

Abstract
Interleukin‐38 (IL‐38) is strongly associated with chronic inflammatory diseases; however, its role in tumorigenesis is poorly understood. We demonstrated that expression of IL‐38, which exhibits high expression in the skin, is downregulated in human cutaneous squamous cell carcinoma and 7,12‐dimethylbenzanthracene/12‐O‐tetradecanoyl phorbol‐13‐acetate‐induced mouse skin tumorigenesis. IL‐38 keratinocyte‐specific knockout mice displayed suppressed skin tumor formation and malignant progression. Keratinocyte‐specific deletion of IL‐38 was associated with reduced expression of inflammatory cytokines, leading to reduced myeloid cell infiltration into the local tumor microenvironment. IL‐38 is dispensable for epidermal mutagenesis, but IL‐38 keratinocyte‐specific deletion reduces proliferative gene expression along with epidermal cell proliferation and hyperplasia. Mechanistically, we first demonstrated that IL‐38 activates the c‐Jun N‐terminal kinase (JNK)/activator protein 1 signal transduction pathway to promote the expression of cancer‐related inflammatory cytokines and proliferation and migration of tumor cells in an IL‐1 receptor‐related protein 2 (IL‐1Rrp2)‐dependent manner. Our findings highlight the role of IL‐38 in the regulation of epidermal cell hyperplasia and pro‐tumorigenic microenvironment through IL‐1Rrp2/JNK and suggest IL‐38/IL‐1Rrp2 as a preventive and potential therapeutic target in skin cancer.
Keywords: IL‐38, IL‐1Rrp2, skin carcinogenesis
Subject Categories: Cancer, Immunology
Decreased inflammation and epidermal cell proliferation in Il‐38 cKO mice suppress skin tumor formation and malignant progression. IL‐1Rrp2/JNK pathway is crucial for IL‐38 mediated expression of inflammatory cytokines and proliferation and migration of tumor cells.
Introduction
Skin cancer is the most common cancer in the United States of America (USA) (Rogers et al, 2010; Ming et al, 2014). Approximately, 2–3 million cases of non‐melanoma skin cancer occur annually worldwide, and its incidence has been rising rapidly over the past several decades (Samarasinghe & Madan, 2012). 7,12‐dimethylbenzanthracene (DMBA)/12‐O‐tetradecanoylphorbol‐13‐acetate (TPA)‐induced skin cancer has remarkable phenotypic and genotypic homology to human cutaneous squamous cell carcinoma (cSCC) development. This model has been widely used to study how exogenous chemically induced inflammation triggers epithelial transformation and promotes subsequent tumor development (Wang et al, 2010b; Cataisson et al, 2012; Modi et al, 2012). The core of skin carcinogenesis lies in the activation of the RAS‐mitogen‐activated protein kinase (MAPK) pathway in early tumor cells and related inflammatory processes that seem to enhance tumor growth (Rundhaug & Fischer, 2010; Cataisson et al, 2012; Hayashi et al, 2015).
The interleukin (IL)‐1 family of cytokines has been widely studied for its pro‐ and anti‐tumor functions in cancer. Several members of this family, such as IL‐1β and IL‐18, have been extensively investigated in cancer to decipher their role in immune cell recruitment as well as tumor cell proliferation, migration, invasion, and metastasis (Guo et al, 2016; Nakamura et al, 2018; Baker et al, 2019). IL‐38 [molecular weight 16.9 kDa, as predicted using ProtParam tool (Yuan et al, 2015)], is the most recently identified member of the IL‐1 family (Bensen et al, 2001; Lin et al, 2001). The main soluble form of IL‐38 is considered to be a secreted ligand, and it is observed mainly in two forms: the full‐length form (1–152 aa) and the truncated form (20–152 aa) (Mora et al, 2016). However, the true nature of IL‐38 is still controversial, and its co‐receptor has not yet been determined. Although several studies have explored the anti‐inflammatory effect of IL‐38, there are some contradictions that require elucidation. Reportedly, IL‐38 can promote the production of several pro‐inflammatory cytokines in different cell types in response to different stimuli (Huard et al, 2021; Mora et al, 2016; van de Veerdonk et al, 2012). As an IL‐1 family receptor antagonist, IL‐38 has been shown to limit IL‐17 production in multiple disease models (Yuan et al, 2016; Boutet et al, 2017; Han et al, 2019). Nevertheless, a recent study has shown that local inflammation was reduced in IL‐38 knockout mice subjected to the experimental autoimmune encephalomyelitis (EAE) model. Increasing evidence suggests that IL‐38 is a crucial element in the pathogenesis of inflammatory autoimmune diseases, such as spondyloarthritis (Xia et al, 2021), psoriasis (Mercurio et al, 2018), psoriatic arthritis (Li et al, 2017), rheumatoid arthritis (Boutet et al, 2016), and systemic lupus erythematosus (Rudloff et al, 2015; Takeuchi et al, 2018). However, few studies to date have investigated the role of IL‐38 in cancer.
In the present study, we aimed to investigate the molecular and cellular targets of IL‐38 during skin tumorigenesis. The study reveals a novel mechanism of IL‐38 in skin cancer and highlights the essential role of keratinocyte‐derived cytokines in cutaneous disease. Our results indicate that IL‐38 keratinocyte‐specific deletion dramatically ameliorates DMBA/TPA‐induced skin tumors accompanied by a reduction in the number of immune cells and expression of cancer‐related inflammatory cytokines. We have also shown that IL‐38 forms a complex with IL‐1 receptor‐related protein type 2 (IL‐1Rrp2) and activates the JNK/AP‐1 signal transduction pathway in an IL‐1Rrp2‐dependent manner. The proliferation and migration of tumor cells and expression of cancer‐related inflammatory cytokines are induced by IL‐38 via an IL‐1Rrp2/JNK‐mediated pathway. Our findings provide in‐depth insights into cytokine functions in cancer and offer a potential new preventive and therapeutic strategy.
Results
Expression of IL‐38 is decreased in human cutaneous squamous cell carcinomas (cSCC) and DMBA/TPA‐induced mouse tumorigenesis
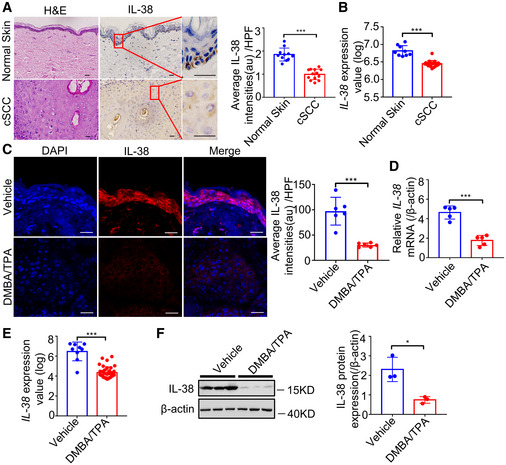
We analyzed IL‐38 expression using the Genotype‐Tissue Expression (GTEx) project (V8 dbGaP Accession phs000424.v8. p2) to determine the landscape of expression of IL‐38. The analysis revealed the lowest expression of IL‐38 in most tissues, while the highest expression was observed in the skin (Fig EV1A). Further, to determine IL‐38 levels in skin tumors, we analyzed the expression of IL‐38 in human skin cancer tissues and found that the protein was significantly lower in cSCC than in normal human skin tissues (Figs 1A and B, and EV1B). Analysis of IL‐38 expression in normal mouse skin and DMBA/TPA‐induced tumors revealed weak levels of IL‐38 in tumors, consistent with findings in human patients (Fig 1C–F). Collectively, these results suggest a possible role of IL‐38 in skin tumorigenesis.
Figure EV1. Expression of IL‐38 in different tissues and the expression of IL‐1 family members in cSCC and DMBA/TPA‐induced mouse tumors.
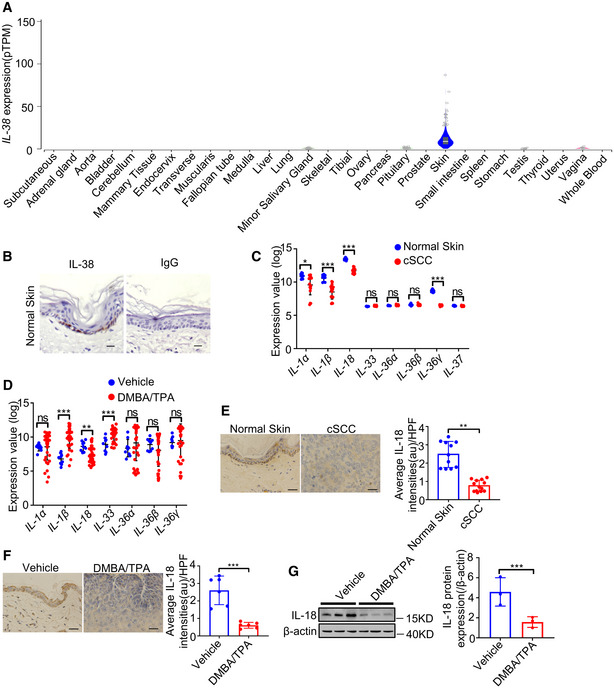
- IL‐38 expression in various tissues sourced from control subjects (n = 6–860) using the GTEx database.
- Representative immunohistochemical staining micrographs of IL‐38 and rabbit IgG in human normal skin. Scale bars represent 200 μm.
- Relative expression of IL‐1 family members in human normal tissues (n = 9) and cSCC (n = 18) were analyzed using Geo Datasets (GSE98767).
- Relative expression of IL‐1 family members in normal tissues (n = 9) and cSCC (n = 38) of mice analyzed using Geo Datasets (GSE63967).
- Representative micrographs of human skin sections stained with anti‐IL‐18 antibody from normal patients (n = 11) and tumors of cSCC patients (n = 13). Scale bars represent 100 μm. The graph shows the quantification of mean IL‐18 expression per high‐powered field in tissues.
- The dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 32 weeks to induce skin tumors. Representative micrographs of mouse normal skin (n = 6) and tumor (n = 6) sections stained with anti‐IL‐18 antibody. Scale bars represent 100 μm. The graph shows the quantification of mean IL‐38 expression in tissues.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test.
Figure 1. IL‐38 is closely related to the occurrence and development of skin tumors.
-
ARepresentative micrographs of human skin sections stained with hematoxylin‐eosin (H&E) (left) and anti‐IL‐38 antibody (right) from normal patients (n = 11) and tumors of cSCC patients (n = 13). Scale bars represent 100 μm. The graph shows the quantification of mean IL‐38 expression per high‐powered field in tissues.
-
BRelative expression of IL‐38 in human normal tissues (n = 9) and cSCC (n = 18) was analyzed using Geo Datasets (GSE98767).
-
C, DThe dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 32 weeks to induce skin tumors. (C) Representative micrographs of mouse normal skin (n = 6) and tumor (n = 6) sections stained with anti‐IL‐38 antibody. The graph shows the quantification of mean IL‐38 expression per high‐powered field in tissues. Scale bars represent 100 μm. (D) Relative expression levels of Il‐38 in normal skin (n = 5) and tumors (n = 5) of mice were quantified using qPCR.
-
ERelative expression levels of Il‐38 in normal tissues (n = 9) and cSCC tissues (n = 38) of mice were analyzed using Geo Datasets (GSE63967).
-
FRepresentative western blot bands indicating IL‐38 in mouse normal skin (n = 3) and DMBA/TPA‐induced tumors (n = 3). The graph shows the quantification of mean IL‐38 expression in tissues.
Data information: Error bars represent the mean ± SD. All data are biological replicates. *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test.
At the same time, we searched the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information (NCBI) and identified multiple IL‐1 family members exhibiting differential expression in skin cancer compared with normal skin searching (Fig EV1C and D). Further, experiments to detect IL‐18 expression in normal human and mouse skin and tumors demonstrated that the expression of IL‐18 decreased in human cutaneous squamous cell carcinoma (cSCC) and DMBA/TPA‐induced mouse tumors compared with its expression in normal skin tissues, which is consistent with the findings in the GEO database (Fig EV1E–G).
