

Abstract
Plasmodium falciparum, the deadliest form of human malaria, remains one of the major threats to human health in endemic regions. Its virulence is attributed to its ability to modify infected red blood cells (iRBC) to adhere to endothelial receptors by placing variable antigens known as PfEMP1 on the iRBC surface. PfEMP1 expression determines the cytoadhesive properties of the iRBCs and is implicated in severe malaria. To evade antibody‐mediated responses, the parasite undergoes continuous switches of expression between different PfEMP1 variants. Recently, it became clear that in addition to antibody‐mediated responses, PfEMP1 triggers innate immune responses; however, the role of neutrophils, the most abundant white blood cells in the human circulation, in malaria remains elusive. Here, we show that neutrophils recognize and kill blood‐stage P. falciparum isolates. We identify neutrophil ICAM‐1 and specific PfEMP1 implicated in cerebral malaria as the key molecules involved in this killing. Our data provide mechanistic insight into the interactions between neutrophils and iRBCs and demonstrate the important influence of PfEMP1 on the selective innate response to cerebral malaria.
Keywords: cerebral malaria, ICAM1, neutrophils, PfEMP1, Plasmodium falciparum
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
As first line of defense against severe malaria, neutrophils recognize specific PfEMP1 molecules on the infected erythrocyte via ICAM‐1. This ligand‐receptor interaction leads to efficient killing of Plasmodium falciparum parasites associated with cerebral malaria.

Introduction
Plasmodium falciparum is the protozoan parasite responsible for the deadliest form of human malaria, which remains one of the major infectious diseases influencing mankind. This parasite infects hundreds of millions of people worldwide, resulting in approximately half a million deaths per year, primarily of young children (WHO, 2016). Plasmodium falciparum replicates within circulating red blood cells of infected individuals, and its virulence is attributed to immune evasion through its ability to modify the red blood cell (RBC) surface.
Plasmodium falciparum like other protozoan and bacterial pathogens can alter the infected host cell surface protein expression, and as a result, vary the antigens that are exposed to the host immune system. The process of antigenic variation involves the variable expression of genes that encode immunodominant surface antigens. These surface antigens frequently play a role in the virulence of the disease, thus linking antigenic variation to pathogenicity (Dzikowski et al, 2006b). Immune evasion of P. falciparum is achieved in two known ways, modified infected red blood cells (iRBCs) adhere to different endothelial receptors found on blood vessel walls, thus avoiding the peripheral circulation and removal by the spleen, and they undergo antigenic variation to prevent host immune recognition of surface antigens. The major antigenic ligands responsible for adherence are members of the P. falciparum Erythrocyte Membrane Protein‐1 (PfEMP1) family (Baruch et al, 1995), antigenically variable proteins that are placed on the surface of iRBC and bind to different host vascular adhesion molecules such as CD36, intercellular adhesion molecule 1 (ICAM‐1), chondroitin sulfate A (CSA), and endothelial protein C receptor (EPCR) (Smith et al, 2001; Janes et al, 2011; Bengtsson et al, 2013; Turner et al, 2013). Sequestration of iRBCs in different organs contributes to life‐threatening manifestations of the disease such as cerebral and pregnancy‐associated malaria (Smith et al, 2013). Therefore, PfEMP1 is considered the main virulence factor of malaria caused by P. falciparum (Pasternak & Dzikowski, 2008). The presence of PfEMP1 on the red cell surface stimulates the antibody response of the host, often successfully clearing the majority of iRBCs from the circulation. However, small subpopulations of parasites switch expression to an alternative PfEMP1 on the surface of iRBCs, thereby avoiding the antibody response and re‐establishing the infection (Deitsch & Dzikowski, 2017). This process is referred to as antigenic variation and is responsible for the persistent nature of the disease as well as the waves of parasitemia frequently observed in P. falciparum infections (Miller et al, 2002).
Over the past decades, significant efforts were invested in understanding immune responses in the context of malaria. In this regard, there have been major advances in our understanding of adaptive immune responses to malaria whereas the role of innate immunity received much less attention. Still, components of innate immunity, including NK cells, macrophages, and monocytes, were shown to play a role in protecting the host against malaria infection (Chua et al, 2013; Wolf et al, 2017). It is somewhat surprising that the role of neutrophils, which are the most abundant of all white blood cells in the human circulation and represent the first line of defense against microbial infections, is understudied in the context of malaria. Neutrophils are phagocytic cells equipped with a wide range of receptors and a variety of antimicrobial weapons. On top of eliminating microbes via phagocytosis, they can also degranulate and deploy neutrophil extracellular traps (NETs) (Amulic & Hayes, 2011; Amulic et al, 2012). These features make neutrophils highly potent scavengers for a variety of pathogens, which suggests that they may play a role in the immune response against malaria infections. Indeed, several studies have shown that TNFα‐stimulated neutrophils have the capacity to phagocytose merozoites as well as intraerythrocytic blood‐stage parasites in vitro (Kumaratilake et al, 1990, 1991, 1992) and in vivo (Wickramasinghe et al, 1987; Bostrom et al, 2017). In addition, hemozoin‐containing neutrophils have been previously reported in clinical isolates (Olivier et al, 2014), and neutrophils were claimed to have the capacity to limit the progression of malaria infection (Trubowitz & Masek, 1968; Sinden & Smalley, 1976; Brown & Smalley, 1981). However, the mechanism by which neutrophils recognize and kill blood‐stage P. falciparum parasites is unknown.
Neutrophils were shown to be able to identify parasite‐derived alterations on the RBC membrane. For example, neutrophils recognize RBCs alterations caused by trypanosome‐secreted microvesicles and eliminate these RBCs from the circulation (Stijlemans et al, 2015). Given the extensive variable modifications induced by P. falciparum parasites on the surface of the iRBC and its association with different adhesion phenotypes that determine some of the most severe manifestations of malaria (Pasternak & Dzikowski, 2008), we were interested to study the possible molecular interaction between neutrophils and iRBCs.
Here, we demonstrate that neutrophils form physical contact with the iRBCs and kill the intracellular stages of malaria parasites. We further show that the interaction between neutrophils and iRBCs is mediated by PfEMP1 on the iRBC surface and ICAM‐1 expressed on neutrophils. In addition, we demonstrate that neutrophils impose strong selective immune pressure on parasite subpopulations expressing PfEMP1 variants associated with cerebral malaria (Lennartz et al, 2017; Adams et al, 2021). Taken together, these data provide novel molecular insights into the mechanisms by which neutrophils contribute to the innate immune response during malaria infection as a selective factor that may influence antigenic expression and protect against severe cerebral manifestations.
Results
Neutrophils interact with P. falciparum iRBCs and kill blood‐stage parasites
Previous studies reported that neutrophils interact with blood‐stage P. falciparum parasites from malaria‐infected individuals (Wickramasinghe et al, 1987; Bostrom et al, 2017). We tested whether naive neutrophils can spontaneously recognize and eliminate iRBCs. Neutrophils were isolated from healthy donors and co‐cultured with iRBCs containing late‐stage NF54 parasites constitutively expressing GFP (GFP+‐NF54). Using bright‐field and fluorescent microscopy, neutrophils were shown to form physical contact with iRBCs and phagocytose the parasites spontaneously (Fig 1A and B). Both schizonts (Fig 1A, upper panel) and trophozoites (Fig 1A, lower panel) were found to be phagocytosed, and blood smears (Fig 1B) corroborate that RBC infected by late‐stage P. falciparum are phagocytosed by neutrophils. Next, we used flow cytometry to assess the extent of neutrophils capacity to interact with RBCs infected with GFP+‐NF54 parasites. Following 10 min of co‐incubation and in the absence of human serum, 30–40% of the neutrophils became GFP+ (Fig 1C middle panel & 1D); opsonization of iRBCs with human serum prior to introducing them into the culture further potentiated this response, with iRBCs interact to about 50–60% of the neutrophils (Fig 1C right panel & 1D), indicating that the response of neutrophils to iRBCs in vivo might be more effective than under controlled culture conditions. Interestingly, the interaction (binding and/or phagocytosis) of the iRBCs did not enhance either apoptosis or necrosis of neutrophils (Fig EV1A).
Figure 1. Human neutrophils interact with and phagocytose P. falciparum‐infected red blood cells.

- Human neutrophils stained for CD66b after incubation with RBCs infected with GFP+ P. falciparum parasites (white arrows). Nuclei were stained with DAPI (blue), neutrophils were stained with CD66b (red), GFP‐expressing parasites (green). Scale bar, 5 µm. The upper and lower panels show two different cells.
