J) Immunofluorescence staining of CD36 and Na+/K+‐ATPase (left), and the quantitation of CD36 colocalization rate (right). (K) BODIPY staining for lipid droplets. (L) Cellular TG contents.
Data information: **P < 0.01. Results are shown as mean ± SD. Student’s t‐test (B, C). One‐way ANOVA (D–F, H‐J, L).
Source data are available online for this figure.
CD36 not only functions as an FAs transporter but also recruits the src family kinase Fyn and Lyn to regulate intracellular signaling pathways, including AMPK and JNK (Samovski et al, 2015; Zhao et al, 2018). We therefore examined whether KLF10 was involved in modulating the association of CD36 with Fyn and Lyn in HepG2 cells (Fig EV3A). KLF10 overexpression markedly increased the protein–protein interaction of CD36 with Fyn and Lyn in HepG2 cells, which was inhibited after 2‐BP treatment (Fig EV3B and C). Protein levels of phosphorylated JNK and TNF‐α were increased in cells overexpressing KLF10, which was markedly suppressed by 2‐BP treatment (Fig EV3D). In addition, KLF10 overexpression decreased the phosphorylation status of AMPK, which was also reversed by 2‐BP treatment (Fig EV3E). Similar results were also observed in HepG2 cells transfected with shRNA targeting zDHHC7 (Fig EV3F–J). Taken together, our results demonstrated that zDHHC7 is required for KLF10‐mediated CD36 palmitoylation and function.
Figure EV3. KLF10 overexpression‐induced CD36/Fyn/Lyn complex formation is dependent on zDHHC7.
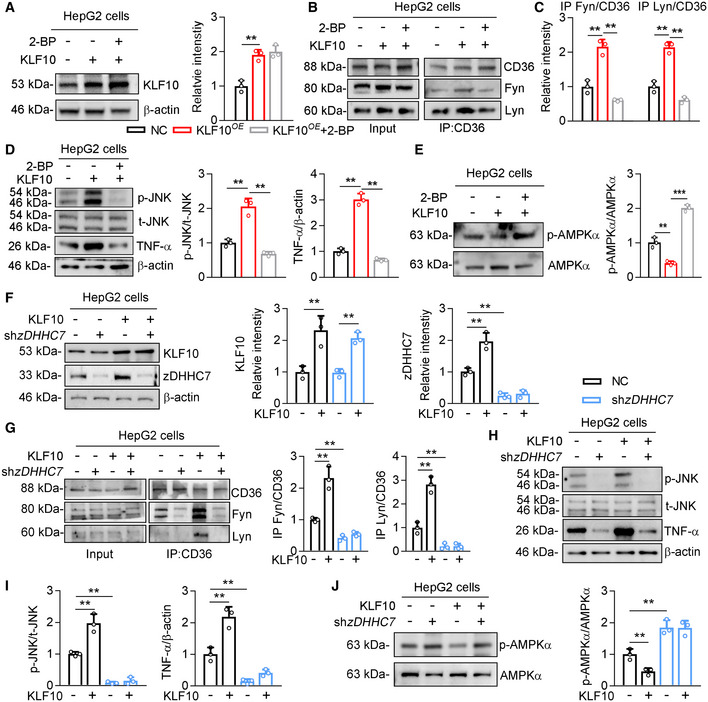
-
A–EHepG2 cells were treated with 2‐BP and/or transfected with lentivirus vectors expressing KLF10. n = 3 independent experiments. (A) Protein levels of KLF10, and its normalization to β‐actin. (B, C) Interactions of CD36 and Fyn/Lyn by co‐immunoprecipitation assays (B), and their quantitation (C). (D) Protein levels of p‐JNK, t‐JNK, and TNF‐α, and their quantitation. (E) Protein level of p‐AMPK and its normalization to t‐AMPK.
-
F–JHepG2 cells were treated with shzDHHC7 and/or transfected with lentivirus vectors expressing KLF10. n = 3 independent experiments. (F) Protein level of KLF10, and its normalization to β‐actin. (G) Interaction of CD36 and Fyn/Lyn by co‐immunoprecipitation assays, and their quantitation. (H) Protein levels of p‐JNK, t‐JNK, and TNF‐α. (I) Quantitation of p‐JNK, and TNF‐α. (J) Protein level of p‐AMPK and its normalization to t‐AMPK.
Data information: **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. One‐way ANOVA (A, C–G, I, J).
Source data are available online for this figure.
CD36 is required for KLF10‐mediated liver damage in NASH mice
To determine whether the pathogenic role of KLF10 in NASH progression is dependent on its regulation of CD36, CD36 WT and knockout (CD36KO ) mice were used. We injected WT and CD36KO mice with AAV‐TBG‐Klf10 to overexpress KLF10 in both groups. Then, mice were administered with WD/CCl4 for 12 weeks to induced NASH progression. As shown in Fig 6A and B, KLF10 was significantly increased and comparable in livers of both WT and CD36KO mice. Besides, hepatic TG contents and liver damage were lower in CD36KO mice, compared with WT mice (Fig 6C–G, column 3 vs. column 1). Notably, KLF10‐induced hepatic inflammation and liver fibrosis were observed in WT mice (Fig 6C and D, column 2 vs. column 1), but were attenuated in CD36KO mice (Fig 6C and D, column 4 vs. column 3). Besides, although body weights were not changed (Fig 6E), KLF10 overexpression increased the liver/body weight ratio (Fig 6E, column 2 vs. column 1), hepatic TG contents (Fig 6F, column 2 vs. column 1), and serum ALT and AST levels (Fig 6G, column 2 vs. column 1) in the WT mice. However, these effects were not observed in the CD36KO mice (Fig 6E–G, column 4 vs. column 3). These results suggest that intact CD36 expression was required for the pathogenic roles of KLF10 in NASH development.
Figure 6. CD36 is required for the pathogenic role of KLF10 in promoting NASH.