IL‐38 keratinocyte‐specific deletion suppresses the development of tumors
To analyze the function of IL‐38 in skin, we crossed mice with floxed alleles of Il‐38 (Il‐38f / f ) with keratin14‐Cre knock‐in mice (K14Cre /+ ‐Il‐38 +/+) to generate mice lacking Il‐38 in keratinocytes (K14Cre /+ ‐Il‐38f / f ) (Fig EV2A–D). Quantitative polymerase chain reaction (qPCR) and western blot analysis confirmed the deletion of Il‐38 in epidermal splits from K14Cre /+ ‐Il‐38f / f mice (Fig EV2E and F). Studies have shown that the epidermis acts as a barrier to prevent water loss and exclude foreign substances and microorganisms (Franzke et al, 2012). Therefore, we analyzed the effect of IL‐38 deletion in keratinocytes on the epidermal barrier in the steady state. Our results showed that keratinocyte‐specific IL‐38 deficiency had no significant effect on the epidermal barrier (Fig EV2G–M). Further, to examine whether IL‐38 has a potential function in tumorigenesis, we applied a two‐stage chemical skin carcinogenesis protocol to generate cutaneous squamous cell carcinoma (cSCC). K14Cre /+ ‐Il‐38f / f mice and control littermates were treated with a single dose of DMBA, followed by weekly application of TPA to allow skin tumor development (Fig 2A). IL‐38 was completely lost or under detectable in DMBA/TPA‐treated skin and DMBA/TPA‐induced tumors of K14Cre /+ ‐Il‐38f / f mice (Fig EV3A–D). Unexpectedly, both the number and volume of tumors were significantly lower in K14Cre /+ ‐Il‐38f / f mice than in Il‐38f / f mice (Fig 2B–D). The first tumors were observed in both Il‐38f / f mice as well as in the K14Cre /+ ‐Il‐38f / f mice 8 weeks after beginning the DMBA/TPA treatment. After 15 weeks, all Il‐38f / f mice developed tumors on their back skin, while approximately 37.5% K14Cre /+ ‐Il‐38f / f mice remained tumor‐free (Fig 2E). In addition, the vast majority of tumors were collected 32 weeks after initiation. Several tumors in Il‐38f / f mice already contained sites of high‐grade dysplasia and foci of microinvasion (Fig 2F; arrow). To further investigate malignant conversion, we performed keratin8 (K8) staining in tumors. Multiple tumors in Il‐38f / f mice were positive for the presence of K8 (Fig 2G). The percentage of tumors that progressed to malignancy was slightly decreased in K14Cre /+ ‐Il‐38f / f mice compared to that in Il‐38f / f mice. Most importantly, the percentage of mice with malignant tumors was significantly lower in IL‐38 keratinocyte‐specific deletion mice (Fig 2H). These results suggest that IL‐38 not only accelerates tumorigenesis, but also the malignant progression of tumors.
Figure EV2. Construction and identification of keratinocytes IL‐38‐specific knockout mice and effect of keratinocyte‐specific IL‐38 deficiency on epidermal barrier.
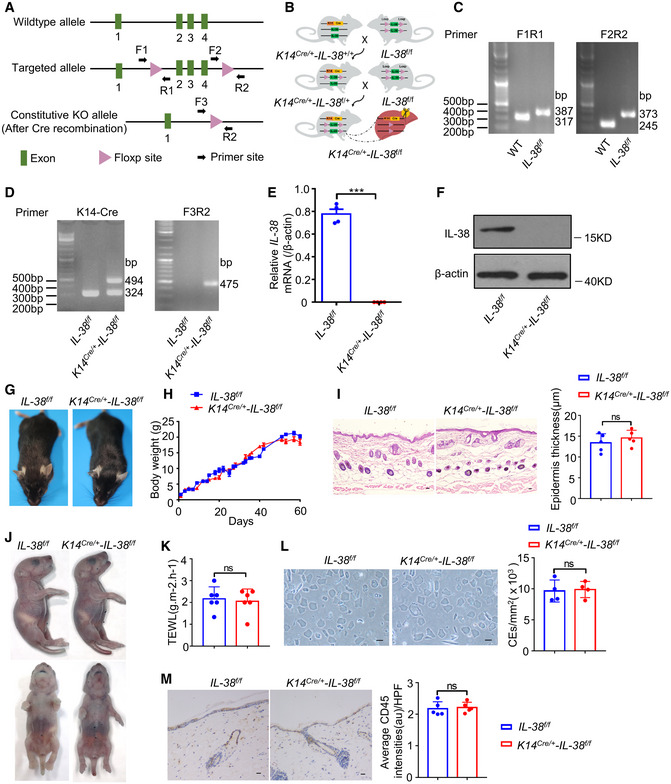
- Construction of Il‐38‐loxP (Il‐38f / f ) mice.
- Typical genetic cross scheme of keratinocyte IL‐38‐specific knockout (K14Cre /+ ‐Il‐38f / f ) mice.
- PCR analysis of WT mice (at 317 and 245 bp) and Il‐38‐loxP mice (at 387 and 373 bp).
- PCR analysis of keratin 14 promoter directing expression of Cre recombinase(left) and deletion of IL‐38 (right).
- Relative expression of IL‐38 in the epidermal splits of Il‐38f / f (n = 4) and K14Cre /+ ‐Il‐38f / f (n = 4) mice.
- Representative western blot bands indicating IL‐38 in the epidermal splits of Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice.
- Appearance of Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice.
- Cumulative body weight changes in Il‐38f / f (n = 12) and K14Cre /+ ‐Il‐38f / f (n = 12) mice in 60 days.
- Representative histological photographs of skin stained with hematoxylin‐eosin (H&E) from Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f (n = 5) mice. Scale bars represent 100 μm. The graph shows the quantification of epidermal thickness.
- Skin barrier‐dependent dye exclusion assay using toluidine blue in Il‐38f / f mice (n = 5) and K14Cre /+ ‐Il‐38f / f littermate (n = 5) at birth.
- TEWL assay measured on ventral surface of newborn Il‐38f / f mice (n = 6) and K14Cre /+ ‐Il‐38f / f littermate (n = 6).
- Representative micrographs of cornified cell envelopes from Il‐38f / f (n = 4) and K14Cre /+ ‐Il‐38f / f (n = 4) mice. Scale bars represent 100 μm. The graph shows the number of cornified cell envelopes per square millimeter.
- Representative immunohistochemical staining micrographs of CD45 in the skin of Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f (n = 5) mice. Scale bars represent 100 μm. The graph shows average intensities of CD45 per high‐powered field.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test.
Figure 2. IL‐38 from keratinocyte‐specific knockout suppressed the progression of DMBA/TPA‐induced cSCC.
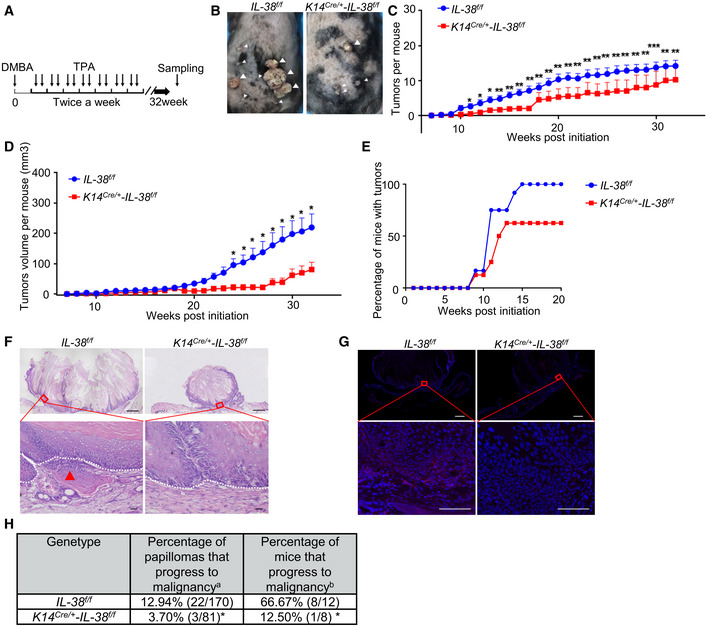
-
ATimeline of DMBA/TPA treatment in two‐stage chemically induced skin tumor development protocol.
-
BRepresentative photographs of Il‐38f / f (n = 12) and K14Cre /+ ‐Il‐38f / f mice (n = 8) treated with DMBA/TPA for 32 weeks. White triangles indicate the tumors on the back of mice.
-
C, DAverage tumor number (C) and volume (D) per mouse from Il‐38f / f (n = 12) and K14Cre /+ ‐Il‐38f / f mice (n = 8) treated with DMBA/TPA.
-
ETumor bearing ratio of Il‐38f / f (n = 12) and K14Cre /+ ‐Il‐38f / f mice (n = 8) treated with DMBA/TPA.
-
FRepresentative histological micrographs stained with hematoxylin‐eosin (H&E) from tumors 32 weeks after initiation. Local micro‐invasive foci (red arrow) were often observed in tumors from K14Cre /+ ‐Il‐38f / f mice (lower panel). Dashed lines indicate the basement membrane. Areas indicated by boxes in the upper pictures are shown at higher magnification in the lower pictures. Top pictures, scale bars represent 800 μm; bottom pictures, scale bars represent 100 μm.
-
GRepresentative micrograph sections stained with K8 from tumors 32 weeks after initiation. Top pictures, scale bars represent 700 μm; bottom pictures, scale bars represent 100 μm.
-
HMalignant conversion rate of tumors 32 weeks after initiation. a = number of carcinomas/total number of tumors. b = number of mice with carcinomas/total number of mice.
Data information: Error bars represent the mean ± SD (C) or mean ± SEM (D). All data are biological replicates. *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test (C and D) or Fisher’s exact test (E and H).
Figure EV3. Expression of IL‐38 and IL‐1Rrp2 in tissues.
-
A–DThe dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 3 weeks to induce the skin inflammation (A and C). (A) Representative immunofluorescent staining micrographs of IL‐38 in the skin of Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice. Scale bars represent 100 μm. (C) Relative expression of IL‐38 in the skin of Il‐38f / f (n = 3) and K14Cre /+ ‐Il‐38f / f (n = 3) mice was detected by western blot. The dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 32 weeks to induce the skin tumors (B and D). (B) Representative immunofluorescent staining micrographs of IL‐38 in the tumors of Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice. Scale bars represent 100 μm. (D) Relative expression of IL‐38 in the tumors of Il‐38f / f (n = 3) and K14Cre /+ ‐Il‐38f / f (n = 3) mice was detected by western blot.
-
ERepresentative immunohistochemical staining micrographs of IL‐1Rrp2 from normal patients (n = 11) and tumors of cSCC patients (n = 13). Scale bars represent 100 μm. The graph shows average intensities of IL‐1Rrp2 per high‐powered field in tissues.
-
FRelative expression of IL‐1Rrp2 in mouse normal skin (n = 3) and DMBA/TPA‐induced tumors (n = 3). The graph shows the quantification of mean IL‐1Rrp2 expression in tissues.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; P values were calculated using Student’s t‐test.