- Giemsa staining of freshly isolated human neutrophils from a healthy donor incubated with iRBCs harboring late‐stage P. falciparum parasites (black arrows). Scale bar, 10 µm.
- Flow cytometry analysis of human neutrophils incubated with opsonized or non‐opsonized GFP+‐late‐stage iRBCs. Free neutrophils were used as control.
- Quantification of GFP+ iRBCs phagocytosed by neutrophils. The phagocytosis was analyzed by flow cytometry (FACS).
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean (SEM). Statistical significance was determined using student t‐test *P ˂ 0.05.
Figure EV1. The impact of phagocytosis on apoptosis and necrosis of PLB985 neutrophils.

-
AAnnexin V and propidium iodide (PI) flow cytometry (FACS) were used to analyze for apoptosis and necrosis of GFP‐ neutrophils that did not phagocytose (left panel) or GFP+ neutrophils that did phagocytose GFP‐expressing P. falciparum iRBCs following a 6‐h co‐incubation. Neutrophils reduce the parasitemia in cultures.
-
BPercentage of luciferase signal reduction during the short killing assay (6 h) of luciferase‐expressing P. falciparum iRBCs with or without neutrophils.
-
CiRBCs used for the short‐term killing assay in (B) were put back into culture and the reduction in parasitemia was measured following 1 and 3 days of co‐culture.
-
DPercentage of parasitemia reduction on days 1 and 3 shown in B.
-
EReduction in parasitemia at day 1 and 3 following neutrophils incubation with iRBC in the presence or absence of catalase. The impact of serum proteins on the neutrophil killing of iRBCs.
-
FShort‐term killing assay of AB+‐serum opsonized (AB+ serum) and non‐opsonized iRBCs (no serum) in the presence of anti‐ICAM1 blocking antibody (mAb 15.2) and anti‐CD11b control antibody.
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean (SEM). Statistical significance was determined using student t‐test *P ˂ 0.05; **P < 0.01.
A key question arising from these observations is whether the interaction between neutrophils and iRBCs leads to parasite killing by neutrophils. To test this, we performed a pulse‐chase experiment where RBCs infected with late‐stage GFP+ parasites were incubated with neutrophils and measured changes in the fraction of GFP+ neutrophils over time. We show that the fraction of GFP+ neutrophils either binding or phagocytosing iRBCs decreased with time (Fig 2A). We repeated this experiment with opsonized iRBCs and found that although the fraction of iRBCs interacting with neutrophils was larger following opsonization (Fig 1C and D), the decrease in the fraction of GFP+ neutrophils by time was like that of neutrophils incubated with non‐opsonized iRBCs (Fig 2A). These data suggest that opsonization can significantly increase the fraction of neutrophils interacting with iRBCs. However, once in contact, opsonized and non‐opsonized iRBCs are cleared at a similar rate.
Figure 2. Killing of P. falciparum‐infected red blood cells by human neutrophils.
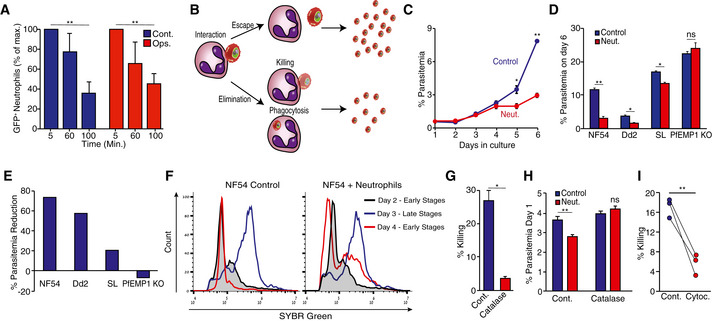
- Interaction between neutrophils and opsonized (red) or non‐opsonized (blue) GFP+ iRBCs by time in a “pulse‐chase” experiment. The percentage of GFP+‐neutrophils following 5 min of co‐culture was set as 100%.
- Model of how a low number of GFP+ neutrophils may be explained by the escape of GFP+ iRBCs (upper part) and how a high number of GFP+ neutrophils might be explained by the elimination (phagocytosis or extracellular killing) of GFP+ iRBCs (lower part) by the neutrophils. The escape of GFP+ iRBCs will allow parasites to propagation, while elimination will impair parasite propagation and cause reduced parasitemia of the culture.
- Parasitemia (percentage %) of NF54 iRBCs cultures with or without daily addition of freshly isolated neutrophils.
- Parasitemia (percentage %) of cultures of RBC infected by different P. falciparum lines (NF54, Dd2, SL, PfEMP1‐KO). The parasites were kept in culture for 6 days in the presence (Neut.) or absence (Cont.) of neutrophils.
- Percentage change in parasitemia following neutrophil challenge of RBCs infected by different parasite lines (NF54, Dd2, SL, PfEMP1‐KO).
- Flow cytometric analysis of the effect of neutrophil challenge on parasite cell cycle progression. Neutrophils were stained with an anti‐CD11b antibody, while iRBCs were labeled with SYBR green. Gating for iRBCs was performed by gating for the SYBR green and CD11b‐negative population, excluding the neutrophils that were SYBR green negative, but CD11b positive.
- Short‐term neutrophil killing of late‐stage NF54‐luciferase‐positive RBCs in the presence (red) or absence (blue) of catalase.
- Parasitemia of cultures of iRBCs challenged with neutrophils in the presence or absence of catalase.
- Short‐term luciferase‐based killing assay using neutrophils from three different donors in the presence or absence of 5 µM cytochalasin D.
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean (SEM). Statistical significance was determined using student t‐test *P ˂ 0.05; **P < 0.01.
The time‐dependent decrease in GFP+ neutrophils could be interpreted as either loss of interaction between the neutrophils and iRBCs (escape) or as parasite elimination by neutrophils (Fig 2B). To discern between these two possibilities and confirm that indeed the interaction between neutrophils and iRBCs leads to parasite killing, we tested whether co‐culturing neutrophils with iRBC limits the increase in parasitemia. To this end, iRBC were cultured with neutrophils for 6 days. Due to the neutrophils’ short life span, we replenished the neutrophils in the culture every 24 h to maintain continuous selective pressure. Our data show that a significant difference in parasitemia may be seen as early as 5 days following the initial introduction of neutrophils to the co‐culture (Fig 2C). We repeated this experiment using three different parasite isolates, two culture adapted lines (NF54 and Dd2) and a recently adapted parasite obtained from a traveler infected in Sierra Leone (SL). In addition, to the adapted parasite lines, neutrophils were incubated with DC‐J, a transgenic line that, when grown in the presence of blasticidin, ceases to express the major surface antigen PfEMP1 (Dzikowski et al, 2006a). The iRBC ratio was measured by flow cytometry daily and showed that incubation with neutrophils significantly reduced the growth rate of the NF54, Dd2, SL lines, while the growth rate of the DC‐J line lacking PfEMP1 expression was unaffected (Fig 2D and E). From this, we reasoned that neutrophils may recognize parasite‐derived surface modifications on iRBCs, and thus would interact better with late‐stage parasites, in which modification of their red cell surface is nearly completed. To test this, we followed the intraerythrocytic development (IDC) of tightly synchronized parasites using DNA staining in flow cytometry to assess how daily neutrophil challenges affect the distribution of different parasite stages. Following the completion of one parasite replication cycle, early stage parasites (day 4, ring stage, low signal population) were obtained in the control culture, which reflects the completion of IDC. However, in cultures which were challenged with neutrophils, we found a significant proportion of parasites that did not progress beyond schizont stage (day 3, schizonts, high signal population). The latter finding most likely reflects the detection of DNA remains from dead schizonts (Fig 2F). These data suggest that the presence of neutrophils prohibited parasite cell cycle completion in a significant fraction of iRBCs. To conclusively determine whether neutrophils kill late‐stage parasites, we co‐cultured luciferase‐expressing NF54 parasites (NF54‐luc (Dahan‐Pasternak et al, 2013; Kolevzon et al, 2014)) with freshly isolated neutrophils. Our data show that neutrophils are capable of killing late‐stage luciferase‐expressing parasites within 6 h and reduce overall parasitemia in the following days (Fig EV1B–D).
