

Abstract
Neurons are highly susceptible to DNA damage accumulation due to their large energy requirements, elevated transcriptional activity, and long lifespan. While newer research has shown that DNA breaks and mutations may facilitate neuron diversity during development and neuronal function throughout life, a wealth of evidence indicates deficient DNA damage repair underlies many neurological disorders, especially age‐associated neurodegenerative diseases. Recently, efforts to clarify the molecular link between DNA damage and neurodegeneration have improved our understanding of how the genomic location of DNA damage and defunct repair proteins impact neuron health. Additionally, work establishing a role for senescence in the aging and diseased brain reveals DNA damage may play a central role in neuroinflammation associated with neurodegenerative disease.
Keywords: DNA damage, DNA damage repair, inflammation, neurodegeneration, neuron
Subject Categories: DNA Replication, Recombination & Repair; Neuroscience
Deficient DNA damage repair underlies neurological disorders and age‐associated neurodegenerative diseases. This review discusses our current understanding of how the genomic location of DNA damage and defunctional repair proteins impact neuron health.

Introduction
Post‐mitotic neurons are the basic cellular unit of the nervous system. Their function controls primary aspects of human physiology, including movement, breathing, and heart rate, as well as higher order processes such as memory and attentional control. However, as a largely non‐renewable resource, neurons must perform these essential tasks while also maintaining their cellular and genomic integrity over many decades of life. To survive the inexorable passage of time, neurons are equipped with accurate and efficient DNA damage response (DDR) pathways. Defective DDR pathways can result in toxic genomic rearrangements, transcriptional dysregulation, and the accumulation of unrepaired lesions (Hoeijmakers, 2009; Madabhushi et al, 2014; Chow & Herrup, 2015; McKinnon, 2017; Tubbs & Nussenzweig, 2017). These insults ultimately push cellular fate toward apoptosis, senescence, or uncontrolled cell division, all of which are hallmarks of age‐associated disease (Hoeijmakers, 2009; Madabhushi et al, 2014; Chow & Herrup, 2015; McKinnon, 2017; Tubbs & Nussenzweig, 2017). We can appreciate the value of neuron viability in particular through the devastating effects of neurodegenerative diseases, which strip individuals of their memories, motor control, and autonomy. As of 2017, neurologic diseases are the third leading cause of death in the United States and the fifth leading cause of death world‐wide (GBD, 2017 US Neurological Disorders Collaborators, 2021; GBD Compare|IHME Viz Hub).
A well‐established link exists between DNA damage and neurodegenerative diseases. In many cases, DNA damage seems to be one of the earliest indicators of neuropathology, suggesting it may be an initiating lesion of toxicity (Chow & Herrup, 2015; Simpson et al, 2015, 2016; Shanbhag et al, 2019). Recently, numerous findings have helped clarify the mechanisms by which DNA damage may mediate neuronal dysfunction. Broadly, these are lessons learned through both DNA damage repair disorders and models of age‐associated neurodegenerative diseases. Additionally, through the advancement of sequencing techniques to map DNA lesions, rearrangements, and mutations, we are just beginning to appreciate the significance of a lesion’s genomic location in relation to its effect on neuronal activity and, ultimately, degeneration (Lodato et al, 2015, 2018; Wei et al, 2016; McConnell et al, 2017; Reid et al, 2021; Rodin et al, 2021; Wu et al, 2021). Finally, while both neuroinflammation and DNA damage are considered hallmarks and mediators of neurodegeneration, the mechanistic relationship between the two has yet to be fully realized. To this end, concepts from senescence cell biology are helping us inform how one might feed into the other (Bussian et al, 2018; Musi et al, 2018; Chow et al, 2019; Zhang et al, 2019; Gillispie et al, 2021). Here, we will cover the recent advances made in each of these subgenres of disease research and how they enhance our understanding of neuronal function and degeneration.
The DNA damage response (DDR)
Our genome continually incurs damage via exogenous agents and endogenous metabolic byproducts. In response to the constant onslaught of genomic lesions, mammalian cells have developed a myriad of DNA damage response (DDR) pathways, each specializing in the detection and correction of a different type of lesion. Although each pathway recruits different proteins and repair enzymes, the basic DDR format remains the same. Lesions are first detected, then they are processed and/or excised by a nuclease. Lastly, a polymerase synthesizes new DNA to replace the missing nucleotides, and a ligase seals the resulting nick together. The following section briefly summarizes DNA lesions and their corresponding DDR pathway.
Single‐Strand Breaks (SSBs)
While many different types of genomic injuries can occur, the majority ultimately manifest in the form of single‐strand breaks (SSBs). This is largely by virtue of reactive oxygen species (ROS), which attack DNA to form oxidized bases and abasic sites (Lindahl & Barnes, 2000; Madabhushi et al, 2014; Tubbs & Nussenzweig, 2017). ROS‐mediated DNA damage is greatest in the nervous system(Nakamura & Swenberg, 1999), likely because neurons exhibit substantial mitochondrial respiration, consuming approximately 20% of the body’s available oxygen(Attwell & Laughlin, 2001). ROS can generate SSBs directly through attacking the DNA backbone or indirectly through the generation of other DNA modifications whose repair requires transient break formation. One of the most abundant ROS‐mediated DNA modifications is 8‐oxo‐7,8‐dihydroguanine (8oxoG), a non‐bulky lesion whose presence can dysregulate gene transcription and whose erroneous repair results in mutagenesis, a major contributor to aging and disease.
Non‐bulky base modifications such as 8oxoG are resolved through base excision repair (BER), wherein a base‐specific glycosylase detects and removes the damaged base, and the backbone is removed by apurinic/apyrimidinic endonuclease 1 (APE1) to generate an intermediate SSB. From here, the SSB can be resolved either through short‐patch SSB repair (sp‐SSBR) or long‐patch SSBR (lp‐SSBR). In short patch SSBR, polymerase β (POLβ) fills in the missing nucleotide and ligase III (LIG3) seals the nick. The alternative lp‐SSBR replaces larger stretches of DNA (2‐13 nucleotides), utilizing flap endonuclease 1 (FEN1), proliferating cell nuclear antigen (PCNA), and polymerase δ/ε (POL δ/ε) to open and replace the broken DNA strand. Ligase I (LIGI) then seals the nick.
In contrast to smaller base lesions, helix‐distorting bulky lesions (which are most commonly caused by UV exposure) are detected by their steric distortion rather than their specific chemical structure. For example, bulky lesions are detected during transcription when their presence stalls RNA polymerase II, which with the help of proteins CSA and CSB (also known as ERCC6 and ERCC8, respectively) initiates transcription‐coupled nucleotide excision repair (TC‐NER). In non‐transcribed or inactive regions of the genome, bulky lesions are detected by the XPC‐RAD23B‐CEN2 complex, which initiates global genomic NER (GG‐NER). Beyond the mechanism of their initial detection, TC‐NER and GG‐NER share the same pathway. The transcription factor complex TFIIH is recruited to the lesion and opens up the DNA, further recruiting other NER repair factors to form a pre‐incision complex. The damaged nucleotide is removed by ERCC1‐XPF and XPG, generating an SSB. New DNA is synthesized by POLβ/δ/ε and then sealed by LIG1 or LIG3.
Apart from ROS, direct SSBs can also be generated by aborted topoisomerase I (TOP1) activity, which occurs when TOP1‐initated breaks meant to relax supercoiled DNA are not resolved. These persisting breaks are termed TOP1 DNA cleavage complexes (Top1cc) (El‐Khamisy et al, 2005). Top1cc accumulation poses a significant threat to the nervous system. First, oxidative DNA damage has been shown to impede Top1cc resolution (Daroui et al, 2004), making neurons particularly sensitized to aborted TOP1 activity. Second, individuals with defunct Tyrosyl‐DNA Phosphodiesterase 1 (TDP1), the SSB repair enzyme responsible for resolving Top1ccs, develop spinocerebellar ataxia with axonal neuropathy (SCAN1). This genetic disease is primarily defined by nervous system deficits such as ataxia, neuropathy, and cerebellar atrophy (Takashima et al, 2002; El‐Khamisy et al, 2005).
Double‐Strand Breaks (DSBs)
While SSBs are the more common form of DNA damage, double‐strand breaks (DSBs) have the higher potential for toxicity. Indeed, it is popularly cited that just one DSB can induce cell cycle arrest and subsequent apoptosis (Huang et al, 1996). However, despite their toxicity, DSBs have also been shown to play important roles in cell physiology. For example, DSBs are required for T‐cell receptor and antibody diversity, chromosomal recombination during meiosis, and in the case of neurons, assist in the expression of immediate early genes (Fig 1) (Suberbielle et al, 2013; Madabhushi et al, 2015; Alt & Schwer, 2018).
Figure 1. Sources of DNA damage in the brain.
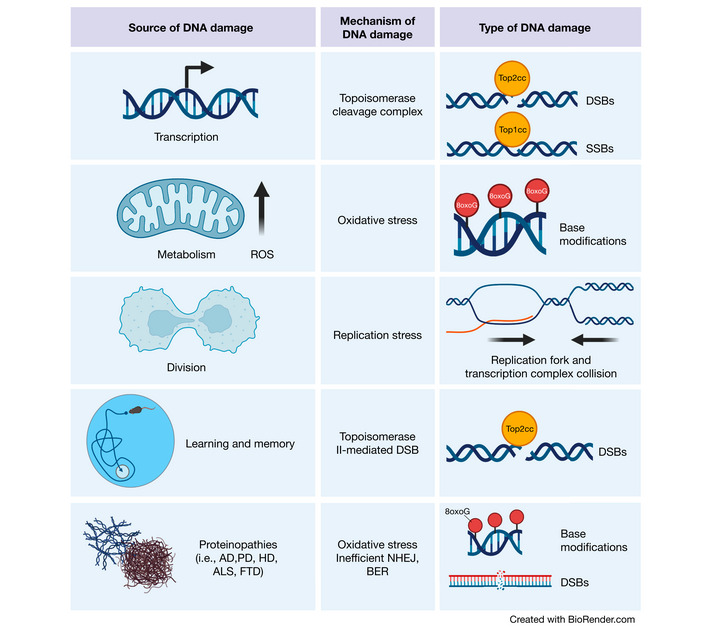
Transcriptional activities can result in topoisomerase cleavage complexes, which lead to the induction of SSBs or DSBs depending upon the topoisomerase in question. Additionally, metabolic activity by mitochondria generate ROS, which can scar DNA bases with oxidative modifications. Although less common in the adult brain, cell division is also a source of DNA damage. Proliferation increases the chance of replication fork and transcription complex collision, thereby inducing DSBs. In the developing brain, this is a particular risk for NPCs, which harbor increased translocations in long genes (where these collisions are most likely to occur) important for neuronal function. Cognitively demanding tasks recruit specific neuronal ensembles whose plasticity is highly dependent upon immediate early gene transcription. Therefore, neurons generate topoisomerase II‐mediated DSBs in response to learning and memory. Finally, the proteins responsible for various neurodegenerative diseases have also been found to play roles in DNA damage detection and repair. (Created with BioRender.com).