IL‐38 promotes inflammation‐driven tumorigenesis in vivo and in vitro
The pathogenesis of skin cancer involves genetic and non‐genetic molecular changes. In particular, important carcinogenic or tumor suppressor changes may increase cell survival and proliferation, impair DNA repair and checkpoint activation, and induce inflammation in vivo (Ming et al, 2014). DMBA is a typical mutagenic agent belongs to polyaromatic hydrocarbons (PAHs) which are accessible to humans and the analogous mutations are also common in human cancers (Sato et al, 2020). Hence, we investigated the DNA damage in keratinocytes 24 h after the treatment of DMBA. However, phospho‐H2AX (γH2AX), which is recruited DNA damage sites (Mah et al, 2010), showed the same frequency staining within basal keratinocytes in DMBA treated skin of Il‐38f / f mice relative to K14Cre /+ ‐Il‐38f / f mice (Fig 3A). To investigate the effect of IL‐38 on Hras mutation in vivo, a quantitative genomic DNA (gDNA) PCR assay was performed to amplify the Hras 61 codon mutations induced by DMBA. The results demonstrated the same frequency of Hras‐mut61 in Il‐38f / f mice as that in K14Cre /+ ‐Il‐38f / f mice (Fig 3B). In addition, DMBA/TPA‐induced skin carcinogenesis has been considered as an example of inflammation‐driven tumorigenesis (Wang et al, 2010b). Many studies have shown that local inflammation and immune cell infiltration are associated with the development of skin cancer (Vahatupa et al, 2019). A recent report suggested that the absence of IL‐38 attenuates local inflammation in an experimental autoimmune encephalomyelitis model (Huard et al, 2021). To explore whether infiltration of immune cells into the skin and the alleviated skin inflammation were related to K14Cre /+ ‐Il‐38f / f mice with the reduction in skin carcinogenesis, we detected immune cells and the levels of pro‐inflammatory chemokines and cytokines in DMBA/TPA‐induced skin tissues. The analyses demonstrated reduced recruitment of Langerhans cells, neutrophils, macrophages, monocytes, and CD4+ T cells in the skin tissues of K14Cre /+ ‐Il‐38f / f mice compared with that in control mice (Figs 3C and EV4K). Moreover, we found that mRNA levels of pro‐inflammatory cytokines, such as Il‐1β, Il‐12, Il‐22, and Tnf‐α, were reduced in K14Cre /+ ‐Il‐38f / f mice compared with control mice. The chemoattractant chemokines Cxcl1, Cxcl2, Ccl17, and Ccl20, which are crucial for the recruitment of inflammatory cells, were also reduced in the skin of K14Cre /+ ‐Il‐38f / f mice compared with control mice (Fig 3D). These data indicate that IL‐38 keratinocyte‐specific deletion could be associated with decreased infiltration of myeloid cells and expression of pro‐inflammatory cytokines and chemokines that attract myeloid cells into the local tumor microenvironment. IL‐38 expression was lower in SCC cell lines than in normal keratinocyte cell lines, consistent with the in vivo results (Fig EV4A and B). Further, to verify the induction of pro‐inflammatory cytokines and chemokines of IL‐38, A431 cells, the well‐known cSCC cell line, were transfected with an expression vector encoding IL‐38 or infected with a lentivirus encoding IL‐38 shRNA cells. We found that the expression and secretion of IL‐38 significantly increased in IL‐38‐transfected cells (Fig EV4C–E) and decreased in IL‐38 shRNA‐infected cells (Fig EV4F–H) compared with control cells. In addition, mRNA expression of inflammatory mediators, such as IL‐1β, IL‐12, IL‐22, TNF‐α, CXCL1, CXCL2, CCL17, and CCL20, was increased in IL‐38‐overexpressing cells (Fig 3E) and decreased in IL‐38‐knockdown cells (Fig 3F). A previous study reported that LPS‐induced IL‐6 production was significantly higher in the presence of IL‐38 (van de Veerdonk et al, 2012). Our findings demonstrated that neither the overexpression nor the knockdown of IL‐38 exerted no significant difference in the expression of IL‐6 compared with its expression in control cells (Fig EV4I and J). These results indicate that IL‐38 promotes inflammation‐driven tumorigenesis in the DMBA/TPA model and in tumor cells.
Figure 3. IL‐38 promotes inflammation‐driven tumorigenesis in vivo and in vitro .
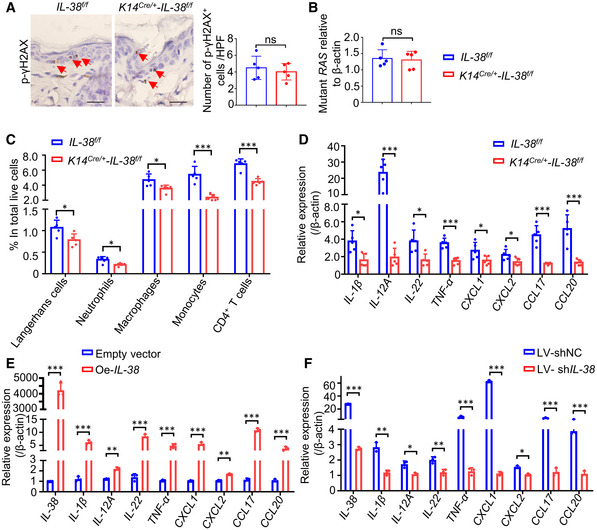
-
ARepresentative immunohistochemical staining micrographs of γH2AX in the skin of Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f (n = 5) mice after treated with DMBA for 24 h. Red triangles indicate the γH2AX+ positive cells. Scale bars represent 100 μm. The graph shows the number of γH2AX+ cells per high‐powered field.
-
BTaqman qPCR analysis of Hras codon‐61 mutations in DMBA‐treated skin of Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f (n = 5) mice after treated with DMBA for 24 h.
-
C, DThe dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 3 weeks to induce the skin inflammation. (C) Percentage of skin‐infiltrating immune cell subsets within total live cells were determined using flow cytometry in DMBA/TPA‐treated Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f mice (n = 5). (D) Relative expression levels of inflammatory mediators in the skin of DMBA/TPA‐treated Il‐38f / f (n = 5) and K14Cre /+ ‐Il‐38f / f mice (n = 5) were quantified using qPCR.
-
E, FRelative expression of inflammatory cytokines in A431 cells after Il‐38 overexpression (E) or knockdown (F) was detected using qPCR.
Data information: Error bars represent the mean ± SD. All data are biological replicates. *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test.
Figure EV4. Expression of IL‐38 and IL‐6 in SCC cell lines and the gating strategy for flow cytometry.
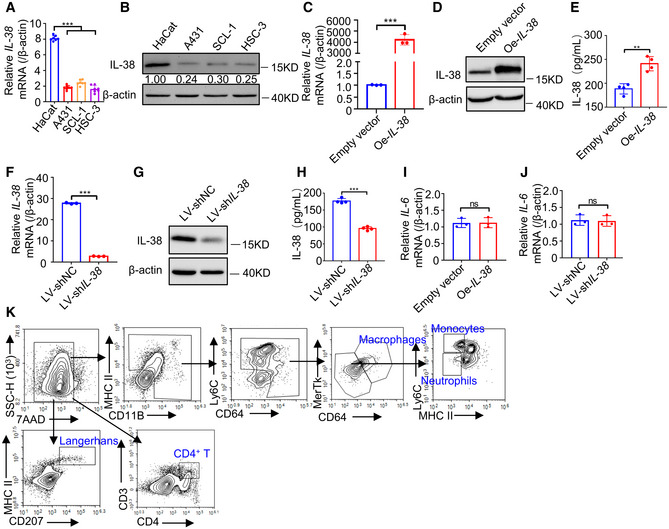
-
ARelative expression of IL‐38 in normal skin cells (n = 7) and SCC cells (n = 7) determined using qPCR.
-
BRelative expression levels of IL‐38 in normal skin and SCC cells were detected using western blot.
-
C–EThe IL‐38 overexpression cell line was constructed by transfecting A431 cells with the pcDNA3.1‐IL‐38 vector expressing IL‐38 and pcDNA3.1 empty vector. (C) Relative expression of IL‐38 was determined using qPCR. (D) Relative expression of IL‐38 was detected using western blotting. (E) The secretion of IL‐38 was measured using ELISA.
-
F–HLentiviruses containing IL‐38 shRNA or Negative Control shRNA were used to infect A431 cells. (F) Relative expression of IL‐38 was determined using qPCR. (G) Relative expression of IL‐38 was detected using western blot. (H) IL‐38 secretion was measured using ELISA.
-
IRelative expression of IL‐6 in IL‐38‐overexpressed A431 cells (n = 3) was determined using qPCR.
-
JRelative expression of IL‐6 in IL‐38‐knockdown A431 cells (n = 3) was determined using qPCR.
-
KThe flow cytometry gating strategy for immune cell detection in DMBA/TPA‐treated skin.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using one‐way ANOVA (A) or Student’s t‐test (C, E, F, and H‐J).
IL‐38 promotes epidermal cancer cell proliferation in vivo and in vitro
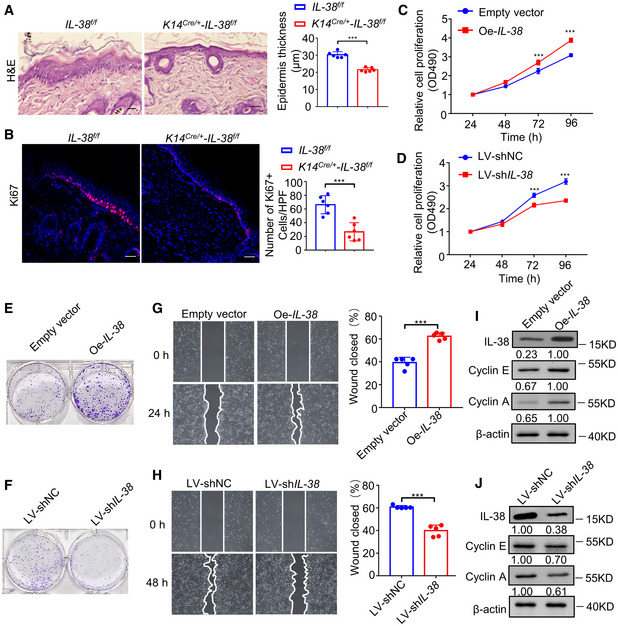
Disorders of cell proliferation are key to tumor formation (Farber, 1995). Since a family member, IL‐1, can stimulate the proliferation of keratinocytes in an autocrine fashion (Kondo, 1999; Jiang et al, 2020), it is possible that IL‐38 also plays a role in accelerating epidermal cancer cell proliferation. To determine whether IL‐38 plays a role in DMBA/TPA‐induced cell proliferation in vivo, we assessed the difference in epidermal thickness and the number of Ki67‐positive epidermal cells in non‐tumor skin from Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice. IL‐38 ablation reduced DMBA/TPA‐induced epidermal hyperplasia (Fig 4A) and the number of Ki67‐positive cells (Fig 4B). In addition, to verify IL‐38 promotion of epidermal cancer cell hyperplasia, we first examined the effect of IL‐38 on cancer cell proliferation and migration. Cell proliferation and migration were enhanced in the cancer cell line A431 with overexpression of IL‐38 (Fig 4C and E, and G) and inhibited if IL‐38 was silenced (Fig 4D and F, and H). Furthermore, we found that overexpression of IL‐38 in A431 cells increased the expression of cyclin E and A (Fig 4I), and treatment with shRNA against IL‐38 decreased their expression (Fig 4J), indicating that IL‐38 may play a role in the regulation of key cell cycle proteins controlling the passage in human cancer cells. These results indicate that IL‐38 promotes epidermal cancer cell proliferation in skin cancer.
Figure 4. IL‐38 promotes the proliferation and migration via mediating key cell cycle proteins of skin cancer cells.
-
A, BThe dorsal hair of normal C57/BL6 mice was shaved and treated with DMBA/TPA twice a week for 3 weeks to induce the skin inflammation. (A) Representative micrograph sections stained with hematoxylin‐eosin (H&E) from the skin of Il‐38f / f (n = 6) and K14Cre /+ ‐Il‐38f / f mice (n = 6) Scale bars represent 100 μm. The graph shows average epidermal thickness. (B) Representative micrograph sections stained with anti‐Ki67 antibody from the skin of Il‐38f / f (n = 6) and K14Cre /+ ‐Il‐38f / f mice (n = 6). Scale bars represent 100 μm. The graph shows average numbers of Ki67+ cells per high‐powered field.
-
C, DProliferation of A431 cells (n = 5) after overexpression (C) or knockdown (D) of IL‐38 was detected using cell proliferation detection reagent.
-
E, FColony‐forming ability of A431 cells after IL‐38 overexpression (E) or knockdown (F) analyzed using the clone formation assay.
-
G, HRepresentative images of the scratch assay (left) and wound closure rate (right) of A431 cells (n = 5) after overexpression (G) or knockdown (H) of IL‐38.
-
I, JRepresentative western blot bands indicating cyclin E and cyclin A in A431 cells after IL‐38 overexpression (I) or knockdown (J).
Data information: Error bars represent the mean ± SD. All data are biological replicates. *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test. NC, negative control; LV, lentivirus; Oe, overexpression.