Although neutrophils are equipped with a wide array of cytotoxic molecules, most of these molecules are antibacterial and as such should not harm eukaryotic malaria parasites. Still, neutrophils can generate a potent‐oxidative burst where cytotoxic reactive oxygen species (ROS) are released into phagosomes and their close vicinity. To determine whether neutrophils use ROS to kill iRBCs, we tested the capacity of freshly isolated neutrophils to kill blood‐stage parasites in the presence or absence of catalase that eliminate neutrophil‐generated H2O2. In the absence of catalase neutrophils killed > 25% of all iRBCs (Fig 2G) and reduced the parasitemia (Fig 2H), while catalase significantly reduced neutrophil cytotoxicity (Fig 2G) and reversed the effect of neutrophils on overall parasitemia (Figs 2H and EV1E). In addition, we found that the inhibition of phagocytosis using cytochalasin D significantly reduces the ability of neutrophils to kill blood‐stage parasites (Fig 2I). Taken together, these data suggest that neutrophils have the capacity to kill blood‐stage P. falciparum parasites through the phagocytosis of iRBCs and targeted oxidative burst. Based on these data, we propose that neutrophils may play an important protective role in the management of infection by killing the malaria parasite and reducing overall parasitemia.
Neutrophils recognize and target iRBC via PfEMP1
PfEMP1 is the major surface antigen expressed on the surface of iRBCs during the second half of the blood stage of the P. falciparum parasite life cycle (Pasternak & Dzikowski, 2008). The observation that, in the absence of PfEMP1 expression, neutrophils do not reduce parasitemia (Fig 2D and E), makes it a prime candidate as a recognition ligand. To validate that PfEMP1 is indeed recognized by neutrophils, we generated GFP‐expressing DC‐J parasites (DC‐J GFP, see methods), providing us with a platform for evaluating their interaction with neutrophils by flow cytometry as described above. Neutrophils were incubated with this parasite line in the presence (wt) or absence (PfEMP1 KO) of PfEMP1 expression and the proportion of GFP+‐neutrophils that became GFP positive was assessed. We found that the proportion of GFP+‐neutrophils was significantly lower when incubated with PfEMP1‐deficient iRBCs (Fig 3A, right panel) compared with those incubated with PfEMP1‐expressing iRBCs (Fig 3A, middle panel) indicating that PfEMP1 contributes to the neutrophil interaction with iRBCs. To further validate this observation, we trypsin‐treated iRBCs to remove all surface proteins including PfEMP1s. Trypsin treatment of wild‐type DC‐J iRBCs reduced the percentage of GFP‐positive neutrophils (Fig 3B) to a similar level of that of both trypsin‐ and non‐trypsin‐treated PfEMP1‐deficient DC‐J‐KO iRBCc (Fig 3B). This points to PfEMP1 as an important surface protein recognized by neutrophils. The approximately 20% GFP+‐neutrophils seen with trypsin‐treated wild‐type DJ‐C and DC‐J knockout (± trypsin‐treated) parasites indicate that either trypsin‐insensitive surface proteins or nonprotein moieties such as sugars may also be recognized by neutrophils. To further investigate the importance of PfEMP1 in the neutrophil‐mediated killing of iRBCs, we generated a luciferase‐expressing DC‐J line. The neutrophil killing of the luciferase‐ and PfEMP1‐expressing DC‐J line (NF54 wt) was significantly higher than the killing of PfEMP1‐KO iRBCs (Fig 3C) further supporting PfEMP1 as a major recognition ligand of neutrophils.
Figure 3. PfEMP1 is the major ligand mediating the interaction and killing of iRBCs by human neutrophils.

- Representative flow cytometric analysis of human neutrophils cultured alone (Control), incubated with late‐stage NF54 parasites (wild type) or PfEMP1 knockout (PfEMP1 KO) GFP+ iRBC.
- Flow cytometric quantification of the effect of trypsin treatment on neutrophil interaction with wild‐type and PfEMP1 KO GFP+ iRBC.
- Luciferase‐based killing assay and neutrophil elimination of late‐stage wild‐type (wt) and PfEMP1ko iRBC.
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean (SEM). Statistical significance was determined using student t‐test **P < 0.01.
Neutrophils ICAM‐1 is essential for iRBCs elimination
The reduced interaction between neutrophils and RBCs infected by knockout parasites points to the importance of PfEMP1. PfEMP1 molecules are known to bind various endothelial host receptors, including ICAM‐1 and EPCR (Bengtsson et al, 2013; Smith et al, 2013; Turner et al, 2013) that are also abundantly expressed on neutrophils (Wang et al, 1997; Sturn et al, 2003). Thus, we hypothesized that these receptors might play a role in mediating the interaction between neutrophils and iRBCs. The short life span of human neutrophils precludes their genetic manipulation. Therefore, to verify this hypothesis, we used the neutrophil‐like PLB985 cell line transduced with ICAM‐1‐ or EPCR‐specific shRNAs. Transducing PLB985 cells with ICAM‐1 shRNAs knocked down the ICAM‐1 expression, while significantly (P < 0.01) increasing the EPCR mRNA level (Fig 4A); similarly, EPCR shRNAs reduced the EPCR expression, while increasing the ICAM‐1 mRNA level sevenfold (Fig 4B), suggesting a possible compensatory mechanism between the two receptors. Using flow cytometric analysis, we confirmed that while ICAM knockdown reduced its expression on the cell surface, EPCR knockdown resulted in surface overexpression of ICAM‐1 (Fig 4C). We then tested how knocking down the neutrophil expression of ICAM‐1 or EPCR affects their interaction with GFP+ parasite‐infected RBCs. We found that the percentage of GFP+‐neutrophils was significantly (P < 0.01) reduced in the ICAM‐1 shRNA‐treated cells compared with neutrophil cells (cont) transduced with control shRNA and thus expressing normal levels of ICAM‐1 (Fig 4D). In contrast, the EPCR knockdown neutrophil cells, in which ICAM‐1 was overexpressed, showed increased neutrophil‐iRBC interaction, that is, a higher percentage of GFP+‐neutrophils (Fig 4D).
Figure 4. Neutrophil ICAM‐1 is an important receptor for their killing ability of iRBCs.

-
A, BqRT‐PCR analysis of the relative expression of ICAM‐1 and EPCR mRNA in non‐treated PLB985 cells (control), ICAM‐1 knockdown (ICAM‐1kd) and EPCR knockdown (EPCRkd) PLB985 cells.
-
CFlow cytometric analysis (FACS) of surface ICAM‐1 (α human mAb, see methods) expression in control non‐treated PLB985, ICAM‐1kd, and EPCRkd PLB985 cells.
-
DFlow cytometry quantification of the percentage of neutrophils that became GFP+ following incubation with GFP+ iRBC of control (untreated PLB985 cells), ICAM‐1kd and EPCRkd PLB985 cells.
-
EShort‐term (6 h) killing assay of NF54‐luciferase iRBCs incubated with PLB985 neutrophils in the presence of anti‐CD11b and anti‐ICAM‐1 blocking (mAb 15.2) and non‐blocking (α human mAb, see methods) antibodies.
-
FInhibition of PLB985 neutrophil (%) phagocytosis of GFP+ iRBCs in the presence of anti‐CD11b and anti‐ICAM‐1 (mAb 15.2) blocking antibodies. The inhibition was quantified by flow cytometry (FACS).
-
GShort‐term (6 h) PLB985 neutrophil killing of NF54‐luciferase iRBCs in the presence or absence of soluble ICAM‐1‐Fc fusion protein (sICAM‐1‐Fc). Soluble RAGE (sRAGE) protein was used as a negative control.
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean. Statistical significance was determined using student t‐test **P < 0.01.
We next used two complementary strategies to conclusively determine if interfering with the PfEMP1‐ICAM‐1 interaction reduces neutrophil‐iRBC contact and killing. First, we show that a monoclonal antibody (mAb 15.2) targeting the PfEMP1‐binding domain of ICAM‐1 (Baratin et al, 2007) significantly (P < 0.01) reduced the ability of neutrophils to kill iRBCs, while this was not the case when using an ICAM‐1 antibody that does not block the PfEMP1‐binding site of ICAM‐1 (Fig 4E). The 15.2 mAb that blocks the ICAM‐1‐PfEMP1 interaction also reduced the neutrophil phagocytosis of iRBCs (Fig 4F). Notably, incubation with AB+ serum did not reduce the inhibitory effect of the anti‐ICAM‐1 blocking antibody (mAb 15.2), indicating that opsonization does not interfere with neutrophil‐iRBC or ICAM‐1‐PfEMP1 interaction (Fig EV1F). As a second approach, we used a soluble form of ICAM‐1 fused to a mutated Fc receptor (sICAM‐1‐FcMut) to compete with the binding of PfEMP1 to neutrophil‐expressed ICAM‐1. Specifically, NF54‐luc‐infected iRBC were incubated for 6 h with naïve human neutrophils in the presence or absence of sICAM‐1‐FcMut. We show that incubation with sICAM‐1‐FcMut significantly reduced the neutrophils’ ability to kill parasites, while incubation with unrelated sRAGE‐FcMut had no effect (Fig 4G). Altogether, these results highlight ICAM‐1 and PfEMP1 as the main mediators of neutrophil interaction with iRBCs, ultimately leading to the killing of P. falciparum.