While replication is likely the primary cause of DSBs in cycling cells, postmitotic neurons presumably incur the majority of their DSBs through transcriptional activity. SSBs may form DSBs through their collision with the transcriptional machinery and replication forks, or through their close proximity to another SSB. DSBs are also directly generated by transcription (Cannan & Pederson, 2016), whereby topoisomerase II (TOP2) induces transient DSBs to relieve torsional stress and facilitate gene expression. These Top2 cleavage complexes (Top2ccs) are usually resolved immediately by Tyrosyl‐DNA Phosphodiesterase 2 (TDP2). Similar to TDP1, mutations in TDP2 result in a rare neurological disease termed spinocerebellar ataxia autosomal recessive 23 (SCAR23), further underscoring the potential toxicity of topoisomerase‐induced DNA damage in the nervous system (Zagnoli‐Vieira et al, 2018; Gómez‐Herreros et al, 2014: 2; Errichiello et al, 2020).
There are two methods of DSB repair: non‐homologous end joining (NHEJ) and homologous recombination (HR). HR is considered an error‐free method of DSB repair by which resected DNA strands utilize their sister chromatid as a template for DNA synthesis. First, the MRN (MRE11, RAD50, NBS1) complex binds to either side of the DSB to facilitate end resection by nucleases and helicases, including C‐terminal binding protein‐interacting protein (CtIP), Exonuclease 1 (EXO1), DNA replication helicase/nuclease 2 (DNA2), Werner syndrome helicase (WRN), and Bloom syndrome helicase (BLM). The resulting ssDNAs are coated by replication protein A (RPA) and RAD51, forming nucleoprotein filaments that invade the sister chromatid to look for sequence homologies. New DNA is then synthesized by a polymerase and ligated with LIG1 or LIG3.
Because HR requires sister chromatids, this pathway can only occur in cycling cells during or following S phase. In contrast, NHEJ operates in all phases of the cell cycle and thus is the only DSB repair pathway available to post‐mitotic cells. In canonical NHEJ, DSBs are first bound on either end by KU70/80 and DNA‐dependent protein kinase (DNA‐PK), then directly ligated back together with Ligase IV (LIG4), X‐Ray Repair Cross Complementing 4 (XRCC4), and XRCC4‐like factor (XLF). In an alternate form of NHEJ (alt‐NHEJ), the broken strands are resected with the same nucleases and helicases used for HR (CtIP, EXO1, DNA2, BLM, WRN), resulting in single‐strand overhangs at either side of the break site. These overhangs then anneal at microhomologies, which are small stretches of complementary DNA, usually 5–20 bp long. Polymerase θ (POLθ) synthesizes new DNA which is then ligated by LIG3. Yet another alternative DSB repair pathway, termed single‐strand annealing (SSA), searches for even larger homologies (> 25 bp). RAD52 mediates the annealing of resected DNA at these larger homologous sequences, and the resulting DNA flaps are excised by ERCC1‐XPF. Both alt‐NHEJ and SSA are inherently error‐prone, as deletions of DNA and translocations must occur to facilitate strand annealing.
SSB and DSB sensing
Following break induction, chromatin is rapidly modified by DNA damage sensors to facilitate the recruitment of DNA repair proteins. Poly(ADP‐ribose) polymerase 1 (PARP1) and Ataxia telangectasia mutated (ATM) are two major sensors of SSBs and DSBs. PARP1 senses both SSBs and DSBs, rapidly generating poly(ADP‐ribose) (PAR) chain scaffolds (PARylation) on itself and other target proteins to recruit DNA repair proteins and relax chromatin at break sites. PARylation by PARP1 recruits X‐Ray Repair Cross Complementing 1 (XRCC1) which is a crucial stabilizer for end‐processing enzymes POLβ and LIG3. PARylation also facilitates MRN recruitment at DSBs. Similarly, following its activation by the MRN complex at DSB sites, the protein kinase ATM phosphorylates numerous downstream substrates such as histone variant H2A.X, checkpoint kinase 2 (CHK2), and p53 to initiate DSB repair, cell cycle arrest, and apoptosis, respectively. In particular, phosphorylation of H2A.X (γH2AX) by ATM is a critical post‐translational modification, flanking DSB sites more than 500 kb upstream and downstream to form γH2AX foci, which function as docking sites for chromatin remodelers and DNA repair proteins.
Neurotoxicity associated with DNA damage
The nervous system is particularly sensitive to loss‐of‐function mutations in DNA repair proteins. The explanation for this sensitivity may lie in the hallmarks of neuronal identity. That is, neurons perform transcriptionally and energetically demanding cellular functions and are post‐mitotic and long‐lived. The consequence of these features is elevated ROS byproducts, exclusion of error‐free HR repair, age‐associated decline in DNA repair enzyme efficiency, and an overall increased chance of somatic mutation. This is not to say that other cell types in the nervous system (i.e., astrocytes, oligodendrocytes, and microglia) are not susceptible to DNA damage. Indeed, DNA damage in glia plays demonstrable roles in neurodegeneration, as we will discuss later. However, compared to neurons, glial cells are replaceable, have lower energy requirements, and, in some cases, are able to re‐enter the cell cycle, thus facilitating DNA repair. Combined, these features reduce the burden of DNA damage toxicity in glia. Therefore, in the following sections, we take a neuro‐centric approach to interpreting the effects of DNA damage on the nervous system.
Functional roles for DNA damage in neuronal activity and development could lead to dysfunction later in life
Another reason why neurons are so susceptible to genomic toxicity stems from the fact that DNA breaks seem to serve a functional role in neuronal activity. Stimulating primary neurons with bicuculline or subjecting mice to fear learning results in the induction of DSBs at the promoters of immediate‐early genes (Fig 1) (Madabhushi et al, 2015; Stott et al, 2021). Even simply introducing a mouse to a new environment is quickly followed by the induction of DSBs in neurons (Suberbielle et al, 2013, 2015). These activity‐induced DSBs are hypothesized to facilitate the expression of immediate early genes through the rapid resolution of topological constraints at their transcription start sites. Previously, to identify the induction of DSBs at immediate‐early gene promoters, researchers have utilized γH2AX chromatin immunoprecipitation (ChIP) sequencing (Madabhushi et al, 2015; Stott et al, 2021), which generates broad peaks associated with DSB detection. More recently developed technologies may help improve the resolution of activity‐associated break induction in neurons (Rybin et al, 2021). For example, a DSB‐mapping technique known as END‐seq was recently used to identify strand breaks in human induced pluripotent stem cell (iPSC)‐derived neurons, resulting in the finding that enhancers are hotspots for SSBs (Canela et al, 2016; Wu et al, 2021). Newer break‐mapping techniques include single nucleotide precision, Break Labeling In Situ and Sequencing (BLISS) for DSBs (Yan et al 2017) and single‐strand break mapping at nucleotide genome level (SSiNGLe) for both SSBs and DSBs (Cao et al, 2019). However, the utility of these techniques has yet to be evaluated, as currently neither has been used to analyze the location of DNA breaks in neurons in physiological or pathological conditions.
Crucially, while DNA breaks may serve a physiological function in learning and memory, their recurrence in neuron regulatory sequences makes these regions extremely vulnerable to mutation and translocation. One can imagine that over time, erroneous DSB repair could lead to mutations that result in transcriptomic dysfunction, which could further manifest at the cellular level as impaired synaptic signaling. In line with this hypothesis, DNA repair mapping reveals that postmitotic neurons do indeed accumulate breaks in regulatory elements associated with neuronal function (Fig 2) (Reid et al, 2021; Wu et al, 2021).
Figure 2. DNA lesions and mutations identified in neural genes and the techniques used to map them.
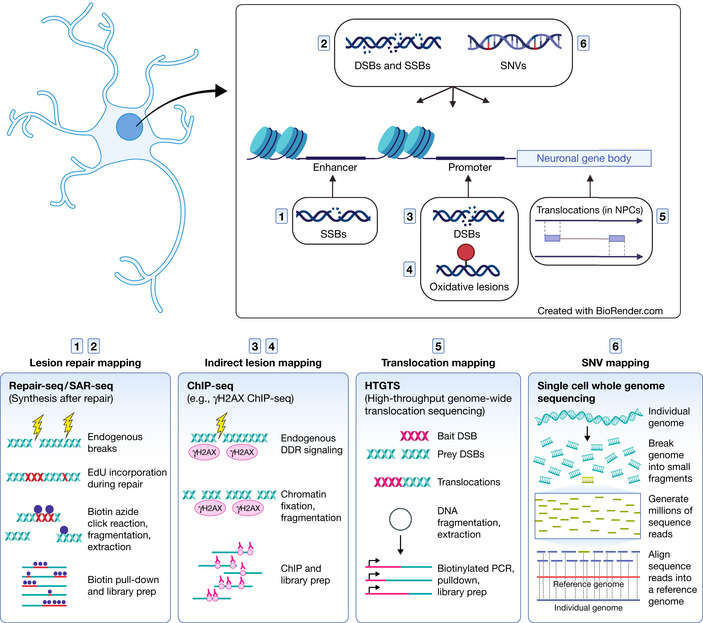
While all cell types incur DNA damage and mutations, neurons in particular are susceptible due to activity‐induced transcription. Immediate‐early genes and other neuronal genes that enable synaptic function are highly transcribed. Accordingly, they accumulate DNA lesions and mutations in their gene body (Wei et al, 2016) and regulatory regions (Lodato et al, 2015; Reid et al, 2021; Wu et al, 2021). The induction of DSBs in the promoters of immediate‐early genes facilitates gene expression (Madabhushi et al, 2015). Over time, these insults may impair neural function (Lu et al, 2004; Lodato et al, 2018; Pao et al, 2020). (Created with BioRender.com).
In addition to postmitotic activity, it is clear that somatic mutations induced by erroneous DNA damage repair or transposable elements are a common feature of neural development, giving rise to neuron diversity through genomic mosaicism (McConnell et al, 2013; Alt & Schwer, 2018; Lodato & Walsh, 2019). While most mutations are likely to be neutral, work has shown that some may underly neurodevelopmental and neurodegenerative disease. For example, work mapping translocations in iPSC‐derived neural precursor cells (NPCs) under replicative stress reveal that DSB hotspots reside in long genes that are important for neuronal function and are risk factors for autism spectrum disorder and schizophrenia (Wei et al, 2016, 2018; Wang et al, 2020). This mapping was accomplished through a technique known as linear amplification‐mediated high‐throughput, genome‐wide, translocation sequencing (LAM‐HTGTS). Using this technique, endogenous DSBs are identified based on their translocation to a “bait” DSB located at a specific region in the genome. Furthermore, a study performing whole exome sequencing of hippocampal tissue from individuals with Alzheimer’s disease (AD) revealed that somatic single nucleotide variations (SNVs) increase with age and are enriched in genes that regulate tau phosphorylation (Park et al, 2019). Accumulation of SNVs at neurodegeneration risk genes could potentially increase risk of disease development.
