

Abstract
Our understanding of human hepatocellular carcinoma (HCC) development and progression has been hampered by the lack of in vivo models. We performed a genetic screen of 10 oncogenes and genetic mutations in Fah‐ablated immunodeficient mice in which primary human hepatocytes (PHHs) are used to reconstitute a functional human liver. We identified that MYC, TP53R249S, and KRASG12D are highly expressed in induced HCC (iHCC) samples. The overexpression of MYC and TP53R249S transform PHHs into iHCC in situ, though the addition of KRASG12D significantly increases the tumorigenic efficiency. iHCC, which recapitulate the histological architecture and gene expression characteristics of clinical HCC samples, reconstituted HCC after serial transplantations. Transcriptomic analysis of iHCC and PHHs showed that MUC1 and FAP are expressed in iHCC but not in normal livers. Chimeric antigen receptor (CAR) T cells against these two surface markers efficiently lyse iHCC cells. The properties of iHCC model provide a biological basis for several clinical hallmarks of HCC, and iHCC may serve as a model to study HCC initiation and to identify diagnostic biomarkers and targets for cellular immunotherapy.
Keywords: biomarkers, hepatocellular carcinoma, humanized mouse models, immunotherapy, oncogenes
Subject Categories: Cancer, Methods & Resources, Signal Transduction
The understanding of hepatocellular carcinoma development and progression is hampered by the lack of in vivo models. This study presents an in situ mouse model of human induced HCC that allows studying HCC initiation and the identification of biomarkers.

Introduction
Primary liver cancer (PLC) has the second‐highest cancer mortality rate worldwide (Broutier et al, 2017). The development of PLC is invariably associated with liver damage caused by hepatitis B virus (HBV) infection, alcohol abuse, or toxin exposure, sequentially resulting in liver cirrhosis, dysplastic lesions, and finally invasive liver carcinoma (Cancer Genome Atlas Research Network. Electronic address wbe & Cancer Genome Atlas Research N, 2017; Gao et al, 2019). Based on its histopathological features, PLC may be mainly classified as HCC, cholangiocarcinoma (CC), hepatoblastoma (HB), and combined hepatocellular‐cholangiocarcinoma (CHC) (Broutier et al, 2017). The cellular origin and molecular genetics of human PLC remain poorly understood due to the lack of suitable models.
Genomic analysis has identified frequently mutated genes, including TP53, KRAS, CTNNB1, IDH1, IDH2, and IL6ST in PLC samples (Totoki et al, 2014; Schulze et al, 2015; Cancer Genome Atlas Research Network. Electronic address wbe & Cancer Genome Atlas Research N, 2017), indicating essential roles of these genes in PLC development. There are six hotspot mutations of TP53 at R175, G245, R248, R249, R273, and R282, identified in various primary and metastatic human cancers. Notably, these TP53 mutants exert their dominant‐negative effects of wild‐type TP53 and possess gains of new functions (Kern et al, 1992; Brosh & Rotter, 2009; Zhao et al, 2019, 2020). TP53R249S is highly associated with HCC and the only hotspot mutant that has been identified among 30% of HCCs that harbor TP53 mutations (Hsu et al, 1991; Hussain et al, 2007; Sladky et al, 2020). Hotspot mutations of KRAS such as KRASG12D and KRASG13D are commonly detected in PLC samples (Hunter et al, 2015). Activation of KrasG12D and deletion of p53 induce the development of HCC and CC in a genetically engineered mouse model (O'Dell et al, 2012). Genomic amplifications in MYC have been identified in 6–25% of primary human cancers (Poon et al, 2006; Schlaeger et al, 2008). Moreover, constitutive overexpression of c‐Myc in the liver of p53‐deficient mice promotes hepatocarcinogenesis (Klocke et al, 2001). These studies reveal that PLC development results from crosstalk of multiple genetic alterations. However, whether these genetic lesions can induce tumorigenesis of PHHs in situ on mouse models has not been investigated.
Humanized mouse models offer considerable advantages in the study of immunology, infectious diseases, regenerative medicine, and cancer (Shultz et al, 2007). Humanized livers have been reconstituted with PHHs in immunodeficient mice with transgenic urokinase plasminogen activator transgenic expression (Tateno et al, 2004) or fumarylacetoacetate hydrolase (Fah) deficiency (Azuma et al, 2007) for toxicity evaluation (Xu et al, 2014). Patient‐derived xenografts (PDX) have been widely used to evaluate the efficacy of antitumor therapies (Kamel‐Reid et al, 1989; Jiang et al, 2016; Lai et al, 2017; Bleijs et al, 2019). However, these approaches have not allowed the cell types from which tumor‐initiating cells originate to be identified or the mechanism by which these tumor‐initiating cells phenotypically/genetically evolve during human disease progression to be assessed. Only a few in vivo models of cancers initiated from primary human cells have been developed. Upon transplantation into immunodeficient mice, primary human hematopoietic cells expressing mixed‐lineage leukemia (MLL) fusion gene generated acute leukemia (Barabe et al, 2007). In addition, both ectopic expression of oncogenes and ablation of tumor suppressor genes successfully induce reprogramming of human‐induced hepatocytes (hiHeps) and primary human intestinal stem cells into malignant cells in culture, although in vitro expansion and selection of organoids before transplantation were required (Hahn et al, 1999; Drost et al, 2015; Sun et al, 2019). However, the direct conversion of human PHHs into HCC in situ has not been established. Here, we screened ten oncogenes and found that overexpression of MYC, TP53R249S, and KRASG12D led to an efficient transformation of PHHs directly into HCC in a humanized liver mouse model. Further analyses confirmed that iHCC recapitulated the immunophenotypic histological architecture, tumor serum marker profile, phenotypes, and gene expression characteristics of HCC.
Results
Engraftment of PHHs in the liver of NSIF mice
To establish a humanized liver mouse model, we transplanted PHHs into NOD/SCID/IL2rg‐/‐/Fah‐/‐ (NSIF) mice, which were generated by targeting the exon 3 of Fah with embryo microinjection of TALEN mRNA (Fig 1A and B) into immunodeficient NOD/SCID/IL2rg‐/‐ (NSI) mice (Ye et al, 2015). Western blotting confirmed that fah was not expressed in NSIF mice (Fig 1C). NSIF mice exhibited liver injury upon withdrawal of nitisinone (NTBC) (Fig 1D), which promotes engraftment of PHHs (Azuma et al, 2007). NSIF mice that did not receive PHH transplantation experienced gradual body weight loss and died approximately 4 weeks after withdrawal of NTBC, whereas the PHH‐transplanted mice survived significantly longer (Fig 1E). The serum ALT and AST levels were significantly reduced in PHH‐transplanted NSIF mice (Fig 1E). The survival duration of PHH‐transplanted NSIF mice were positively correlated with the repopulation efficiencies of PHHs in the liver and the serum level of human ALB (Fig 1F and G, and Appendix Table S1). Next, immunohistochemistry (IHC) and immunofluorescence (IF) staining confirmed that the engrafted PHHs were morphologically similar to human hepatocytes and expressed human FAH, CK8/18, and ALB (Fig 1G and H). To trace PHHs in vivo, we overexpressed enhanced green fluorescent protein (EGFP) and luciferase (Luc) in PHHs and transplanted the cells into NSIF mice via an intrasplenic injection (Fig 2A and B). Bioluminescence imaging (BLI) using an IVIS Spectrum system showed that PHHs were specifically engrafted into the liver but not other organs in NSIF mice (Fig 2C and D). These data indicate NSIF mice could be used to generate humanized liver models.
Figure 1. Establishment of Fah knockout NSI mice for the humanized liver mouse model.
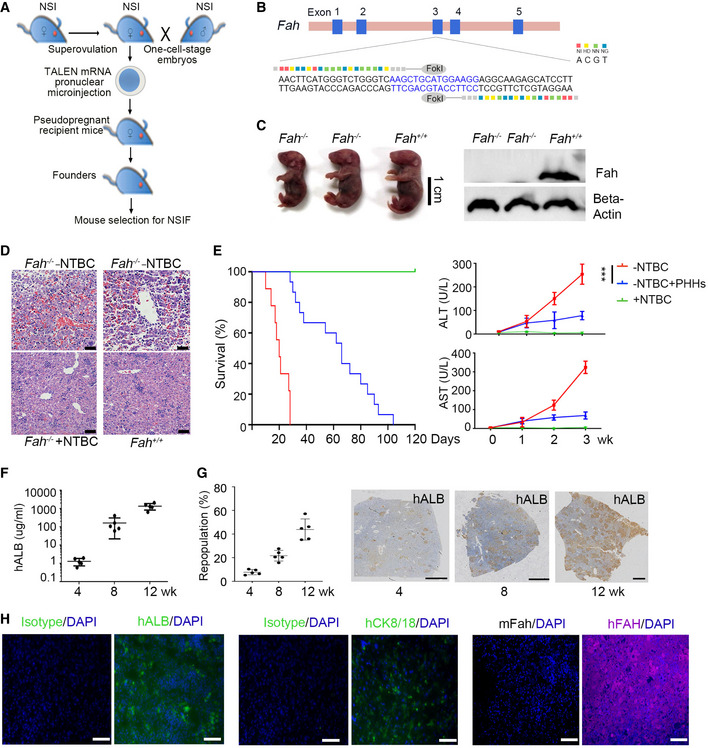
- Schematic outline of the pronuclear microinjection procedure. In brief, female NSI mice were superovulated with PMSG and hCG and then mated with male NSI mice. Fertilized eggs were collected from NSI mice with a vaginal plug. TALEN mRNA pairs were injected into the cytoplasm of pronuclear‐stage mouse embryos. The injected embryos were then transferred into pseudopregnant surrogate mothers. The mouse pups were genotyped. The mouse was drawn by the authors (Z. J. and P. Li) using Adobe Photoshop.
- DNA‐binding sequences and the spacer region (in blue) of TALENs targeting the exon 3 of Fah.
- NSI mice homozygous for the Fah knockout displayed a lethal neonatal phenotype (left). Fah expression was detected in the livers of homozygous NSI mice with Fah knockout and Fah intact NSI mice by Western blotting (right). Cropped blots are shown.
- Histological staining of livers from Fah‐deficient (NSIF) with and without NTBC treatment and Fah intact (NSI) mice without NTBC treatment. Scale bars, 50 μm.
- (Left) Kaplan–Meier survival curves of PHH‐infused NSIF mice (in blue, n = 15) and NSIF mice without PHHs (in red, n = 9) after NTBC withdrawal and NTBC‐treated NSIF mice (in green, n = 10); (Right) Serum ALT and AST levels in NSIF mice (+NTBC, n = 5), moribund NSIF mice (−NTBC, n = 5) and PHH‐engrafted (8‐week‐old) mice (−NTBC, n = 5) at indicated time points post‐NTBC withdrawal; data are presented as the mean ± SD, and statistical significance was determined by two‐way ANOVA with Sidak's multiple comparisons test and defined as ***P ≤ 0.001.