Neutrophils impose strong selective pressure against ICAM‐1‐binding iRBCs expressing PfEMP1 associated with cerebral malaria
Our data indicate that neutrophil ICAM‐1 interacts with PfEMP1 on the surface of iRBCs and that this interaction is required for iRBC killing. These data led us to hypothesize that neutrophils may selectively eliminate iRBCs expressing a subset of PfEMP1 with ICAM‐1‐binding properties. To test this hypothesis, we performed a number of experiments using NF54 iRBCs preselected to express PFD1235w (PF3D7_0425800) an ICAM‐1‐binding PfEMP1 associated with severe and cerebral malaria (Jensen et al, 2004; Joergensen et al, 2010; Bengtsson et al, 2013; Adams et al, 2021). Using antibody‐selected iRBCs transcribing PFD1235w (Fig 5A, panel I and II) and transfected with a luciferase‐reporter plasmid, we found a significant (P < 0.01) increased neutrophil killing of PFD1235w‐expressing iRBCs compared to the killing of nonselected iRBCs surface expressing other PfEMP1 variants (Fig 5B). In addition, neutrophils showed a significantly (P < 0.01) increased interaction with ICAM‐1‐binding, that is, PFD1235w‐expressing iRBCs than iRBCs not selected for binding to ICAM‐1 (Fig 5C). The differences in neutrophil–iRBC interaction and killing were also reflected in the differences in parasitemia of the two parasite populations when cultured in the presence of neutrophils for 5 additional days (Fig 5D). From these data, we concluded that neutrophils are indeed more efficient in killing iRBCs expressing ICAM‐1‐binding PfEMP1. To determine the impact of neutrophils on the antigenic repertoire of iRBCs, we cultured ICAM‐1‐selected iRBCs with or without neutrophils and assessed the var gene transcription of the parasites. iRBC cultures not exposed to neutrophils did not show any change in var gene expression, and PFD1235w remained the dominant transcript (Fig 5E, top panel), while neutrophil‐exposed cultures showed no to low transcription of this var gene (Fig 5E, bottom panel). From this, we conclude that neutrophils impose a strong selective pressure on iRBCs expressing ICAM‐1‐binding PfEMP1 such as PFD1235w.
Figure 5. Neutrophils select against iRBC expression of ICAM‐1‐binding PfEMP1.

- Steady‐state mRNA levels of NF54 var genes measured by qRT‐PCR. The NF54 line was transfected with a luciferase expression vector and selected for ICAM‐1‐binding. PFD1235w/PF3D7_0425800 is the transcriptionally dominant var gene expressed by this line. Steady‐state mRNA levels of each individual var gene are presented as relative copy number to the housekeeping gene arginyl‐tRNA synthetase (PFL0900c). II. Immunofluorescence image of PFD1235w surface expressed on ICAM‐1 selected NF54‐luciferase iRBCs. The punctuate PfEMP1 pattern was detected using an anti‐PFD1235w antibody.
- Short‐term (6 h) neutrophil killing of unselected NF54 and antibody‐selected PFD1235w‐expressing parasite lines.
- Flow cytometric quantification of neutrophil interaction with MitoTracker (APC+)‐stained NF54 iRBCs and antibody‐selected PFD1235w‐expressing iRBC.
- Percent reduction in parasitemia of NF54 and antibody‐selected PFD1235w‐expressing parasites after 5 days of co‐culture with neutrophils, compared to unchallenged parasites.
- var gene transcription profiles measured by qRT‐PCR of antibody‐selected PFD1235w‐expressing parasite line cultured in the absence (upper panel) or presence (lower panel) of neutrophils. Steady‐state mRNA levels of each individual var gene was calculated as in A. I.
Data information: Results represent the average of three biological replicates (n = 3) ± standard error of the mean. Statistical significance was determined using student t‐test **P < 0.01. scale bar = 2 µm.
Discussion
In recent years, mounting evidence has implicated components of the innate arm of the human immune system as important defense mechanisms against malaria infections. For example, NK cells were shown to produce pro‐inflammatory cytokines in malaria infection and kill iRBCs either directly or via antibody‐dependent cell‐mediated cytotoxicity (ADCC) (Wolf et al, 2017). Similarly, monocytes and macrophages play a role in the antimalaria immune response via secretion of cytokines and elimination of iRBCs through cytokine secretion or ADCC. In addition, these cells are large enough and can eliminate iRBCs via phagocytosis (Chua et al, 2013). Surprisingly, although neutrophils are the most abundant leukocyte in human circulation and have well‐characterized roles in eliminating pathogenic infections, little is known about their role in malaria (Ley et al, 2018). The capacity of neutrophils to phagocytose merozoites and gametocytes in vitro has been demonstrated previously (Trubowitz & Masek, 1968; Sinden & Smalley, 1976). Additionally, neutrophils were shown to respond to malaria parasites by generating reactive oxygen species (Salmon et al, 1986), and by limiting the growth of malaria parasites in vitro (Brown & Smalley, 1981). Recently, it was shown that neutrophils accumulate in the intervillous space in the placenta during pregnancy‐associated malaria (Bostrom et al, 2017). However, the mechanisms by which neutrophils interact and eliminate intracellular blood‐stage parasites were thus far not elucidated.
Here, we show that neutrophils recognize iRBCs via their surface‐expressed ICAM‐1, and PfEMP1 the main antigen expressed on the infected red blood cell surface. Once neutrophils and iRBCs interact, the neutrophils can clear approximately 30% of the P. falciparum‐infected RBCs culture within < 2 h.
Plasmodium falciparum have the capacity to alternate between the expressions of PfEMP1 variants that bind different receptors. The identification of neutrophil ICAM‐1 as an iRBC recognition receptor would suggest that neutrophils may exhibit improved killing efficiency against specific parasite subpopulations that express ICAM‐1‐binding PfEMP1 variants. This was supported by our demonstration of neutrophils preferentially interacting with and killing iRBCs expressing PFD1235w, this protein is a group A PfEMP1 that facilitates dual receptor binding to human endothelial ICAM‐1 and EPCR (Lennartz et al, 2017). The increased ICAM‐1 expression by brain endothelial cells, as well as the cell swelling and damage to the blood–brain barrier seen during cerebral malaria has been associated such group A PfEMP1s (Lennartz et al, 2017; Tuikue Ndam et al, 2017; Adams et al, 2021). Interestingly, activated neutrophils were associated with cerebral malaria even though neutrophils were rarely seen in brain microvasculature autopsy samples (Feintuch et al, 2016). However, a recent postmortem examination demonstrated that NETosis was strongly associated with iRBC sequestration in retinal capillaries of children who died from cerebral malaria (Knackstedt et al, 2019). It is worth noting that iRBCs containing early‐stage parasites remain in the circulation until PfEMP1 is transported onto the red cell surface leading to cytoadherence of late‐stage iRBCs. Thus, the primary interactions of neutrophils and iRBCs expressing ICAM‐1‐binding PfEMP1s may happen already in the circulation prior to iRBCs sequestering in the brain microvasculature. Thus, the strong selective immune pressure imposed by neutrophils against these specific parasite populations suggests that neutrophils play an important protective role as the first line of defense against severe and cerebral malaria.
Our findings, using naïve neutrophils in culture, aligns with a previous reports suggesting that even though TNFα stimulation enhances the ability of neutrophils to kill blood‐stages parasites, it is not obligatory (Kumaratilake et al, 1990). Moreover, the data in this study indicate that even though human neutrophils mediated killing of P. falciparum‐infected RBCs is enhanced by TNFα stimulation, neutrophils have a significant capacity to eliminate blood‐stage parasites even without any further stimulation. Interestingly, similar clearance rates were obtained in opsonized and non‐opsonized parasites suggesting that the rate‐limiting step for iRBC elimination is the recognition by neutrophils rather than the killing per se. This clearance indeed translated into a significant decrease in the growth rate of the cultured parasite. Clearly, the neutrophils’ ability to kill intraerythrocytic stage parasites is not sufficient to completely clear the infection, but is likely to contribute to the control of the infection by the adaptive immune effector mechanisms that kicks in later.