DNA repair syndromes as proxies for aging and neurodegenerative disease
Some of the most long‐standing evidence for the role of DNA damage in aging and neurodegeneration stems from inheritable DNA damage disorders, which frequently present with neurologic abnormalities. With the exception of AOA5, which seems to have manifested exclusively in adults so far (Hoch et al, 2017: 1; O’Connor et al, 2018: 1), the majority of DNA damage disorders typically present in early childhood. Why then would these disorders support the hypothesis that DNA damage plays critical roles in aging and neurodegeneration? First, as discussed in the previous section, we must acknowledge the pivotal role of DNA damage repair in neurodevelopment, a period in which the rapid proliferation of NPCs results in profound transcriptional and replication‐associated DNA damage. Deficient DNA damage repair in these vulnerable NPCs results in developmental abnormalities such as microcephaly, which is a feature of many DNA damage disorders. However, many DNA damage disorders are also defined by age‐associated pathologies such as progressive brain atrophy and peripheral neuropathy. In these cases, it is likely that post‐mitotic neurons are bearing the brunt of the DNA repair deficit. Thus, whether the resulting pathologies of a DNA repair deficit are developmental or age‐associated likely depends on both the brain cell type composition at the time and the selective vulnerabilities of different neuronal subtypes to different repair deficits. Finally, the pathogenic load of a loss‐of‐function mutation accelerates the development of age‐associated pathology, which may account for a DNA damage disorder presenting in childhood rather than later in life. In contrast, an individual devoid of DNA repair mutations must experience the progressive stress of aging (i.e. damage accumulation, oxidative stress, declining DNA repair enzyme efficiency) in order to recapitulate the pathogenic load of a DNA damage disorder. In the following subsections, we highlight two recently developed models of DNA damage disorders (one driven by mutant XRCC1, and the other by mutant ATM, and APTX) that have clarified mechanisms of DNA damage‐mediated neuronal dysfunction.
Neurological diseases caused by SSBR mutations
To date, loss‐of‐function mutations in SSBR proteins have manifested exclusively as neurodegenerative syndromes. Ataxia with Oculomotor Apraxia types 1 and 4 (AOA1, AOA4) are caused by mutations in DNA end‐processing enzymes Aprataxin (APTX) and Polynucleotide Kinase 3′‐Phosphatase (PNKP) respectively. As their names denote, both are progressive neurodegenerative diseases typified by cerebellar atrophy, ataxia, and oculomotor apraxia. SCAN1, caused by defective TDP1, is a similar neurodegenerative syndrome additionally characterized by peripheral neuropathy (Takashima et al, 2002; El‐Khamisy et al, 2005). More recently, mutations have been identified in XRCC1 (Hoch et al, 2017; O’Connor et al, 2018: 1), the protein that complexes with and stabilizes all of these end‐processing enzymes. Defective XRCC1 manifests in individuals as Ataxia with Oculomotor apraxia Type 5 (AOA5), another slow‐progressing neurodegenerative disease.
If SSBs are left unrepaired in cycling cells, they can form DSBs upon collision with DNA replication complexes (Ryan et al, 1991). Cycling cells with defective SSBR can access error‐free HR during cell division to repair the resulting DSBs. However, post‐mitotic neurons are not equipped with this alternate method of repair. This may explain why diseases of SSBR are exclusive to the nervous system; neurons are not able to mitigate SSB accumulation without the presence of a functioning SSBR. To this point, recent investigations into how mutant XRCC1 confers neuropathy have helped clarify the mechanisms by which defective SSBR could be neurotoxic. First, the study of patient fibroblasts from an individual with AOA5 revealed that in the absence of XRCC1, PARP1 becomes hyperactive, producing excessive amounts of poly(ADP‐ribose) (Hoch et al, 2017). Unchecked PARP1 activity can induce cell death by progressive NAD+/ATP depletion and Parthanatos, a cell death signaling pathway triggered by excessive poly(ADP‐ribose) (David et al, 2009). PARP1 hyperactivity was further demonstrated through conditional deletion of XRCC1 in the mouse brain, which resulted in progressive cerebellar degeneration, ataxia, seizure‐like activity, and dysregulated presynaptic calcium signaling in the hippocampus (Hoch et al, 2017; Komulainen et al, 2021).
The mechanisms by which PARP1 hyperactivity could mediate neurotoxicity and dysregulated presynaptic calcium signaling have been explored in an additional pair of recent publications. First, in the absence of XRCC1, PARP1 was found “trapped” at break intermediates produced during BER, thus impeding access of repair factors POLβ and LIG3 (Demin et al, 2021). This indicates XRCC1 is a crucial regulator of PARP1 activity. Second, PARP1 hyperactivity in XRCC1‐deficient cells was shown to suppress transcription through the recruitment of ubiquitin protease USP3, leading to excessive deubiquitination of histone substrates (Adamowicz et al, 2021). Suppressed transcription may account for the dysregulated calcium signaling observed in neurons from XRCC1 Nes‐Cre mice, specifically through the suppression of genes that regulate calcium homeostasis. To this point, a separate publication revealed that iPSC‐derived neurons accumulate SSBs at enhancers regulating neuronal activity (Wu et al, 2021). These SSB hotspots were identified through genome‐wide mapping of DNA damage repair, dubbed SAR‐seq (Synthesis After Repair), whereby EdU incorporation into break sites serves as a molecular landmark for break repair, and END‐seq (Wu et al, 2021). SSB accumulation at enhancers regulating neuronal activity provides a tempting mechanistic explanation for the neurodegenerative hallmarks of SSBR syndromes.
In contrast to mutations in the SSBR pathways, mutations that dysregulate the NER pathway are additionally characterized by symptoms occurring outside of the nervous system. For example, the hallmark feature of Xeroderma Pigmentosum, caused by XP gene mutations, is skin peeling and crusting due to the skin cells' inability to repair bulky DNA modifications caused by UV exposure. Only about 20–30% of XP individuals develop progressive neurodegeneration (Nouspikel, 2008). Furthermore, individuals with Cockayne Syndrome (CS), who are diagnosed based on delayed development, light sensitivity, and progeria, or Trichothiodystrophy (TTD), whose hallmark feature is brittle hair, can present with neurodevelopmental defects such as microcephaly, dysmyelination, and intellectual disability (Diderich et al, 2011). Notably, while postmitotic neurons are able to repair bulky DNA modifications in both the template and non‐template strand of transcribed genes, global NER is naturally attenuated in non‐transcribed regions of the genome (Nouspikel & Hanawalt, 2000; Nouspikel, 2008). Combined with the added stressor of NER mutations, this may account for the neurodegenerative phenotypes observed in XP, CS, and TTD.
Neurological diseases caused by DSBR mutations
One of the most well‐known DDR syndromes is Ataxia telangectasia (AT), which is caused by mutations in ATM kinase. Individuals with AT exhibit profound immune deficiency and increased cancer susceptibility as well as progressive cerebellar atrophy, which results in ataxia by early childhood (McKinnon, 2012). Mutations in the MRN complex also produce syndromes with similar clinical phenotypes. Specifically, defective MRE11 causes AT‐like disorder, which results in cerebellar atrophy, and defective NBS1 causes Nijmegen Breakage Syndrome, which results in microcephaly (Madabhushi et al, 2014; McKinnon, 2017).
While murine knockout of ATM recapitulates the immune deficits observed in AT patients, these mutant mice seem to exhibit only mild ataxia and cerebellar atrophy (Barlow et al, 1996; Kuljis et al, 1997). This has been a major limiting factor toward teasing out the molecular mechanisms of AT‐related neurodegeneration. Nevertheless, studies utilizing ATM knockout mice have still revealed important roles for ATM in neuron health. First, abnormalities in lysosomal storage have been observed in Purkinje cells of ATM knockout mice, suggesting a pre‐degenerative increase in cellular stress (Barlow et al, 2000). Additionally, enriched Top1ccs are also observed in the cerebellum, similar to what is observed in TDP1 knockout mice, suggesting ATM also plays a role in topoisomerase‐I mediated break sensing and repair (Katyal et al, 2007, 2014). Finally, glial ATM seems to play crucial roles in neuronal health as well. Specifically, cerebellar cultures grown from ATM knockout mice exhibit disrupted network synchrony, which is rescued by culture with wild‐type astrocytes (Kanner et al, 2018).
Notably, the development of a new mouse model of AT harboring both ATM nonsense mutation and APTX knockout was found to better recapitulate the neurologic deficits observed in AT (Perez et al, 2021). While progressive cerebellar atrophy and ataxia were not observed in mice with individual mutations in either ATM or APTX, the combination of both mutations lowered the threshold for neuronal genomic instability, resulting in neurological deficits (Perez et al, 2021). This indicates that at least for murine models of AT, the ATM knockout alone is fairly well tolerated, and additional genomic stress is needed to bring about toxicity in neurons (Tal et al, 2018). A follow‐up study utilizing this new mouse model explores an intriguing explanation for why certain neuronal populations seem selectively vulnerable to neurodegeneration observed in AT (and other DNA damage syndromes for that matter) (Kwak et al, 2021). While both cerebellar neuron subtypes (Purkinje and granule cells) experience DNA break repair deficiencies in the ATM/APTX double mutant mouse, ATAC‐seq (assay for transposase‐accessible chromatin using sequencing) revealed that Purkinje cells harbor uniquely open regions of chromatin that were perturbed by aberrant RNA splicing and subsequent R‐loop formation (a three‐stranded structure formed by an RNA:DNA template hybrid and the non‐template DNA strand) (Kwak et al, 2021). The consequent disruption in Purkinje cell gene expression ultimately results in cerebellar atrophy and ataxia.
DNA damage in neurodegenerative disease
In addition to observations made from genetic disorders, work characterizing the molecular pathophysiology of age‐associated neurodegenerative diseases further implicates DNA damage in brain aging and disease. In the following subsections, we highlight recent publications that explore the effects of DNA damage in the context of age‐associated neurodegenerative disease, as well as recently developed DNA lesion mapping methodologies that may help us better define regions of the neuronal genome that are vulnerable to lesion accumulation and repair (Rybin et al, 2021).
Oxidative DNA damage
Increased oxidative DNA damage is observed in brain tissue from aged individuals (Mecocci et al, 1993) and patients with AD (Nunomura et al, 2001; Lovell & Markesbery, 2007; Weissman et al, 2007), Parkinson’s disease (PD) (Alam et al, 1997), Huntington’s disease (HD) (Browne et al, 1997), and amyotrophic lateral sclerosis (ALS) (Ferrante et al, 1997; Bogdanov et al, 2000). This is also accompanied by an age‐associated decline in BER efficiency (Weissman et al, 2007; Xu et al, 2008; Sykora et al, 2015). Recently, a mechanism for the age and disease‐associated increase in 8oxoG has been proposed. Specifically, the histone deacetylase HDAC1 has been shown to be critical for the repair of age‐associated 8oxoG accumulation by increasing the activity of the DNA glycosylase OGG1 (Pao et al, 2020). In fact, the pharmacological activation of HDAC1 was shown to mitigate oxidative lesion accumulation in both aged mice and 5XFAD mice (mice expressing human APP and PSEN1 with five familial AD mutations), and improve cognition in 5XFAD mice (Pao et al, 2020). Importantly, the location of such oxidized bases has proven to be an important mediator of their neurotoxicity. Specifically, the aging brain contains increased 8oxoG at the guanine‐rich promoters of genes regulating synaptic function, resulting in their transcriptional suppression (Fig 2) (Lu et al, 2004). 8oxoG is capable of repressing gene transcription through many mechanisms, including blocking transcription factor binding (Ghosh & Mitchell, 1999; Moore et al, 2016), recruiting chromatin remodelers that result in the methylation of gene promoters(Wang et al, 2018b; Xia et al, 2017, 4), and erroneous repair through BER, which could result in single nucleotide variations (SNVs). Accordingly, single‐cell whole genome sequencing has revealed that somatic SNVs located near neuronal genes increase with age in human neurons, presumably due to errors in BER, NER, and transcription‐associated repair (Fig 2) (Lodato et al, 2015, 2018). It is possible that at the population level, this accumulated genomic diversity in postmitotic neurons could result in neuronal dysfunction.