IL‐38 forms a complex with IL‐1Rrp2 and activates JNK/AP‐1 signal transduction pathway in an IL‐1Rrp2‐dependent manner
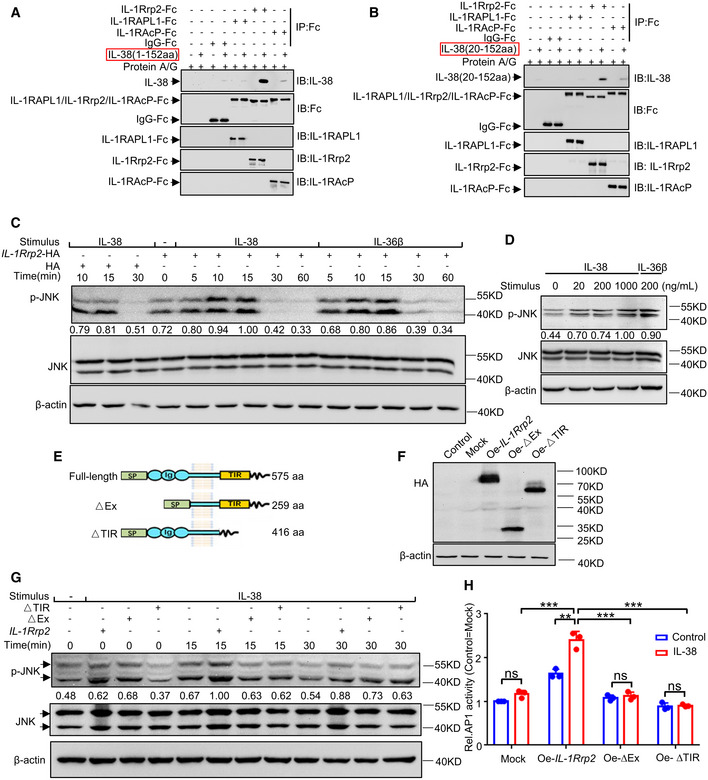
IL‐38 has pro‐inflammatory or anti‐inflammatory functions depending on its form, concentration, external stimuli, and local environmental background (van de Veerdonk et al, 2012; Xie et al, 2019). More experimental data are needed to explore the specific role and functional signaling pathways of IL‐38. We hypothesized that IL‐38 promotes tumor development via IL‐1 receptor family members. We analyzed a panel of candidates from the IL‐1 receptor family, including IL‐1Rrp2, IL‐1RAPL1, and IL1RAcp, for their association with IL‐38. To examine the interaction of IL‐38 with IL‐1Rrp2, IL‐1RAPL1, and IL1RAcp, IL‐38 (1–152aa) or IL‐38 (20–152aa) precipitated via protein A/G magnetic beads in the presence of IL‐1Rrp2‐Fc, IL‐1RAPL1‐Fc, IL1RAcp‐Fc, and IgG‐Fc fusion proteins, experiments were performed using western blot analysis and antibodies against IL‐38, Fc, IL‐1Rrp2, IL‐1RAPL1, and IL1RAcp. As shown in Fig 5A and B, IL‐38 (1–152 aa) and IL‐38 (20–152 aa) bound to IL‐1Rrp2‐Fc but did not bind to the other immobilized receptors. To investigate whether IL‐38 specifically exerts a biological effect dependent on the IL‐1Rrp2 pathway, we tested whether IL‐38 could activate JNK via this receptor. HEK293T cells were transfected with either a mock mammalian expression vector or an expression vector encoding IL‐1Rrp2. IL‐38 stimulation of 293T cells transfected with IL‐1Rrp2 but not transfected with vector alone led to phosphorylation of JNK with peak phosphorylation occurring at 15 min, which was similar to IL‐36β (Fig 5C). To better define the activation of the pathway leading to JNK by IL‐38, dose–response studies were carried out for this molecule. 293T cells were transfected with IL‐1Rrp2 and stimulated with IL‐38 at various concentrations and IL‐36β for 15 min. We found that IL‐38 dose‐dependently activated the JNK pathway (Fig 5D). Moreover, to identify the domains of IL‐1Rrp2 required for its interaction with the receptor complexes, several IL‐1Rrp2 deletion mutants were generated, including ∆Ex (lacking the extracellular Ig domain with deletion of amino acids 20–335) and ∆TIR (lacking the TIR domain with deletion of amino acids 381–539) (Fig 5E). The 293T cells transfected with the HA‐tagged full‐length or deletion mutants of IL‐1Rrp2 (Fig 5F) were either untreated or treated with IL‐38 for the indicated times, followed by western blot analysis with antibodies against p‐JNK, JNK, and β‐actin. Whereas overexpression of full‐length IL‐1Rrp2 significantly activated JNK upon IL‐38 stimulation, mutant IL‐1Rrp2 inhibited IL‐38‐induced JNK activation (Fig 5G). IL‐38 stimulates the activation of JNK, and AP‐1 acts as a major downstream effector of JNK (Hammouda et al, 2020). Therefore, we explored which IL‐1Rrp2 was essential for IL‐38 induced AP‐1 activation in 293T cells. Our results indicated that 293T cells transfected with the HA‐tagged full‐length IL‐1Rrp2 induced a more pronounced AP‐1 activation upon IL‐38 stimulation when compared to cells transfected with a mock mammalian expression vector. In contrast, deletion of the extracellular Ig domain (∆Ex) and the intracellular TIR domain (∆TIR) abolished the ability of IL‐38 to activate AP‐1 (Fig 5H). Taken together, these results strongly suggest that IL‐38 is able to activate the JNK/AP‐1 signal transduction pathway in an IL‐1Rrp2‐dependent manner and that both the extracellular Ig domain and the intracellular TIR domain are important for activation of IL‐38 signaling.
Figure 5. IL‐38 binds to IL‐1Rrp2 to activate JNK/AP‐1 pathway.
-
A, BPull‐down assay of IL‐38 (1–152 aa) (A) or IL‐38 (20–152 aa) (B) with IL‐1Rrp2‐Fc, IL‐1RAPL1‐Fc, IL1RAcp‐Fc, and IgG‐Fc fusion proteins.
-
CCell extracts prepared from 293T cells transfected with a mock mammalian expression vector (left) or an expression vector encoding IL‐1Rrp2 (right) were probed using anti‐phospho‐JNK, anti‐JNK, or anti‐β‐actin antibodies after stimulation with IL‐38 (200 ng/ml) or IL‐36β (200 ng/ml) at different times.
-
DCell extracts prepared from 293T cells transfected with an expression vector encoding IL‐1Rrp2 were probed using anti‐phospho‐JNK, anti‐JNK, or anti‐β‐actin antibodies after stimulation with IL‐38 at different concentrations.
-
ESchematic structures of full‐length IL‐1Rrp2 and its deletion mutants. Lengths are indicated in amino acids (aa). SP, signal peptide; Ig, immunoglobulin domain; TIR, TIR domain.
-
FCell extracts prepared from 293T cells transfected with HA‐tagged full‐length or deletion mutants of IL‐1Rrp2, were probed using anti‐HA or anti‐β‐actin antibodies. Oe, overexpression.
-
GCell extracts prepared from 293T cells transfected with full‐length or deletion mutants of IL‐1Rrp2 were probed using anti‐phospho‐JNK, anti‐JNK, or anti‐β‐actin antibodies after stimulation with IL‐38 (200 ng/ml) at different times.
-
HAfter transfection with the full‐length or deletion mutants of IL‐1Rrp2 overexpression plasmid, 293T cells were treated with IL‐38 (200 ng/ml) for an additional 6 h and AP‐1 activity was measured using a luciferase reporter assay.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using Student’s t‐test.
IL‐38 promotes the expression of cancer‐related inflammatory cytokines and proliferation and migration of tumor cell through IL‐1Rrp2/JNK
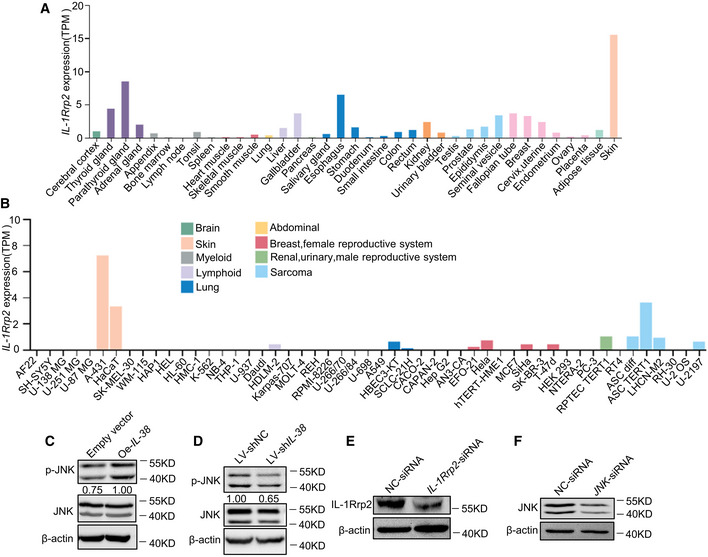
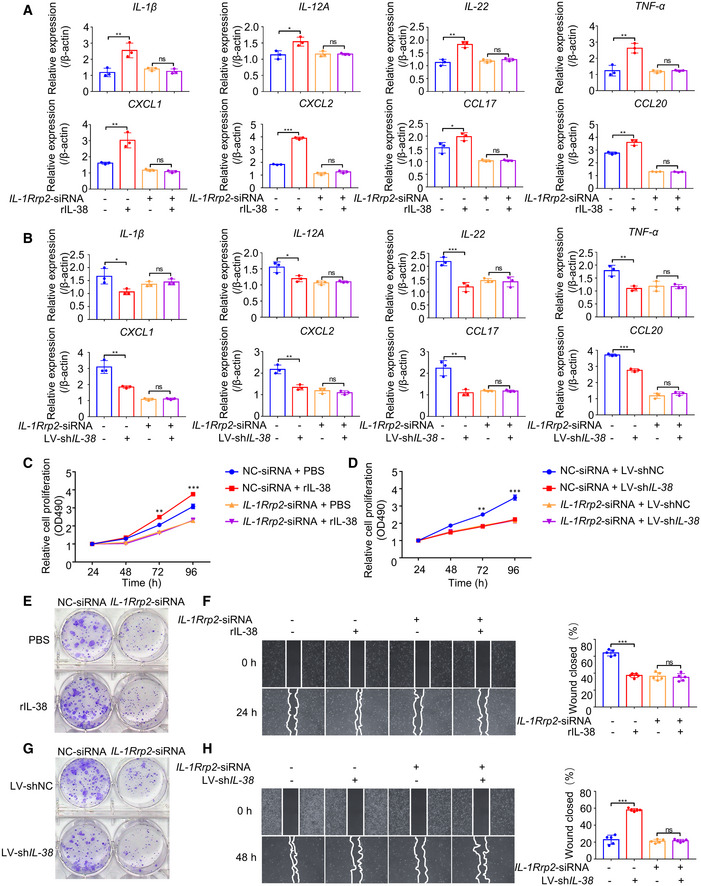
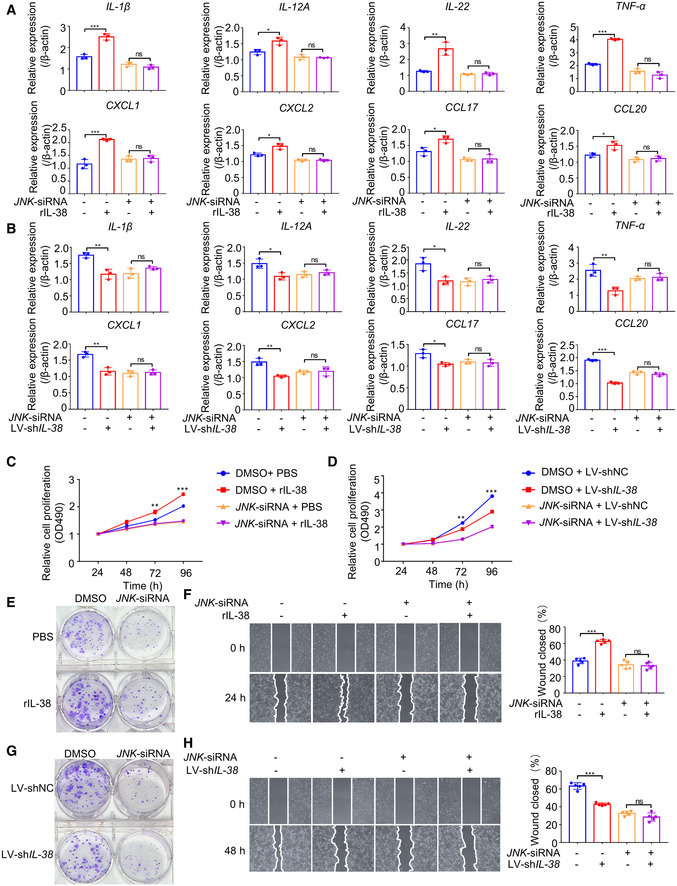
The activation of IL‐1Rrp2 can stimulate the production of IL‐22, leading to the proliferation and recovery of epithelial cells and secretion of inflammatory cytokines (Queen et al, 2019). The JNK signaling pathway mediates various cellular processes, including cell proliferation, survival, and migration, and regulates the production of inflammatory cytokines/chemokines by keratinocytes and the recruitment of immune cells in various diseases (Hammouda et al, 2020). To test whether IL‐38 promotes proliferation and migration of tumor cells and the expression of cancer‐related inflammatory cytokines through IL‐1Rrp2/JNK, we first examined the expression of IL‐1Rrp2 in various tissues and cell lines. According to the Human Protein Atlas Database, we found that IL‐1Rrp2 is highly expressed in the skin (Fig EV5A) and multiple human skin‐related cells (Fig EV5B). IL‐1Rrp2 expression showed no significant difference between normal skin and tumors (Fig EV3E and F). We found that overexpression of IL‐38 in A431 cells promoted JNK phosphorylation (Fig EV5C), while knockdown of IL‐38 inhibited JNK phosphorylation (Fig EV5D). To demonstrate whether IL‐1Rrp2/JNK is important for IL‐38 to stimulate the expression of inflammatory cytokines in tumor cells, A431 cells were transfected with small interfering RNAs (siRNAs), designed specifically to silence IL‐1Rrp2 (Fig EV5E) or JNK (Fig EV5F), followed by recombinant IL‐38 stimulation or treatment with shRNA against IL‐38. Surprisingly, we found that the expression of inflammatory cytokines in A431 cells increased after recombinant IL‐38 stimulation, and decreased in IL‐38‐knockdown cells, whereas neither the recombinant IL‐38 stimulation nor treatment with shRNA against IL‐38 affected the production of inflammatory cytokines in cells with IL‐1Rrp2 (Fig 6A and B) or JNK (Fig 7A and B) knockdown. Furthermore, we found that recombinant IL‐38 can promote tumor cell proliferation and migration, and treatment with shRNA against IL‐38 suppressed cell proliferation and migration, but both had no effect on tumor cells with IL‐1Rrp2 (Fig 6C–H) or JNK (Fig 7C–H) knockdown. These results indicate that IL‐38 promotes the expression of cancer‐related inflammatory cytokines and the proliferation and migration of tumor cells depend on the IL‐1Rrp2/JNK signaling pathway.