In the current study, we focused on the function of normal density neutrophils (NDN) obtained from healthy donors. This subpopulation of neutrophils represents malaria‐naïve, non‐inflammatory neutrophils, and provided us with the opportunity to study the initial interaction between neutrophils and iRBCs. It is likely that circulating neutrophil subpopulations may have diverse phenotypic responses to iRBCs similar to the phenotypic plasticity of neutrophils demonstrated in cancer (Sagiv et al, 2015). Nonetheless, our observations provide novel insight into the role played by neutrophils in P. falciparum malaria infection. We demonstrate that neutrophils use both phagocytosis and ROS production to kill blood‐stage parasites, but the exact killing mechanism and the possible involvement of NETosis still remain to be established. Interestingly, the interaction between neutrophils and iRBCs parallels the interaction between iRBCs and the endothelium that also depending on parasite phenotype involves PfEMP1 and ICAM‐1 (Fig 6). It is well documented that cytoadhesion triggers local inflammation (van der Heyde et al, 2006; Schofield, 2007), and it is therefore plausible that neutrophil interaction with iRBCs occurs not only in the circulation but also at the site of iRBCs sequestration. The fact that the same receptor employed by P. falciparum to both cytoadhere and avoid removal by the spleen is also utilized by neutrophils to kill iRBCs, may represent an additional aspect of host–pathogen co‐evolution not looked at previously.
Figure 6. The interaction of iRBC with endothelium and neutrophils.

Infected RBCs can interact with both endothelial cells and neutrophils. Left panel, iRBCs adhere to the endothelium as a strategy for escaping removal by the spleen. The binding of iRBC to the endothelial cell is mediated by PfEMP1 and different endothelial receptors, for example, ICAM‐1, CSA, CD36, EPCR. Right panel, neutrophil surface expresses ICAM‐1 and binds to PfEMP1 on iRBC. This results to extracellular killing or the phagocytosis and killing of the iRBC (not shown)—see also Fig 2B.
As it appears, not all neutrophils actively engage in iRBC interaction (see Fig 1C) suggesting the possible existence of different neutrophil subtypes, with different roles in malaria infection. The concept that neutrophils are not a homogenous population of cells but actually consist of specialized subsets has been demonstrated in various clinical conditions ranging from cancer (Sagiv et al, 2015) to periodontal disease (Fine et al, 2016). Our results agree with these findings and suggest that neutrophil functional heterogeneity may also be relevant in response to other infectious pathogens.
Trypsin treatment of iRBCs and removal of surface PfEMP1 did not completely abolish the interaction between neutrophils and the infected RBCs. It is known that not all PfEMP1 are trypsin sensitive (Ghumra et al, 2012), and therefore, it is likely that other parasite proteins on the iRBC surface (such as rif, stevor, and pfmc‐2 M) are also semiresistant and might still be left to interact with the neutrophils (Kyes et al, 1999; Dzikowski et al, 2006b; Lavazec et al, 2006, 2007; Niang et al, 2009, 2014; Bachmann et al, 2015; Ch'ng et al, 2017; Saito et al, 2017). Similarly, knocking down neutrophil ICAM‐1 expression using shRNA did not completely abolish the interaction between the two cell types. Neutrophils express several cell surface receptors for pathogen and inflammatory sensing (e.g., G‐protein‐coupled receptors, Fc‐receptors, Toll‐like receptors, and C‐type lectins (Futosi et al, 2013)), some of which might contribute to their ability to kill iRBCs. This is supported by our finding that opsonization of iRBCs by addition of AB+ serum led to increased neutrophil killing of the infected red cells pointing to an involvement of Fc‐receptors on the neutrophils. While the role of other neutrophil receptors for iRBC recognition was not the scope of this paper, our data emphasize the importance of neutrophil ICAM‐1 for their killing of iRBC expressing a specific subset of PfEMP1. Further detailed investigation of the interactions between neutrophils and other parasite surface ligands is required to comprehensively understand how neutrophils function in malaria infection.
Materials and Methods
Parasites and cell cultures
All parasites used were derivatives of the NF54, Dd2, and a field isolate from Sierra Leone (SL). The DC‐J line growing on blasticidin as previously described (Dzikowski et al, 2006a) was used to create the PfEMP1 KO line and DC‐J GFP+ line following transfection with GFP expression vector as described below. For luciferase assays, we used NF54‐luc parasites which were previously described (Salazar et al, 2012). Parasite lines were cultivated at 5% (v/v) hematocrit in RPMI medium 1640, 0.5% (v/v) Albumax II (Invitrogen), 0.25% sodium bicarbonate, and 0.1 mg/ml gentamicin. Parasites were incubated at 37°C in an atmosphere of 5% (v/v) oxygen, 5% (v/v) carbon dioxide, and 90% (v/v) nitrogen. NF54 parasite line expressing the PFD1235w var gene was selected using antibodies against DBLβ_D4 domains of specific ICAM‐1‐binding PfEMP1s (PFD1235w) as described (Bengtsson et al, 2013).
The human myeloid leukemia cell line PLB‐985 (a generous gift from Dr. Borko Amulic) was cultured in RPMI‐1640 medium supplemented with 10% FCS, 2 mM l‐glutamine, 100 units/ml penicillin, and 100 µg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2 in air. Cell cultures were passaged two to three times a week to maintain a cell density of 2 × 105–106 cells/ml. ICAM‐1kd PLB985 cells were generated by lentiviral transduction with ICAM‐1‐specific shRNAs from Sigma (TRCN0000372478). EPCRkd PLB985 cells were generated by lentiviral transduction with EPCR‐specific shRNAs from Sigma (TRCN0000300553). Control cells were transduced an empty vector (pLKO). For granulocytic differentiation, exponentially growing PLB‐985 cells at a starting density of 2 × 105/ml were cultured in RPMI‐1640 medium supplemented with 0.5% DMF and 0.5% FCS for 6 days. The medium was changed once on day 3 during the differentiation period.
Parasite transfections and selections
For neutrophil–iRBC interaction assays, the DC‐J and NF54 parasite lines (Dzikowski et al, 2006a) were transfected with pHgfpTIDH plasmids that constitutively express GFP fused to an unrelated exogenous protein (tet repressor). This construct was made by replacing the luciferase sequence in pHLIRH expression vector (Epp et al, 2008) with the tet‐gfp fusion using HindIII and BamHI. Parasites were transfected as previously described (Wu et al, 1995; Deitsch et al, 2001). For luciferase‐killing assays, the DC‐J parasite line was transfected with phLI1055Dh plasmid to constitutively express luciferase (Amit‐Avraham et al, 2015). Stable transfectants carrying plasmids with hDHFR‐selectable marker were selected with 4 nM WR99210. Selection for PfEMP1‐null expression in the transgenic line DC‐J was done using 2 μg/ml blasticidin.
Neutrophil purification
Normal density neutrophils (NDN) were isolated as previously described (Sagiv et al, 2016). In brief, heparinized blood (20 U/ml) collected from healthy donors was mixed with an equal volume of Dextran 500 (3% in saline) and incubated for 30 min at room temperature. The leukocyte‐rich supernatant was layered on top of Histopaque 1077 (Sigma) and centrifuged at 400 g for 30 min. Neutrophils were collected in the pellet fraction and were resuspended in 20 ml 0.2% NaCl for 30 s to remove contaminating erythrocytes. Isotonicity was restored by the addition of 20 ml 1.6% NaCl. Neutrophils were then washed three times in PBS. Neutrophil purity and viability were determined visually and were consistently > 98%. All blood donors provided written informed consent in accordance with the Declaration of Helsinki. The medical ethics committee of the Hadassah‐Hebrew University Medical Center approved the used protocol.
Late‐stage iRBC isolation
Parasite cultures were synchronized using Percoll/sorbitol gradient centrifugation as previously described (Dzikowski et al, 2006a). Briefly, iRBCs were layered on a step gradient of 40%/70% (v/v) Percoll containing 6% (w/v) sorbitol. The gradients were then centrifuged at 12,000 g for 20 min at room temperature. Tightly synchronized, late‐stage parasites were recovered from the 40%/70% interphase, washed twice with complete culture media and counted.
Parasite staining for flow cytometry interaction assays
MitoTracker Red CMXRos (ThermoFisher M7512) dye was dissolved in DMSO at a concentration of 1 mM and stored at −20°C until use. A 5‐μM working solution was prepared with culture media prior to staining tightly synchronized late‐stage iRBCs. Approximately, 106 iRBCs were resuspended in 100 μl of 5 μM CMXRos and incubated at 37°C for 30 min. iRBCs were washed twice with growth media to remove unbound dye.