Aberrant DNA repair may also directly mediate the severity of neurodegenerative disorders like HD, whose toxicity is derived from the CAG trinucleotide expansion of the Huntingtin gene. Specifically, age‐dependent CAG expansion occurs in the brains of individuals with HD, gradually increasing the toxicity of the Huntington protein in the striatum. Notably, CAG expansion has been found to be dependent upon DNA repair proteins such as OGG1, MLH1, and MSH2, whose detection of oxidized or mismatched bases in these repetitive elements could potentially elicit erroneous repair, leading to CAG expansion (Kovtun et al, 2007; Bettencourt et al, 2016; Pinto et al, 2013; Manley et al, 1999; Lee et al, 2017, 1). Gene‐wide association studies have further identified SNPs in DNA maintenance genes that influence the age of onset in this disease (Lee et al, 2015; Moss et al, 2017). Therefore, erroneous DNA repair mechanisms also directly influence HD severity.
To further understand how DNA lesions accumulate in the brain, multiple sequencing techniques are now available that map these DNA lesions genome‐wide. Previously, studies have approximated the location of 8oxoG lesions through OGG1 ChIP‐seq (Hao et al, 2018), which is a somewhat indirect method as it captures lesion detection rather than the lesion itself. Newer antibody or biotin‐conjugate methods have also been developed to directly target 8oxoG (Ding et al, 2017; Amente et al, 2019); however, these techniques only permit a window of resolution of around 150 nucleotides. To this end, single‐nucleotide resolution DNA lesion sequencing techniques are now being developed that could better define oxidative hotspots in the neuron genome. One such sequencing technique, Click‐Code‐seq, utilizes BER excision enzymes and click chemistry to specifically tag 8oxoG (Wu et al, 2018). Another sequencing technique, Nick‐seq, is capable of detecting a variety of DNA modifications based on the enzyme or chemistry used to first convert the DNA modification to a strand break (Cao et al, 2020). It will be interesting to see if these new technologies can be applied to models of neurodegeneration as well as the postmortem human brain.
SSBs and DSBs
A plethora of studies document increased levels of both SSBs and DSBs in AD, HD, PD, and ALS (Mullaart et al, 1990; Adamec et al, 1999; McKinnon, 2013, 2017; Madabhushi et al, 2014; Alt & Schwer, 2018; Shanbhag et al, 2019; Thadathil et al, 2021), and their accumulation seem to correlate with important milestones in disease progression. Specifically, increased DDR foci are observed in neurons from individuals with mild cognitive impairment (MCI) and AD compared to aged counterparts (Shanbhag et al, 2019). Similarly, DSB marker proteins correlate with cognitive impairment in individuals with low levels of amyloid and tau pathology (Simpson et al, 2015). More recently, increased ROS and DSBs have also been documented in neurons derived directly from sporadic AD patient fibroblasts, indicating that age‐dependent features of genomic instability are recapitulated in human in vitro models of disease (Mertens et al, 2021). These findings have led to the hypothesis that DNA strand breaks may be a contributing factor to disease progression (Chow et al, 2019).
As previously discussed, one mechanism of DNA break‐mediated neurodegeneration may stem from PARP1 hyperactivity. Increased PARP1 activation and elevated poly(ADP‐ribose) has been observed in numerous neurodegenerative disorders and their associated animal models (Thapa et al, 2021; Mao & Zhang 2022). For example, oxidative damage‐induced poly(ADP‐ribose) mediates α‐synuclein aggregation and consequent neurodegeneration in PD (Kam et al, 2018), facilitates the formation of cytoplasmic TDP‐43 foci in ALS (McGurk et al, 2018a, 2018b), and can mediate neuroinflammatory activity in AD (Kauppinen et al, 2011). In combination with studies utilizing XRCC1‐deficient neurons, these findings have motivated the development of PARP inhibitors as a potential therapeutic for a variety of nervous system disorders (Thapa et al, 2021).
In addition to PARP1 activity, many genes that encode the building blocks of neurodegenerative proteinopathies have been found to play a role in DDR as well. For example, the genes whose mutations can cause ALS or frontotemporal dementia (fused in sarcoma (FUS) and transactive response DNA binding protein 43 (TDP‐43)) are involved in SSB and DSB detection and repair. FUS is an RNA/DNA binding protein that has been found to facilitate DSB repair through its colocalization with HDAC1 (Wy et al, 2013, 1). Furthermore, phosphorylation of FUS occurs in response to DSB detection by proteins ATM and DNA‐PK (Gardiner et al, 2008; Deng et al, 2014), and FUS activates and recruits the XRCC1‐LIG3 complex to sites of oxidative DNA damage in a PARP1‐dependent manner (Naumann et al, 2018; Wang et al, 2018a). Recently, TDP‐43 was also found to be directly involved in NHEJ through its recruitment and stabilization of the XRCC4‐LIG4 complex at DSBs (Guerrero et al, 2019; Mitra et al, 2019). The Huntington protein may also play a role in DNA damage detection, potentially serving as a scaffold for ATM at sites of oxidized DNA (Maiuri et al, 2017), and ATXN3 and PNKP at sites of transcription‐coupled DNA break repair (Gao et al, 2019). Finally, α‐synuclein, whose cytoplasmic aggregation is a hallmark of PD, is able to bind double‐strand DNA and facilitate NHEJ in homeostasis (Schaser et al, 2019). However, toxic aggregation of α‐synuclein in the nucleus or cytosol elicits the accumulation of DSBs (Vasquez et al, 2017; Milanese et al, 2018). Tau may also play a role in DSB repair as well, as its deletion in the mouse brain leads to the accumulation of DSB foci in neurons (Violet et al, 2014; Mansuroglu et al, 2016).
While these findings have firmly established DNA breaks as a mechanism of neurodegenerative disease, the mechanisms by which break location may contribute to disease progression are not well described. To this end, genome‐wide profiling of DNA breaks in post‐mitotic neurons have revealed that their locations are likely major mediators of their toxicity. As described earlier, the use of nucleoside analog incorporation to map DNA repair has led to the observation that postmitotic neurons harbor SSBs and other forms of breaks in the regulatory regions of genes that modulate synaptic plasticity and neuronal function (Reid et al, 2021; Wu et al, 2021). It follows that while these breaks may be functionally relevant, over time, their erroneous repair could lead to disruptive translocations, mutations, and genomic instability.
Neuroinflammation associated with DNA damage
In addition to DNA damage, neuroinflammation has emerged as a core feature and mechanism of neurodegenerative disease. Indeed, many of the risk genes associated with AD mediate their effects through microglia, the brain‐resident macrophages (Glass et al, 2010; Heneka et al, 2015; Nott et al, 2019). More recently, researchers have identified senescent‐like brain cells in murine models of AD and tauopathy, and their removal via senolytic drugs seems to mitigate pathology and improve cognition (Bussian et al, 2018; Musi et al, 2018; Zhang et al, 2019; Ogrodnik et al, 2021). Thus, senescence is now considered as a potential driver of neuroinflammation and neurodegeneration. Importantly, while not well‐studied in the context of the degenerating brain, the mechanistic link between DNA damage and senescence has already been defined in great detail (d’Adda di Fagagna, 2008; Miyamoto, 2011; Li & Chen, 2018). Below, we discuss recent mechanistic insights between DNA damage and neuroinflammation, and how DNA damage may influence neurodegeneration through the activation of senescent‐like signaling from different brain cell types.
DNA damage induces senescence
DNA damage is a potent activator of inflammatory signaling and senescence (Coppé et al, 2008; Rodier et al, 2009; Brzostek‐Racine et al, 2011; Härtlova et al, 2015). Initially defined by the permanent cessation of cell division and evasion of apoptosis, senescence is an age‐associated cellular state that is thought to contribute to organismal aging, cancer, and more recently, neurodegeneration (Di Micco et al, 2021). Importantly, removal of senescent cells promotes healthy aging in mice, making senescence‐associated inflammatory signaling an attractive target of therapeutic intervention for aging and age‐related disease (Tilstra et al, 2012; Childs et al, 2015; Baker et al, 2016; Ogrodnik et al, 2021). Senescent cells can be identified by a variety of factors, one being the secretion of pro‐inflammatory cytokines (termed senescence‐associated secretory phenotype (SASP)) to modulate their microenvironment. Initially, induction of persistent DDR signaling in cell cultures including fibroblasts (Coppé et al, 2008; Rodier et al, 2009), bone marrow‐derived macrophages (BMDMs) (Härtlova et al, 2015), and primary monocytes (Brzostek‐Racine et al, 2011) was found to elicit the SASP (Coppé et al, 2008). This secretory profile was found to be at least partially ATM‐dependent, implying the importance of DSB repair specifically in immune activation (Rodier et al, 2009; Brzostek‐Racine et al, 2011). Previous work with AT fibroblasts had shown that the canonical immune transcription factor NF‐kB is activated in an ATM‐dependent manner following treatment with a radiomimetic, ionizing radiation, or topoisomerase inhibitor (Lee et al, 1998; Piret et al, 1999; Li et al, 2001). This suggested NF‐kB was critical for the expression of pro‐inflammatory signaling molecules following DNA damage. Subsequent studies confirmed that following DSB induction, activated ATM couples with a number of protein intermediates leading to its cytoplasmic transport and downstream activation of NF‐kB (Hinz et al, 2010; Wu et al, 2010; Miyamoto, 2011). In addition to inducing the expression of inflammatory signaling proteins, NF‐kB also exerts anti‐apoptotic activity through the expression of caspase inhibitors such as FLIP, XIAP, and c‐XIAP. This directly counteracts pro‐apoptotic activity mediated by DNA damage‐induced p53 signaling. Thus, the interplay between NF‐kB and p53 strongly influence cellular fate following DNA damage accumulation (Karin & Lin, 2002).