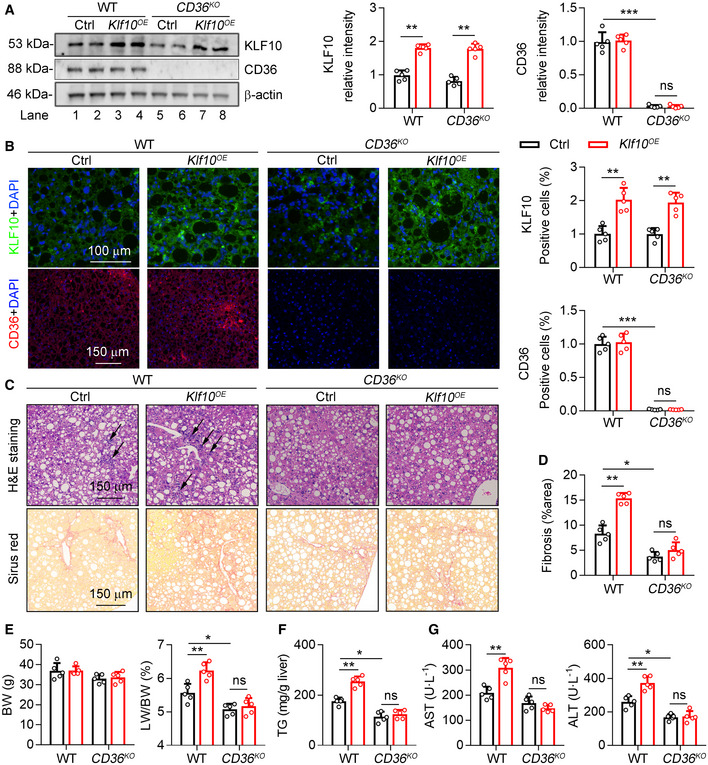
-
A–G6‐week‐old male CD36 wild‐type (WT) or knockout mice (CD36KO) were administered with AAV‐GFP (Ctrl) or AAV‐KLF10 (KLF10OE) through tail vein injection, and then kept on WD/CCl4 for 12 weeks. n = 5 per group. (A) Protein levels of KLF10 and CD36 in the liver (left), and their normalization to β‐actin (right). (B) Representative immunofluorescence staining of KLF10 and CD36 (left), and the quantitative results (right). (C) H&E and Sirius red staining of liver sections. (D) Quantitative results of Sirius red staining. (E) Body weights and liver/body weight ratio. (F) Hepatic TG contents. (G) Serum AST and ALT levels.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. One‐way ANOVA (A, B, D–G).
Source data are available online for this figure.
CD36 palmitoylation is required for KLF10 to promote the plasma membrane localization of CD36
We next administered CD36KO mice with AAV‐CD36 or AAV‐mCD36 to re‐express two types of CD36 proteins through tail vein injection. The mCD36 is the mutant form at its palmitoylation sites. Meanwhile, mice were administered with AAV‐Klf10 or AAV‐GFP and then placed on WD/CCl4 for 12 weeks to induce NASH. CD36 protein was absent in livers of CD36KO mice, and significantly increased after AAV‐Cd36 or AAV‐mCd36 injection (Fig EV4A and B). WT CD36 overexpression induced liver injury compared with CD36 KO mice, as evidenced by a significant elevation of serum ALT and AST, hepatic TG contents, liver/body weight ratio, and fibrosis area (Fig 7A–D, column 2 vs. column 1). Besides, there is a significant difference in NASH outcomes between mice with CD36 and mCD36 overexpression in the absence of KLF10 overexpression (Fig 7A–D, column 4 vs. column 2), suggesting that palmitoylation of CD36 is an important step in NASH progression. More importantly, hepatic lipid accumulation and liver injury were dramatically increased in mice injected with CD36 OE and Klf10OE (Fig 7A–D, column 3 vs. column 2). However, Klf10OE has little synergistic effects in mice injected with mCD36 OE (Fig 7A–D, column 5 vs. column 4). Furthermore, the CD36 distributions on plasma membranes of hepatocytes and its palmitoylation were higher in mice injected with CD36 OE and Klf10OE (Fig 7E–I, column 2 vs. column 1), compared with mice injected with mCD36 OE and Klf10OE (Fig 7E–I, column 4 vs. column 3).
Figure EV4. Inhibition of CD36 palmitoylation attenuated KLF10‐mediated lipid accumulation.
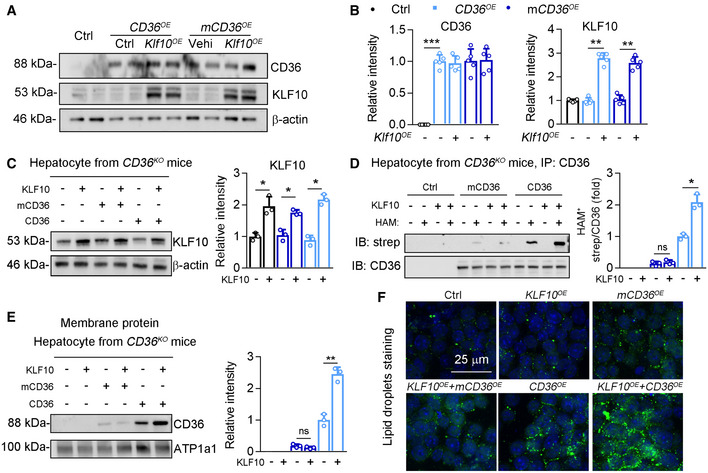
-
A, BProtein levels of CD36 and KLF10 (A), and their normalizations to β‐actin (B). n = 5. Results represent three independent experiments. The experimental strategy has been described in the Fig 7.
-
C–FMouse primary hepatocytes were isolated from CD36KO mice and then transfected with lentivirus‐wt‐Cd36, lentivirus‐mCd36, and lentivirus ‐Klf10 for 24 h. n = 3 independent experiment. (C) Protein level of KLF10 (left) and its normalization to β‐actin (right). (D) Protein levels of palmitoylated CD36. (E) CD36 protein expression in the plasma membrane fractions, and its normalized to ATP1a1. (F) BODIPY staining for lipid droplets in hepatocytes.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. One‐way ANOVA (B–E).
Source data are available online for this figure.
Figure 7. CD36 palmitoylation is required for KLF10‐mediated NASH progression.