Neutrophil–iRBC interaction assay and opsonization
Primary neutrophils or differentiated PLB985 cells were incubated with fluorescent late‐stage iRBC either expressing GFP or stained using MitoTracker as described, at a 10:1 ratio at 37°C for different time periods. Samples were washed, and the extent of neutrophils–iRBC interaction (% fluorescent neutrophils) was determined using flow cytometry. Opsonization of iRBCs was performed by culturing iRBCs with AB human serum (Sigma) for 30 min at 37°C. To assess ligand–receptor specificity to this interaction, we performed these assays using anti‐Cd11b antibody (Biolegend Cat # 101211, 10 μl/ml) and a non‐PfEMP1‐blocking anti‐ICAM‐1 antibody (Biolegend Cat # 322702, 10 μl/ml) as negative controls. An anti‐ICAM‐1 monoclonal antibody (15.2) that blocks the PfEMP1‐binding site (Thermofisher, MA180910, 10 μl/ml) was used as blocking antibody as described (Baratin et al, 2007). All antibodies were incubated with iRBCs for 30 min at room temperature in culture media prior to flow cytometry interaction assays. To inhibit phagocytosis, neutrophils were pre‐incubated with 5 µM cytochalasin D (Sigma‐Aldrich), an actin polymerization inhibitor, for 1 h at 37°C prior to the addition of fluorescent iRBCs.
AnnexinV/PI staining
Cells were washed twice with PBS and then incubated with 2 μl Annexin V‐Alexa Fluor 647 (Life technologies A23204) in 40 μl Annexin V buffer (150 mM NaCl, 4 mM KCl, 1.5 mM CaCl2, 10 mM Hepes) for 15 min. Samples were then supplemented with 400 μl of Annexin V with Propidium Iodide (Fluka 70335, 1 μl/sample) and read by FACS.
Immunofluorescent staining
Immunofluorescent staining was performed as described before (Fastman et al, 2018) with few modifications. Briefly, following the co‐culture of neutrophils and iRBC, samples were washed and stained with mouse anti‐CD66b (BioLegend Cat # 305112, 1:200). Samples were then washed, cyto‐centrifuged, and fixed using a fresh fixative solution (4% paraformaldehyde (EMS) and 0.0075% glutaraldehyde (EMS) in PBS). Fixed samples were treated with 0.1% Triton‐X100 (Sigma) in PBS and blocked using CAS‐Block (Life Technologies Cat # 008120). Cells were then incubated with a rabbit anti‐GFP (Invitrogen Cat # A11122, 1:250), washed and incubated with Alexa Fluor 568 goat anti‐Mouse (Abcam Cat # ab175473, 1:500) and Alexa Fluor 488 goat anti‐rabbit (Molecular Probes Cat #A11034, 1:250) secondary antibodies. Polyclonal IgG antibodies against the ICAM‐1‐binding domain (DBLβ_D4) of the PFD1235w PfEMP1 were used for surface labeling as described (Joergensen et al, 2010). Samples were washed and mounted in Fluoroshield mounting medium with DAPI (abcam), covered with cover slips and imaged. Fluorescent images were obtained using a Plan Apo λ 100× oil NA = 1.5, WD = 130 μm lens on a Nikon Eclipse Ti‐E microscope equipped with a CoolSNAP Myo CCD camera. Images were processed using the NIS‐Elements AR (4.40 version) software.
Growth inhibition of parasite co‐cultured with neutrophils
Parasite cultures were synchronized as described above and late stages were counted by flow cytometry. Approximately 106 parasites were cultured in 100‐μl uninfected RBCs, resulting in a parasitemia of ~1%. Human neutrophils were isolated as described above and 106 cells were added to the culture every 24 h for 5 consecutive days. Parasitemia was evaluated every 24 h by flow cytometry. For each experiment, neutrophils from the same donor were used for the 5 consecutive days. Growth inhibition assays were repeated at least three times.
Luciferase‐based killing assay
Luciferase‐expressing parasite cultures were synchronized and late‐stage parasites were put back into culture as described. After 20 h, uninfected RBCs were lysed using Streptolysin O (Sigma) activated with 100 mM DTT. Isolated rings were washed three times and returned to the culture without uninfected RBCs. After 20 h, isolated iRBCs were collected and plated in 100 μl RPMI‐1640 with 2% FCS in 96 wells (1 × 106/well) and 106 purified neutrophils were added in a 100‐μl volume. Following 6‐h incubation, samples were lysed using saponin, centrifuged and the supernatant was discarded. The pellet was then lysed using 50 μl Bright‐GLO (Promega E2620) lysis buffer. Luciferase activity was measured following addition of 50 μl Bright‐GLO luciferase substrate, using Tecan F200 microplate luminescence reader. Extent of killing was determined by the ratio between parasites alone and parasites co‐cultured with neutrophils. Killing assays were repeated at least three times. As we obtained similar results with both autologous and allogenic red blood cells, neutrophils and red blood cells were not donor matched in all experiments.
Evaluation of culture parasitemia
The level of parasitemia was evaluated by flow cytometry. 50 µl samples taken from the parasite cultures were washed in PBS and incubated 30 min with 1:10,000 SYBR Green I DNA stain (Life Technologies). Since neutrophils have DNA as well, distinguishing neutrophils was done by adding anti‐CD11b‐APC antibody (Biolegend 301309) 1:400 in parallel to the SYBR Green staining. APC+ cells were excluded from the analysis. The fluorescence profiles of infected erythrocytes were measured on CytoFLEX (Beckman Coulter) and analyzed by the CytExpert software.
RNA extraction and cDNA synthesis
RNA was extracted from synchronized parasite cultures at 20–24 h after percoll/sorbitol gradient centrifugation. RNA was extracted with the TRIZOL LS Reagent® as described (Kyes et al, 2000) and purified on PureLink column (Invitrogen) according to manufacturer's protocol. Isolated RNA was then treated with DNase I (TaKaRa) to degrade contaminating gDNA. cDNA synthesis was performed from 500 ng total RNA with PrimeScript™ RT Reagent Kit (TaKaRa) as described by the manufacturer.
Real‐time RT‐qPCR
Steady‐state mRNA levels of the entire var gene family were measured by RT‐qPCR reactions using a primer set designated to detect transcripts of all var gene in the NF54 genome (Salanti et al, 2003) with few modifications (Frank et al, 2007). Transcript copy numbers were determined using the formula 2−ΔΔCT as described in the Applied Biosystems User Bulletin 2 using NF54 gDNA as the calibrator. Specifically, relative copy number was calculated as two exponential negative ((Ct target gene in cDNA—Ct reference gene in cDNA)‐(Ct target gene in gDNA—Ct target gene in gDNA)).
Soluble protein preparation
For soluble receptor expression, 4T1 cells were infected with viral particles prepared from tet‐inducible pLV_TRE_RFP vector (kindly provided by Prof. Eli Keshet, The Hebrew University of Jerusalem) expressing the respective genes, and mRFP‐positive cells were sorted using BD FACSARIA III cell sorter. Soluble receptor expression was induced by adding 1 μg/ml doxycycline (Sigma) to the cells the day before the assay. sICAM‐1‐Fc was prepared by amplifying the extracellular ICAM‐1 domain from neutrophil cDNA using Phusion Flash High‐Fidelity PCR master mix. The PCR fragment was inserted into the pLV_TRE_mRFP vector. The mutant Fc fragment of human IgG1 that does not bind Fc receptors, and as such will not trigger antibody‐dependent cell‐mediated cytotoxicity (Stanietsky et al, 2009), was prepared by amplifying the Fc fragment of the CSI‐Ig (Fc mut)‐IRES‐puro plasmid kindly provided by Prof. Ofer Mandelboim (The Hebrew University of Jerusalem) using Phusion Flash High‐Fidelity PCR master mix. The mutant Fc fragment was inserted into the pLV_TRE_mRFP vector. Soluble RAGE was used as a negative control and prepared as previously described (Sionov et al, 2019).
Lentiviral infection
2.5 × 106 293T cells seeded the day before in 10 ml DMEM+10% heat‐inactivated FCS were transfected with 20 μg of the respective lentiviral vectors, 15 μg of pCMV‐ΔR8.91 gag‐pol, and 5 μg VSV‐G (pMD2.G) using the calcium phosphate DNA precipitation method. For MigR1‐luc retroviral vector, pCL‐Eco was used as gag‐pol instead of ΔR8.91. On the following day, the medium was replaced, and viral supernatant was collected after 24–48 h and 0.45 μm filtrated. 4T1 cells were incubated in the filtrated viral supernatant in the presence of 8 μg/ml polybrene (Sigma) for 24 h. After 5–7 days, mRFP+ cells were sorted using BD FACSARIA III cell sorter. Pooled sorted cells were used for the experiments.
Statistical analysis
For experiments comparing differences between groups, we used Student’s t‐tests. Differences were considered significant when P < 0.05. Data are presented as mean ± SEM.