Interestingly, a subsequent study revealed that DNA damage can also evoke type‐I interferon signaling in AT cells due to the accumulation of cytosolic DNA fragments, suggesting activation of inflammatory signaling via DNA damage can occur independent of the DSB DDR (Härtlova et al, 2015). Rather, this pathway is initiated through the detection of self‐DNA in the cytosol, the source of which can be DNA damage (Dou et al, 2017; Erdal et al, 2017; Glück et al, 2017; Yang et al, 2017), micronuclei rupture (Harding et al, 2017; Mackenzie et al, 2017), deficient nuclease activity (Ahn et al, 2012; Gao et al, 2015), or de‐repression of retrotransposable elements (De Cecco et al, 2019). In addition to genomic DNA, mitochondrial DNA (mtDNA) is also a major source of innate immune activation (Luna‐Sánchez et al, 2021; Lin et al, 2022). Cytosolic self‐DNA are sensed by the nucleic acid sensor cyclic GMP‐AMP (cGAMP) synthase (cGAS) which produces the second messenger cGAMP upon binding to free‐floating DNA. The production of cGAMP in turn activates Stimulator of IFN Genes (STING). The activation of STING mediates a number of downstream signaling cascades, including the activation of both IRF3 and NF‐kB, thus resulting in the expression of interferons and pro‐inflammatory cytokines (Abe & Barber, 2014; Dunphy et al, 2018; Li & Chen, 2018). Therefore, DNA damage has direct molecular links to senescence‐associated inflammatory signaling, both through DDR pathway activation and the mis‐localization of DNA itself (Fig 3).
Figure 3. DNA damage initiates inflammatory signaling through DDR and cGAS‐STING. ATM‐NF‐kB pathway.
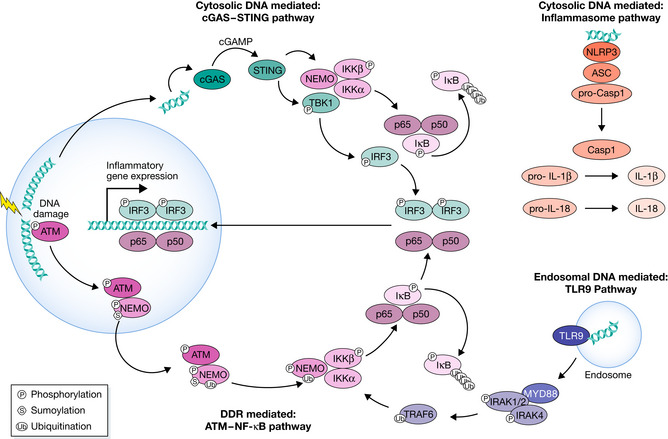
The activation of ATM by DSBs leads to its coupling with NEMO in the nucleus. NEMO is phosphorylated by ATM and SUMOylated by PIASy in a PARP1‐dependent manner, which is not shown here. These modifications lead to NEMO monoubiquitination, and the ATM‐NEMO complex is transported to the cytoplasm. Here, NEMO partners with IKKb and IKKa to form the active Inhibitor of KappaB Kinase (IKK) complex. The IKK phosphorylates IkB, allowing NF‐kB to be transported to the nucleus. The most common form of NF‐kB is the heterodimer p50‐p65, shown here. The phosphorylation of IkB leads to its polyubiquitination and subsequent degradation. cGAS‐STING pathway: DSBs result in the leakage of self‐DNA into the cytosol, which is sensed by cGAS. cGAS generates second messenger cyclic GAMP. cGAMP binds to STING, which activates TANK‐binding kinase 1 (TBK1), which in turn activates IFN Regulatory Factor 3 (IRF3). Homodimerized IRF3 transports to the nucleus and activates the expression of inflammatory genes. STING also facilitates the formation of the IKK complex, which phosphorylates IkB to activate NF‐kB. TLR9 pathway: Endosomal double‐strand DNA is bound by TLR9, activating MyD88, which interacts with and activates IRAK1,2,and 4. IRAK1 and 4 dissociate from MyD88 and activate TRAF6. TRAF6 ubiquitinates NEMO, a member of the IKK complex that results in NF‐kB translocation into the nucleus. Inflammasome pathway: NLRP3 detects cytosolic DNA, leading to the assembly of the NLRP3, ASC, pro‐Caspase I inflammasome. pro‐Caspase I autoproteolytically matures to functional Caspase I, which cleaves pro‐IL1b and pro‐IL18 to generate functional IL1b and IL18.
Additional mechanisms exist to elicit inflammatory gene transcription following the detection of cytosolic nucleic acids (Miller et al, 2021). This includes TLR9 (Toll‐like receptor 9), which binds to double‐stranded DNA engulfed in endosomes, particularly bacterial DNA or mtDNA. This recruits adaptor protein MYD88 (myeloid differentiation primary response 88) to activate NFκB and stimulate the expression of inflammatory cytokines. An additional mechanism of cytosolic DNA‐mediated inflammation is through the NLRP3 inflammasome, which senses cell stressors (such as cytosolic DNA) to activate the cleavage of pro‐1L1b and pro‐IL18 via Caspase I to elicit their secretion. In particular, microglial NLRP3 has been shown to be a major driver of amyloid beta and tau toxicity in AD, suggesting its activation has important implications for disease progression (Halle et al, 2008; Venegas et al, 2017; Ising et al, 2019).
Interestingly, work characterizing the neurodevelopmental defects observed in the neuroinflammatory disorder Aicardi‐Goutières Syndrome (AGS), a type I interferonopathy, indicates that DNA damage can mediate cellular toxicity through multiple different mechanisms in the same disease. AGS is caused by mutations in genes that regulate nucleic acid metabolism. For example, deficits in RNASEH2, an enzyme responsible for removing ribonucleotides from DNA and resolving R‐loops, comprise more than half of AGS diagnoses (Crow & Manel, 2015). Accordingly, RNASEH2 murine models of AGS reveal that CGAS‐STING sensing of micronuclei is the cause for type I IFN signaling in this disease (Mackenzie et al, 2016, 2017; Pokatayev et al, 2016). However, recent work investigating the cause of AGS neurotoxicity revealed that it is the DDR‐mediated activation of p53, not CGAS‐STING, that causes neuron cell death, although increased micronuclei and R‐loops were observed in astrocytes (Aditi et al, 2021). This indicates that different phenotypes of DNA damage response deficits are mediated by different pathways, which may have preferential function in different cell types.
Senescence‐associated inflammation in brain aging and neurodegeneration
It has since been shown that removal of senescent cells in the brain mitigates neurofibrillary tangle (NFT) burden, amyloid burden, neuronal loss, and cognitive decline in mouse models of tauopathy (Bussian et al, 2018; Musi et al, 2018), AD (Zhang et al, 2019), PD (Chinta et al, 2018), and normal aging (Ogrodnik et al, 2021). Notably, each of these studies attribute different cell types as the primary sources of senescence. For example both microglia (Bussian et al, 2018), astrocytes (Bussian et al, 2018), and neurons (Musi et al, 2018) have been found to display senescent‐like phenotypes in Tau P301S mice, and have been proposed as drivers of cognitive decline and neurodegeneration. Additionally, senescent astrocytes have been identified as modulators of PD pathology (Chinta et al, 2018). Senescent‐like phenotypes have also been identified in oligodendrocyte precursor cells (OPCs) in APP/PS1 mice, and their removal has been shown to mitigate amyloid load and cognitive decline (Zhang et al, 2019). Finally, removal of senescent microglia from aged mice has been found to improve cognition (Ogrodnik et al, 2021). In total, these findings place senescence as an encouraging target for therapeutic intervention in neurodegenerative disease, but bring into question how senescence presents in different brain cell types, and how this might affect the progression of various diseases.
Studies have also made more direct links between DNA damage, neuroinflammation, and neurotoxicity. For example, ATM‐deficient microglia have been shown to accumulate cytosolic DNA, thus activating the cGAS‐STING pathway to elicit pro‐inflammatory and neurotoxic signals (Quek et al, 2017; Song et al, 2019). Interestingly, sequencing the cytosolic DNA fragments from these microglia revealed that most were derived from the nuclear DNA as intergenic repetitive elements, suggesting that the source of cytosolic DNA may not be random (Song et al, 2021). Recent single‐nucleus RNA sequencing of AT cerebella reveals that microglial activation likely precedes neuronal degeneration, further emphasizing the role of microglia in ATM‐mediated neuropathology (Lai et al, 2021).
The role of neuronal DNA damage in neuroinflammation
Notably, the concept of a senescent‐like phenotypes in neurons has been particularly controversial, presumably because senescence was initially defined in the context of cycling cells (Gillispie et al, 2021; Sah et al, 2021). Nevertheless, observations of senescent‐like phenotypes in neurons have been reported for quite some time. As early as 2012, the term “senescence” was used to describe pathological features of neurons in the aged mouse brain, including increased DSBs, lipid peroxidation, and senescent‐associated β‐galactosidase staining (Jurk et al, 2012). Here, DSBs were considered the major driver of these senescence‐associated phenotypes because knock‐out of p21, a protein activated by p53 to initiate DSB‐induced cell cycle arrest, was able to mitigate their enrichment. Interestingly, re‐inspection of older transcriptional data also revealed similar enrichment in AD neurons. In a 2006 study, neurons with high levels of tau pathology or no tau pathology were isolated from AD brain tissue and transcriptionally characterized (Dunckley et al, 2006). This dataset was later re‐analyzed to show that neurons with high levels of tau were enriched for signatures of DNA damage and senescence (Dunckley et al, 2006; Musi et al, 2018). Senescent‐like phenotypes in neurons have also been noted in other models of aging and neurodegeneration (Chow et al, 2019; Moreno‐Blas et al, 2019).
Because DNA damage accumulates in neurons early on in AD, and because it is also a primary driver of senescence, it is worth hypothesizing that DNA lesions mediate neurodegeneration at least in part through neuronal senescence, although senescence in other cell types clearly also plays important roles in neurodegeneration. Nevertheless, future research will have to dissect how different cell types react to senescent neurons, and how this may facilitate the development of neurodegenerative disease. Importantly, as DNA damage is a well‐established source of inflammatory signaling, this uncovers a previously unstudied aspect of neuronal response to DNA damage. Compared to microglia and astrocytes, the capacity for inflammatory signaling in neurons has received little attention. Indeed, the protein machinery required to detect cytosolic DNA such as STING is reported to be low in neurons compared to glial cells (Mathur et al, 2017; Song et al, 2019). However, work examining the immune response to neurotropic viral infections reveals neuron‐derived inflammatory signaling is a critical feature of the antiviral response (Klein et al, 2005; Chakraborty et al, 2010; Di Liberto et al, 2018). The detection of viral nucleic acids utilizes the same cGAS‐STING pathway as that described for detection of self‐DNA within the cytosol of senescent cells, suggesting that the mechanism of innate immune signaling in DSB‐bearing neurons may be of some significance. Indeed, observations of neuron‐derived immune signaling at early stages of neurodegenerative disease indicate that this may play a role in disease progression (Dutta et al, 2020; Welikovitch et al, 2020). Notably, neuronal cGAS‐STING has been shown to be activated in models of HD and ALS, suggesting that toxic protein aggregates may stimulate DNA release (mitochondrial or genomic) to initiate NFκB activation in neurons (Sharma et al, 2020; Yu et al, 2020).