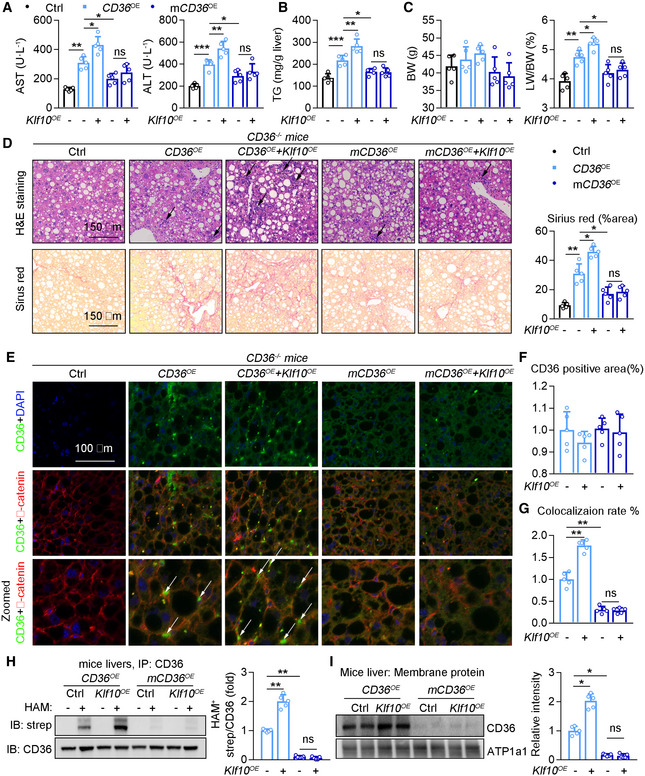
-
A–I6‐week‐old male CD36 knockout mice were administered with AAV‐TBG‐wildtype‐CD36, wildtype‐CD36 plus KLF10, palmitoylation site mutant CD36, mutant‐CD36 plus KLF10 through tail vein injection, and then kept on WD/CCl4 for 12 weeks. n = 5 per group. (A) Serum AST and ALT levels. (B) Hepatic TG contents. (C) Body weights and liver/body weight ratio. (D) H&E and Sirius red staining of liver sections (left), and quantitation of Sirius red staining (right). (E) Immunofluorescence staining of CD36 and β‐catenin. (F) Quantitation of CD36 positive area. (G) Colocalization rate of CD36 and β‐catenin. (H) Protein levels of palmitoylated CD36 in mouse livers (left), and its quantitative results (right). (I) CD36 protein expression in the plasma membrane fractions, and its normalization to ATP1a1.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. One‐way ANOVA (A–D, F‐I).
Source data are available online for this figure.
Next, mouse primary hepatocytes were isolated from CD36KO mice and then transfected with lentiviruses containing WT CD36, mCD36, or Klf10 (Fig EV4C). When compared with WT‐CD36 OE hepatocytes, higher levels of CD36 palmitoylation, cellular TG contents, and CD36 localization on plasma membranes were observed in hepatocytes expressing wt‐CD36 OE and Klf10OE (Fig EV4D–F, column 6 vs. column 5). In contrast, these effects were comparable between hepatocytes expressing mCD36 OE and mCD36 OE plus Klf10OE (Fig EV4D–F, column 4 vs. column 3). Taken together, these in vivo and in vitro findings suggest that the ability of CD36 palmitoylation is required for KLF10 in the regulation of hepatic lipid retention and liver injury.
Hepatocyte‐KLF10 deficient mice are protected from diet‐induced NASH pathogenesis
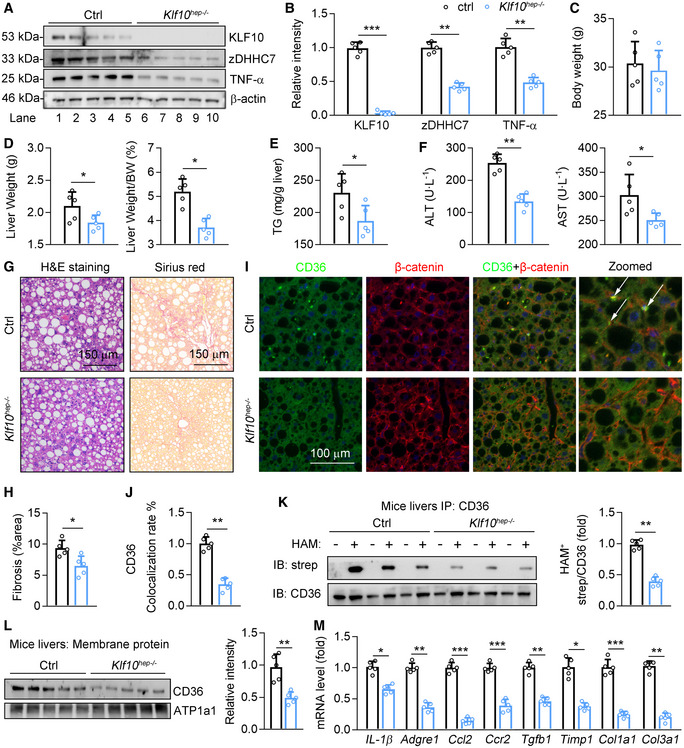
Finally, we generated hepatocyte‐specific KLF10 knockout mice (Klf10hep −/−) by crossing Klf10 fl/fl mice to mice bearing an Albumin‐Cre transgene (Fig 8A). Klf10hep −/− mice and control WT littermates (Ctrl) were treated with WD/CCl4 to induce NASH progression. As a result, the protein levels of zDHHC7 and TNF‐α were significantly decreased in livers of Klf10hep −/− mice, compared with those in control mice (Fig 8A and B). Loss of Klf10 did not affect body weight in mice (Fig 8C), but markedly decreased liver weight and liver/body weight ratio (Fig 8D). Besides, hepatic TG contents and serum ALT and AST levels were reduced in Klf10hep −/− mice (Fig 8E and F). H&E and Sirius red staining showed that Klf10‐deficient mice are protected from inflammatory cells infiltration and liver fibrosis (Fig 8G and H). The distributions of CD36 on hepatocyte plasma membranes were reduced in Klf10hep −/− mice than control mice (Fig 8I and J). Furthermore, IP‐ABE results indicated that palmitoylated CD36 protein was significantly decreased in the livers of Klf10hep −/− mice (Fig 8K). In agreement, the abundance of CD36 in plasma membrane of hepatocytes was significantly decreased in Klf10hep −/− mice (Fig 8L). Moreover, expression levels of genes involved in the hepatic inflammation and fibrosis were dramatically downregulated in Klf10hep −/− mice (Fig 8M). Collectively, hepatocyte‐specific Klf10 depletion reduced CD36 palmitoylation and the distribution of CD36 on hepatocyte plasma membranes, leading to improvements on the hepatic TG accumulation and liver injury.
Figure 8. Liver‐specific KLF10‐deficient mice are resistant to NASH pathogenesis.