- Human ALB levels in the sera of PHH‐engrafted NSIF mice (n = 5) were measured by ELISA at indicated time points post‐NTBC withdrawal, data are presented as the mean ± SD. N‐numbers refer to biological replicates.
- (Left) Quantification of the repopulation efficiencies of PHHs in livers estimated by hALB‐positive staining at indicated time points post‐transplantation. Data are presented as the mean ± SD. N‐numbers refer to biological replicates (n = 5); (Right) Representative immunohistochemical staining of hALB in the livers of NSIF mice at indicated time points post‐PHH transplantation. Scale bars, 1,000 μm.
- Engraftment of PHHs into NSIF mouse livers was detected immunostaining for human ALB, CK8/18, and FAH. Scale bars, 50 μm.
Source data are available online for this figure.
Figure 2. Bioluminescence imaging of humanized liver mice.
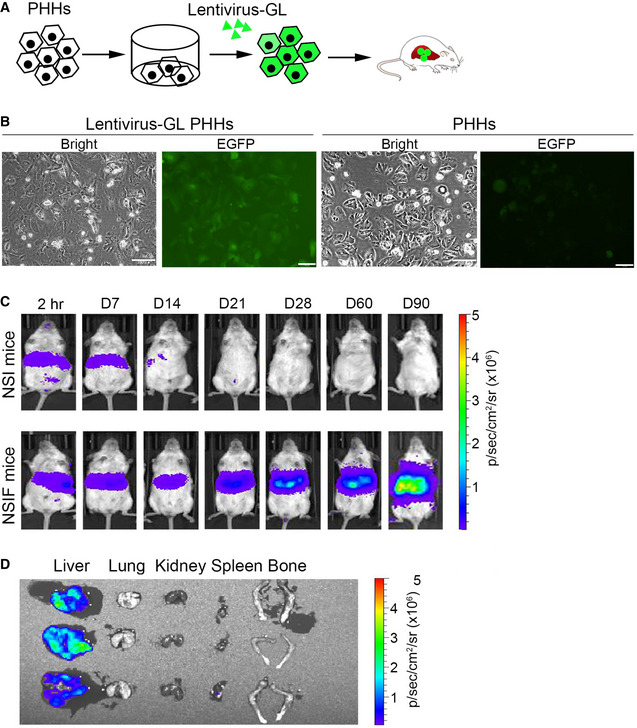
- Schematic illustration of PHHs labeled via EGFP and luciferase (Lentivirus‐GL) and intrasplenically injected into NSIF mice.
- PHHs were transduced with/without lentiviruses containing EGFP and luciferase. Scale bar, 100 μm.
- Bioluminescence signals were monitored in NSI and NSIF mice (n = 3) infused with EGFP‐Luc transduced PHHs at the indicated time points post‐PHH infusion. Representative images of a NSI and a NSIF mouse transduced with PHHs are shown.
- Bioluminescence signals were detected in the livers but not in other tissues of NSIF mice that were transplanted with EGFP‐Luc‐labeled PHHs (n = 3).
Screening oncogenes contribute to the transformation of PHHs into HCC
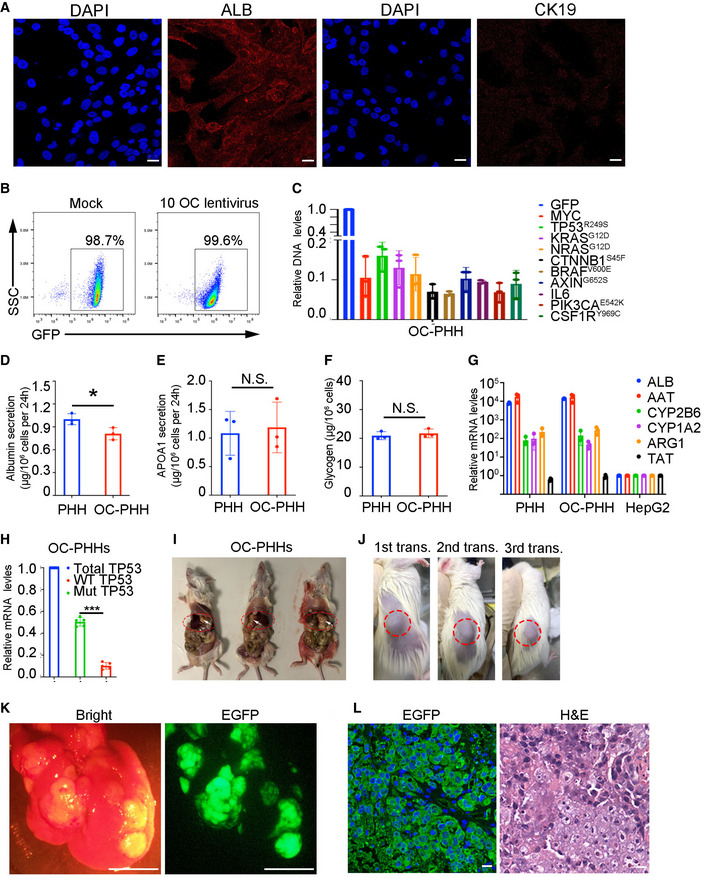
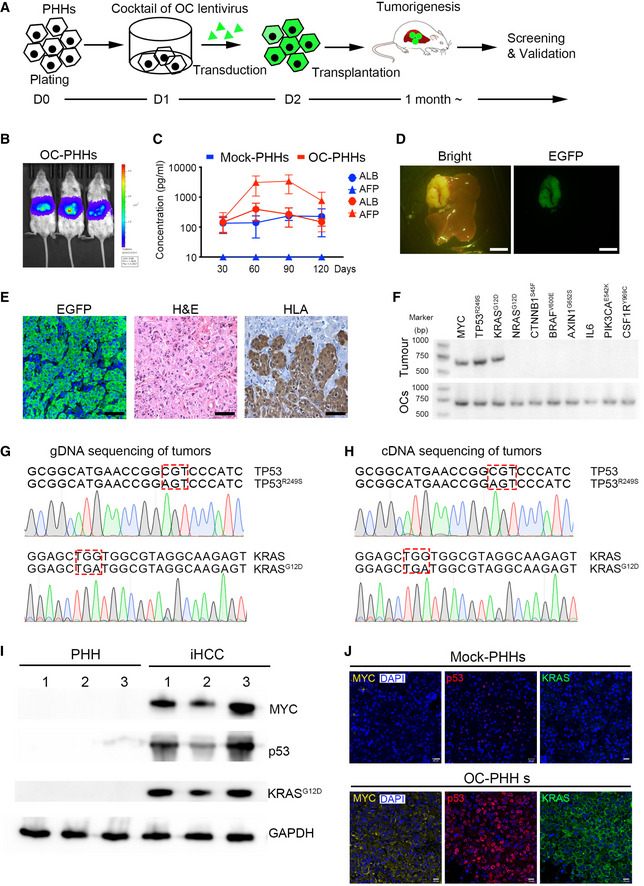
To identify oncogenes that were able to induce reconstituted PHHs to transform into tumor cells in situ, we selected 10 oncogenes or oncogenic mutants as oncogenic candidates (OC), including MYC, TP53R249S, KRASG12D, NRASG12D, CTNNB1S45F, BRAFV600E, AXIN1G652S, IL6, PIK3CAE542K , and CSF1RY969C , based on their contributions to HCC development identified in previous studies (Schulze et al, 2015; Bailey et al, 2018; Llovet et al, 2018). The expression levels of KRAS, NRAS, and AXIN1 were positively correlated with poor prognosis in HCC patients according to The Cancer Genome Atlas (TCGA) database analysis (Appendix Fig S1A). We cloned these oncogenes and oncogenic mutants individually into a lentiviral vector containing the EGFP sequence (Appendix Fig S1B). PHHs from healthy donors highly expressed Albumin (ALB) but barely expressed CK19, a bile duct/progenitor marker (Zhang et al, 2018) (Fig EV1A). PHHs were transduced with a cocktail of lentiviruses containing 10 OC and lentiviruses encoding EGFP and luciferase as a reporter with similar transduction efficiencies higher than 97% (Fig EV1B and Appendix Table S2) and were then transplanted into NSIF mice via splenic injection on day 2 (Fig 3A). The qPCR results showed that the copy numbers of these 10 oncogenic lentiviruses in PHHs were similar (Fig EV1C). The functions of PHHs were not severely affected by the transduction of oncogenes, as both OC‐transduced PHHs and unmodified PHHs produced Albumin and APOA, stored glycogen, and expressed hepatocyte‐associated genes, including ALB, AAT, CYP2B6, CYP1A2, ARG1, and TAT at similar levels (Fig EV1D–G). Notably, ectopic expression of mutant TP53 was significantly higher than endogenous TP53 (WT) (Fig EV1H). After 4 months of transplantation, genetically modified PHHs significantly engrafted in the liver of NSIF mice (Fig 3B). The serum levels of AFP, an HCC marker (Sun et al, 2019), increased, while ALB concentrations did not change 3 months post‐OC transduced‐PHHs infusion (Fig 3C). In contrast, AFP was not detectable in NSIF mice that were transplanted with EGFP only‐transduced PHHs (mock group) (Fig 3C). Three to 6 months post‐transplantation, 8 out of the 30 NSIF mice transplanted with OC‐transduced PHHs showed sporadic sickness and were necropsied. Tumors were detected in the recipients of OC‐transduced PHHs but not in the mock group (Figs 3D and EV1I, and Appendix Table S2). These induced HCC (iHCC) were derived from human cells, as they expressed EGFP and HLA (Fig 3E). Single‐cell suspensions from these tumors reconstituted tumors in immunodeficient NSI mice after both serial subcutaneous transplantation (Fig EV1J) and orthotopic injection (Fig EV1K and L). To identify the oncogenic candidates that contributed to the tumorigenicity of PHHs in vivo, we performed PCR to amplify lentiviral sequences that integrated into the genomic DNA of the primary iHCC from the OC group mice. We found that all eight tumor samples contained copies of MYC and TP53R249 , and seven out of eight (87.5%) tumor samples contained copies of KRASG12D but not any other candidates (Fig 3F and Appendix Table S2). The DNA sequencing results confirmed that mutant forms of TP53R249S and KRASG12D were integrated into the iHCC genomes (Fig 3G) and were expressed (Fig 3H). Consistently, MYC, p53, and KRASG12D were highly expressed in iHCC samples but not in humanized livers reconstituted with mock‐PHHs from NSIF mice (Fig 3I and J). In addition, none of the 9 NSIF mice that were transplanted with mock‐PHHs from three different donors developed tumors (Fig 3J and Appendix Table S2). Those data indicate that humanized liver models are suitable for inducing tumorigenesis.