Intriguingly, the inflammatory transcription factor NF‐kB also plays neuron‐specific roles in learning and memory. Inhibition of NF‐kB signaling specifically in neurons impairs synaptic plasticity and synaptogenesis (O’Mahony et al, 2006; Boersma et al, 2011). Furthermore, suppression of neuronal NF‐kB in vivo renders neurons more susceptible to kainic acid‐induced neurotoxicity (Fridmacher et al, 2003). These data suggest that on top of its function as a pro‐survival and pro‐inflammatory transcription factor, NF‐kB is also critical for homeostatic neuronal function (Kaltschmidt & Kaltschmidt, 2009). Thus, dysregulated NF‐kB signaling in neurons could be a key mechanism linking DNA damage accumulation, altered synaptic function, and disease progression.
Conclusions
DNA damage has long been associated with the aging brain and neurodegeneration; however, the exact mechanisms by which DNA lesions drive these processes are unclear. The utilization of murine models of DNA damage and DNA break mapping techniques have allowed us to identify how DNA damage may regulate the expression of genes essential for neuronal function, and how this might lead to dysfunction later in life. While these are mechanisms specific to neurons, emerging evidence also suggests that many cell types in the brain, including astrocytes, microglia, oligodendrocytes, and neurons, may mediate the cytotoxicity of DNA damage through senescence‐associated signaling. However, it remains to be seen whether DNA damage itself is the main inducer of senescence in each of these cell types. Regardless, it is clear that DNA damage can elicit neuron dysfunction broadly through two distinct mechanisms. First, the location of the lesion has a significant impact on transcriptional mechanisms required for normal cell function. Second, the downstream signaling pathways of lesion detection, whether they be through DDR or cytosolic nucleic acid sensing, can elicit apoptotic or inflammatory signaling that lead to neurotoxicity.
In need of answers.
Are neuron subtypes differentially vulnerable to DNA damage? The wide range of activity and metabolic demands encompassed by neuronal subtypes may predispose certain populations to genomic toxicity. Single cell sequencing techniques are likely to play a crucial role in dissecting the differential responses to lesion accumulation.
What is the functional role of DNA damage‐mediated senescence in neurons? It will be important to determine if neurons harboring DNA damage play meaningful roles in age‐associated neuroinflammation and neurodegeneration.
How does lesion and mutation accumulation in neuronal genes result in neurotoxicity? Numerous studies reveal scars of DNA damage are most abundant around genes important for synaptic function. However, it is unclear how these mutations modify transcription. While this technology is still in its infancy, simultaneously profiling of the genome and transcriptome at single‐cell resolution would clarify the functional readout of these mutations.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
EMBO reports (2022) 23: e54217.
References
- Abe T, Barber GN (2014) Cytosolic‐DNA‐mediated, STING‐dependent proinflammatory gene induction necessitates canonical NF‐κB activation through TBK1. J Virol 88: 5328–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamec E, Vonsattel JP, Nixon RA (1999) DNA strand breaks in Alzheimer’s disease. Brain Res 849: 67–77 [DOI] [PubMed] [Google Scholar]
- Adamowicz M, Hailstone R, Demin AA, Komulainen E, Hanzlikova H, Brazina J, Gautam A, Wells SE, Caldecott KW (2021) XRCC1 protects transcription from toxic PARP1 activity during DNA base excision repair. Nat Cell Biol 23: 1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aditi , Downing SM, Schreiner PA, Kwak YD, Li Y, Shaw TI, Russell HR, McKinnon PJ (2021) Genome instability independent of type I interferon signaling drives neuropathology caused by impaired ribonucleotide excision repair. Neuron 109: 3962–3979.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN (2012) STING manifests self DNA‐dependent inflammatory disease. Proc Natl Acad Sci USA 109: 19386–19391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8‐Hydroxyguanine levels in substantia Nigra. J Neurochem 69: 1196–1203 [DOI] [PubMed] [Google Scholar]
- Alt FW, Schwer B (2018) DNA double‐strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 71: 158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amente S, Di Palo G, Scala G, Castrignanò T, Gorini F, Cocozza S, Moresano A, Pucci P, Ma B, Stepanov I et al (2019) Genome‐wide mapping of 8‐oxo‐7,8‐dihydro‐2’‐deoxyguanosine reveals accumulation of oxidatively‐generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res 47: 221–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, A. Saltness R, Jeganathan KB, Verzosa GC, Pezeshki A et al (2016) Naturally occurring p16Ink4a‐positive cells shorten healthy lifespan. Nature 530: 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D et al (1996) Atm‐deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171 [DOI] [PubMed] [Google Scholar]
- Barlow C, Ribaut‐Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle A‐M, Mariani J et al (2000) ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA 97: 871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt C, Hensman‐Moss D, Flower M, Wiethoff S, Brice A, Goizet C, Stevanin G, Koutsis G, Karadima G, Panas M et al (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann Neurol 79: 983–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MCH, Dresselhaus EC, Biase LMD, Mihalas AB, Bergles DE, Meffert MK (2011) A requirement for nuclear factor‐κB in developmental and plasticity‐associated synaptogenesis. J Neurosci 31: 5414–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Brown RH, Matson W, Smart R, Hayden D, O’Donnell H, Flint Beal M, Cudkowicz M (2000) Increased oxidative damage to DNA in ALS patients. Free Radic Biol Med 29: 652–658 [DOI] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, Macgarvey U, Baik MJ, Berger SC, Muquit MMK, Bird ED, Beal MF (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41: 646–653 [DOI] [PubMed] [Google Scholar]
- Brzostek‐Racine S, Gordon C, Scoy SV, Reich NC (2011) The DNA damage response induces IFN. J Immunol 187: 5336–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau‐dependent pathology and cognitive decline. Nature 562: 578–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, Nussenzweig A (2016) DNA breaks and end resection measured genome‐wide by end sequencing. Mol Cell 63: 898–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannan WJ, Pederson DS (2016) Mechanisms and consequences of double‐strand DNA break formation in chromatin. J Cell Physiol 231: 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao BO, Wu X, Zhou J, Wu H, Liu L, Zhang Q, DeMott MS, Gu C, Wang L, You D et al (2020) Nick‐seq for single‐nucleotide resolution genomic maps of DNA modifications and damage. Nucleic Acids Res 48: 6715–6725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Salazar‐García L, Gao F, Wahlestedt T, Wu C‐L, Han X, Cai YE, Xu D, Wang F, Tang LU et al (2019) Novel approach reveals genomic landscapes of single‐strand DNA breaks with nucleotide resolution in human cells. Nat Commun 10: 5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Nazmi A, Dutta K, Basu A (2010) Neurons under viral attack: victims or warriors? Neurochem Int 56: 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, van Deursen JM (2015) Cellular senescence in aging and age‐related disease: from mechanisms to therapy. Nat Med 21: 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, Rajagopalan S, Limbad C, Madden DT, Campisi J et al (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep 22: 930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow H, Herrup K (2015) Genomic integrity and the ageing brain. Nat Rev Neurosci 16: 672–684 [DOI] [PubMed] [Google Scholar]
- Chow H‐M, Shi M, Cheng A, Gao Y, Chen G, Song X, So RWL, Zhang J, Herrup K (2019) Age‐related hyperinsulinemia leads to insulin resistance in neurons and cell‐cycle‐induced senescence. Nat Neurosci 22: 1806–1819 [DOI] [PubMed] [Google Scholar]
- Coppé J‐P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P‐Y, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLOS Biol 6: e301 [Google Scholar]
- Crow YJ, Manel N (2015) Aicardi‐Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F (2008) Living on a break: cellular senescence as a DNA‐damage response. Nat Rev Cancer 8: 512–522 [DOI] [PubMed] [Google Scholar]
- Daroui P, Desai SD, Li T‐K, Liu AA, Liu LF (2004) Hydrogen peroxide induces topoisomerase I‐mediated DNA damage and cell death *. J Biol Chem 279: 14587–14594 [DOI] [PubMed] [Google Scholar]
- David KK, Andrabi SA, Dawson TM, Dawson VL (2009) Parthanatos, a messenger of death. Front Biosci‐Landmark 14: 1116–1128 [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD et al (2019) L1 drives IFN in senescent cells and promotes age‐associated inflammation. Nature 566: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demin AA, Hirota K, Tsuda M, Adamowicz M, Hailstone R, Brazina J, Gittens W, Kalasova I, Shao Z, Zha S et al (2021) XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol Cell 81: 3018–3030.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Holler CJ, Taylor G, Hudson KF, Watkins W, Gearing M, Ito D, Murray ME, Dickson DW, Seyfried NT et al (2014) FUS is phosphorylated by DNA‐PK and accumulates in the cytoplasm after DNA damage. J Neurosci 34: 7802–7813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Liberto G, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, Steinbach K, Vincenti I, Klimek B, Lingner T et al (2018) Neurons under T cell attack coordinate phagocyte‐mediated synaptic stripping. Cell 175: 458–471.e19 [DOI] [PubMed] [Google Scholar]
- Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F (2021) Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 22: 75–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diderich K, Alanazi M, Hoeijmakers JHJ (2011) Premature aging and cancer in nucleotide excision repair‐disorders. DNA Repair 10: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Fleming AM, Burrows CJ (2017) Sequencing the mouse genome for the oxidatively modified base 8‐Oxo‐7,8‐dihydroguanine by OG‐Seq. J Am Chem Soc 139: 2569–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z et al (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, LaFleur BJ, Coon KD, Brown KM, Caselli R et al (2006) Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging 27: 1359–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, Unterholzner L (2018) Non‐canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF‐κB signaling after nuclear DNA damage. Mol Cell 71: 745–760.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta K, Thammisetty SS, Boutej H, Bareil C, Julien J‐P (2020) Mitigation of ALS pathology by neuron‐specific inhibition of nuclear factor kappa B signaling. J Neurosci 40: 5137–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW (2005) Defective DNA single‐strand break repair in spinocerebellar ataxia with axonal neuropathy‐1. Nature 434: 108–113 [DOI] [PubMed] [Google Scholar]
- Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ (2017) A prosurvival DNA damage‐induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev 31: 353–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errichiello E, Zagnoli‐Vieira G, Rizzi R, Garavelli L, Caldecott KW, Zuffardi O (2020) Characterization of a novel loss‐of‐function variant in TDP2 in two adult patients with spinocerebellar ataxia autosomal recessive 23 (SCAR23). J Hum Genet 65: 1135–1141 [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH Jr, Beal MF (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem 69: 2064–2074 [DOI] [PubMed] [Google Scholar]
- Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israël A, Mémet S (2003) Forebrain‐specific neuronal inhibition of nuclear factor‐κB activity leads to loss of neuroprotection. J Neurosci 23: 9403–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li X‐D, Chen X, Li Q‐Z, Wight‐Carter M, Chen ZJ (2015) Activation of cyclic GMP‐AMP synthase by self‐DNA causes autoimmune diseases. Proc Natl Acad Sci USA 112: E5699–E5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, Yuan S, Dickey AS, Choudhary S, Ashizawa T et al (2019) Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 8: e42988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M, Toth R, Vandermoere F, Morrice NA, Rouse J (2008) Identification and characterization of FUS/TLS as a new target of ATM. Biochem J 415: 297–307 [DOI] [PubMed] [Google Scholar]
- GBD 2017 US Neurological Disorders Collaborators (2021) Burden of neurological disorders across the US from 1990–2017: a global burden of disease study. JAMA Neurol 78: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD Compare | IHME Viz Hub, authors.