Human data
Informed consent was obtained from all subjects and that the experiments conformed to the principles set out in the WMA Declaration of Helsinki for research number 0091‐17‐HMO.
Author contributions
Tamir Zelter: Conceptualization; Investigation; Methodology; Writing—original draft. Jacob Strahilevitz: Conceptualization; Investigation. Karina Simantov: Investigation; Methodology. Olga Yajuk: Investigation. Yvonne Mary Adams: Investigation; Methodology; Writing—review & editing. Anja Jensen: Conceptualization; Supervision; Funding acquisition; Methodology; Writing—original draft; Writing—review & editing. Ron Dzikowski: Conceptualization; Supervision; Funding acquisition; Methodology; Writing—original draft; Writing—review & editing. Zvi Granot: Conceptualization; Supervision; Funding acquisition; Methodology; Writing—original draft; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
RD, ZG, TZ, and ARJ conceived of the study; TZ, RD, ZG, JS, KS, OY, YA, and ARJ were involved in the methodology and investigation of the study; RD and ZG supervised and wrote the original draft.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Acknowledgements
ZG is supported by Israel Science Foundation (ISF) Grant 405/18, the Israel Cancer Research Fund, the Deutsche Forschungsgemeinschaft (DFG), and the Rosetrees Trust. ZG is also supported by the Samuel and Isabel Friedman Chair in Experimental Medicine. This work was supported partially by the Israeli Academy for Science, Israel Science Foundation (ISF) Grant 1523/18 and in part by European Research Council (erc.europa.eu) Consolidator Grant 615412 (to R.D.). RD is also supported by the Dr. Louis M. Leland and Ruth M. Leland Chair in Infectious Diseases. ARJ is supported by the Lundbeck Foundation (R313‐2019‐322). The Danish Agency for Higher Education and Science International Network Programme supported the collaboration between ARJ and RD (0192‐00058B). We are thankful to Dr. Borko Amulic for kindly providing us with the human myeloid leukemia PLB‐985 cell line.
EMBO reports (2022) 23: e53641.
Contributor Information
Ron Dzikowski, Email: rond@ekmd.huji.ac.il.
Zvi Granot, Email: zvikag@ekmd.huji.ac.il.
Data availability
No large primary datasets have been generated and deposited.
References
- Adams Y, Olsen RW, Bengtsson A, Dalgaard N, Zdioruk M, Satpathi S, Behera PK, Sahu PK, Lawler SE, Qvortrup K et al (2021) Plasmodium falciparum erythrocyte membrane protein 1 variants induce cell swelling and disrupt the blood‐brain barrier in cerebral malaria. J Exp Med 218: e20201266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit‐Avraham I, Pozner G, Eshar S, Fastman Y, Kolevzon N, Yavin E, Dzikowski R (2015) Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum . Proc Natl Acad Sci USA 112: E982–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30: 459–489 [DOI] [PubMed] [Google Scholar]
- Amulic B, Hayes G (2011) Neutrophil extracellular traps. Curr Biol 21: R297–298 [DOI] [PubMed] [Google Scholar]
- Bachmann A, Scholz JA, Janssen M, Klinkert MQ, Tannich E, Bruchhaus I, Petter M (2015) A comparative study of the localization and membrane topology of members of the RIFIN, STEVOR and PfMC‐2TM protein families in Plasmodium falciparum‐infected erythrocytes. Malar J 14: 274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratin M, Roetynck S, Pouvelle B, Lemmers C, Viebig NK, Johansson S, Bierling P, Scherf A, Gysin J, Vivier E et al (2007) Dissection of the role of PfEMP1 and ICAM‐1 in the sensing of Plasmodium falciparum‐infected erythrocytes by natural killer cells. PLoS One 2: e228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Taraschi TF, Howard RJ (1995) Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 82: 77–87 [DOI] [PubMed] [Google Scholar]
- Bengtsson A, Joergensen L, Rask TS, Olsen RW, Andersen MA, Turner L, Theander TG, Hviid L, Higgins MK, Craig A et al (2013) A novel domain cassette identifies Plasmodium falciparum PfEMP1 proteins binding ICAM‐1 and is a target of cross‐reactive, adhesion‐inhibitory antibodies. J Immunol 190: 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom S, Schmiegelow C, Abu Abed U, Minja DTR, Lusingu J, Brinkmann V, Honkpehedji YJ, Loembe MM, Adegnika AA, Mordmuller B et al (2017) Neutrophil alterations in pregnancy‐associated malaria and induction of neutrophil chemotaxis by Plasmodium falciparum . Parasite Immunol 39: 12433 [DOI] [PubMed] [Google Scholar]
- Brown J, Smalley ME (1981) Inhibition of the in vitro growth of Plasmodium falciparum by human polymorphonuclear neutrophil leucocytes. Clin Exp Immunol 46: 106–109 [PMC free article] [PubMed] [Google Scholar]
- Ch’ng J‐H, Sirel M, Zandian A, del Pilar Quintana M, Chun Leung Chan S, Moll K, Tellgren‐Roth A, Nilsson I, Nilsson P, Qundos U et al (2017) Epitopes of anti‐RIFIN antibodies and characterization of rif‐expressing Plasmodium falciparum parasites by RNA sequencing. Sci Rep 7: 43190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CL, Brown G, Hamilton JA, Rogerson S, Boeuf P (2013) Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol 29: 26–34 [DOI] [PubMed] [Google Scholar]
- Dahan‐Pasternak N, Nasereddin A, Kolevzon N, Pe'er M, Wong W, Shinder V, Turnbull L, Whitchurch CB, Elbaum M, Gilberger TW et al (2013) PfSec13 is an unusual chromatin associated nucleoporin of Plasmodium falciparum, which is essential for parasite proliferation in human erythrocytes. J Cell Sci 126: 3055–3069 [DOI] [PubMed] [Google Scholar]
- Deitsch KW, Driskill CL, Wellems TE (2001) Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res 29: 850–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch KW, Dzikowski R (2017) Variant gene expression and antigenic variation by malaria parasites. Annu Rev Microbiol 71: 625–641 [DOI] [PubMed] [Google Scholar]
- Dzikowski R, Frank M, Deitsch K (2006a) Mutually exclusive expression of virulence genes by malaria parasites is regulated independently of antigen production. PLoS Pathog 2: e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzikowski R, Templeton TJ, Deitsch K (2006b) Variant antigen gene expression in malaria. Cell Microbiol 8: 1371–1381 [DOI] [PubMed] [Google Scholar]
- Epp C, Raskolnikov D, Deitsch KW (2008) A regulatable transgene expression system for cultured Plasmodium falciparum parasites. Malar J 7: 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fastman Y, Assaraf S, Rose M, Milrot E, Basore K, Arasu BS, Desai SA, Elbaum M, Dzikowski R (2018) An upstream open reading frame (uORF) signals for cellular localization of the virulence factor implicated in pregnancy associated malaria. Nucleic Acids Res 46: 4919–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feintuch CM, Saidi A, Seydel K, Chen G, Goldman‐Yassen A, Mita‐Mendoza NK, Kim RS, Frenette PS, Taylor T, Daily JP (2016) Activated neutrophils are associated with pediatric cerebral malaria Vasculopathy in Malawian children. MBio 7: e1300–e1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, Cherney D, Glogauer M (2016) Distinct oral neutrophil subsets define health and periodontal disease states. J Dent Res 95: 931–938 [DOI] [PubMed] [Google Scholar]
- Frank M, Dzikowski R, Amulic B, Deitsch K (2007) Variable switching rates of malaria virulence genes are associated with chromosomal position. Mol Microbiol 64: 1486–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futosi K, Fodor S, Mocsai A (2013) Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17: 1185–1197 [DOI] [PubMed] [Google Scholar]
- Ghumra A, Semblat J‐P, Ataide R, Kifude C, Adams Y, Claessens A, Anong DN, Bull PC, Fennell C, Arman M et al (2012) Induction of strain‐transcending antibodies against Group A PfEMP1 surface antigens from virulent malaria parasites. PLoS Pathog 8: e1002665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE (2006) A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol 22: 503–508 [DOI] [PubMed] [Google Scholar]