Figure EV5. Expression of IL‐1Rrp2 in tissues and cell lines and the effect of IL‐38 on JNK activation in A431 cell line.
-
A, BRelative expression of IL‐1Rrp2 in human normal tissues (A) or cell lines (B) based on TPM values. Data were obtained from the Human Protein Atlas Dataset available from proteinatlas.org.
-
CCell extracts prepared from A431 cells transfected with a mock mammalian expression vector (left) or an expression vector encoding IL‐38 (right), were blotted with anti‐phospho‐JNK, anti‐JNK, or anti‐β‐actin antibodies.
-
DCell extracts prepared from A431 cells infected with lentiviruses containing Negative Control shRNA (left) or IL‐38 shRNA (right) were probed using anti‐phospho‐JNK, anti‐JNK, or anti‐β‐actin antibodies.
-
EThe expression of IL‐1Rrp2 in A431 cells transfected with IL‐1Rrp2‐siRNA was detected using western blot.
-
FThe expression of JNK in A431 cells transfected with JNK‐siRNA was detected using western blot.
Figure 6. IL‐38 promotes the expression of inflammatory mediators and proliferation and migration of skin tumor cells dependent on IL‐1Rrp2.
-
A, BRelative expression levels of inflammatory mediators were detected using qPCR after posttranscriptional silencing of IL‐1Rrp2 by RNA interference, followed by rIL‐38 (200 ng/ml) stimulation (A) or IL‐38 knockdown (B).
-
C, DCell proliferation detection reagents were used to examine the effect of recombinant IL‐38 (200 ng/ml) protein (C) or IL‐38 knockdown (D) on A431 cell proliferation after IL‐1Rrp2 interference.
-
E–HCell cloning formation assays were used to examine the effect of recombinant IL‐38 (200 ng/ml) protein stimulation (E) or IL‐38 knockdown (G) on A431 cell proliferation after IL‐1Rrp2 interference. Representative images of the scratch assay (left) and wound closure rate (right) of A431 cells (n = 5) treated with recombinant IL‐38 (200 ng/ml) (F) or knockdown of IL‐38 (H) after IL‐1Rrp2 interference.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using one‐way ANOVA (A, B, F and H) or Student’s t‐test (C and D).
Figure 7. IL‐38 promotes the expression of inflammatory mediators and proliferation and migration of skin tumor cells in skin tumor cells dependent on JNK.
-
A, BRelative expression levels of inflammatory mediators were detected using qPCR after posttranscriptional silencing of JNK by RNA interference, followed by rIL‐38 (200 ng/ml) stimulation (A) or IL‐38 knockdown (B).
-
C, DCell proliferation detection reagents were used to examine the effect of recombinant IL‐38 (200 ng/ml) protein (C) or IL‐38 knockdown (D) on A431 cell proliferation after JNK interference.
-
E–HCell cloning formation assays were used to examine the effect of recombinant IL‐38 (200 ng/ml) protein stimulation (E) or IL‐38 knockdown (G) on A431 cell proliferation after JNK interference. Representative images of the scratch assay (left) and wound closure rate (right) of A431 cells (n = 5) treated with recombinant IL‐38 (200 ng/ml) (F) or knockdown of IL‐38 (H) after JNK interference.
Data information: Error bars represent the mean ± SD. All data are biological replicates. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; P values were calculated using one‐way ANOVA (A, B, F and H) or Student’s t‐test (C and D).
Discussion
IL‐38 is highly expressed in inflammatory tissues to mediate an excessive inflammatory response (van de Veerdonk et al, 2012; Xie et al, 2020). However, the function of IL‐38 in tumors remains largely unknown. There is little information about how IL‐38 influences the pathogenesis of cSCC, including its development, progression, and prognosis. Here, we provide evidence that IL‐38 promotes cSCC progression by regulating the pro‐tumorigenic microenvironment and epidermal cell hyperplasia in an IL‐1Rrp2‐dependent manner. The role of other IL‐1 family members, such as IL‐18 and IL‐33, in melanoma has been investigated in mouse models extensively. IL‐18 mRNA expression was found to be significantly lower in melanoma tissues than normal tissues (Gil & Kim, 2019), and IL‐18 was reported to promote the growth of B16F10 melanoma cells (Cho et al, 2000) and enhance the ability of melanoma cells to migrate via the generation of ROI and the MAPK pathway (Jung et al, 2006), which is consistent with our findings. The high expression of IL‐33 was found to associate with better overall survival in melanoma patients (Wagner et al, 2020). The role of IL‐33 in melanoma has been observed both its pro‐ and anti‐cancer effects, which may be due to the timing and dosage of IL‐33 administration, and the specificity of IL‐33 (Gao et al, 2013, 2015; Long et al, 2018; Jevtovic et al, 2020; Schuijs et al, 2020). Our data and those of previous studies indicate that IL‐1 family members play an important role in the progression of skin cancer.
In our study, for the first time, we investigated the expression pattern of IL‐38 protein in the skin tissues of cSCC patients and DMBA/TPA‐induced mouse tumors. We found that the expression of IL‐38 in skin tumor tissues was lower than that in normal tissues, which is consistent with the results in non‐small cell lung cancer and colorectal cancer (Wang et al, 2018; Chen et al, 2020). A decrease in IL‐38 suggests that it might play an inhibitory role in skin tumors. However, we found that IL‐38 promoted the pathogenesis of skin tumors in vivo and in vitro. Considering that IL‐38 can potentially be expressed by cells other than keratinocytes, we performed experiments to detect the expression of IL‐38 in DMBA/TPA‐treated skin and DMBA/TPA‐induced tumors of Il‐38f / f and K14Cre /+ ‐Il‐38f / f mice. We observed complete loss of IL‐38 from skin lysates in DMBA/TPA‐treated skin and DMBA/TPA‐induced tumors in K14Cre /+ ‐Il‐38f / f mice (Fig EV3A and D). To detect whether this contradiction was caused by IL‐1Rrp2 expression, we performed experiments to detect IL‐1Rrp2 expression in skin versus tumor. Our results showed that IL‐1Rrp2 expression did not differ significantly between normal skin and tumors (Fig EV3E and F). Similar apparent contradictions were observed for IL‐33 and IL‐37, the other two IL‐1 family members. IL‐33 expression was also inversely correlated with the stages of human lung cancers, but IL‐33 enhances lung cancer progression by selecting more malignant cells in the tumor microenvironment (Akimoto et al, 2016). The expression of IL‐37 is higher in oral squamous cell carcinoma (OSCC) than in normal controls (Volpe et al, 1997). However, recombinant IL‐37b‐treated cells showed decreased production of LPS‐stimulated IL‐6, TNF‐α, and IL‐1β, which have been reported to promote malignant transformation and tumor aggression in oral cancer (Volpe et al, 1997; Wang et al, 2010a; Yoshida et al, 2012; Akimoto et al, 2016). Moreover, many studies have determined that IL‐38 is increased in multiple diseases, while increased IL‐38 inhibited the progression of inflammatory bowel disease, myocardial infarction, and type 2 diabetes, and blockade of IL‐38 promoted the progression of acute respiratory syndrome, and fatal sepsis (Xu et al, 2018; Chai et al, 2020; Liu et al, 2020; Wei et al, 2020; Xie et al, 2020). Our data and those of previous studies indicate that the differential expression of a biomarker observed in clinical data may not conclusively demonstrate the real role of the biomarker in pathological processes without corresponding experiments in animal models.
At present, the regulation of IL‐38, including its expression, function, and molecular signaling pathways, is still unclear. Primarily, IL‐38 is considered an anti‐inflammatory cytokine that can suppress the production of several pro‐inflammatory cytokines in different cell types in response to activation by different stimuli (Pan et al, 2021; Xia et al, 2021); however, this issue remains controversial. For example, recombinant full‐length IL‐38 increased the level of IL‐6 in LPS‐stimulated dendritic cells (DCs) or human macrophages (Mora et al, 2016; van de Veerdonk et al, 2012). A similar phenomenon was observed with IL‐1β stimulation. Full‐length IL‐38 significantly increased IL‐6 production in human macrophages after stimulation with IL‐1β. Van de Veerdonk and colleagues reported that IL‐38‐induced inhibition of IL‐22 and IL‐17 expression in human peripheral blood mononuclear cells (PBMCs) produced a dose‐dependent response that differed from the classic IL‐1Ra induction. Specifically, the production of IL‐22 and IL‐17 was inhibited at low concentrations of IL‐38, but increased the production of IL‐22 by 39% at higher concentrations (van de Veerdonk et al, 2012). Recently, it has been reported that IL‐38 may have an N‐terminal cleavage mechanism whose truncated form can regulate the secretion of cytokines IL‐6 and IL‐8 by IL‐1β‐stimulated macrophages through the JNK/AP‐1 signaling pathway, which relies on IL‐1RAPL1, which is the orphan receptor of the IL‐1 family (Hussein, 2005; Paoloni et al, 2009; Coste et al, 2010). In general, IL‐38, a new member of the IL‐1 family, as an important immunoregulatory cytokine, might exert similar but different inhibitory or activation functions on IL‐36Ra depending on the IL‐1Rrp2/ IL‐1RACP/ IL‐1RAPL1 mediated signaling pathway. The specific mechanism remains to be studied further. In our study, we found that IL‐38 can combine with IL‐1Rrp2 to activate JNK (Fig 5A–G), and the activation of JNK promotes the expression of pro‐inflammatory cytokines and chemokines in tumor cells, leading to the recruitment of immune cells, promoting the abnormal proliferation of tumor cells, and further aggravating the disease. As previously reported by Han et al, IL‐38 binds to IL‐1RAPL1 on skin γδT cells (Han et al, 2019). Mora et al also reported that IL‐38 was able to bind IL‐1RAPL1, but IL‐1Rrp2 showed a significantly higher affinity for IL‐38 than IL‐1RAPL1 (Mora et al, 2016). Moreover, IL‐1RAPL1 shares high amino acid sequence identity in the extracellular domain with IL‐1RAcP (30.3%) (Yoshida et al, 2012). Therefore, IL‐1RAPL1 is more likely to act as a co‐receptor for receptors of IL‐1 family members, such as IL‐1RAcP. A previous report has shown that IL‐1Rrp2 mRNA was most strongly expressed in keratinocytes, which was at least tenfold more than in bone marrow‐derived dendritic cells (BMDCs), splenic CD4+ T cells, bone marrow‐derived macrophages, and bone marrow‐derived neutrophils, whereas IL‐1Rrp2 mRNA was not detected in CD8+ and B cells (Vigne et al, 2011), which is similar to the findings reported by Foster et al (2014). In our results, we also found that IL‐1Rrp2 was highly expressed in the skin (Fig EV5A) and multiple human skin‐related cells, especially in the skin cancer cell line (A431) (Fig EV5B). In this regard, IL‐1Rrp2 meets the requirements of a stimulatory immune checkpoint for skin cancer.