-
A–MAlbumin‐cre mice were crossed with KLF10 flox/flox to generate hepatocyte‐specific KLF10 knockout mice (KLF10 hep‐/‐ ). Then, 8‐week‐old male KLF10 hep‐/‐ mice and KLF10 flox/flox were treated with WD/CCl4 for 12 weeks. KLF10 flox/flox mice were used as the control (Ctrl). n = 5 per group. (A, B) Protein levels of KLF10, zDHHC7, and TNF‐α (A), and their normalization to β‐actin (B). (C) Body weights. (D) Liver weights and liver/body weight ratio. (E) Hepatic TG contents. (F) Serum ALT and AST levels. (G) H&E staining and Sirius red staining of liver sections. (H) Quantitative results of Sirius red staining. (I) Immunofluorescence staining of CD36 and β‐catenin. The arrows represent the colocalization of CD36 and β‐catenin. (J) Quantitative results of colocalization rate of CD36 and β‐catenin. (K) Protein levels of palmitoylated CD36 in mouse livers. (L) CD36 protein expression in the plasma membrane fractions from mouse livers, and its normalization to ATP1a1. (M) Relative mRNA level of genes involved in the liver inflammation and fibrosis.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Results are shown as mean ± SD. Student’s t‐test (B–F, H, J–M).
Source data are available online for this figure.
Discussion
Hepatic lipid accumulation and inflammation are key features of NASH. However, the pathogenic cross‐roads linking FAs metabolism, lipotoxicity, and hepatic inflammation remain unclear. Here, our results indicate that KLF10 is an important regulator of the plasma membrane localization of CD36 in the liver. Mechanistically, KLF10 increases CD36 palmitoylation through transcriptional activation of zDHHC7, which in turn promotes CD36 membrane localization. Furthermore, loss of CD36 or palmitoylation site‐mutated CD36 inhibited KLF10 overexpression‐induced liver injury. In addition, hepatocyte‐specific depletion of KLF10 improved symptoms of NASH in a WD/CCl4‐induced NASH model. Collectively, these results indicate that KLF10 in hepatocyte may promote the development and progression of NASH through activation of zDHHC7‐CD36 palmitoylation axis. However, we cannot rule out the possibility that KLF10 may exert its pathogenic roles in NASH progression through other transcriptional targets. For instance, we also found that Gadd45g was upregulated by KLF10 overexpression or in the livers of NASH mice (Dataset EV1). This protein has been shown to regulate cytokine expression in T cells (Schmitz, 2013). Whether KLF10 could promote hepatic inflammatory response in the development of NASH through Gadd45g remains to be determined. Therefore, further studies are still needed to explore more transcriptional targets of KLF10 in the regulation of NASH progression. Our transcriptome screening may provide a valuable resource to investigate this issue.
Interestingly, whole‐body KLF10‐deficient mice displayed enhanced liver injury and fibrosis priming upon methionine‐ and choline‐deficient (MCD) diet challenging (Leclère et al, 2020). Besides, KLF10 expression could be induced by carbohydrate response element‐binding protein (ChREBP) upon glucose stimulation in mouse hepatocytes (Iizuka et al, 2011). KLF10 knockout mice exhibited significant hepatic steatosis, inflammation, and liver injury upon high sucrose diet feeding (Lee et al, 2020). T cell‐specific KLF10 knockout mice were susceptible to diet‐induced obesity, insulin resistance, and fatty liver (Wara et al, 2020). Therefore, we speculate that the role of KLF10 in NASH progression might be cell or tissue‐specific. Furthermore, KLF10 displayed a circadian expression pattern in mouse livers, and depletion of hepatocyte KLF10 altered hepatic circadian transcriptome, leading to exacerbated high sugar consumption‐associated metabolic effects (Guillaumond et al, 2010; Ruberto et al, 2021). However, another study reported that hepatic KLF10 expression is elevated in the obese and diabetic mice (Yang et al, 2017). Overexpression of KLF10 in the liver of C57BL/6J mice increased blood glucose levels and impaired glucose tolerance, while hepatic KLF10 knockdown in db/db and diet‐induced obese mice decreased blood glucose levels and improved glucose tolerance (Yang et al, 2017). Although the reasons behind these inconsistent results remain unclear, the feeding and treatment conditions are different between these studies. High sugar consumption usually leads to simple steatosis, whereas WD plus CCl4 treatment was used in our study to explore the potential function of KLF10 in the steatosis‐to‐NASH progression. Thus, KLF10 may play a diverse role in metabolic liver diseases in the context of different physiological and pathological conditions.
Palmitoylation is the covalent attachment of palmitate to cysteine residues of proteins. This post‐translational modification can increase the lipophilicity of the modified proteins, thereby regulating subcellular distribution and altering protein function (Rocks et al, 2010; Yang et al, 2018). Palmitoylation is catalyzed by a large family of DHHC‐containing palmitoyl acyltransferases, which contains 7 members in yeast and more than 20 in mammals (Mitchell et al, 2006). It has been reported that the localization of CD36 on the plasma membrane of hepatocytes was markedly increased in NASH patients compared to patients with simple steatosis (Zhao et al, 2018). However, the regulatory mechanisms of CD36 palmitoylation in hepatocytes remain unclear. zDHHC7 has been shown to regulate TH17 cell differentiation by palmitoylating STAT3 and promoting its membrane recruitment and phosphorylation (Zhang et al, 2020). Additionally, zDHHC7 is the principal palmitoyl‐acyltransferase for glucose transporter 4 and regulates it membrane translocation in adipose tissue and skeletal muscle (Du et al, 2017). However, to the best of our knowledge, we for the first time demonstrate the role of zDHHC7 in the regulation of hepatic lipid metabolism. To be noted that, Wang et al (2019) reported that knockdown of zDHHC7 did not affect palmitoylation of CD36 in HEK293T cells. They further identified that zDHHC4 and zDHHC5 are required for CD36 palmitoylation in adipocytes (Wang et al, 2019). We speculate that the palmitoyl acyltransferases for target proteins might be cell‐ or tissue‐specific. For instance, zDHHC5 was shown to facilitate STAT3 palmitoylation in oligodendrocytes (Ma et al, 2022), while only overexpression of zDHHC7 and zDHHC3 could increase the palmitoylation level of STAT3 in HEK293T cells (Zhang et al, 2020).