Figure EV1. iHCC derived from PHHs by oncogenic candidate transduction.
-
ACommercial cryopreserved PHHs (PHH1, PHH4, and PHH5) were seeded in 6‐well plates and immunoassayed with antibodies that recognize ALB and KRT19. Scale bars, 20 μm.
-
BRepresentative FACS analysis of PHHs that were transduced with a cocktail of 10 OC lentiviruses (right) or mock lentivirus (left).
-
CqPCR assays measuring the transduction efficiencies of individual oncogenes using the genomic DNA samples of OC‐transduced PHHs. Relative copy numbers of DNA of oncogenes to the DNA of GFP. Data are presented as the mean ± SD (n = 3). N‐numbers refer to biological replicates.
-
DThe levels of albumin secretion in PHHs and OC‐transduced PHHs were measured by ELISA (n = 3), P = 0.0378 with unpaired t‐test, *P ≤ 0.05. Data are presented as the mean ± SD. N‐numbers refer to biological replicates.
-
EThe levels of APOA1 secretion in PHHs and OC‐transduced PHHs were measured by ELISA (n = 3), P > 0.05 with unpaired t‐test, data are presented as the mean ± SD, N.S. indicates non‐statistical significance. N‐numbers refer to biological replicates.
-
FGlycogen storage was quantified by colorimetric in PHHs and OC‐transduced PHHs (n = 3 ), P > 0.05 with unpaired t‐test, data are presented as the mean ± SD. N.S. indicates non‐statistical significance. N‐numbers refer to biological replicates.
-
GqPCR results of expression levels of hepatic genes in PHHs, OC‐transduced PHHs, and HepG2 cells on day 2 post‐transduction in culture (n = 3), data are presented as the mean ± SD. N‐numbers refer to biological replicates.
-
HThe expression levels of endogenous (WT) and mutant (Mut) TP53 in OC‐transduced PHHs (n = 5, P < 0.0001 with one‐way ANOVA by Tukey's multiple comparison test), ***P ≤ 0.001, data are presented as the mean ± SD. N‐numbers refer to biological replicates.
-
IMice with advanced intrahepatic tumor exhibited swollen and ascites‐containing abdomens, allowing detection of tumor burden by palpation. Tumors were pointed with white arrows.
-
J–LPrimary iHCC samples were harvested from mice from the OC group and were transplanted. 1 × 106 iHCC cells were subjected for serial transplantations into NSI mice subcutaneously or splenically. Tumors were reconstituted post‐three rounds of subcutaneous transplantations of iHCC (J); Bright‐field and EGFP imaging of an explanted liver from a tumor‐bearing NSI mouse post splenic injection of iHCC cells. Scale bars, 0.5 cm (K); The anti‐GFP immunofluorescence and H&E staining of tumor dissection from a tumor‐bearing NSI mouse post splenic transplantation of iHCC. Scale bars, 20 μm (L).
Figure 3. Screening of oncogenes involved in the transformation of PHHs into HCC cells.
-
ASchematic representation showing the process for induction of in situ HCC after lentiviral transduction of PHHs with a cocktail of 10 OC lentiviruses.
-
BBioluminescence imaging of three recipient NSIF mice (n = 3) that had been transplanted with OC‐transduced PHHs 4 months ago. The colors from blue to red indicate intrahepatic bioluminescence signal strength 4 months post‐transplantation of OC transduced PHHs.
-
CALB and AFP levels in sera from NSIF mice that were infused with OC‐transduced PHHs (OC‐PHHs) or EGFP‐transduced PHHs (mock‐PHHs) were measured by ELISA at indicated time points. (n = 5, the data are presented as the mean ± SD values. Two out of five mice died before 120 days post‐transplantation in the OC‐PHHs group. The levels of ALB and AFP in the rest three mice were measured.).
-
DBright and EGFP imaging of an explanted liver of a mouse from the OC group revealed an advanced intrahepatic tumor, Scale bars, 0.5 cm.
-
EAnti‐GFP immunofluorescence (left), H&E staining (center), anti‐HLA‐I IHC staining (right) images of tumor dissection from the OC group. Scale bars, 50 μm.
-
FRepresentative PCR amplification of MYC, TP53R249S, KRASG12D, NRASG12D, CTNNB1S45F, BRAFV600E, AXIN1G652S, IL6, PIK3CAE542K and CSF1RY969C in the genomic DNA of dissected primary tumors harvested from NSIF mice transplanted with OC‐PHHs (Tumor) and their corresponding overexpression vector plasmids as positive controls (OCs).
-
G, HGenomic DNA (gDNA) and cDNA of tumor samples were obtained for PCR amplification using forward primers that recognize TP53R249S or KRASG12D and reverse primers specific for EGFP. DNA sequencing confirmed mutations of TP53 and KRAS in the genomic DNA (G) and mRNA (H) of dissected primary tumors harvested from tumor‐bearing mice. Red squares indicate mutant codes in TP53R249S and KRASG12D .
-
IExpression of MYC, p53, and KRASG12D was detected in the iHCC samples (n = 3, biological replicates) by Western blotting. Cropped blots are shown.
-
JImmunofluorescence analysis of MYC, p53, and KRAS expression in iHCC samples derived from OC‐PHHs and normal humanized livers that were reconstituted with mock‐PHHs. Scale bars, 20 μm.
Source data are available online for this figure.
MYC, TP53, and KRAS corporately transform PHHs into HCC in vivo
To validate whether the combination of MYC, TP53R249S, and KRASG12D can transform PHHs into iHCC, we transduced PHHs with a mixture of lentiviruses overexpressing MYC, TP53R249S, and KRASG12D (MTK) and intrasplenically infused them into NSIF mice. Six months later, 63.6% (14 out of 22 mice) of the recipient mice developed iHCC. Tumorigenic rate of MTK‐transduced PHHs is significantly higher than that of 16.6% (2 out of 12 mice) MT‐transduced PHHs (Fig 4A and Table 1). While tumors were not detected in the 9 NSIF mice that were transplanted with PHHs transduced only with EGFP (Table 1 and Appendix Table S2). In addition, colonies derived from single cells of different tumors, such as iHCC1‐1 and iHCC2‐1 contained all MYC, TP53R249S , and KRASG12D (Fig 4B), suggesting that individual iHCC cells expressed all the three OCs. The H&E‐stained sections of orthotropic iHCC samples presented heterogeneous morphologies ranging from solid/compact to pseudoglandular rosette structures of HCC, in contrast to the homogeneous structure of normal human hepatocytes (Fig EV2). Tumors derived from MTK‐transduced PHHs expressed HLA, ALB, and HCC markers, including AFP and GPC3 (Figs 4C and EV3A). Notably, iHCC samples (iHCC1‐1 and iHCC3‐1) expressed cytokeratin‐19 (CK19) and EpCAM, which are expressed in hepatic progenitors (Rao et al, 2008) and are considered as poor prognostic markers (Chan et al, 2014; Govaere et al, 2014; Llovet et al, 2015) in HCC patients (Figs 4C and EV3A). To investigate whether wild‐type TP53 affects tumorigenesis in PHHs that overexpress MYC, TP53R249S, and KRASG12D, we inactivated TP53 by CRISPR/Cas9 in MTK‐transduced PHHs and orthotopically transplanted these genetically modified PHHs into NSIF mice for tumor watch (Fig EV3B and C). Tumors were detected in three out of four recipient mice (Fig EV3D). These results demonstrate that overexpression of TP53R249S, MYC, and KRASG12D induced PHHs with WT p53 inactivation to transform into iHCC in vivo.
Figure 4. Generation of genetically defined in situ humanized liver cancers.
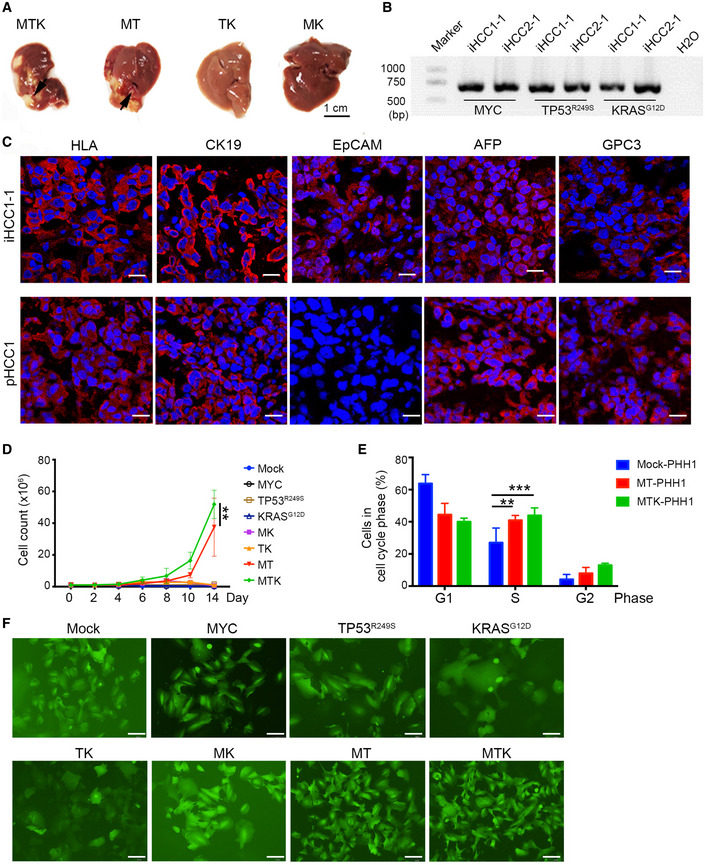
- Representative images of in situ liver carcinomas derived from PHHs transduced with a combination of MYC, TP53R249S, and KRASG12D (MTK, n = 22) or a combination of MYC and TP53R249S (MT, n = 12) in NSIF mice. No tumors were detected in any of the mice injected with PHHs that were transduced with TP53R249 and KRASG12D (TK, n = 9) or PHHs that were transduced with MYC and KRASG12D (MK, n = 9). Scale bar, 1 cm.
- Genomic DNA of colonies that were derived from single cells of iHCC1‐1 and iHCC2‐1 tumor samples were extracted for PCR amplification of MYC, TP53R249S, and KRASG12D .
- Immunofluorescence analysis of the human cell marker HLA, the ductal/CC markers KRT19 and EpCAM, and the HCC markers AFP and GPC3 in a iHCC sample from NSIF mice transplanted with MTK‐transduced PHHs from the donor PHH1 (iHCC1‐1) and a clinical patient HCC sample (pHCC1). Nuclei were counterstained with DAPI. Scale bars, 20 μm.