In conclusion, this study reveals a new key cytokine mechanism that can amplify and maintain chronic inflammation and cell proliferation in skin cancer. In addition, our data enhance our understanding of IL‐38 and highlight the role of IL‐38 in cancer. These findings will assist in the development of new strategies for cancer prevention and treatment.
Materials and Methods
Animals
Il38‐floxed (C57BL/6J‐Il1f10em1cyagen) mice on a C57BL/6 background were purchased from Cyagen Biosciences Inc. Krt14‐Cre mice (STOCK Tg[Krt14‐cre]1Amc/J) mice were purchased from the Nanjing Biomedical Research Institute of Nanjing University (J004782). C57BL/6 mice (8–12 weeks old) were purchased from Vital River Laboratory Animal Technology Co., Ltd. The animals were housed under the following controlled conditions: 12‐h light/12‐h dark cycles, temperature stabilized at 25 ± 1°C, and free access to water and food. The animal experimental protocol was approved by the Animal Experiment Ethics Committee of Sichuan University. The experimental procedures were based on the ethical guidelines for the care and use of laboratory animals of the National Institutes of Health (https://grants.nih.gov/grants/olaw/guide‐for‐the‐care‐and‐use‐of‐laboratory‐animals.pdf) and the International Association for the Study of Pain (IASP). We made every effort to reduce the number of animals involved in the experiments and reduce their suffering. After weaning, the Mouse Direct PCR Kit (Bimake) was used to extract genomic DNA from tail biopsies for PCR detection and genotyping using gene‐specific primers (Appendix Table S1).
Human subjects
This study followed the principles of the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report and was approved by the Ethics Committee of the West China Hospital, Sichuan University (Chengdu, Sichuan, China). Written informed consent was obtained from all study participants prior to the study. All patients were clinically and pathologically confirmed, and there were no other autoimmune diseases, immunodeficiency diseases, or tumors. None of the patients received radiotherapy, chemotherapy, cryotherapy, or laser treatment before sample collection. We collected tumors from 13 cSCC patients (7 males and 6 females) and healthy skin from 11 donors (6 males and 5 females). Fresh skin samples were immediately fixed in 4% paraformaldehyde in phosphate‐buffered saline (PBS; ZSGB‐BIO) for paraffin embedding.
Cell lines
Human embryonic kidney (HEK293T) cells and human cSCC (A431) cells were purchased from ATCC (CRL‐11268; CRL‐1555). HEK293T or A431 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% (v:v) fetal bovine serum (FBS; Gibco), 100 U/ml penicillin G, and 0.1 mg/ml streptomycin sulfate (Gibco). The cells were cultured at 37°C with 5% CO2. None of the cells were contaminated with mycoplasma.
DMBA/TPA skin cancer model
For chemical skin carcinogenesis, 50 μg DMBA (Sigma‐Aldrich) was dissolved in 200 μl acetone and applied to the back skin of 6–8‐week‐old mice one week after shaving. One week later, 5 μg TPA (Sigma‐Aldrich) in 200 μl acetone was applied twice a week for up to 32 weeks. Mice were examined weekly, and tumor numbers and sizes were measured when the tumor diameter was greater than 1 mm. At the end of the experiment, the back skin and tumors were used for histology, immunohistochemistry, immunofluorescence, flow cytometry, RNA, and protein isolation.
RNA isolation and quantitative reverse transcription PCR
According to the manufacturer's protocol, TRIzol (ThermoFisher Scientific) was used to extract total RNA from cells or mouse tissues. Gel electrophoresis was performed to determine the integrity of the extracted total RNA. Reverse transcription was performed using the PrimeScript RT kit with gDNA Eraser (Takara). cDNA was generated at 37°C for 15 min and 85°C for 5 s, and qPCR analysis was performed on the obtained cDNA (20 ng) with TB Green™ Premix Ex Taq™ II (Takara) following the manufacturer’s instructions. The relative expression between samples was determined using the 2−ΔΔCt formula. The melting curves were confirmed to ensure the amplification of a single product. All primers were synthesized by Chengdu Qing Ke Zi Xi Biotechnology Co., Ltd., and the primer sequences are shown in Appendix Tables S2 and S3.
Western blotting
The samples derived from cells or tissues were lysed, separated using SDS–PAGE gels (Beyotime Institute of Biotechnology), and then transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore). For western blotting analysis, the proteins were incubated overnight with the following primary antibodies: human IL‐38 (Abcam, ab180898; 2 µg/ml), mouse IL‐38 (R&D Systems, MAB7774; 2 µg/ml), IL‐18 (Abcam, ab71495; 2 µg/ml), β‐actin (CST, 4970; 1:1,000 dilution), JNK (CST, 9252; 1:1,000 dilution), p‐JNK (CST, 4668; 1:1,000 dilution), Cyclin E (CST, 20808; 1:1,000 dilution), Cyclin A (R&D Systems, AF5999; 0.5 µg/ml), HA (CST, 3724; 1:1,000 dilution), human IL‐1Rrp2 (R&D Systems, AF872; 0.1 µg/ml), mouse IL‐1Rrp2 (LSBio, LS‐B10135; 2 µg/ml), IL‐1RAPL1 (R&D Systems, MAB975; 1 µg/ml), IL‐1RAcp (Santa Cruz Biotechnology, sc‐376872; 1:500 dilution), and IgG‐Fc (Sino Biological, SSA001; 1:10,000 dilution). The primary antibodies were labeled using goat anti‐rabbit, rabbit anti‐goat, or goat anti‐mouse antibodies conjugated to horseradish peroxidase (HRP) (Invitrogen, A27036 and ZSGB‐BIO; 1:10,000 dilution) and further detected using ECL reagents (Merck Millipore, WBKLS0500). The band intensity in the image was quantified using ImageJ (National Institutes of Health), which included only the band intensity in the linear range.
Enzyme‐linked immunosorbent assay (ELISA)
To detect the levels of IL‐38, cell culture supernatants were collected and IL‐38 levels were measured using the Human IL‐38/IL‐1F10 ELISA kit (Neobioscience) according to the manufacturer’s instructions.
Hematoxylin and eosin (H&E) staining
Normal skin and skin tumors were fixed in 4% paraformaldehyde in PBS, sectioned, embedded in paraffin, and stained with hematoxylin and eosin (H&E) for histopathological examination. Images were captured using an Olympus BX600 microscope (Olympus Corporation, Tokyo, Japan) and SPOT Flex camera (Olympus Corporation, Tokyo, Japan) and analyzed using ImagePro Plus (version 6.0, Media Cybernetics) software. The thickness of the epithelium and the degree of malignancy of the tumor in the independent areas were evaluated.
Immunohistochemistry
Normal skin and skin tumors were fixed in 4% paraformaldehyde, and the fixed sections were incubated in 3% H2O2 solution and protected from light for 10–15 min. Afterward, the sections were incubated with 5% normal goat serum at room temperature for 30 min to block non‐specific antibody binding. Subsequently, the sections were stained with IL‐38 (Abcam, ab180898; 1:200 dilution), γH2AX (Invitrogen, PA5‐77995; 1:500 dilution), IL‐1Rrp2 (LSBio, LS‐B10135; 10 µg/ml), human IL‐18 (Abcam, ab243091; 1:1,000 dilution), and mouse IL‐18 (Abcam, ab71495; 1 µg/ml) primary antibodies. The slides were then rinsed, incubated with a biotin‐conjugated secondary antibody for 30 min, and incubated with horseradish peroxidase streptavidin (HRP Streptavidin) for 30 min (ZSGB‐BIO). The sections were developed using a 3,3ʹ‐diaminobenzidine (DAB) substrate kit (ZSGB‐BIO, ZLI‐9017), and hematoxylin was used for reverse staining. Images were captured using an Olympus BX600 microscope and SPOT Flex camera. ImagePro Plus was used to further quantify the DAB intensity and the number of γH2AX‐positive cells in the image.
Immunofluorescence analysis
Tissue biopsies were directly embedded in optimal cutting temperature (OCT) compound, and the frozen sections were fixed in cold methanol‐acetone (1:1) for 15 min, permeabilized with 0.3% Triton X‐100 (Sigma‐Aldrich, X100) in PBS for 15 min, blocked with 5% bovine serum albumin (BSA; Sigma‐Aldrich, B2064) in PBS for 30 min, and then incubated overnight at 4°C with primary antibodies against IL‐38 (Abcam, ab180898; 5 µg/ml), rabbit IgG (Abcam, ab37415; 5 µg/ml), Keratin‐8 (Abcam, ab53280; 1:300 dilution) and Ki67 (Abcam, ab16667; 1:200 dilution). TRITC‐conjugated donkey anti‐rabbit antibodies (A16040; 1:1,000 dilution, 4 μg/ml) were used as secondary reagents (purchased from Invitrogen). Nuclear counterstaining was performed with 4ʹ,6‐diamidino‐2‐phenylindole (DAPI; Sigma‐Aldrich, D9542). Images were analyzed using a Leica DM RXA2 confocal microscope controlled by Leica Microsystems confocal software (version 2.61 Build 1537; Leica Microsystems, Wetzlar, Germany). ImageJ (National Institutes of Health) was used to quantify the fluorescence intensities and the number of Ki67‐positive cells of the images.
Skin barrier function assays
The rate of transepidermal water loss (TEWL) from the skin of newborn mice was determined using a GPSkin evaporimeter (GPOWER, South Korea). Euthanized newborn mice were immediately dehydrated by incubation (1 min each) in 25%, 50%, and 75% methanol/PBS followed by 1 min in 100% methanol, then rehydrated with the same series of methanol solutions (1 min incubations), washed for 1 min in PBS, and subsequently stained for 10 min in 0.1% toluidine blue in water. The cells were photographed immediately after destaining in PBS. A defined area of murine dorsal skin (25 mm2) was boiled in isolation buffer (20 mM Tris–HCl, pH 7.5, 5 mM EDTA, 10 mM DTT, and 2% SDS) under vigorous shaking for 40 min. After centrifugation, the cornified cell envelopes (CEs) were washed twice in isolation buffer and imaged using an Olympus BX600 microscope and SPOT Flex camera.
Isolation of genomic DNA
Full‐thickness murine dorsal skin was removed, and the subcutaneous fat was removed by scraping with closed curved scissors. The dorsal skin was incubated in 0.5 M ammonium thiocyanate for 20 min at 37°C to allow separation of the epidermis from the dermis. The gDNA of the epidermis was isolated using the TIANamp Genomic DNA Kit (Qiagen) according to the manufacturer’s protocol. Isolated tissue gDNA was analyzed using a NanoDrop‐2000 (Thermo Scientific) to determine purity and concentration.