It has been well established that the function of CD36 as an FFA transporter mainly depends on its localization on the plasma membrane (Goldberg et al, 2009). Translocation of CD36 to the plasma membrane has also been described in cardiac muscle cells as well as platelets and pneumocytes (Luiken et al, 2002, 2002,2002, 2002; Chabowski et al, 2013). In hepatocytes, prolonged exposure to FFAs not only increased total expression of CD36, but also triggered its translocation to the plasma membranes (Chabowski et al, 2013). In particular, in histologically normal livers, CD36 is weakly detected in the cytoplasm of hepatocytes, whereas it is markedly expressed at the plasma membrane in NAFLD patients (Miquilena‐Colina et al, 2011; Zhao et al, 2018). On the other hand, blocking the palmitoylation of CD36 reduces its distribution in hepatocyte plasma membranes and protects mice from nonalcoholic steatohepatitis (Zhao et al, 2018). In the current study, we provided compelling evidence demonstrating that KLF10 promotes CD36 palmitoylation and distribution on the hepatocellular plasma membrane. Our results showed that the distribution of CD36 on the plasma membrane of hepatocytes is remarkably increased after KLF10 AAV infection and decreased in hepatic KLF10‐deficient mice. Besides, KLF10‐induced fatty acid metabolic dysregulation and inflammation could be attenuated by inhibition of CD36 palmitoylation. Together, our results strengthen the understanding of molecular mechanisms in regulating CD36 function in hepatocytes.
In conclusion, we demonstrate that enhanced KLF10 expression induces zDHHC7 to facilitate CD36 translocation to the plasma membrane of hepatocytes, resulting in hepatic lipid accumulation and inflammation. The imbalance between FAs intake and consumption and the activation of the inflammatory response contribute to NASH development. Conversely, selectively inhibition of KLF10 in hepatocytes can reduce zDHHC7 expression, thereby decreasing CD36 translocation to plasma membrane, and improving liver injury in diet‐induced NASH mice. Thus, the present study provides evidence showing that hepatic KLF10 plays an important role in NASH pathogenesis. Targeting hepatic KLF10 or zDHHC7 may offer a new therapeutic strategy for the treatment of NASH and related metabolic liver diseases.
Materials and Methods
Animal studies
All animal care and experimental protocols for in vivo studies were conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH (NIH publication no. 85‐23, revised 1996). Sample size for animal studies was calculated based on a survey of data from published research or preliminary studies. No mice were excluded for statistical analysis. C57BL/6J mice, CD36KO mice (B6/JGpt‐Cd36em11Cd1894 /Gpt, Strain No. T010474), KLF10 flox/flox (B6/JGpt‐Klf10em1Cflox /Gpt, Strain No. T018201), and Albumin‐Cre mice (B6/JGpt‐H11 em1Cin(Alb‐iCre) /Gpt, Strain No. T003814) based on C57BL/6J background were obtained from Gempharmatech Co., Ltd. (Nanjing, Jiangsu, China). These mice were maintained in SPF units of the Animal Center of Shenzhen People's Hospital with a 12‐h light cycle from 8 a.m. to 8 p.m., 23 ± 1°C, 60–70% humidity. Mice were allowed to acclimatize to their housing environment for 7 days before experiments. At the end of experiments, all mice were anesthetized and euthanized in a CO2 chamber followed by collection of liver tissues and blood samples. To induce NASH pathogenesis, 8‐week‐old male C57BL/6J mice were fed a WD containing 21.1% fat, 41% Sucrose, and 1.25% Cholesterol by weight (Teklad diets, Cat#: TD. 120528) and a high sugar solution (23.1 g/l d‐fructose (Sigma‐Aldrich, Cat#: G8270) and 18.9 g/l d‐glucose (Sigma‐Aldrich, Cat#: F0127)). CCl4 (Sigma‐Aldrich, Cat#: 289116‐100ML) at a dose of 0.32 µg/g of body weight was injected intraperitoneally once/week. To induce simple steatosis (NAFL), 8‐week‐old male C57BL/6J mice were fed with a high‐fat diet (research diets, Cat#: D12492) for 12 weeks. Adeno‐associated virus type 9 (GV388, Shanghai GeneChem Company) containing KLF10 (1.99E + 13 v.g./ml), WT CD36 (1.1E + 13 v.g./ml), and palmitoylation sites‐mutated CD36 (mCD36, 1.45E + 13 v.g./ml) were administered into mice through tail vein injection. The animal protocol was approved by the Animal Care Committees of Jinan University (No. 20200331‐14) and performed in compliance with the ARRIVE guidelines.
Liver specimens from humans
Age‐matched male liver biopsy‐proven NAFL and NASH patients were recruited and diagnosed in the Department of Endocrinology, Shenzhen People's Hospital. Liver biopsies were preserved in formalin for histology and immunostaining analysis. Written informed consent was obtained from each patient. This study was approved by the Ethics Committee of Shenzhen People's Hospital (No. NFEC‐2018‐127), and conducted in accordance with the 1975 Declaration of Helsinki.
Cell culture and transfection
HepG2 cells (CLS Cat# 300198/p2277_Hep‐G2, American Type Culture Collection) were cultured in DMEM medium containing 10% fetal bovine serum, 50 mg/ml penicillin/streptomycin, and 2 mmol/l glutamine. The ability of the cells to take up fatty acids was measured using a commercial kit (Sigma‐Aldrich, Catalog no. MAK156) according to the manufacturer’s protocol. A CD36 variant was generated using PCI‐CD36 as a template and the mutation primers with a multiple site‐directed mutagenesis Kit (QuickChange II XL, Agilent Technologies), in which the cysteine (Cys‐3, 7) of WT CD36 were replaced with alanine, and the cysteine (Cys‐464, 466) were replaced with serine (Zhao et al, 2018). The lentivirus constructs including GV367 empty vector, WT CD36 (1E+8 TU/ml), palmitoylation site‐mutated CD36 (mCD36; 1E+9 TU/ml), and KLF10 (GV492; 5.0E+8 TU/ml) were provided by GeneChem Company (Shanghai, China).
Primary hepatocyte isolation
Mouse primary hepatocytes were isolated using a modified two‐step collagenase perfusion technique. Briefly, the liver specimen was cannulated under sterile conditions and flushed once with 50 ml washing buffer containing 2.5 mM EGTA (Sigma‐Aldrich). This was followed by perfusion with 30 ml digestion buffer containing 0.03% w/v collagenase (Roche Diagnostics) allowing recirculation of the perfusate. The resulting cell suspension was poured through a 100 μM Cell Strainer and centrifuged with subsequent washing of the cell pellet using Serum‐free medium (50 g, 2 min, 4°C). Cells were then re‐suspended in William’s medium E (Biochrom AG) containing 10% fetal bovine serum. Cell number and viability were determined by the Trypan blue exclusion test. Hepatocytes were cultured using six‐well plates precoated with a single layer of rat tail collagen. Cells were seeded at a concentration of 2.5 × 106 viable cells per well. Sixteen to eighteen hours later, culture medium were changed to remove dead and nonadherent cells.