- Growth curves of PHHs after transduction with various combinations of MYC (M), TP53R249S (T) and KRASG12D (K) in culturing medium supplemented with hHGF (10 ng/ml), hEGF (5 ng/ml), and WNT3a (5 ng/ml), P = 0.0038 for MTK‐ versus MT‐transduced PHH by two‐way ANOVA with Tukey's multiple comparison test; **P = 0.0038 for MTK‐ versus MT‐transduced PHH by two‐way ANOVA with Tukey's multiple comparison test; data are presented as the mean ± SD. N‐numbers refer to biological replicates (n = 3).
- Cell cycle distribution analysis of mock‐PHHs, MTK‐transduced PHHs, and MT‐transduced PHHs on day 10. **P = 0.0034 for MT‐transduced PHHs versus mock‐PHHs and ***P = 0.0009 for MTK‐transduced PHHs versus mock‐PHHs by two‐way ANOVA with Tukey's multiple comparison test; data are presented as the mean ± SD. N‐numbers refer to biological replicates (n = 3).
- Morphology of PHHs transduced with various combinations of MYC (M), TP53R249S (T), and KRASG12D (K) at day 10. EGFP positivity indicates transduction of oncogenic candidates: scale bars, 100 μm.
TABLE 1.
Validation of the combination of MYC, TP53R249S, and KRASG12D for transforming PHHs into HCC.
|
Hepatocytes a OC b |
PHH1 (AKB) | PHH2 (XSM) | PHH3 (ANG) | In total d |
|---|---|---|---|---|
| MTK | 6/10 c | 4/6 | 4/6 | 14/22 |
| MT | 1/6 | 1/3 | 0/3 | 2/12 |
| TK | 0/3 | 0/3 | 0/3 | 0/9 |
| MK | 0/3 | 0/3 | 0/3 | 0/9 |
| Mock | 0/3 | 0/3 | 0/3 | 0/9 |
Hepatocytes: PHHs from three donors, including PHH1 (AKB, female, 39 years old), PHH2 (XSM, female, 59 years old), and PHH3 (ANG, male, 3 months old), were purchased from Bioreclamation IVT (Baltimore, MD, USA) and were used in the experiment.
OC: A cocktail of lentivirus containing different combinations of oncogenic candidates (M for MYC, T for TP53R249S, and K for KRASG12D) were transduced into PHHs from different donors. Mock: lentivirus containing EGFP only.
6/10: The frequency of mice that developed iHCC in the group where PHHs from different donors (PHH1~PHH3) were transduced with indicated combination of oncogenic candidates. For example, 6 out of 10 mice, in which PHHs from the donor PHH1 were transduced with MTK and were transplanted, were observed bearing tumors.
In total: ***P < 0.001 for MTK‐ versus MT/Mock‐transduced PHH by one‐way ANOVA with Tukey's multiple comparison test; data are presented as the mean ± SD.
Figure EV2. Morphologic characteristics of iHCC.
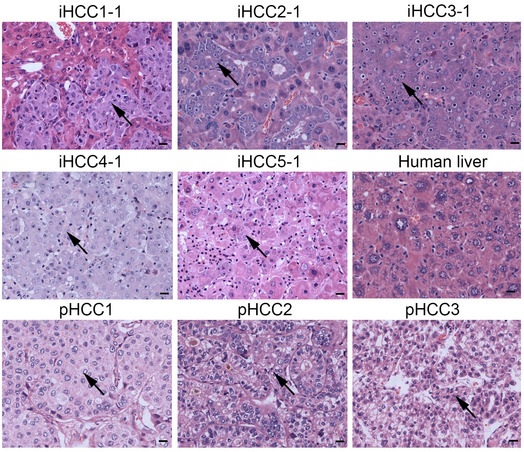
Representative H&E‐stained sections of iHCC tissues (iHCC1‐5) from tumor‐bearing mice transplanted with PHHs from 5 different donors (PHH1~5) transduced with the combination of MYC, TP53R249S , and KRASG12D overexpression lentiviruses, normal human liver tissue (Human liver), and primary HCC tissues from three different patients (pHCC1~3). Tumor cells are pointed with black arrows. Scale bar, 20 μm.
Figure EV3. iHCC samples express HCC markers.
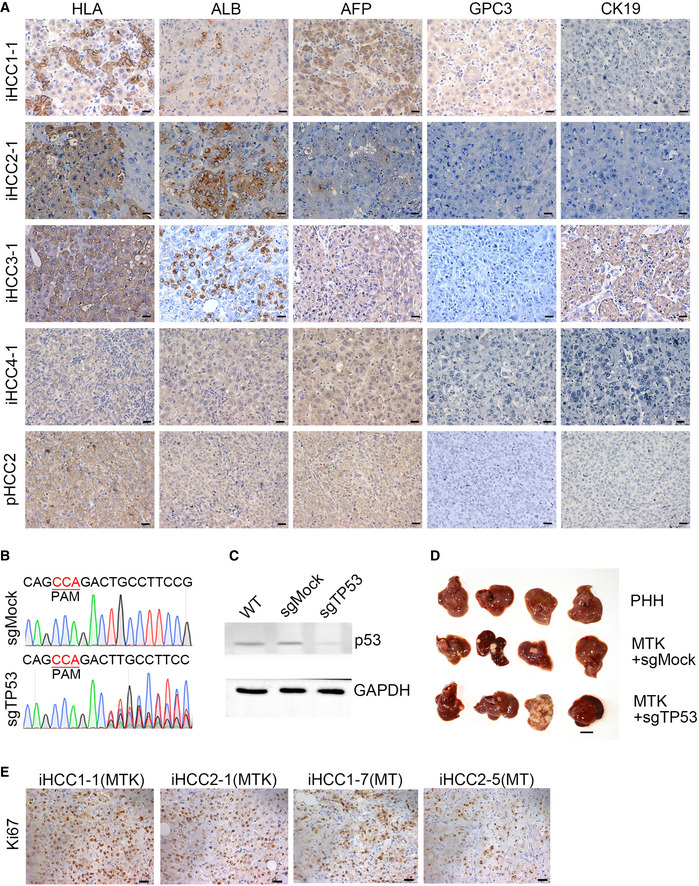
- Representative IHC images of four iHCC samples (iHCC1‐1, iHCC2‐1, iHCC3‐1, and iHCC4‐1) from NSIF mice transplanted with MTK‐transduced PHHs from four different PHH donors (PHH1~4) and an HCC tissue from a patient (pHCC2). Anti‐HLA‐ABC, anti‐ALB, anti‐AFP, anti‐GPC3 and anti‐KRT19 antibodies were used. Scale bars, 20 μm.
- DNA sequencing confirmed mutations of TP53 in the genomic DNA of sgTP53 transduced PHH cells.
- Expression of p53 was detected in the PHH cells transduced with sgMock or sgTP53 by Western blotting.
- Representative images of in situ liver carcinomas derived from PHHs transduced with a combination of MTK with or without deletion of TP53 in NSIF mice. 3 out of 4 mice harbored tumor in both MTK+ sgMock and MTK+ sgTP53 groups after 11 weeks (n = 4 per group). N‐numbers refer to biological replicates.
- Representative IHC images Ki67 staining of liver sections from NSIF mice transplanted with PHHs that were transduced with the combinations of MYC/TP53R249S/KRASG12D (MTK) or MYC/TP53R249S (MT) lentiviruses. Scale bars, 20 μm.
Source data are available online for this figure.
We next sought to determine the minimal oncogene profile required for the HCC transformation by transducing PHHs with two combinations of lentiviruses expressing MYC, TP53R249S, KRASG12D, and transplanting these PHHs into NSIF mice. Tumors were detected in only 16.6% (2 out of 12 mice) of recipient mice transplanted with PHHs overexpressing MYC and TP53R249S (MT group). Still, they were absent in both the TP53R249S and KRASG12D (TK) combinational group and the MYC and KRASG12D (MK) combinational group (Fig 4A, and Table 1, and Appendix Table S3). Moreover, MTK‐transduced tumor samples contained more Ki67+ cells than MT‐transduced iHCC samples (Fig EV3E), suggesting that RAS signaling promotes the proliferation of tumor cells in iHCC. In culture, 1 million PHHs were transduced with different combinations of oncogenic lentiviruses leading on day 2, to 0.8 ± 0.1 million transduced PHHs (Fig 4D). MTK‐ and MT‐transduced PHHs started to thrive on day 6, while MYC‐, TP53R249S‐, KRASG12D‐, MK‐, and TK‐transduced PHHs did not expand in culture (Fig 4D). Consistent with the in vivo results, PHHs transduced with the MTK oncogene combination proliferated faster than MT‐transduced cells (Fig 4D). Cell cycle analysis results showed that the MTK‐ and MT‐transduced PHH populations had significantly more proliferative cells (S‐phase) than the mock‐transduced PHH population (Fig 4E). Furthermore, MT‐ and MTK‐transduced PHHs showed a squamous morphology compared to the flattened, senescent morphology seen in cultures of other transduced PHHs (Fig 4F). Taken together, these results suggest that MYC and TP53R249S are indispensable for inducing PHHs to transform into HCC cells and that RAS signaling further promotes this transformation. Thus, we established a humanized mouse model of iHCC via cotransfection of MYC, TP53R249S, and KRASG12D for the following experiments.
iHCC recapitulates the transcriptional profiles and hallmarks of HCC
We next compared the global gene expression profiles of iHCC to those of corresponding parental PHHs using RNA‐sequencing analysis. Principal component analysis (PCA) showed that iHCC from various donors were clustered together and were separated from PHHs of their corresponding donors (Fig 5A). The analysis of relative transcript abundance of 17,667 genes revealed that hepatic transcription factors, including FOXA2, FOXA3, HNF4A, and ATF5, were highly expressed in PHHs, while tumor‐associated genes (MYC, TP53, and KRAS) were upregulated in iHCC cells (Fig 5B and Dataset EV1). Moreover, gene ontology (GO) analysis confirmed that the differentially expressed genes (DEGs) enriched in iHCC were associated mainly with the cell cycle, chromosome segregation, telomere maintenance, and cellular responses to DNA damage. In contrast, DEGs enriched in PHHs were associated with liver metabolic functions (Fig 5C). The heatmap shows that iHCC cells expressed both HCC/hepatocytes markers (Trerotoli et al, 2009) (AFP, GPC3, MYC, and ALB) and hepatic progenitor markers (EPCAM, KRT18, KRT19) (Fig 5D). Interestingly, genes related to epithelial‐to‐mesenchymal transition (EMT), including TWIST1, VIM, and SNAI1, were upregulated in iHCC cells compared to the corresponding PHHs (Fig 5D), consistent with the mesenchymal morphology of iHCC cells and epithelial morphology of PHHs (Fig 4F). Karyotype analysis revealed that compared to PHHs, iHCC cells had numerical aberrations, including loss and trisomy of chromosomes (Appendix Fig S2). Notably, copy number gains of chromosome 1, commonly identified in HCC samples from patients(Kusano et al, 1999), were also detected in iHCC2‐1 and iHCC3‐1 samples (Appendix Fig S2). These results indicate that iHCC exhibit upregulation of oncogenes, downregulation of hepatic transcription factors, and genomic instability.