Hras cloning and mutant‐specific real‐time PCR
Mutant‐specific primers 5′‐CTAAGCCTGTTGTTTTGCAGGAC and 3′‐CATGGCACTATACTCTTCTA and a custom TaqMan probe 5′‐6FAM‐CGGAAACAGGTGGTCAT‐MGB‐3′ were designed as part of a novel real‐time PCR assay (Nelson et al, 1992; Modi et al, 2012) to quantify the number of DMBA‐induced codon‐61 Hras CAA‐>CTA mutations. DNA was diluted to 50 ng/µl in fresh double‐distilled water (ddH2O), and 6.75 µl was aliquoted into a 96‐well plate (total 337.5 ng DNA/well). The primers used were single‐stranded DNA oligonucleotides diluted in ddH2O to a concentration of 10 µM. The 6′‐FAM‐conjugated probe was shipped pre‐diluted to a concentration of 5 µM. For Taqman probe‐based detection of product, a Mastermix was made with the following volumes per well; (i) 12.25 µl 2× Premix Ex Taq™ (Takara), (ii) 2.25 µl forward primer, (iii) 2.25 µl reverse primer, and (iv) 1.50 µl probe. TaqMan real‐time PCR was performed on a Roche LightCycler® 96. Relative expression between samples was determined using the 2−ΔΔCt method.
Isolation of DMBA/ TPA‐treated skin cells
For skin inflammation, 50 μg of DMBA (Sigma‐Aldrich) dissolved in 200 μl acetone was applied on the back skin of 6–8‐week‐old mice one week after shaving. One week later, 5 μg TPA (Sigma‐Aldrich) in 200μL acetone was applied twice a week for up to 2 weeks. To obtain single‐cell suspensions from dorsal skin, 2 × 3 cm sections of skin samples were incubated in 5 ml RPMI medium (Gibco) containing 500 μg/ml Liberase (Roche) for approximately 1.75 h at 37°C, chopped with sharp scissors, and incubated for an additional 15 min with 0.1 mg/ml DNase (Roche). A single‐cell suspension of dorsal skin was obtained by mechanical dissociation with a gentleMACS dissociator (Miltenyi Biotech, Bergisch Gladbach, Germany), followed by filtration through 40 and 70 μm cell strainers. Cells were then washed once with PBS.
Flow cytometry
To obtain single‐cell suspensions from dorsal skin, 2 × 3 cm sections of skin samples were incubated in 5 ml RPMI medium (Gibco) containing 500 μg/ml Liberase (Roche) for approximately 1.75 h at 37°C, chopped with sharp scissors, and incubated for an additional 15 min with 0.1 mg/ml DNase (Roche). A single‐cell suspension of dorsal skin was obtained by mechanical dissociation with a gentleMACS dissociator (Miltenyi Biotech, Bergisch Gladbach, Germany), followed by filtration through 40 μm and 70 μm cell strainers. Cells were then washed once with PBS. Flow cytometry was performed using the NovoCyte flow cytometer and ACEA NovoExpress™ software (ACEA Biosciences, San Diego, CA, USA). The single‐cell suspensions were stained with the following antibodies: CD11B‐APC (17‐0112‐82), MHC II‐FITC (11‐5322‐81), CD64‐PE/Cyanine7 (139314), Ly6C‐APC/Fire™ 750 (108456), MerTk‐PE (151506), CD207‐PE (144204), CD3‐APC/Cyanine7 (100222), and CD4‐PE (100408). Antibodies were purchased from eBioscience and BioLegend and used at 1:100 dilution.
Pull‐down assays
Human IL‐1Rrp2‐Fc (R&D Systems), human IL‐1RAcp‐Fc (Sino Biological), human IL‐1RAPL1‐Fc (Sino Biological), or human IgG‐Fc (Sino Biological) fusion proteins (1.5 µM each) were bound to 1.5 µM IL‐38 (Adipogen) by incubating overnight at 4°C. Afterward, Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific) were added to the complexes and incubated for 1 h at room temperature to form the immune complex. The magnetic beads were washed five times with a wash buffer (Thermo Fisher Scientific). Bound proteins were eluted from the magnetic beads by incubation at room temperature in elution buffer (Thermo Fisher Scientific). Twenty‐five percent of the eluted proteins were detected by western blotting.
Luciferase reporter assays
One day prior to transfection, 2 × 105 cells were seeded in 12‐well plates with 5% CO2 at 37°C overnight. Cells were transiently transfected with 2 µg AP‐1 luciferase reporter (Promega) and 0.2 µg Renilla luciferase control vector pRL‐TK (YouBio) using X‐tremeGENE™ HP DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. After 24 h, the growth medium was replaced, IL‐38 was added for another 6 h, and the luciferase protein activity was measured using the Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity in the lysate. The background obtained from the mock‐transfected cells was subtracted from each experimental value.
siRNA and shRNA mediated silencing of gene expression
A431 cells were transfected with 5 pmol IL‐1Rrp2 siRNA (Dharmacon) using DharmaFECT transfection reagent, or JNK‐siRNA (CST) using Lipofectamine™ RNAiMAX reagent (Invitrogen) for 48 h according to the manufacturer’s instructions. Human shRNA‐IL‐38 lentivirus (IL‐38 shRNA, 5ʹ‐GCAGACCAGAAGGCTCTATAC‐3ʹ) and negative control lentivirus (NC shRNA) were obtained from Shanghai GenePharma Co., Ltd. Lentiviruses were produced in A431 cells according to the manufacturer’s instructions. The A431 cells were incubated with the lentivirus particles, and puromycin (Thermo Fisher Scientific; 3 μg/ml) was used to obtain cells stably expressing the lentiviral construct two‐day post‐infection.
Transfection of recombinant vector
HEK293T cells were maintained in DMEM (Gibco) supplemented with 10% heat‐inactivated FBS (Gibco), 100 U/ml penicillin G, and 0.1 mg/ml streptomycin sulfate (Gibco). One day prior to transfection, 4 × 105 cells were seeded in 6‐well plates with 5% CO2 at 37°C overnight. Cells were transiently transfected with 2 µg pcDNA3.1‐IL‐38 (Genewiz), pCMV3‐IL‐1Rrp2‐HA (Genewiz), pCMV3‐IL‐1Rrp2 ∆Ex‐HA (Genewiz), and pCMV3‐IL‐1Rrp2 ∆TIR‐HA (Genewiz) using X‐tremeGENE™ HP DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. Cells stably expressing these proteins were selected with G418 (Gibco; 200 μg/ml) or Hygromycin B (Sigma‐Aldrich; 200 μg/ml) two‐day post‐transfection.
Cell proliferation assay
CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS; Promega) was used to observe and compare cell proliferation ability. A431 cells were plated in 96‐well plates at a density of 4 × 103 cells per well. After seeding, the cell proliferation was assessed. The cells were incubated for 4 h in 20 µl of MTS at 37°C. The optical density (OD) at 490 nm was determined using a microplate reader. The difference in cell proliferation in each group was calculated.
Colony formation assay
A431 cells were digested with trypsin (Gibco) and resuspended in single‐cell suspensions. A total of 500 cells from each group were cultured in 6‑well plates for 14 days. The cells were then fixed with 4% paraformaldehyde in PBS and stained with crystal violet staining solution (Beyotime) for 10 min, followed by rinsing with distilled water.
Cell migration assay
Cell migration ability was calculated using a wound healing assay. A431 cells were plated on 6‐well plates at a concentration of 5 × 105 cells/well and allowed to form a confluent monolayer for 24 h. Cells were incubated in serum‐free media containing 10 μg/ml mitomycin C (MedChemExpress) for 1 h to completely inhibit cell proliferation. The monolayer was scratched with a sterile pipette tip (200 µl), washed with serum‐free medium to remove floating and detached cells, and photographed (0, 24, and 48 h) using an inverted fluorescence microscope (Olympus, Tokyo, Japan).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 7 software. The percentage of mice with tumor, the malignant conversion rate, and the percentage of mice with malignant tumors were analyzed using Fisher’s exact test. The Student’s t‐test was used to compare two groups and the analysis of variance (ANOVA) test was used to compare three or more groups. Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, and ***P < 0.001.
Author contributions
Hong Zhou: Conceptualization; Software; Investigation; Methodology; Writing—original draft; Project administration; performed the experiments. Qixiang Zhao: Investigation; Methodology. Chengcheng Yue: Investigation; Project administration. Jiadong Yu: Investigation; Project administration. Huaping Zheng: Investigation; Project administration. Jing Hu: Investigation; Project administration. Zhonglan Hu: Investigation; Project administration. Haozhou Zhang: Investigation. Xiu Teng: Investigation. Xiao Liu: Investigation. Xiaoqiong Wei: Investigation. Yuxi Zhou: Investigation. Fanlian Zeng: Investigation. Yan Hao: Investigation. Yawen Hu: Investigation. Xiaoyan Wang: Investigation. Fulei Zhao: Investigation. Chen Zhang: Investigation. Linna Gu: Investigation. Wenling Wu: Resources. Yifan Zhou: Resources. Kaijun Cui: Resources. Nongyu Huang: Resources. Wei Li: Resources. Zhen Wang: Writing—review & editing. Jiong Li: Resources; Funding acquisition; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
HZho and JL conceptualized the study and designed the methodology. HZho, QZ, CY, JY, HZhe, XT, XL, ZH, XW, FZh, YHa, YHu, XW, FZh, CZ, LG, JH, and JL performed the experiments. WW, YiZ, KC, NH, YuZ, and WL provided key reagents. HZho, ZW, QZ, CY, and JL wrote the manuscript with input from all the authors.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81673061 to Jiong Li, 81472650 to Jiong Li, 31271483 to Jiong Li, and 81573050 to Li Wei), and the National Science and Technology Major Project (2018ZX09303006‐001‐006 to Jiong Li and 2019ZX09201004‐003 to Jiong Li).
EMBO reports (2022) 23: e53791.
Contributor Information
Zhen Wang, Email: wangzhen@scu.edu.cn.
Jiong Li, Email: lijionghh@scu.edu.cn.
Data availability
The datasets produced in this study are available in the following databases: RNA‐Seq data: GTEx Portal phs000424.v8.p2 (https://www.gtexportal.org/home/gene/ENSG00000136697%20). RNA‐Seq data: Gene Expression Omnibus GSE98767 (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE98767); GSE63967 (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE63967). RNA‐Seq data: Human Protein Atlas (https://www.proteinatlas.org/ENSG00000115598‐IL1RL2/tissue; https://www.proteinatlas.org/ENSG00000115598‐IL1RL2/cell#gene_informationExpression).