Detection of CD36 protein palmitoylation
Protein palmitoylation was assessed by immunoprecipitation and acyl‐biotin exchange (IP‐ABE) as previously described (Brigidi & Bamji, 2013). Briefly, total proteins were extracted using a lysis buffer with protease inhibitors and N‐ethylmaleimide. After precipitating CD36 protein with anti‐CD36 antibody (Novus) and magnetic beads (Millipore), samples were re‐suspended with stringent buffer. Then, 0.5 ml lysis buffer (pH 7.2) was added to all hydroxylamine (HAM) samples. All samples were rotated at room temperature for 50 min, and then mixed with Biotin‐BMCC buffer, and rotated for 50 min at 4°C. Proteins were eluted from the beads by boiling in sample buffer and the cleared supernatants were resolved by SDS–PAGE and electro‐transferred to PVDF membranes (Millipore). For detecting palmitoylated CD36, membranes were incubated with HRP‐conjugated anti‐streptavidin (abcam, Cat#: ab7403) for 1 h at 37°C. For detecting total CD36, membranes were incubated with anti‐CD36 (Novus) and subsequent HRP‐conjugated secondary antibody (Zsbio).
Isolation and analysis of cellular plasma membrane fractions
Cellular plasma membrane was isolated using a commercial Kit (Abcam) according to the manufacturer’s instruction. Briefly, cells were collected and homogenized in Homogenize Buffer Mix. The homogenates were then centrifuged at 700 g for 10 min to remove the nuclear pellet. The supernatants from the original 700 g spin were centrifuged at 10,000 g for 30 min at 4°C, collected, and designated ‘‘cytoplasm extracts”. The pellet was re‐suspended in Upper and Lower Phase solution to separate plasma membrane fractions from cellular organelle membrane. The suspension was mixed thoroughly and centrifuged at 1,000 g for 10 min at 4°C. Upper phase solution was collected and diluted with five volumes of water. Then, the mixture was centrifuged at 14,000 g at 4°C for 20 min. The plasma membrane fraction pellet was dissolved in 0.5% Triton X‐100.
Lipid droplet staining and measurement of fluorescent signals
For lipid droplet staining, cells were fixed with paraformaldehyde and stained with 400 lM Bodipy 493/503 for 30 min. All images were captured using a Zeiss microscope. A fluorescent counter was also used to measure the relative fluorescent units of lipid droplet staining.
RNA sequencing
A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. The clustering of the index‐coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3‐cBot‐HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform, and 150 bp paired‐end reads were generated.
Quantitative real‐time PCR
Total RNAs were extracted by Trizol (Invitrogen) and dissolved in an appropriate amount of RNase water. The cDNA was obtained by a reverse transcription kit purchased from New England Biolab (lpswich, MA, USA). qRT–PCR was performed using the ABI StepOnePlus™ Real‐time PCR system (Applied Biosystem) with specific primers (Table EV1). The relative mRNA level of target genes was analyzed using the equation 2−ΔΔCT. β‐actin was used as a housekeeping gene for analysis.
Western blots
Liver tissues or cells were lysed or homogenized in the lysis buffer (sigma‐Aldrich; St. Louis, MO, USA), and total protein obtained according to the classical protocol. 1:1,000 diluted fresh primary antibody or anti‐β‐actin antibody (1:5,000) in PBS containing 1% fresh dry fat‐free milk, and 1:5,000 diluted fresh HRP‐conjugated anti‐rabbit or mouse IgG in PBS containing 1% fresh dry fat‐free milk. Western blotting experiments were performed using antibodies for KLF10 (Abcam, Cat#: ab73537), CD36 (Abcam, Cat#: ab133625), zDHHC7 (Novus, Cat#: NBP2‐94487), TNF‐α (Abcam, Cat#: ab183218), Fyn (Novus, Cat#: NB500‐517), Lyn (Abcam, Cat#: ab32398), t‐JNK (Novus, Cat#: MAB2076), p‐JNK (Abcam, Cat#: ab124956), AMPK (Abcam, Cat#: ab32047), and p‐AMPK (Abcam, Cat#: ab23875). After capturing, the density of image was quantified with Image J software (National Institutes of Health, Bethesda, MD, USA) by a person who was blinded to the experiment. Levels of proteins of interest were normalized to β‐actin or ATP1a.
ELISA
Serum AST and ALT concentrations were measured using commercial ELISA kits (Abcam) according to the manufacturer’s protocol. Hepatic lipids were extracted from livers using a mixture of chloroform and methanol. Hepatic triglyceride (TG) contents were measured by a commercial kit (Jiancheng Bioengineering, Nanjing, China) and normalized by liver weights.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed with a commercial kit from Millipore following the manufacturer’s instructions with the primers listed in Table EV2. Briefly, liver tissues were lysed in lysis buffer and sonicated (15 s on and 90 s off, repeated 8 times). After precipitation with Agarose A for 30 min, the fragmented DNA was pulled down with KLF10 antibody or IgG and then subjected to amplification by qPCR.
Dual luciferase reporter assay and site‐directed mutagenesis
The full‐length human zDHHC7 gene was amplified by PCR from HepG2 cells cDNA. Six fragments (−3,923 to −471, −3,253 to −471, −2,743 to −471, −2,064 to −471, and −1,496 to −471) of the zDHHC7 promoter were cloned by PCR and inserted into pGL3 luciferase vector using the primers listed in Table EV3. Point mutations in the zDHHC7 promoter (−3,049 to −3,039) were generated by the Fast Mutagenesis System (TransGen) with the primers listed in Table EV3. All constructs were confirmed by DNA sequencing analysis. For dual luciferase reporter gene assays, HepG2 cells were transfected with zDHHC7 promoter plasmids and KLF10, as well as Renilla luciferase. Firefly and Renilla luciferase activities were then measured by a dual luciferase reporter gene system (Promega, Madison, WI, USA).