Figure 5. Genetically defined iHCCs recapitulate the transcriptional profiles of human HCC.
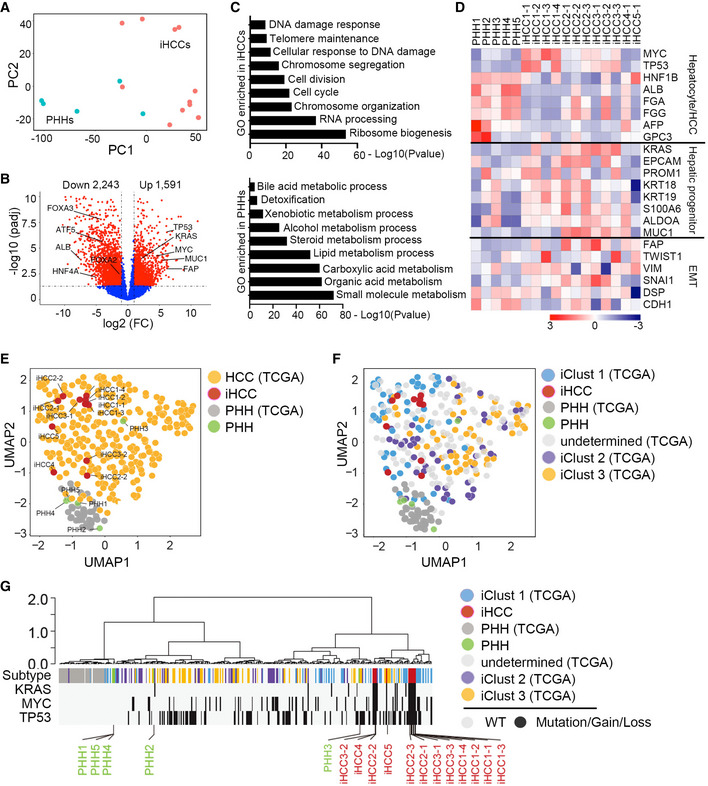
- PHHs from five donors (PHH1 (AKB, female, 39 years old), PHH2 (XSM, female, 59 years old), PHH3 (ANG, male, 3 months old), PHH4 (HVN, male, 33 years old), and PHH5 (QBU, male, 50 years old)) and their corresponding iHCC cells were used for RNA‐seq analysis. Four iHCC samples (iHCC1‐1~iHCC1‐4) from the donor PHH1, three iHCC samples (iHCC2‐1~iHCC2‐3) from the donor PHH2, three iHCC samples (iHCC3‐1~iHCC3‐3) from the donor PHH3, one iHCC sample (iHCC4‐1) from the donor PHH4, and an iHCC sample (iHCC5‐1) from the donor PHH5 were used. The PCA plot shows samples plotted in two dimensions using their projections onto the first two principal components (PC1 and PC2). Each data point represents one sample (blue, PHHs; red, iHCC). PC1 was correlated with the type of sample (iHCCs versus PHHs).
- Volcano plot of RNA‐seq data showing genes with differential expression (1,591 upregulated and 2,243 downregulated genes, fold change ≥ 2 and P value ≤ 0.05) in iHCC cells compared to PHHs. TP53, KRAS, MYC, MUC1, and AFP were highly expressed in iHCC cells, whereas hepatocyte signature markers, including FOXA2, FOXA3, ATF5, HNF4A, and ALB were highly expressed in PHHs.
- Gene ontology (GO) analysis of the pathways enriched with DEGs (fold change ≥ 2 and P value ≤ 0.05) in iHCC cells and PHHs. Genes associated with telomere maintenance and the cell cycle were highly enriched in iHCC cells (top), while genes associated with biomacromolecule metabolisms were highly enriched in PHHs (bottom).
- Heat map analysis of the log2(RPKM) values of selected genes found to be highly expressed or downregulated in PHHs and iHCC cells. These genes were classified into three categories based on their functions.
- UMAP visualization of iHCC and clinical HCC samples from the TCGA‐LIHC database based on their gene expression profiles.
- UMAP visualization of iHCC and three subtypes of clinical HCC samples from the TCGA‐LIHC database (iClust 1–3, TCGA‐LIHC) based on their gene expression profiles.
- Unsupervised clustering analysis of iHCC and clinical HCC samples in relative to normal PHHs. The distribution of molecular attributes (TP53 and KRAS mutation, MYC amplification) of iHCC, clinical HCC samples, and PHHs were displayed.
We then investigated whether iHCC recapitulate molecular profiles of HCC clinical samples. We compared the global transcription profiles of iHCC and PHHs based on our RNA‐seq analysis to those of HCC samples containing normal and mutated TP53 and amplified MYC and normal hepatocytes, which were available in TCGA database (TCGA‐LIHC, 374 HCC and 50 normal samples). Uniform Manifold Approximation and Projection (UMAP) analysis showed that iHCCs and HCC samples shared similar gene expression profiles and were different from those of PHHs from our samples and from the database (Fig 5E). In addition, HCC samples from the database were further classified into three major subtypes (iClust1‐3). iClust1 is considered more aggressive with robust proliferation and less differentiation than iClust2 and iClust3 (Cancer Genome Atlas Research Network. Electronic address wbe & Cancer Genome Atlas Research N, 2017). UMAP and dendrogram analysis show that iHCC samples resembled the iClust1 subtype in respect of transcriptional profiles (Fig 5F) and TP53/MYC/KRAS mutations (Fig 5G). In addition, GO analysis confirmed that the differentially expressed genes (DEGs) enriched in iHCCs and iClust 1 samples were associated mainly with cellular responses to DNA damage, telomere activity, and cell cycle, compared to that of iClust 2 and iClust 3 HCC (Fig EV4A). Though KRAS mutations have not been reported in HCC tumors that harbor p53 mutations or MYC amplification in the TCGA database, we have identified that 126 tumors with TP53 mutations, 140 tumors with mutations in the RTK/RAS/PI3K signaling pathways (KRAS, MET, PIK3CA, PTEN, NF1, and NRAS), and 197 tumors with the MYC/WNT pathways (CTNNB1, AXIN1, APC, and MYC) from 348 HCC tumors obtained from the TCGA database (Fig EV4B and Dataset EV2). Therefore, while KRAS mutations are not frequent, we believe that other modes of RTK activation can substitute for these mutations. In addition, 17 HCC samples contain MYC amplification and TP53 mutations, and 6 HCC samples harbor TP53 mutations and RAS expression upregulation. Furthermore, 35 HCC samples carry mutations in TP53, the RTK/RAS/PI3K and WNT/MYC pathways (Dataset EV2). Taken together, these analyses suggest this iHCC model represents an iClust1 subtype of HCC samples with similar transcription profiles and mutation profiles in TP53, RTK/RAS/PI3K and WNT/MYC pathways.
Figure EV4. Mutational signatures in liver hepatocellular carcinoma(TCGA‐LIHC).
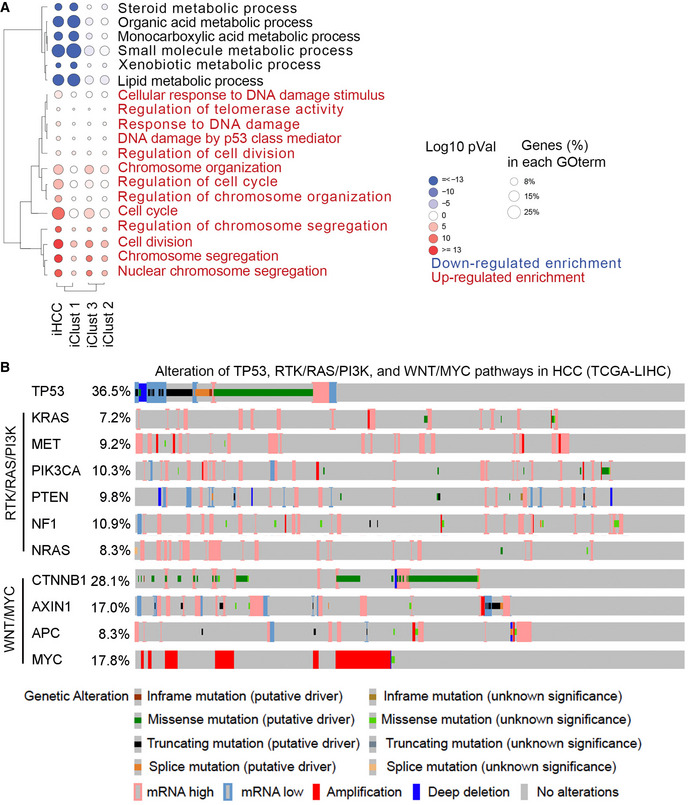
- Gene ontology (GO) analysis of the pathways enriched with DEGs (fold change ≥ 2 and P value≤ 0.05) in iHCC cells and clinical patient HCC (iClust 1, iClust 2 and iClust 3).
- Alteration signatures of TP53 mutations, and mutations in the RTK/RAS/PI3K and WNT/MYC pathways in clinical HCC samples obtained from the TCGA‐LIHC database.
The iHCC model enables the identification of therapeutic targets for HCC
As iHCC derived from NSIF mice have not been edited by adaptive immunity, we used them to identify biomarkers of early HCC as potential immunotherapeutic targets. Based on RNA‐seq analysis of iHCC cells and PHHs (Fig 5B), we determined that 55 genes encoding cell surface markers were upregulated in iHCC cells compared to PHHs (Dataset EV3). We then selected the top 20 genes that were highly expressed in iHCC cells (Fig 6A). With GEPIA analysis, four out of the top 20 genes (SLC34A2, FBN2, FOLR1, and SLC39A10) were associated with poor prognosis as evidenced by their prognostic value in cohorts of primary HCC patients according to the TCGA datasets (Menyhart et al, 2018) (TCGA‐LIHC) (Fig EV5A and Dataset EV3). We then focused on FAP and MUC1, as they are associated with HCC progression (Zou et al, 2018; Kasprzak & Adamek, 2019) and have been used as targets for CAR‐T cell therapy for cardiac fibrosis (Aghajanian et al, 2019), breast cancer, and lung cancer (Wei et al, 2017; Zhou et al, 2019). FAP was highly expressed in 4 of the 5 donor‐derived iHCC, and MUC1 expression was detected in 3 of the 5 donor‐derived iHCC but not in normal livers (Fig EV5B and C). We then generated anti‐FAP and anti‐MUC1 CAR‐T (CARFAP‐T and CARMUC1‐T) cells by transducing human T cells with third‐generation CAR molecules incorporating CD28‐ and TLR2‐derived costimulatory domains (Lai et al, 2018) (Fig EV5D and E). CARFAP‐T and CARMUC1‐T cells efficiently lysed iHCC2‐1 cells with high FAP and MUC1 expression in vitro, respectively (Fig 6B). To evaluate the antitumor effects of CARFAP‐T and CARMUC1‐T cells in vivo, we subcutaneously transplanted iHCC tumor tissues into NSI mice and intravenously transferred 5 × 106 CAR‐T cells or mock T cells on day 10 post‐tumor transplantation. Tumors were efficiently suppressed in xenografts that were injected with CARFAP‐T and CARMUC1‐T cells but not in the mock T or PBS groups (Fig 6C).