- Ghosh R, Mitchell DL (1999) Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res 27: 3213–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillispie GJ, Sah E, Krishnamurthy S, Ahmidouch MY, Zhang B, Orr ME (2021) Evidence of the cellular senescence stress response in mitotically active brain cells‐implications for cancer and neurodegeneration. Life Basel Switz 11: 153 [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T‐W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A (2017) Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Herreros F, Schuurs‐Hoeijmakers JHM, McCormack M, Greally MT, Rulten S, Romero‐Granados R, Counihan TJ, Chaila E, Conroy J, Ennis S et al (2014) TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet 46: 516–521 [DOI] [PubMed] [Google Scholar]
- Guerrero EN, Mitra J, Wang H, Rangaswamy S, Hegde PM, Basu P, Rao KS, Hegde ML (2019) Amyotrophic lateral sclerosis‐associated TDP‐43 mutation Q331K prevents nuclear translocation of XRCC4‐DNA ligase 4 complex and is linked to genome damage‐mediated neuronal apoptosis. Hum Mol Genet 28: 2459–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐β. Nat Immunol 9: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Qi T, Pan L, Wang R, Zhu B, Aguilera‐Aguirre L, Radak Z, Hazra TK, Vlahopoulos SA, Bacsi A et al (2018) Effects of the stimuli‐dependent enrichment of 8‐oxoguanine DNA glycosylase1 on chromatinized DNA. Redox Biol 18: 43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Härtlova A, Erttmann SF, Raffi FAM, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA et al (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti‐microbial innate immunity. Immunity 42: 332–343 [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss‐Coray T, Vitorica J, Ransohoff RM et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Stilmann M, Arslan SÇ, Khanna KK, Dittmar G, Scheidereit C (2010) A cytoplasmic ATM‐TRAF6‐cIAP1 module links nuclear DNA damage signaling to ubiquitin‐mediated NF‐κB activation. Mol Cell 40: 63–74 [DOI] [PubMed] [Google Scholar]
- Hoch NC, Hanzlikova H, Rulten SL, Tétreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA et al (2017) XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541: 87–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JHJ (2009) DNA damage, aging, and cancer. N Engl J Med 361: 1475–1485 [DOI] [PubMed] [Google Scholar]
- Huang LC, Clarkin KC, Wahl GM (1996) Sensitivity and selectivity of the DNA damage sensor responsible for activating p53‐dependent G1 arrest. Proc Natl Acad Sci USA 93: 4827–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira‐Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575: 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Jill Saffrey M, Cameron K et al (2012) Postmitotic neurons develop a p21‐dependent senescence‐like phenotype driven by a DNA damage response. Aging Cell 11: 996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C (2009) NF‐κB in the nervous system. Cold Spring Harb Perspect Biol 1: a001271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam T‐I, Mao X, Park H, Chou S‐C, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R et al (2018) Poly(ADP‐ribose) drives pathologic α‐synuclein neurodegeneration in Parkinson’s disease. Science 362: eaat8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner S, Goldin M, Galron R, Ben Jacob E, Bonifazi P, Barzilai A (2018) Astrocytes restore connectivity and synchronization in dysfunctional cerebellar networks. Proc Natl Acad Sci USA 115: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF‐κB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Katyal S, El‐Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ (2007) TDP1 facilitates chromosomal single‐strand break repair in neurons and is neuroprotective in vivo . EMBO J 26: 4720–4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, Zhao J, Russell HR, Petrini JHJ, Nitiss JL et al (2014) Aberrant topoisomerase‐1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat Neurosci 17: 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Suh SW, Higashi Y, Berman AE, Escartin C, Won SJ, Wang C, Cho S‐H, Gan L, Swanson RA (2011) Poly(ADP‐ribose)polymerase‐1 modulates microglial responses to amyloid β. J Neuroinflammation 8: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS (2005) Neuronal CXCL10 directs CD8+ T‐cell recruitment and control of west Nile virus encephalitis. J Virol 79: 11457–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komulainen E, Badman J, Rey S, Rulten S, Ju L, Fennell K, Kalasova I, Ilievova K, McKinnon PJ, Hanzlikova H et al (2021) Parp1 hyperactivity couples DNA breaks to aberrant neuronal calcium signalling and lethal seizures. EMBO Rep 22: e51851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT (2007) OGG1 initiates age‐dependent CAG trinucleotide expansion in somatic cells. Nature 447: 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis RO, Xu Y, Aguila MC, Baltimore D (1997) Degeneration of neurons, synapses, and neuropil and glial activation in a murine Atm knockout model of ataxia–telangiectasia. Proc Natl Acad Sci USA 94: 12688–12693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YD, Shaw TI, Downing SM, Tewari A, Jin H, Li Y, Dumitrache LC, Katyal S, Khodakhah K, Russell HR et al (2021) Chromatin architecture at susceptible gene loci in cerebellar Purkinje cells characterizes DNA damage‐induced neurodegeneration. Sci Adv 7: eabg6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Kim J, Jeffries AM, Tolles A, Chittenden TW, Buckley PG, Yu TW, Lodato MA, Lee EA (2021) Single‐nucleus transcriptomic analyses reveal microglial activation underlying cerebellar degeneration in Ataxia Telangiectasia. 10.1101/2021.09.09.459619 [PREPRINT] [DOI]
- Lee J‐M, Chao MJ, Harold D, Abu Elneel K, Gillis T, Holmans P, Jones L, Orth M, Myers RH, Kwak S et al (2017) A modifier of Huntington’s disease onset at the MLH1 locus. Hum Mol Genet 26: 3859–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐M, Wheeler V, Chao M, Vonsattel J, Pinto R, Lucente D, Abu‐Elneel K, Ramos E, Mysore J, Gillis T et al (2015) Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162: 516–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S‐J, Dimtchev A, Lavin MF, Dritschilo A, Jung M (1998) A novel ionizing radiation‐induced signaling pathway that activates the transcription factor NF‐κB. Oncogene 17: 1821–1826 [DOI] [PubMed] [Google Scholar]
- Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y, Karin M, Rotman G (2001) ATM is required for IκB kinase (IKK) activation in response to DNA double strand breaks *. J Biol Chem 276: 8898–8903 [DOI] [PubMed] [Google Scholar]
- Li T, Chen ZJ (2018) The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215: 1287–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Liu N, Qin Z, Wang Y (2022) Mitochondrial‐derived damage‐associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin 10.1038/s41401-022-00879-6 [DOI] [Google Scholar]
- Lindahl T, Barnes DE (2000) Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65: 127–134 [DOI] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN et al (2018) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359: 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Walsh CA (2019) Genome aging: somatic mutation in the brain links age‐related decline with disease and nominates pathogenic mechanisms. Hum Mol Genet 28: R197–R206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X et al (2015) Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Markesbery WR (2007) Oxidative DNA damage in mild cognitive impairment and late‐stage Alzheimer’s disease. Nucleic Acids Res 35: 7497–7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao S‐Y, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883–891 [DOI] [PubMed] [Google Scholar]
- Luna‐Sánchez M, Bianchi P, Quintana A (2021) Mitochondria‐induced immune response as a trigger for neurodegeneration: a pathogen from within. Int J Mol Sci 22: 8523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaitė Ž, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B et al (2016) Ribonuclease H2 mutations induce a cGAS/STING‐dependent innate immune response. EMBO J 35: 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C‐A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A et al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning A, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao P‐C et al (2015) Activity‐induced DNA breaks govern the expression of neuronal early‐response genes. Cell 161: 1592–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai L‐H (2014) DNA damage and its links to neurodegeneration. Neuron 83: 266–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon‐Mom WMC, Truant R (2017) Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet 26: 395–406 [DOI] [PubMed] [Google Scholar]
- Manley K, Shirley TL, Flaherty L, Messer A (1999) Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 23: 471–473 [DOI] [PubMed] [Google Scholar]
- Mansuroglu Z, Benhelli‐Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A, Delattre L, Loyens A, Talahari S, Bégard S et al (2016) Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep 6: 33047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Zhang G (2022) The role of PARP1 in neurodegenerative diseases and aging. FEBS J 289: 2013–2024. [DOI] [PubMed] [Google Scholar]
- Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, Mistry KN, Do D, Marsh SE, Abud EM et al (2017) Activation of the STING‐dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96: 1290–1302.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing‐Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM et al (2013) Mosaic copy number variation in human neurons. Science 342: 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes‐Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D et al (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: the brain somatic mosaicism network. Science 356: eaal1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Gomes E, Guo L, Mojsilovic‐Petrovic J, Tran V, Kalb RG, Shorter J, Bonini NM (2018a) Poly(ADP‐Ribose) prevents pathological phase separation of TDP‐43 by promoting liquid demixing and stress granule localization. Mol Cell 71: 703–717.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Mojsilovic‐Petrovic J, Van Deerlin VM, Shorter J, Kalb RG, Lee VM, Trojanowski JQ, Lee EB, Bonini NM (2018b) Nuclear poly(ADP‐ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun 6: 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol Mech Dis 7: 303–321 [Google Scholar]
- McKinnon PJ (2013) Maintaining genome stability in the nervous system. Nat Neurosci 16: 1523–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ (2017) Genome integrity and disease prevention in the nervous system. Genes Dev 31: 1180–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF (1993) Oxidative damage to mitochondrial DNA shows marked age‐dependent increases in human brain. Ann Neurol 34: 609–616 [DOI] [PubMed] [Google Scholar]
- Mertens J, Herdy JR, Traxler L, Schafer ST, Schlachetzki JCM, Böhnke L, Reid DA, Lee H, Zangwill D, Fernandes DP et al (2021) Age‐dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 28: 1533–1548.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese C, Cerri S, Ulusoy A, Gornati SV, Plat A, Gabriels S, Blandini F, Di Monte DA, Hoeijmakers JH, Mastroberardino PG (2018) Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, Adams PD (2021) Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184: 5506–5526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, Gao J, Pandey A, Taylor JP, Kraemer BC et al (2019) Motor neuron disease‐associated loss of nuclear TDP‐43 is linked to DNA double‐strand break repair defects. Proc Natl Acad Sci USA 116: 4696–4705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S (2011) Nuclear initiated NF‐κB signaling: NEMO and ATM take center stage. Cell Res 21: 116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SPG, Kruchten J, Toomire KJ, Strauss PR (2016) Transcription factors and DNA repair enzymes compete for damaged promoter sites*. J Biol Chem 291: 5452–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Blas D, Gorostieta‐Salas E, Pommer‐Alba A, Muciño‐Hernández G, Gerónimo‐Olvera C, Maciel‐Barón LA, Konigsberg M, Massieu L, Castro‐Obregón S (2019) Cortical neurons develop a senescence‐like phenotype promoted by dysfunctional autophagy. Aging 11: 6175–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, Durr A, Mead S, Holmans P, Jones L et al (2017) Identification of genetic variants associated with Huntington’s disease progression: a genome‐wide association study. Lancet Neurol 16: 701–711 [DOI] [PubMed] [Google Scholar]
- Mullaart E, Boerrigter METI, Ravid R, Swaab DF, Vijg J (1990) Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol Aging 11: 169–173 [DOI] [PubMed] [Google Scholar]