References
- Akimoto M, Hayashi JI, Nakae S, Saito H, Takenaga K (2016) Interleukin‐33 enhances programmed oncosis of ST2L‐positive low‐metastatic cells in the tumour microenvironment of lung cancer. Cell Death Dis 7: e2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KJ, Houston A, Brint E (2019) IL‐1 family members in cancer; two sides to every story. Front Immunol 10: 1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensen JT, Dawson PA, Mychaleckyj JC, Bowden DW (2001) Identification of a novel human cytokine gene in the interleukin gene cluster on chromosome 2q12‐14. J Interferon Cytokine Res 21: 899–904 [DOI] [PubMed] [Google Scholar]
- Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, Morel F, Lecron JC, Rolli‐Derkinderen M, Bourreille A et al (2016) Distinct expression of interleukin (IL)‐36alpha, beta and gamma, their antagonist IL‐36Ra and IL‐38 in psoriasis, rheumatoid arthritis and Crohn's disease. Clin Exp Immunol 184: 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet MA, Najm A, Bart G, Brion R, Touchais S, Trichet V, Layrolle P, Gabay C, Palmer G, Blanchard F et al (2017) IL‐38 overexpression induces anti‐inflammatory effects in mice arthritis models and in human macrophages in vitro . Ann Rheum Dis 76: 1304–1312 [DOI] [PubMed] [Google Scholar]
- Cataisson C, Salcedo R, Hakim S, Moffitt BA, Wright L, Yi M, Stephens R, Dai RM, Lyakh L, Schenten D et al (2012) IL‐1R‐MyD88 signaling in keratinocyte transformation and carcinogenesis. J Exp Med 209: 1689–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai YS, Lin SH, Zhang M, Deng L, Chen Y, Xie K, Wang CJ, Xu F (2020) IL‐38 is a biomarker for acute respiratory distress syndrome in humans and down‐regulates Th17 differentiation in vivo. Clin Immunol 210: 108315 [DOI] [PubMed] [Google Scholar]
- Chen F, Zhang F, Tan Z, Hambly BD, Bao S, Tao K (2020) Interleukin‐38 in colorectal cancer: a potential role in precision medicine. Cancer Immunol Immunother 69: 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D, Song H, Kim YM, Houh D, Hur DY, Park H, Yoon D, Pyun KH, Lee WJ, Kurimoto M et al (2000) Endogenous interleukin‐18 modulates immune escape of murine melanoma cells by regulating the expression of Fas ligand and reactive oxygen intermediates. Cancer Res 60: 2703–2709 [PubMed] [Google Scholar]
- Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, Eychene A, Lebecque S, Renno T (2010) Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest 120: 3663–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber E (1995) Cell proliferation as a major risk factor for cancer: a concept of doubtful validity. Can Res 55: 3759–3762 [PubMed] [Google Scholar]
- Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, Ward NL, Johnston A (2014) IL‐36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol 192: 6053–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner‐Tuderman L, Blobel CP (2012) Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand‐dependent terminal keratinocyte differentiation. J Exp Med 209: 1105–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Li X, Zhang L, Bai L, Dong W, Gao K, Shi G, Xia X, Wu L, Zhang L (2013) Transgenic expression of IL‐33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett 335: 463–471 [DOI] [PubMed] [Google Scholar]
- Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, Lu J, Qin W, Qi Y, Xie F et al (2015) Tumoral expression of IL‐33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol 194: 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil M, Kim KE (2019) Interleukin‐18 Is a prognostic biomarker correlated with CD8(+) T cell and natural killer cell infiltration in skin cutaneous melanoma. J Clin Med 8: 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Fu S, Zhang J, Liu B, Li Z (2016) Targeting inflammasome/IL‐1 pathways for cancer immunotherapy. Sci Rep 6: 36107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammouda MB, Ford AE, Liu Y, Zhang JY (2020) The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 9: 857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Mora J, Huard A, da Silva P, Wiechmann S, Putyrski M, Schuster C, Elwakeel E, Lang G, Scholz A et al (2019) IL‐38 ameliorates skin inflammation and limits IL‐17 production from gammadelta T cells. Cell Rep 27: 835–846.e835 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Momoi Y, Tanuma N, Kishimoto A, Ogoh H, Kato H, Suzuki M, Sakamoto Y, Inoue Y, Nomura M et al (2015) Abrogation of protein phosphatase 6 promotes skin carcinogenesis induced by DMBA. Oncogene 34: 4647–4655 [DOI] [PubMed] [Google Scholar]
- Huard A, Do HN, Frank AC, Sirait‐Fischer E, Fuhrmann D, Hofmann MCJ, Raue R, Palmer G, Brune B, de Bruin N et al (2021) IL‐38 ablation reduces local inflammation and disease severity in experimental autoimmune encephalomyelitis. J Immunol 206: 1058–1066 [DOI] [PubMed] [Google Scholar]
- Hussein MR (2005) Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol 32: 191–205 [DOI] [PubMed] [Google Scholar]
- Jevtovic A, Pantic J, Jovanovic I, Milovanovic M, Stanojevic I, Vojvodic D, Arsenijevic N, Lukic ML, Radosavljevic GD (2020) Interleukin‐33 pretreatment promotes metastatic growth of murine melanoma by reducing the cytotoxic capacity of CD8(+) T cells and enhancing regulatory T cells. Cancer Immunol Immunother 69: 1461–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Tsoi LC, Billi AC, Ward NL, Harms PW, Zeng C, Maverakis E, Kahlenberg JM, Gudjonsson JE (2020) Cytokinocytes: the diverse contribution of keratinocytes to immune responses in skin. JCI Insight 5: e142067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung MK, Song HK, Kim KE, Hur DY, Kim T, Bang S, Park H, Cho DH (2006) IL‐18 enhances the migration ability of murine melanoma cells through the generation of ROI and the MAPK pathway. Immunol Lett 107: 125–130 [DOI] [PubMed] [Google Scholar]
- Kondo S (1999) The roles of keratinocyte‐derived cytokines in the epidermis and their possible responses to UVA‐irradiation. J Investig Dermatol Symp Proc 4: 177–183 [DOI] [PubMed] [Google Scholar]
- Li J, Liu L, Rui W, Li X, Xuan D, Zheng S, Yu Y, Zhang J, Kong N, Zhu X et al (2017) New interleukins in psoriasis and psoriatic arthritis patients: the possible roles of interleukin‐33 to interleukin‐38 in disease activities and bone erosions. Dermatology 233: 37–46 [DOI] [PubMed] [Google Scholar]
- Lin H, Ho AS, Haley‐Vicente D, Zhang J, Bernal‐Fussell J, Pace AM, Hansen D, Schweighofer K, Mize NK, Ford JE (2001) Cloning and characterization of IL‐1HY2, a novel interleukin‐1 family member. J Biol Chem 276: 20597–20602 [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen T, Zhou F, Mu D, Liu S (2020) Interleukin‐38 increases the insulin sensitivity in children with the type 2 diabetes. Int Immunopharmacol 82: 106264 [DOI] [PubMed] [Google Scholar]
- Long A, Dominguez D, Qin L, Chen S, Fan J, Zhang M, Fang D, Zhang Y, Kuzel TM, Zhang B (2018) Type 2 innate lymphoid cells impede IL‐33‐mediated tumor suppression. J Immunol 201: 3456–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah LJ, El‐Osta A, Karagiannis TC (2010) gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24: 679–686 [DOI] [PubMed] [Google Scholar]
- Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, Gubinelli E, Mazzanti C, Cavani A, Ruvo M et al 2018) IL‐38 has an anti‐inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti‐IL‐17A treatment. Cell Death Dis 9: 1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming M, Han W, Zhao B, Sundaresan NR, Deng CX, Gupta MP, He YY (2014) SIRT6 promotes COX‐2 expression and acts as an oncogene in skin cancer. Cancer Res 74: 5925–5933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi BG, Neustadter J, Binda E, Lewis J, Filler RB, Roberts SJ, Kwong BY, Reddy S, Overton JD, Galan A et al (2012) Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science 335: 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora J, Schlemmer A, Wittig I, Richter F, Putyrski M, Frank AC, Han Y, Jung M, Ernst A, Weigert A et al (2016) Interleukin‐38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol 8: 426–438 [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kassem S, Cleynen A, Chretien ML, Guillerey C, Putz EM, Bald T, Forster I, Vuckovic S, Hill GR et al (2018) Dysregulated IL‐18 Is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell 33: 634–648.e5 [DOI] [PubMed] [Google Scholar]
- Nelson MA, Futscher BW, Kinsella T, Wymer J, Bowden GT (1992) Detection of mutant Ha‐ras genes in chemically initiated mouse skin epidermis before the development of benign tumors. Proc Natl Acad Sci USA 89: 6398–6402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Wang M, Chen X, Chen Y, Ai S, Wang M, Su W, Liang D (2021) Elevated IL‐38 inhibits IL‐23R expression and IL‐17A production in thyroid‐associated ophthalmopathy. Int Immunopharmacol 91: 107300 [DOI] [PubMed] [Google Scholar]
- Paoloni M, Davis S, Lana S, Withrow S, Sangiorgi L, Picci P, Hewitt S, Triche T, Meltzer P, Khanna C (2009) Canine tumor cross‐species genomics uncovers targets linked to osteosarcoma progression. BMC Genom 10: 625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen D, Ediriweera C, Liu L (2019) Function and regulation of IL‐36 signaling in inflammatory diseases and cancer development. Front Cell Dev Biol 7: 317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers HW, Weinstock MA, Harris AR, Hinckley MR, Feldman SR, Fleischer AB, Coldiron BM (2010) Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146: 283–287 [DOI] [PubMed] [Google Scholar]
- Rudloff I, Godsell J, Nold‐Petry CA, Harris J, Hoi A, Morand EF, Nold MF (2015) Brief report: interleukin‐38 exerts antiinflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol 67: 3219–3225 [DOI] [PubMed] [Google Scholar]
- Rundhaug JE, Fischer SM (2010) Molecular mechanisms of mouse skin tumor promotion. Cancers 2: 436–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarasinghe V, Madan V (2012) Nonmelanoma skin cancer. J Cutan Aesthet Surg 5: 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Fujimura T, Hidaka T, Lyu C, Tanita K, Matsushita S, Yamamoto M, Aiba S (2020) Possible roles of proinflammatory signaling in keratinocytes through aryl hydrocarbon receptor ligands for the development of squamous cell carcinoma. Front Immunol 11: 534323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijs MJ, Png S, Richard AC, Tsyben A, Hamm G, Stockis J, Garcia C, Pinaud S, Nicholls A, Ros XR et al (2020) ILC2‐driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat Immunol 21: 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Seki T, Kobayashi N, Sano K, Shigemura T, Shimojo H, Matsumoto K, Agematsu K (2018) Analysis of serum IL‐38 in juvenile‐onset systemic lupus erythematosus. Mod Rheumatol 28: 1069–1072 [DOI] [PubMed] [Google Scholar]
- Vahatupa M, Pemmari T, Junttila I, Pesu M, Jarvinen TAH (2019) Chemical‐Induced skin carcinogenesis model using Dimethylbenz[a]Anthracene and 12‐O‐Tetradecanoyl Phorbol‐13‐Acetate (DMBA‐TPA). J Vis Exp 10.3791/60445 [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, Joosten LA, van der Meer JW, Hao R, Kalabokis V et al (2012) IL‐38 binds to the IL‐36 receptor and has biological effects on immune cells similar to IL‐36 receptor antagonist. Proc Natl Acad Sci USA 109: 3001–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigne S, Palmer G, Lamacchia C, Martin P, Talabot‐Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE et al (2011) IL‐36R ligands are potent regulators of dendritic and T cells. Blood 118: 5813–5823 [DOI] [PubMed] [Google Scholar]
- Volpe F, Clatworthy J, Kaptein A, Maschera B, Griffin AM, Ray K (1997) The IL1 receptor accessory protein is responsible for the recruitment of the interleukin‐1 receptor associated kinase to the IL1/IL1 receptor I complex. FEBS Lett 419: 41–44 [DOI] [PubMed] [Google Scholar]
- Wagner M, Ealey KN, Tetsu H, Kiniwa T, Motomura Y, Moro K, Koyasu S (2020) Tumor‐derived lactic acid contributes to the paucity of intratumoral ILC2s. Cell Rep 30: 2743–2757.e5 [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang S, Li L, Liu X, Mei K, Wang X (2010a) Structural insights into the assembly and activation of IL‐1beta with its receptors. Nat Immunol 11: 905–911 [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang W, Wu T, Chu H (2018) Reduced interleukin‐38 in non‐small cell lung cancer is associated with tumour progression. Open Biol 8: 180132 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang L, Yi T, Zhang W, Pardoll DM, Yu H (2010b) IL‐17 enhances tumor development in carcinogen‐induced skin cancer. Cancer Res 70: 10112–10120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Lan Y, Zhong Y, Yu K, Xu W, Zhu R, Sun H, Ding Y, Wang Y, Zeng Q (2020) Interleukin‐38 alleviates cardiac remodelling after myocardial infarction. J Cell Mol Med 24: 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia HS, Liu Y, Fu Y, Li M, Wu YQ (2021) Biology of interleukin‐38 and its role in chronic inflammatory diseases. Int Immunopharmacol 95: 107528 [DOI] [PubMed] [Google Scholar]
- Xie C, Yan W, Quan R, Chen C, Tu L, Hou X, Fu Y (2020) Interleukin‐38 is elevated in inflammatory bowel diseases and suppresses intestinal inflammation. Cytokine 127: 154963 [DOI] [PubMed] [Google Scholar]
- Xie L, Huang Z, Li H, Liu X, Zheng S, Su W (2019) IL‐38: a new player in inflammatory autoimmune disorders. Biomolecules 9: 345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Lin S, Yan X, Wang C, Tu H, Yin Y, Cao J (2018) Interleukin 38 protects against lethal sepsis. J Infect Dis 218: 1175–1184 [DOI] [PubMed] [Google Scholar]
- Yoshida T, Shiroshima T, Lee SJ, Yasumura M, Uemura T, Chen X, Iwakura Y, Mishina M (2012) Interleukin‐1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J Neurosci 32: 2588–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Li Y, Pan X, Peng X, Song G, Jiang W, Gao Q, Li M (2016) IL‐38 alleviates concanavalin A‐induced liver injury in mice. Int Immunopharmacol 40: 452–457 [DOI] [PubMed] [Google Scholar]
- Yuan X, Peng X, Li Y, Li M (2015) Role of IL‐38 and its related cytokines in inflammation. Mediators Inflamm 2015: 807976 [DOI] [PMC free article] [PubMed] [Google Scholar]