Figure 6. Genetically defined iHCC as a platform for validation of potential therapeutic targets.
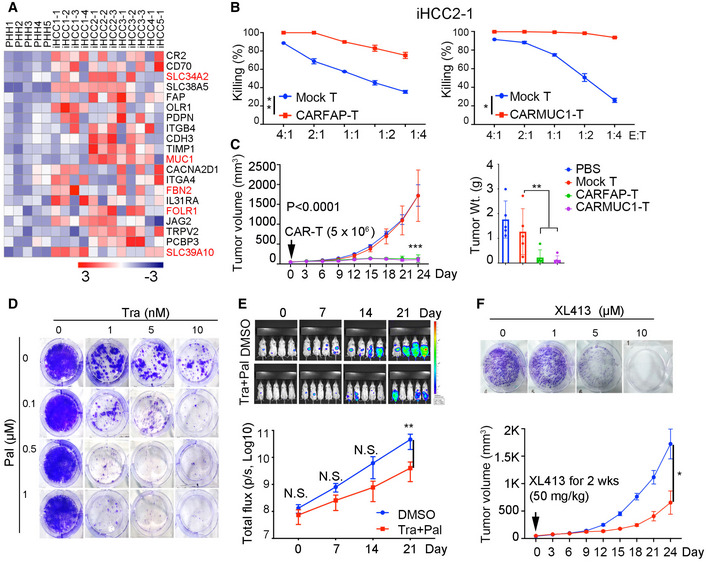
- Heat map shows the top 20 genes encoding cell surface markers found to be highly expressed in iHCC samples compared to the corresponding PHHs. Within these 20 genes, seven genes highlighted in red were selected because their expression was correlated with poor prognosis of HCC patients based on TCGA‐HCC (TCGA‐LIHC) analysis.
- Representative cytotoxicity CARFAP‐T and CARMUC1‐T cells assessed by coincubation with iHCC2‐1 cells. P = 0.0038 (CARFAP‐T versus mock T), and P < 0.0478 (CARMUC1‐T versus mock‐T). P values were determined by paired t‐test, **P ≤ 0.05. Cells were seeded at E: T ratios ranging from 4:1 to 1:4; data are presented as the mean ± SD. N‐numbers refer to biological replicates (n = 3).
- Growth of iHCC in xenografts (Left) and tumor weights of iHCC dissected from xenografts (Right) (n = 5), the xenografts were treated with mock, CARFAP‐, or CARMUC1‐T cells when tumors reach 50 mm3 volume. ***P ≤ 0.001 (tumor volume of CARFAP‐/CARMUC1‐T versus mock T) with two‐way ANOVA with Tukey's multiple comparison test. **P ≤ 0.01 (tumor weight of CARFAP‐/CARMUC1‐T versus mock T) with one‐way ANOVA with multiple comparison test. Data are presented as the mean ± SD. N‐numbers refer to biological replicates.
- Clonogenic assay of iHCC1‐1 cells treated with a MEK inhibitor (trametinib) and/or a CDK4/6 inhibitor (palbociclib); Representative images were shown based on three biological replicates.
- Luciferase and EGFP marked iHCC1‐1 cells (1 × 106) were intrasplenically injected into NSI mice on day 0. iHCC‐bearing mice were treated with DMSO (mock group, n = 4), or trametinib (3 mg/kg body weight) plus palbociclib (150 mg/kg body weight, n = 5) for 2 weeks (Daily oral gavage of drugs from Day 3 to Day 17). Top, on days 0, 7, 14, and 21, bioluminescence imaging was conducted. Bottom, quantification of the total flux analyzed by in vivo bioluminescence imaging of luciferase activity. P = 0.0057 on day 21 by two‐way ANOVA with Sidak's multiple comparison test, **P ≤ 0.01. Data are presented as the mean ± SD. N‐numbers refer to biological replicates.
- Clonogenic assays (Top) of iHCC1‐1 cells treated with XL413. The data are the representative of three biological replications. (Bottom) Growth curves of iHCC in xenografts (n = 5) treated with PBS or XL413 at 50 mm3 tumor volume. P = 0.0486 (XL413 versus PBS group) by two‐way ANOVA with Sidak's multiple comparison test at Day 24, *P ≤ 0.05. Data are presented as the mean ± SD. N‐numbers refer to biological replicates.
Figure EV5. Cell surface markers associated with poor prognosis and the design of CAR molecules.
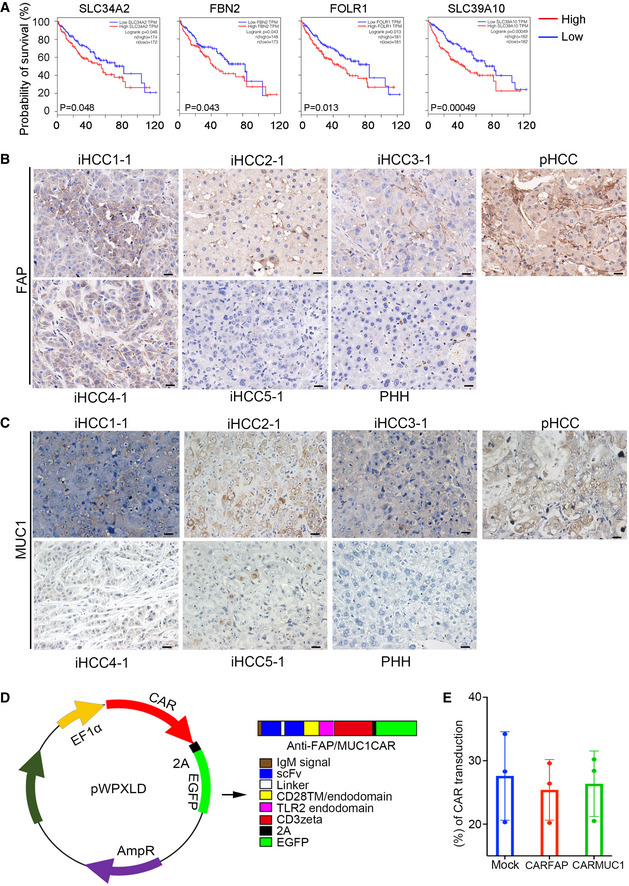
-
AKaplan‐Meier analysis of the TCGA‐HCC (TCGA‐LIHC) cohorts based on the expression levels of SLC34A2, FBN2, FOLR1, and SLC39A10 in the cohort samples (n = 364, high expression in red, low expression in black, the number of patients were indicated in figures, Statistical significance was determined using a log‐rank test).
-
B, CRepresentative IHC staining of FAP and MUC1 in a normal liver (PHH), primary HCC (pHCC) and five MTK‐transduced iHCC tissues (iHCC1‐1, iHCC2‐1, iHCC3‐1, iHCC4‐1, and iHCC5‐1) that were derived from five different donors (PHH1~5). Scale bars, 20 μm.
-
DDesign of the pWPXLD‐CAR vector. pWPXLD‐CAR contains an anti‐FAP/MUC1 single‐chain fragment variation (scFv), CD28 transmembrane and endodomain, TLR2 endodomain, CD3ζ domain, and EGFP.
-
EThe transduction efficiency of pWPXLD‐CAR is shown. Data are presented as the mean ± SD. N‐numbers refer to biological replicates.
Recent studies have demonstrated that the combination of the MEK inhibitor trametinib (Tra) and the CDK4/6 inhibitor palbociclib (Pal) leads to efficient tumor control through the induction of retinoblastoma (RB) protein‐mediated cellular senescence in KRAS‐ and TP53‐ mutant cancers (Ruscetti et al, 2018), revealing that the induction of senescence may constitute a strategy for the treatment of TP53 mutant liver cancers (Wang et al, 2019) or pancreatic cancers(Ruscetti et al, 2020). We thus evaluated the efficacy of the combination treatment of trametinib and palbociclib against iHCC with KRAS and TP53 mutations. Palbociclib alone did not affect the growth of iHCC cells in vitro, whereas trametinib repressed iHCC cell expansion (Fig 6D). Of note, the combination of trametinib and palbociclib was more effective than trametinib alone for inducing senescence, as shown by the staining of SA‐β‐gal (Ruscetti et al, 2020), in iHCC cells (Appendix Fig S3A). In addition, the combination treatment with trametinib and palbociclib suppressed the growth of iHCC in the xenograft model (Fig 6E). We further evaluated the efficacy of XL413, a CDC7 kinase inhibitor that specifically induces senescence in HCC samples (Wang et al, 2019) in the iHCC model. Of interest, XL413 inhibited the growth of iHCC in culture and in xenografts (Fig 6F) and induced senescence in iHCC (Appendix Fig S3B). Taken together, these results indicate that iHCC is a model for the identification of therapeutic targets and evaluation of anticancer drug efficacy for HCC.
Discussion
Genetically engineered mouse models are powerful tools to study PLC. Ectopic expression of Myc or KrasG12D with p53 depletion induces mouse hepatocytes to transform into HCC or ICC (Klocke et al, 2001; O'Dell et al, 2012). However, mouse and human hepatocytes are different in numerous aspects (Odom et al, 2007). Therefore, humanized PLC models are in need for the identification of therapeutic targets and evaluation of antitumor drug efficacy. Though human liver cancer organoids have been established recently (Sun et al, 2019), the organoids are derived from human‐induced hepatocytes (hiHeps) that are reprogrammed from human fibroblasts rather than PHHs (Huang et al, 2014). Furthermore, organoid models lack a tumor microenvironment. Here, we first demonstrated that human PHHs can be transformed into iHCC cells in situ upon overexpression of MYC, TP53R249S, and KRASG12D, providing us a model for investigating the initiation and progression of human HCC. iHCC mimics the histological architecture, tumor serum marker profile, and gene expression characteristics of human HCC. However, Ki‐67, a marker of proliferation (Sales‐Gil et al, 2021), was highly expressed in iHCC, which is not common in primary human HCC, possibly indicating the aggressiveness of iHCC. To our knowledge, it is the first in vivo model of orthotopic transformation of human PHHs into HCC cells. Therefore, humanized HCC models are better than murine HCC models for the identification of therapeutic targets and evaluation of anti‐tumor drug efficacy. Of course, iHCC models have some disadvantages compared to murine spontaneous HCC models, such as the lack of an immune system.