Histological analysis
Mouse tissues were preserved with 4% paraformaldehyde solution, dehydrated, and embedded in paraffin. The sections (4 μm) were used for histological analysis. H&E and Sirus red staining were performed as standard instructions. Positive cells were morphometrically quantified with image processing software (Image J) by a technician who blinded to the experiments.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde for 30 min at room temperature, washed with PBS for three times, permeabilized with 0.5% Triton X‐100 for 20 min, and blocked with 10% of goat serum in PBS for 1 h at room temperature. Cells were then incubated with anti‐CD36 (1: 100, Abcam, Cat#: ab23680), anti‐TGN38 (M‐290, Sc‐33784, Santa Cruz), anti‐Na+/K+‐ATPase antibody (1: 100, Abcam, Cat#: ab167390), or zDHHC7 antibody (1: 100, Novus, Cat#: NBP2‐32486) overnight at 4°C followed by washing with PBS for three times. Cells were incubated with Alexa Fluor 488 goat anti‐mouse or Alexa Fluor 594 goat anti‐rabbit IgG secondary antibody (1:200; Thermo Fisher Scientific) for 1 h at room temperature followed by washing with PBS for three times. Cells were then stained with DAPI and washed with PBS before cover slides were mounted. Images were taken using a LEICA confocal microscopy. Liver paraffin sections were fixed with 4% paraformaldehyde for 30 min and then permeabilized with 0.5% Triton X‐100 in PBS for 10 min. After washing in PBS for 5 min, liver sections were blocked with 10% of goat serum in PBS for 1 h at room temperature, and then incubated with CD36 (1: 100; Abcam, Cat#: ab23680), and β‐catenin (1: 100; Abcam, Cat#: ab32572) overnight at 4 %. Nuclei staining was done with DAPI at 2 ng/μl for 5 min. Images were obtained on a Leica Microscope.
Statistical analysis
All of data were generated from at least three independent experiments. Each value is presented as mean ± SD. All the raw data were initially subjected to a normal distribution and analysis by 1‐sample K‐S of nonparametric test with SPSS 22.0 software. For animal and cellular experiments, a two‐tailed unpaired Student’s t‐test was performed to compare two groups. One‐way ANOVA followed by the Tukey’s post‐hoc test was used to compare more than two groups. Correlation coefficient was calculated using Spearman correlation test. To avoid bias, all statistical analyses were performed blind. Statistical significance was indicated at *P < 0.05, **P < 0.01, and ***P < 0.001.
Author contributions
Shu Yang: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Writing—original draft. Lijing Jia: Conceptualization; Resources; Data curation; Formal analysis; Supervision; Funding acquisition; Project administration; Writing—review and editing. Jiaqing Xiang: Data curation; Formal analysis; Investigation; Methodology. Guangyan Yang: Data curation; Formal analysis; Investigation; Methodology. Shanhu Qiu: Formal analysis; Methodology. Lin Kang: Formal analysis; Methodology. Peilin Zheng: Formal analysis; Methodology. Zhen Liang: Conceptualization; Formal analysis; Supervision; Project administration. Yan Lu: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
SY: Conceptualization; Investigation; Writing—original draft. LJ: Conceptualization; Supervision; Funding acquisition; Writing—original draft. JX: Investigation. GY: Investigation. SQ: Methodology. LK: Methodology. PZ: Methodology. ZL: Supervision; Project administration. YL: Conceptualization; Funding acquisition; Supervision; Project administration; writing—review & editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work was supported by grants from the Shenzhen Science and technology R & D Foundation (No. KCXFZ202002011010445), the National Natural Science Foundation of China (No. 82000824), China Postdoctoral Science Foundation (No. 2020M683179), the Program of Shanghai Academic Research Leader, and the Basic Research of Science and Technology Innovation Action Plan by Shanghai Municipal Science and Technology Committee (Nos. 21XD1423400 and 21JC1401300).
EMBO reports (2022) 23: e54229.
Contributor Information
Zhen Liang, Email: liang.zhen@szhospital.com.
Yan Lu, Email: lu.yan2@zs-hospital.sh.cn.
Data availability
RNA‐Seq data: Gene Expression Omnibus GSE197053 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197053) and GSE197054 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE197054).
References
- Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min H‐K, Mirshahi F et al (2016) A diet‐induced animal model of non‐alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 65: 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T et al (2012) Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61: 416–426 [DOI] [PubMed] [Google Scholar]
- Brigidi G, Bamji S (2013) Detection of protein palmitoylation in cultured hippocampal neurons by immunoprecipitation and acyl‐biotin exchange (ABE). J vis Exp 50031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabowski A, Żendzian‐Piotrowska M, Konstantynowicz K, Pankiewicz W, Mikłosz A, Łukaszuk B, Górski J (2013) Fatty acid transporters involved in the palmitate and oleate induced insulin resistance in primary rat hepatocytes. Acta Physiol 207: 346–357 [DOI] [PubMed] [Google Scholar]
- Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G (2011) Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 301: G825–G834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA (2000) Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275: 32523–32529 [DOI] [PubMed] [Google Scholar]
- Cui A, Fan H, Zhang Y, Zhang Y, Niu D, Liu S, Liu Q, Ma W, Shen Z, Shen L et al (2019) Dexamethasone‐induced Krüppel‐like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J Clin Invest 129: 2266–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Murakami S, Sun Y, Kilpatrick C, Luscher B (2017) DHHC7 palmitoylates glucose transporter 4 (Glut4) and regulates Glut4 membrane translocation. J Biol Chem 292: 2979–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ (2018) Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24: 908–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg I, Eckel R, Abumrad N (2009) Regulation of fatty acid uptake into tissues: lipoprotein lipase‐ and CD36‐mediated pathways. J Lipid Res 50: S86–S90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray S, Wang B, Orihuela Y, Hong E‐G, Fisch S, Haldar S, Cline GW, Kim JK, Peroni OD, Kahn BB et al (2007) Regulation of gluconeogenesis by Krüppel‐like factor 15. Cell Metab 5: 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaumond F, Gréchez‐Cassiau A, Subramaniam M, Brangolo S, Peteri‐Brünback B, Staels B, Fiévet C, Spelsberg TC, Delaunay F, Teboul M (2010) Kruppel‐like factor KLF10 is a link between the circadian clock and metabolism in liver. Mol Cell Biol 30: 3059–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hames KC, Vella A, Kemp BJ, Jensen MD (2014) Free fatty acid uptake in humans with CD36 deficiency. Diabetes 63: 3606–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PN, Fan L, Sweet DR, Jain MK (2019) The krüppel‐like factors and control of energy homeostasis. Endocr Rev 40: 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Takeda J, Horikawa Y (2011) Krüppel‐like factor‐10 is directly regulated by carbohydrate response element‐binding protein in rat primary hepatocytes. Biochem Biophys Res Commun 412: 638–643 [DOI] [PubMed] [Google Scholar]