- Janes JH, Wang CP, Levin‐Edens E, Vigan‐Womas I, Guillotte M, Melcher M, Mercereau‐Puijalon O, Smith JD (2011) Investigating the Host binding signature on the Plasmodium falciparum PfEMP1 protein family. PLoS Pathog 7: e1002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ATR, Magistrado P, Sharp S, Joergensen L, Lavstsen T, Chiucchiuini A, Salanti A, Vestergaard LS, Lusingu JP, Hermsen R et al (2004) Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J Exp Med 199: 1179–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joergensen L, Bengtsson DC, Bengtsson A, Ronander E, Berger SS, Turner L, Dalgaard MB, Cham GKK, Victor ME, Lavstsen T et al (2010) Surface co‐expression of two different PfEMP1 antigens on single Plasmodium falciparum‐infected erythrocytes facilitates binding to ICAM1 and PECAM1. PLoS Pathog 6: e1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt SL, Georgiadou A, Apel F, Abu‐Abed U, Moxon CA, Cunnington AJ, Raupach B, Cunningham D, Langhorne J, Krüger R et al (2019) Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol 4: aaw0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolevzon N, Nasereddin A, Naik S, Yavin E, Dzikowski R (2014) Use of peptide nucleic acids to manipulate gene expression in the malaria parasite Plasmodium falciparum . PLoS One 9: e86802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaratilake LM, Ferrante A, Rzepczyk CM (1990) Tumor necrosis factor enhances neutrophil‐mediated killing of Plasmodium falciparum . Infect Immun 58: 788–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaratilake LM, Ferrante A, Rzepczyk C (1991) The role of T lymphocytes in immunity to Plasmodium falciparum. Enhancement of neutrophil‐mediated parasite killing by lymphotoxin and IFN‐gamma: comparisons with tumor necrosis factor effects. J Immunol 146: 762–767 [PubMed] [Google Scholar]
- Kumaratilake LM, Ferrante A, Jaeger T, Rzepczyk CM (1992) Effects of cytokines, complement, and antibody on the neutrophil respiratory burst and phagocytic response to Plasmodium falciparum merozoites. Infect Immun 60: 3731–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyes S, Pinches R, Newbold C (2000) A simple RNA analysis method shows var and rif multigene family expression patterns in Plasmodium falciparum . MolBiochemParasitol 105: 311–315 [DOI] [PubMed] [Google Scholar]
- Kyes SA, Rowe JA, Kriek N, Newbold CI (1999) Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum . Proc Natl Acad Sci USA 96: 9333–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavazec C, Sanyal S, Templeton TJ (2006) Hypervariability within the Rifin, Stevor and Pfmc‐2TM superfamilies in Plasmodium falciparum . Nucleic Acids Res 34: 6696–6707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavazec C, Sanyal S, Templeton TJ (2007) Expression switching in the stevor and Pfmc‐2TM superfamilies in Plasmodium falciparum . MolMicrobiol 64: 1621–1634 [DOI] [PubMed] [Google Scholar]
- Lennartz F, Adams Y, Bengtsson A, Olsen RW, Turner L, Ndam NT, Ecklu‐Mensah G, Moussiliou A, Ofori MF, Gamain B et al (2017) Structure‐guided identification of a family of dual receptor‐binding PfEMP1 that is associated with cerebral malaria. Cell Host Microbe 21: 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD (2018) Neutrophils: new insights and open questions. Sci Immunol 3: aat4579 [DOI] [PubMed] [Google Scholar]
- Miller LH, Baruch DI, Marsh K, Doumbo OK (2002) The pathogenic basis of malaria. Nature 415: 673–679 [DOI] [PubMed] [Google Scholar]
- Niang M, Bei A, Madnani K, Pelly S, Dankwa S, Kanjee U, Gunalan K, Amaladoss A, Yeo K, Bob N et al (2014) STEVOR is a Plasmodium falciparum erythrocyte binding protein that mediates merozoite invasion and rosetting. Cell Host Microbe 16: 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niang M, Yan Yam X, Preiser PR (2009) The Plasmodium falciparum STEVOR multigene family mediates antigenic variation of the infected erythrocyte. PLoS Pathog 5: e1000307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Van Den Ham K, Shio MT, Kassa FA, Fougeray S (2014) Malarial pigment hemozoin and the innate inflammatory response. Front Immunol 5: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak ND, Dzikowski R (2008) PfEMP1: an antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum . Int J Biochem Cell Biol 41: 1463–1466 [DOI] [PubMed] [Google Scholar]
- Sagiv J, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov R et al (2015) Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep 10: 562–573 [DOI] [PubMed] [Google Scholar]
- Sagiv JY, Voels S, Granot Z (2016) Isolation and characterization of Low‐ vs. high‐density neutrophils in cancer. Methods Mol Biol 1458: 179–193 [DOI] [PubMed] [Google Scholar]
- Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, Shida K, Palacpac NMQ, Itagaki S, Iwanaga S et al (2017) Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature 552: 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanti A, Staalsoe T, Lavstsen T, Jensen AT, Sowa MP, Arnot DE, Hviid L, Theander TG (2003) Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A‐adhering Plasmodium falciparum involved in pregnancy‐associated malaria. Mol Microbiol 49: 179–191 [DOI] [PubMed] [Google Scholar]
- Salazar E, Bank EM, Ramsey N, Hess KC, Deitsch KW, Levin LR, Buck J (2012) Characterization of Plasmodium falciparum adenylyl cyclase‐beta and its role in erythrocytic stage parasites. PLoS One 7: e39769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon D, Vilde JL, Andrieu B, Simonovic R, Lebras J (1986) Role of immune serum and complement in stimulation of the metabolic burst of human neutrophils by Plasmodium falciparum . Infect Immun 51: 801–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield L (2007) Intravascular infiltrates and organ‐specific inflammation in malaria pathogenesis. Immunol Cell Biol 85: 130–137 [DOI] [PubMed] [Google Scholar]
- Sinden RE, Smalley ME (1976) Gametocytes of Plasmodium falciparum: phagocytosis by leucocytes in vivo and in vitro. Trans R Soc Trop Med Hyg 70: 344–345 [DOI] [PubMed] [Google Scholar]
- Sionov RV, Fainsod‐Levi T, Zelter T, Polyansky L, Pham CT, Granot Z (2019) Neutrophil cathepsin G and tumor cell rage facilitate neutrophil anti‐tumor cytotoxicity. Oncoimmunology 8: e1624129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Gamain B, Baruch DI, Kyes S (2001) Decoding the language of var genes and Plasmodium falciparum sequestration. Trends Parasitol 17: 538–545 [DOI] [PubMed] [Google Scholar]
- Smith JD, Rowe JA, Higgins MK, Lavstsen T (2013) Malaria's deadly grip: cytoadhesion of Plasmodium falciparum‐infected erythrocytes. Cell Microbiol 15: 1976–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H et al (2009) The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA 106: 17858–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stijlemans B, Cnops J, Naniima P, Vaast A, Bockstal V, De Baetselier P, Magez S (2015) Development of a pHrodo‐based assay for the assessment of in vitro and in vivo erythrophagocytosis during experimental trypanosomosis. PLoS Negl Trop Dis 9: e0003561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturn DH, Kaneider NC, Feistritzer C, Djanani A, Fukudome K, Wiedermann CJ (2003) Expression and function of the endothelial protein C receptor in human neutrophils. Blood 102: 1499–1505 [DOI] [PubMed] [Google Scholar]
- Trubowitz S, Masek B (1968) Plasmodium falciparum: phagocytosis by polymorphonuclear leukocytes. Science 162: 273–274 [DOI] [PubMed] [Google Scholar]
- Tuikue Ndam N, Moussiliou A, Lavstsen T, Kamaliddin C, Jensen ATR, Mama A, Tahar R, Wang CW, Jespersen JS, Alao JM et al (2017) Parasites causing cerebral falciparum malaria bind multiple endothelial receptors and express EPCR and ICAM‐1‐binding PfEMP1. J Infect Dis 215: 1918–1925 [DOI] [PubMed] [Google Scholar]
- Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JEV, Avril M, Brazier AJ, Freeth J, Jespersen JS, Nielsen MA et al (2013) Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 498: 502–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Sexton DM, Redmond HP, Watson RW, Croke DT, Bouchier‐Hayes D (1997) Intercellular adhesion molecule‐1 (ICAM‐1) is expressed on human neutrophils and is essential for neutrophil adherence and aggregation. Shock 8: 357–361 [DOI] [PubMed] [Google Scholar]
- WHO (2016) World Malaria Report, 2016. [PREPRINT]
- Wickramasinghe SN, Phillips RE, Looareesuwan S, Warrell DA, Hughes M (1987) The bone marrow in human cerebral malaria: parasite sequestration within sinusoids. Br J Haematol 66: 295–306 [DOI] [PubMed] [Google Scholar]
- Wolf AS, Sherratt S, Riley EM (2017) NK cells: uncertain allies against malaria. Front Immunol 8: 212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sifri CD, Lei HH, Su XZ, Wellems TE (1995) Transfection of Plasmodium falciparum within human red blood cells. Proc Natl Acad Sci USA 92: 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]