Based on previous reports (Schulze et al, 2015; Bailey et al, 2018; Llovet et al, 2018), we selected ten candidates for overexpression in PHHs and identified enrichment of MYC, TP53R249S, and KRASG12D in tumors harvested from the livers of xenografted mice. Since individual oncogenes such as MYC and RAS fails to induce the transformation of PHHs (Sun et al, 2019), we did not transduce individual oncogenes into PHHs in this study. However, it is worthy of performing a genetic screen for various combinations of these oncogenes that can transform PHHs into iHCC in the future. TP53R249S, frequently detected in human HCC related to hepatitis B infection and aflatoxin B1, gains its function via MYC activation upon CDK4 phosphorylation at Serine 249 and consequent PIN1 binding (Hussain et al, 2007; Liao et al, 2017). Though the loss of heterozygosity of TP53 in HCC patient samples was not uncommon (Hsu et al, 1994; Zhu et al, 2004), mutant TP53 was able to inhibit the activity of WT TP53 (Kern et al, 1992). The mechanistic basis for the cooperation among these oncoproteins has been elucidated. Mutant TP53 enhances KRASG12D activity through modulating RNA splicing in pancreatic ductal adenocarcinoma (Escobar‐Hoyos et al, 2020). Mutant KRAS and endogenous wild‐type p53 were shown to induce cellular senescence that limits liver cancer development in pre‐malignant hepatocytes, depending on an intact CD4+ T cell‐mediated adaptive immune response (Kang et al, 2011; Mehta et al, 2021), which is absent in our iHCC model. Although the combination of MYC, TP53R249S, and KRASG12D was uncommon in human HCC, iHCC cells derived from MTK‐transduced PHHs can be used to investigate further crosstalk among MYC, TP53R249S, and KRASG12D in human HCC. In addition, new mechanisms underlying how TP53R249S, MYC, and KRASG12D cooperatively transform primary human hepatocytes (PHHs) into HCC need to be addressed in future studies.
Whole‐genome sequencing has identified numerous genetic mutations and greatly enforced our understanding of human HCC (Cancer Genome Atlas Research Network. Electronic address wbe & Cancer Genome Atlas Research N, 2017). However, most of these mutations have not been characterized as to whether they can drive the initiation of HCC. We identified MYC, TP53R249S, and KRASG12D out of the ten oncogenes with the capacity of inducing human PHHs into HCC in vivo. Due to robust lentiviral transduction efficiency in human PHHs, our iHCC humanized mouse model constitutes a platform for performing high‐throughput genetic screening to distinguish the driver mutations detected in patient samples by whole‐genome sequencing that contribute to the transformation of PHHs to HCC cells in the numerous genetic lesions. Nevertheless, reconstitution protocol needs to be further optimized for improving in vivo repopulation of PHHs (Azuma et al, 2007). In addition to gain‐of‐function, we can also perform loss‐of‐function genetic screening using the CRISPR/Cas9 system to identify new tumor suppressor genes in HCC using our model in future research (Michels et al, 2020).
Most patients with HCC present with advanced disease, and treatment options are limited (Llovet et al, 2003). CAR‐T/NK cell therapy is a promising immunotherapeutic strategy for treating multiple refractory blood malignancies. Nevertheless, further advances are required for treating solid tumors with CAR‐T/NK cells (Schnalzger et al, 2019). One challenge is to identify safe and effective tumor antigens in solid tumors. There are four antigens as CAR‐T targets for treating HCC with limited success: glypican‐3 (GPC3)(Jiang et al, 2016; Shi et al, 2020) (NCT03198546, NCT02395250, NCT03146234), α‐fetoprotein (AFP) (Liu et al, 2017) (NCT 03349255), CD147 (Tseng et al, 2020), and CD133 (Dai et al, 2020) (NCT02541370). It is thus urgent to detect biomarkers of early‐stage HCC. We found that iHCC in our humanized mice expressed MUC1 and FAP, positively correlated with poor survival prognosis (Zou et al, 2018; Kasprzak & Adamek, 2019). MUC1 and FAP have been identified as CAR‐T targets for treating non‐small cell lung cancer (Wei et al, 2017) (NCT03198052) and cardiac fibrosis respectively, but not for HCC treatment. Therefore, our model may serve as a tool for identification of biomarkers and targets of early‐stage HCC.
Materials and Methods
Generation of NSIF mice
All experimental protocols were performed in accordance with the institutional guidelines of the China Council on Animal Care and approved by the Ethics Committee of Animal Experiments at Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences (GIBH). All mice were bred and maintained in specific pathogen‐free (SPF)‐grade cages and provided autoclaved food and water. NSI and ICR mouse strains were used as embryos and foster mothers, respectively. Female NSI mice (8–10 weeks of age) were superovulated by an intraperitoneal injection of 5 IU of PMSG (Sigma) and 5 IU of hCG (C1063, Sigma) at 48‐h intervals. The superovulated female mice were mated with male NSI mice, and the fertilized embryos were collected from the oviducts. TALEN mRNAs were injected into the cytoplasm of fertilized eggs displaying recognizable pronuclei in M2 medium using a piezo‐driven micromanipulator. A 25 ng/μl sample of Fah‐TALEN mRNAs was injected. After a 24‐h incubation at 37°C, the surviving embryos were selected and transferred into the oviducts of pseudopregnant foster mothers. Editing of the Fah gene was analyzed in the resulting offspring.
Recombinant vectors and viral packaging
The cDNA of each oncogene (MYC, TP53R249S, KRASG12D, NRASG12D, CTNNB1S45F, BRAFV600E, AXIN1G652S, IL6, PIK3CAE542K , and CSF1RY969C ) or third‐generation anti‐FAP and anti‐MUC1 CAR vectors incorporating the CD28 and TLR2 costimulatory endodomains were cloned into the second‐generation lentiviral vector pWPXLD. sgRNA targeting TP53 was cloned into LentiCRISPR v2 (addgene 52961) vector (Appendix Table S4). Lentiviral particles were produced in HEK‐293T cells following polyethyleneimine (PEI) (49553‐93‐7, Polysciences, Inc., USA)‐mediated transfection with the pWPXLD‐based transfer plasmid and the packaging plasmids psPAX2 and pMD2.G. Lentivirus‐containing supernatant was harvested at 48 and 72 h post‐transfection and filtered through a 0.45 µm filter.
Cells and culture conditions
PHH were cultured in DMEM/F‐12 medium with 10% heat‐inactivated FBS (Gibco, Grand Island, NY, USA), 10 mM HEPES, 2 mM glutamine (Gibco, Grand Island, NY, USA), and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA) supplemented with hHGF (10 ng/ml, Cat#100‐39, Peprotech, NJ, USA), hEGF (5 ng/ml, Cat#AF‐100‐15, Peprotech), and WNT3a (5 ng/ml, Cat#315‐20). iHCC were cultured in DMEM/F‐12 (Gibco, Grand Island, NY, USA), supplemented with 10% heat‐inactivated FBS (Gibco, Grand Island, NY, USA), 10 mM HEPES, 2 mM glutamine (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA). All cells were cultured at 37°C in an atmosphere of 5% carbon dioxide.
Transduction of hepatocytes and tracking of hepatocyte engraftment
Primary human hepatocytes from five different donors (PHH1 (AKB, female, 39 years old), PHH2 (XSM, female, 59 years old), PHH3 (ANG, male, 3 months old), PHH4 (HVN, male, 33 years old) and PHH5 (QBU, male, 50 years old)) were purchased from Bioreclamation IVT (Baltimore, MD, USA). These cells were transduced with a lentivirus which expresses an oncogene and EGFP or luciferase as indicated in the experiment. Twenty‐4 h after transduction, hepatocytes were cultured in DMEM/F‐12 medium supplemented with hHGF (10 ng/ml, Cat#100‐39, Peprotech), hEGF (5 ng/ml, Cat#AF‐100‐15, Peprotech), and WNT3a (5 ng/ml, Cat#315‐20, Peprotech). The transduction efficiency was determined by fluorescence‐activated cell sorting (FACS). EGFP‐ and luciferase‐positive hepatocytes (0.5 × 106 cells/mouse) were transplanted into the spleens of NSIF mice. Hepatocyte engraftment was evaluated using a cooled charge‐coupled device (CCD) camera system (IVIS 100 Series Imaging System, Xenogen, Alameda, CA, USA). Mice were intraperitoneally injected with 75 mg/kg D‐luciferin firefly potassium salt and imaged 5 min later with auto‐exposure time. The total and average emissions were quantified using Living Image software (Xenogen, Alameda, CA, USA). For oncogene screening and validation, viral titers in the mixture were adjusted to the same multiplicity of infection (MOI = 5.0) to obtain a transduction rate of > 90% on day 2 after infection and 0.5 million GFP‐positive transduced hepatocytes were transplanted into NSIF mice via intrasplenic injection. For the MEK inhibitor (Trametinib, T2125, Targetmol) and a CDK4/6 inhibitor (Palbociclib, T1785, Targetmol) treatment assay, MYC, TP53R249S, and KRASG12D cells that were further transduced with the EGFP and luciferase lentiviral expression vectors (2 × 106 cells per mouse) were injected intrasplenically into 6‐ to 8‐week‐old immunodeficient NSI mice. Mice were randomly assigned to treatment with vehicle or trametinib (3 mg/kg body weight, daily gavage)/palbociclib (150 mg/kg body weight, daily gavage) 5 days per week for 2 weeks. On days 0, 7, 14, and 21, bioluminescence imaging was conducted. For XL413 (S7547, Selleck) treatment, iHCC cells were subcutaneously injected into NSI mice. Xenografted mice were treated with PBS or XL413 (50mg/kg for 2 weeks) at 50 mm3 tumor volume. The tumor volume was calculated as (length × width2)/2.
Transplantation of human hepatocytes
All animal experiments were performed in accordance with institutional regulations. Primary human liver cells (0.5–1 million) were transplanted into the spleens of NSIF mice after the withdrawal of NTBC‐containing drinking water (Azuma et al, 2007). The NTBC concentration was gradually decreased (3.5 mg/l, days 0 to 2; 1.75 mg/l, days 3 and 4; and 0.8 mg/l, days 5 and 6), and NTBC was then completely withdrawn 1 week after transplantation. NTBC was readministered for 5 days, 3–4 weeks later when a transplanted mouse had lost > 20% of its pretransplant weight. The animals' body weights were monitored twice weekly after transplantation. The surviving recipient mice were euthanized for collection of blood and liver samples.