- Kennedy D, Kuchibhotla S, Westfall K, Silverstein R, Morton R, Febbraio M (2011) A CD36‐dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc Res 89: 604–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclère PS, Rousseau D, Patouraux S, Guérin S, Bonnafous S, Gréchez‐Cassiau A, Ruberto AA, Luci C, Subramaniam M, Tran A et al (2020) MCD diet‐induced steatohepatitis generates a diurnal rhythm of associated biomarkers and worsens liver injury in Klf10 deficient mice. Sci Rep 10: 12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Oh AR, Lee HY, Moon YA, Lee HJ, Cha JY (2020) Deletion of KLF10 leads to stress‐induced liver fibrosis upon high sucrose feeding. Int J Mol Sci 22: 331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ma X, Xu L (2020) The roles of zinc finger proteins in non‐alcoholic fatty liver disease. Liver Res 4: 35–39 [Google Scholar]
- Lindenmeyer CC, McCullough AJ (2018) The natural history of nonalcoholic fatty liver disease‐an evolving view. Clin Liver Dis 22: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Friedman SL, Shulman GI (2021) Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184: 2537–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiken JJFP, Arumugam Y, Bell RC, Calles‐Escandon J, Tandon NN, Glatz JFC, Bonen A (2002) Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am J Physiol Endocrinol Metab 283: E612–E621 [DOI] [PubMed] [Google Scholar]
- Luiken J, Koonen D, Willems J, Zorzano A, Becker C, Fischer Y, Tandon N, Van Der Vusse G, Bonen A, Glatz J (2002) Insulin stimulates long‐chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 51: 3113–3119 [DOI] [PubMed] [Google Scholar]
- Ma Y, Liu H, Ou Z, Qi C, Xing R, Wang S, Han Y, Zhao TJ, Chen Y (2022) DHHC5 facilitates oligodendrocyte development by palmitoylating and activating STAT3. Glia 70: 379–392 [DOI] [PubMed] [Google Scholar]
- Miquilena‐Colina ME, Lima‐Cabello E, Sanchez‐Campos S, Garcia‐Mediavilla MV, Fernandez‐Bermejo M, Lozano‐Rodriguez T, Vargas‐Castrillon J, Buque X, Ochoa B, Aspichueta P et al (2011) Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non‐alcoholic steatohepatitis and chronic hepatitis C. Gut 60: 1394–1402 [DOI] [PubMed] [Google Scholar]
- Mitchell D, Vasudevan A, Linder M, Deschenes R (2006) Protein palmitoylation by a family of DHHC protein S‐acyltransferases. J Lipid Res 47: 1118–1127 [DOI] [PubMed] [Google Scholar]
- Rada P, González‐Rodríguez Á, García‐Monzón C, Valverde ÁM (2020) Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis 11: 802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Gerauer M, Vartak N, Koch S, Huang Z‐P, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B et al (2010) The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141: 458–471 [DOI] [PubMed] [Google Scholar]
- Ruberto AA, Gréchez‐Cassiau A, Guérin S, Martin L, Revel JS, Mehiri M, Subramaniam M, Delaunay F, Teboul M (2021) KLF10 integrates circadian timing and sugar signaling to coordinate hepatic metabolism. Elife 10: e65574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samovski D, Sun J, Pietka T, Gross R, Eckel R, Su X, Stahl P, Abumrad N (2015) Regulation of AMPK activation by CD36 links fatty acid uptake to β‐oxidation. Diabetes 64: 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz I (2013) Gadd45 proteins in immunity. Adv Exp Med Biol 793: 51–68 [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Tabas I, Pajvani UB (2020) Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158: 1913–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima M, Ogawa W, Hayashi K, Inoue H, Kinoshita S, Okamoto Y, Sakaue H, Wataoka YU, Emi A, Senga Y et al (2010) Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes 59: 1608–16015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao N, Wagner S, Lublin D (1996) CD36 is palmitoylated on both N‐ and C‐terminal cytoplasmic tails. J Biol Chem 271: 22315–22320 [DOI] [PubMed] [Google Scholar]
- Thorne R, Ralston K, de Bock C, Mhaidat N, Zhang X, Boyd A, Burns G (2010) Palmitoylation of CD36/FAT regulates the rate of its post‐transcriptional processing in the endoplasmic reticulum. Biochim Biophys Acta 1803: 1298–1307 [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou H‐I, Hoshida Y et al (2018) A simple diet‐ and chemical‐induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69: 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hao J‐W, Wang XU, Guo H, Sun H‐H, Lai X‐Y, Liu L‐Y, Zhu M, Wang H‐Y, Li Y‐F et al (2019) DHHC4 and DHHC5 facilitate fatty acid uptake by palmitoylating and targeting CD36 to the plasma membrane. Cell Rep 26: 209–221 [DOI] [PubMed] [Google Scholar]
- Wara AK, Wang S, Wu C, Fang F, Haemmig S, Weber BN, Aydogan CO, Tesmenitsky Y, Aliakbarian H, Hawse JR et al (2020) KLF10 Deficiency in CD4+ T Cells Triggers Obesity, Insulin Resistance, and Fatty Liver. Cell Rep 33: 108550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng S‐Y, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, Regen T, Molokanova O, Chen T, Bopp T et al (2018) IL‐4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. EBioMedicine 29: 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Vijayakumar A, Kahn BB (2018) Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol 19: 654–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chen Q, Sun L, Zhang H, Yao L, Cui X, Gao Y, Fang F, Chang Y (2017) KLF10 transcription factor regulates hepatic glucose metabolism in mice. Diabetologia 60: 2443–2452 [DOI] [PubMed] [Google Scholar]
- Ye D, Yang K, Zang S, Lin Z, Chau H‐T, Wang Y, Zhang J, Shi J, Xu A, Lin S et al (2016) Lipocalin‐2 mediates non‐alcoholic steatohepatitis by promoting neutrophil‐macrophage crosstalk via the induction of CXCR2. J Hepatol 65: 988–997 [DOI] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15: 11–20 [DOI] [PubMed] [Google Scholar]
- Zhang H, Chen QI, Yang M, Zhu B, Cui Y, Xue Y, Gong N, Cui A, Wang M, Shen L et al (2013) Mouse KLF11 regulates hepatic lipid metabolism. J Hepatol 58: 763–770 [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhou L, Xu Y, Yang M, Xu Y, Komaniecki G, Kosciuk T, Chen X, Lu X, Zou X et al (2020) A STAT3 palmitoylation cycle promotes T17 differentiation and colitis. Nature 586: 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zhang C, Luo X, Wang P, Zhou W, Zhong S, Xie Y, Jiang Y, Yang P, Tang R et al (2018) CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non‐alcoholic steatohepatitis. J Hepatol 69: 705–717 [DOI] [PubMed] [Google Scholar]
- Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX, Zhang P, She ZG, Zhu L, Cai J, Li H (2019) Unexpected rapid increase in the burden of NAFLD in China from 2008 to 2018: a systematic review and meta‐analysis. Hepatology 70: 1119–1133 [DOI] [PubMed] [Google Scholar]