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, Orr ME (2018) Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17: e12840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura J, Swenberg JA (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res 59: 2522–2526 [PubMed] [Google Scholar]
- Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, Naujock M, Günther R, Jin M, Stanslowsky N et al (2018) Impaired DNA damage response signaling by FUS‐NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 9: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y et al (2019) Brain cell type–specific enhancer–promoter interactome maps and disease ‐ risk association. Science 366: 1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T (2008) Nucleotide excision repair and neurological diseases. DNA Repair 7: 1155–1167 [DOI] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC (2000) Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol Cell Biol 20: 1562–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S et al (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: 759–767 [DOI] [PubMed] [Google Scholar]
- O’Connor E, Vandrovcova J, Bugiardini E, Chelban V, Manole A, Davagnanam I, Wiethoff S, Pittman A, Lynch DS, Efthymiou S et al (2018) Mutations in XRCC1 cause cerebellar ataxia and peripheral neuropathy. J Neurol Neurosurg Psychiatry 89: 1230–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu S, Kwon H, LeFevour A, Chakraborty‐Sett S, Greene WC (2006) NF‐κB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol 26: 7283–7298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, Weigand BM, Patel AD, Pirtskhalava T, Inman CL et al (2021) Whole‐body senescent cell clearance alleviates age‐related brain inflammation and cognitive impairment in mice. Aging Cell 20: e13296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao P‐C, Patnaik D, Watson LA, Gao F, Pan L, Wang J, Adaikkan C, Penney J, Cam HP, Huang W‐C et al (2020) HDAC1 modulates OGG1‐initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat Commun 11: 2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Lee J, Jung ES, Kim M‐H, Kim IB, Son H, Kim S, Kim S, Park YM, Mook‐Jung I et al (2019) Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat Commun 10: 3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez H, Abdallah MF, Chavira JI, Norris AS, Egeland MT, Vo KL, Buechsenschuetz CL, Sanghez V, Kim JL, Pind M et al (2021) A novel, ataxic mouse model of ataxia telangiectasia caused by a clinically relevant nonsense mutation. Elife 10: e64695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St. Claire J, Panigrahi GB, Hou C, Holloway K, Gillis T et al (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome‐wide and candidate approaches. PLoS Genet 9: e1003930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret B, Schoonbroodt S, Piette J (1999) The ATM protein is required for sustained activation of NF‐κB following DNA damage. Oncogene 18: 2261–2271 [DOI] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ (2016) RNase H2 catalytic core Aicardi‐Goutières syndrome‐related mutant invokes cGAS‐STING innate immune‐sensing pathway in mice. J Exp Med 213: 329–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek H, Luff J, Cheung K, Kozlov S, Gatei M, Lee CS, Bellingham MC, Noakes PG, Lim YC, Barnett NL et al (2017) A rat model of ataxia‐telangiectasia: evidence for a neurodegenerative phenotype. Hum Mol Genet 26: 109–123 [DOI] [PubMed] [Google Scholar]
- Reid DA, Reed PJ, Schlachetzki JCM, Nitulescu II, Chou G, Tsui EC, Jones JR, Chandran S, Lu AT, McClain CA et al (2021) Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science 372: 91–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppé J‐P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin RE, Dou Y, Kwon M, Sherman MA, D’Gama AM, Doan RN, Rento LM, Girskis KM, Bohrson CL, Kim SN et al (2021) The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra‐deep whole‐genome sequencing. Nat Neurosci 24: 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan AJ, Squires S, Strutt HL, Johnson RT (1991) Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res 19: 3295–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybin MJ, Ramic M, Ricciardi NR, Kapranov P, Wahlestedt C, Zeier Z (2021) Emerging technologies for genome‐wide profiling of DNA breakage. Front Genet 11: 1785 [Google Scholar]
- Sah E, Krishnamurthy S, Ahmidouch MY, Gillispie GJ, Milligan C, Orr ME (2021) The cellular senescence stress response in post‐mitotic brain cells: cell survival at the expense of tissue degeneration. Life 11: 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E et al (2019) Alpha‐synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep 9: 10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A, Rissman RA, Masliah E, Mucke L (2019) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol Commun 7: 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Rajendrarao S, Shahani N, Ramírez‐Jarquín UN, Subramaniam S (2020) Cyclic GMP‐AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc Natl Acad Sci USA 117: 15989–15999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Matthews FE, Shaw PJ, Heath PR, Brayne C, Garwood C, Higginbottom A, Wharton SB & MRC Cognitive Function and Ageing Neuropathology Study Group (2015) A neuronal DNA damage response is detected at the earliest stages of Alzheimer’s neuropathology and correlates with cognitive impairment in the Medical Research Council’s Cognitive Function and Ageing Study ageing brain cohort. Neuropathol Appl Neurobiol 41: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Minett T, Matthews FE, Heath PR, Shaw PJ, Goodall E, Garwood CJ, Ratcliffe LE, Brayne C et al (2016) Neuronal DNA damage response‐associated dysregulation of signalling pathways and cholesterol metabolism at the earliest stages of Alzheimer‐type pathology. Neuropathol Appl Neurobiol 42: 167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Aw JTM, Ma F, Cheung MF, Leung D, Herrup K (2021) DNA repair inhibition leads to active export of repetitive sequences to the cytoplasm triggering an inflammatory response. J Neurosci 41: 9286–9307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Ma F, Herrup K (2019) Accumulation of cytoplasmic DNA Due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J Neurosci 39: 6378–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott RT, Kritsky O, Tsai L‐H (2021) Profiling DNA break sites and transcriptional changes in response to contextual fear learning. PLoS One 16: e0249691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Djukic B, Evans M, Kim DH, Taneja P, Wang X, Finucane M, Knox J, Ho K, Devidze N et al (2015) DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 6: 8897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L (2013) Physiologic brain activity causes DNA double‐strand breaks in neurons, with exacerbation by amyloid‐β. Nat Neurosci 16: 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Misiak M, Wang Y, Ghosh S, Leandro GS, Liu D, Tian J, Baptiste BA, Cong W‐N, Brenerman BM et al (2015) DNA polymerase β deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res 43: 943–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MAM, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M et al (2002) Mutation of TDP1, encoding a topoisomerase I–dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32: 267–272 [DOI] [PubMed] [Google Scholar]
- Tal E, Alfo M, Zha S, Barzilai A, De Zeeuw CI, Ziv Y, Shiloh Y (2018) Inactive Atm abrogates DSB repair in mouse cerebellum more than does Atm loss, without causing a neurological phenotype. DNA Repair 72: 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thadathil N, Delotterie DF, Xiao J, Hori R, McDonald MP, Khan MM (2021) DNA double‐strand break accumulation in Alzheimer’s disease: evidence from experimental models and postmortem human brains. Mol Neurobiol 58: 118–131 [DOI] [PubMed] [Google Scholar]
- Thapa K, Khan H, Sharma U, Grewal AK, Singh TG (2021) Poly (ADP‐ribose) polymerase‐1 as a promising drug target for neurodegenerative diseases. Life Sci 267: 118975 [DOI] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N et al (2012) NF‐κB inhibition delays DNA damage–induced senescence and aging in mice. J Clin Invest 122: 2601–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez V, Mitra J, Hegde PM, Pandey A, Sengupta S, Mitra S, Rao KS, Hegde ML (2017) Chromatin‐bound oxidized α‐synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson’s disease. J Alzheimers Dis 60: S133–S150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira‐Saecker A, Schwartz S, Santarelli F, Kummer MP et al (2017) Microglia‐derived ASC specks cross‐seed amyloid‐β in Alzheimer’s disease. Nature 552: 355–361 [DOI] [PubMed] [Google Scholar]
- Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E et al (2014) A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 8: 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Guo W, Mitra J, Hegde PM, Vandoorne T, Eckelmann BJ, Mitra S, Tomkinson AE, Van Den Bosch L, Hegde ML (2018a) Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat Commun 9: 3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wei P‐C, Lim CK, Gallina IS, Marshall S, Marchetto MC, Alt FW, Gage FH (2020) Increased neural progenitor proliferation in a hiPSC model of autism induces replication stress‐associated genome instability. Cell Stem Cell 26: 221–233.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Hao W, Pan L, Boldogh I, Ba X (2018b) The roles of base excision repair enzyme OGG1 in gene expression. Cell Mol Life Sci 75: 3741–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P‐C, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, Schwer B (2016) Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell 164: 644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P‐C, Lee C‐S, Du Z, Schwer B, Zhang Y, Kao J, Zurita J, Alt FW (2018) Three classes of recurrent DNA break clusters in brain progenitors identified by 3D proximity‐based break joining assay. Proc Natl Acad Sci USA 115: 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman L, Jo D‐G, Sørensen MM, de Souza‐Pinto NC, Markesbery WR, Mattson MP, Bohr VA (2007) Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 35: 5545–5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welikovitch LA, Carmo SD, Maglóczky Z, Malcolm JC, Lőke J, Klein WL, Freund T, Cuello AC (2020) Early intraneuronal amyloid triggers neuron‐derived inflammatory signaling in APP transgenic rats and human brain. Proc Natl Acad Sci USA 117: 6844–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, McKeague M, Sturla SJ (2018) Nucleotide‐resolution genome‐wide mapping of oxidative DNA damage by click‐code‐seq. J Am Chem Soc 140: 9783–9787 [DOI] [PubMed] [Google Scholar]
- Wu W, Hill SE, Nathan WJ, Paiano J, Callen E, Wang D, Shinoda K, van Wietmarschen N, Colón‐Mercado JM, Zong D et al (2021) Neuronal enhancers are hotspots for DNA single‐strand break repair. Nature 593: 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z‐H, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V (2010) ATM and NEMO‐dependent ELKS ubiquitination coordinates TAK1‐mediated IKK activation in response to genotoxic stress. Mol Cell 40: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wy W, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Ir Mackenzie IRA, Huang EJ, Tsai LH (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16: 1383–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, Nie Y, Cai YI, Zhang YW, Yu L‐R et al (2017) CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 31: 653–668.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Herzig M, Rotrekl V, Walter CA (2008) Base excision repair, aging and health span. Mech Ageing Dev 129: 366–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L et al (2017) BLISS is a versatile and quantitative method for genome‐wide profiling of DNA double‐strand breaks. Nat Commun 8: 15058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ (2017) cGAS is essential for cellular senescence. Proc Natl Acad Sci USA 114: E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C‐H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, De Nardo D et al (2020) TDP‐43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183: 636–649.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagnoli‐Vieira G, Bruni F, Thompson K, He L, Walker S, de Brouwer APM, Taylor R, Niyazov D, Caldecott KW (2018) Confirming TDP2 mutation in spinocerebellar ataxia autosomal recessive 23 (SCAR23). Neurol Genet 4: e262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M et al (2019) Senolytic therapy alleviates Aβ‐associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22: 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]