Measurement of human ALB, AFP, APOA1, and glycogen
Small amounts of blood were collected weekly from the left saphenous vein with a heparinized blood capillary tube. The human ALB concentration was measured with a Human Albumin ELISA Quantitation Kit (E80‐129, Bethyl Laboratories). The AFP concentration was measured with an AFP kit (EHAFP, Invitrogen, Thermo Fisher) according to the corresponding manufacturer's protocol. To determine the levels of stored glycogen, 1 × 106 cells were collected. Human albumin was measured using the Human albumin ELISA quantitation set. Glycogen storage was measured with a Glycogen assay kit (MAK016, Sigma‐Aldrich) and human APOA1 was measured using a APOA1 detection kit (SEKH00903, Solarbio) according to the manufacturers' instructions.
Quantitative real‐time PCR
mRNA was extracted from cells with TRIzol reagent (15596026, Thermo Fisher) and reverse transcribed into cDNA using the PrimeScriptTM RT reagent Kit (Takara, Kasatsu, Japan). All reactions were performed with TransStart Tip Green qPCR SuperMix (TransGene, Beijing, China) on a Bio‐Rad CFX96 real‐time PCR machine (Bio‐Rad, Hercules, CA), using the primers shown in Appendix Table S4. Delta CT calculations were relative to β‐actin and corrected for PCR efficiencies.
Histological and immunofluorescence assays
Tissue samples were fixed with 4% formalin, embedded in paraffin, sectioned at a thickness of 4 μm, and stained with H&E. Images were acquired with a microscope (Leica DMI6000B, Leica Microsystems, Wetzlar, Germany). Paraffin sections were used for immunofluorescence (IF) and immunohistochemical (IHC) staining. A primary antibody (1:100–1:2,000) was added to the tissue sections on slides and incubated overnight at 4°C. Alexa Fluor 647‐conjugated goat anti‐mouse (1:2,000) and Alexa Fluor 568/488‐conjugated goat anti‐rabbit (1:2,000) antibodies were added and incubated for 1 h at room temperature. DAPI was used to stain nuclei, and the prepared slides were analyzed using a Zeiss LSM800 microscope. Anti‐HLA Class I ABC antibody (ab70328), Anti‐Cytokeratin 19 antibody (ab76539), Anti‐c‐Myc antibody (ab32072), Anti‐p53 antibody (ab32389), Anti‐KRAS antibody (ab275876), Anti‐EpCAM antibody (ab223582), Anti‐AFP antibody (ab169552), Anti‐GPC3 antibody (ab95363), Anti‐MUC1 antibody (ab109185), and anti‐FAP antibody (ab207178) were purchased from Abcam.
RNA sequencing
mRNA from primary PHH or iHCC was prepared according to the TruSeqTM RNA Sample Preparation Guide, and sequencing was performed on BGISEQ‐500 (BGI, Wuhan, China). Sequencing reads were mapped to the human RefSeq‐RNA reference sequence using the FANSe2 algorithm. Reads mapped with tophat2 were associated with genes using the custom Perl scripts that allowed no more than 2 unmapped bases. Cufflinks (version 2.1.1) was used to identify reads consistent with the annotated genes. These genes were quantified using the reads per kilobase million (RPKB) method. For small genes (< 200 bps), a minimum of 10 mappable reads were required. Mappable reads were imported into the DEGseq software package to calculate up‐ or downregulation of genes.
Integrative clustering of iHCC and clinical HCC using iCluster
We used publicly available data provided by the TCGA Research Network (http://xena.ucsc.edu) to perform hierarchical clustering and GSEA analyses. The RNA sequencing data of TCGA‐HCC (374 HCC samples and 50 normal samples) were downloaded from TCGA. To understand the subgroups formed by integrating various molecular platforms of iHCC, we utilized iCluster, which formulates the problem of subgroup discovery as a joint multivariate regression of multiple data types with reference to a set of common latent variables that represent the underlying tumor subtypes (Mo et al, 2013; Cancer Genome Atlas Research Network. Electronic address wbe & Cancer Genome Atlas Research N, 2017).
DNA extraction and sequence analysis
DNA was extracted from samples using a HiPure Tissue DNA Mini Kit (D3121, Magen, China). In brief, samples of approximately 10 mg were digested in 250 µl of ATL buffer supplemented with proteinase K overnight at 55°C and were then treated with RNase for 30 min at 37°C. Genomic DNA was subjected to PCR amplification, and mutations were identified by direct sequencing. To avoid amplification of endogenous genes, we ensured that the PCR primer pairs flanked the introns in the 10 oncogenes (Appendix Table S4).
Western blot assays
Western blotting was performed following a routine protocol. The hiHep organoids were lysed in RIPA lysis buffer (supplemented with NaF (1:100), Na3VO4 (1:200), P8340 (1:100)) and an equal amount of total protein lysates were separated by 6–15% SDS–PAGE and transferred to a PVDF membrane (Millipore). The membrane was blocked with 5% (w/v) reagent‐grade non‐fat milk and incubated overnight with primary antibodies (MYC (5605, CST), KRASG12D (14429, CST,), p53(48818, CST)) at 4°C followed by secondary antibody incubation for 1 h. The protein bands were visualized using Luminata Western ECL substrate (Sigma).
Long‐term cell proliferation assays (colony formation assays)
Cells were seeded into 6‐well plates (1 × 104 cells per well) and cultured with/without drugs as indicated for 10–14 days. The medium was changed twice weekly. Cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet (in water) (Lin et al, 2020).
SA‐β‐gal staining
SA‐β‐gal activity was assessed using a Senescence β‐Galactosidase Staining Kit (C0602, Beyotime, China) according to the manufacturer's instructions. iHCC cells were plated in a 6‐well plate (1 × 105 cells per well) and treated with a MEK inhibitor (trametinib, T2125, Targetmol), a CDK4/6 inhibitor (palbociclib, T1785, Targetmol), and XL413 as indicated. SA‐β‐gal‐positive cells (senescent cells) were identified as blue‐stained cells under standard light microscopy.
Cell cycle analysis
PHHs transduced with the MYC/TP53R249S/KRASG12D combination were harvested by trypsinization on day 10. Cells were washed two times with PBS, fixed at −20°C in 70% ethanol for 12 h and stained in 300 μl of propidium iodide (final concentration of 50 mg/ml) and 0.1% Triton X‐100 at 37°C for 15 min. The distribution of cells in different phases of the cell cycle was analyzed by flow cytometry (Agilent NovoCyte, USA).
Isolation, transduction, and expansion of primary human T lymphocytes
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy adult donors using Lymphoprep (Stem Cell Technologies, Vancouver, Canada). T cells were negatively selected from PBMCs with a MACS Pan T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and activated using microbeads coated with anti‐human CD3, anti‐human CD2, and anti‐human CD28 antibodies (Miltenyi Biotec, Bergisch Gladbach, Germany) at a bead: cell ratio of 1:2 and a density of 2.5 × 106 cells/ml for 2 days in RPMI‐1640 medium supplemented with 10% heat‐inactivated fetal bovine serum (FBS), 100 IU/ml recombinant human IL‐2, 10 mM HEPES, 2 mM glutamine and 1% penicillin/streptomycin. On day 2 post‐activation, T cells were transfected with CAR vector lentiviral supernatants in the presence of 8 μg/ml polybrene at an MOI of 2.0 (Sigma‐Aldrich, St. Louis, USA). Twelve hours after transfection, T cells were cultured in fresh medium containing IL‐2 (300 U/ml); subsequently, fresh medium was added every 3 days to maintain cell density within the range of 0.5–1 × 106 cells/ml. Healthy PBMC donors provided informed consent to use their samples for research purposes, and all procedures were approved by the Research Ethics Board of GIBH.
Xenograft models and in vivo assessment
Animal experiments were performed in the Laboratory Animal Center of GIBH, and all animal procedures approved by the Animal Welfare Committee of GIBH. All protocols were approved by the relevant Institutional Animal Care and Use Committee (IACUC). NSI mice were maintained in specific pathogen‐free (SPF)‐grade cages and provided autoclaved food and water. Mice were randomized into experimental groups of ≥ 5. Direct injection of indicated tumor cells in 200 μl PBS was performed to establish subcutaneous (flank) tumors. At the indicated time for each experiment, 5 × 106 transduced human T (GFP+ or CAR+) cells in 200 μl PBS were adoptively transferred to tumor‐bearing mice systemically by tail vein injection. Peripheral blood was obtained by retro‐orbital bleeding. Tumors were measured every 3 days with a caliper. Tumor volume was calculated using the following equation: (length × width2)/2.
Statistical analysis
Kaplan–Meier's analysis of the correlation between candidate genes expression and overall survival was acquired from the GEPIA analysis tool (http://gepia.cancer‐pku.cn/index.html), which was based on datasets from The Cancer Genome Atlas (TCGA). Data are presented as the mean ± SD/SEM values, and significance was evaluated by Student's t‐test or ANOVA as indicated. Differences with P < 0.05 were considered statistically significant.
Author contributions
Zhiwu Jiang: Conceptualization; Resources; Formal analysis; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review & editing. Lin Cheng: Conceptualization; Formal analysis; Investigation; Methodology. Zhiping Wu: Data curation; Investigation; Methodology. Linfu Zhou: Resources; Formal analysis; Methodology. Haitao Wang: Software; Formal analysis; Methodology. Qilan Hong: Software; Formal analysis; Methodology. Qiting Wu: Resources; Methodology. Youguo Long: Resources; Methodology. Yunlin Huang: Investigation; Methodology. Gaoqi Xu: Methodology. Yao Yao: Resources; Supervision; Funding acquisition; Validation; Methodology. Zhaoyang Tang: Resources; Validation; Investigation; Methodology. Zhenfeng Zhang: Resources; Formal analysis; Methodology; Project administration. Lili Yang: Resources; Validation; Methodology. Wei Luo: Resources; Methodology. Jie Yang: Resources; Investigation; Methodology. Likun Gong: Resources; Investigation; Methodology. Pentao Liu: Resources; Supervision; Validation; Investigation; Methodology. Xinwen Chen: Resources; Validation; Methodology. ShuZhong Cui: Resources; Validation; Investigation. Qi Zhang: Resources; Validation; Methodology. Yinxiong Li: Conceptualization; Resources; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review & editing. Peng Li: Conceptualization; Resources; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization, ZJ and PLi; Methodology, ZJ, LC, ZW, LZ, HW, QH, GX, and PLi; Investigation, ZJ, LC, ZW and PLi; Validation, YH, GX, YY, and LG; Formal Analysis, ZJ, and LC; Resources, QW, LZ, YLo, ZT, ZZ, WL, JY, SC and QZ; Writing—Original Draft, ZJ and PLi; Writing—Review and Editing, YLi, PLiu, ZJ, LY, and PLi; Supervision, YLi, PLiu, and XC; Project Administration, YLi. PLi; Funding Acquisition, ZJ, YY, JY, YL, and PLi.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Source Data for Figure 1C
Source Data for Figure 3I
Acknowledgements
We thank Professor Jean Paul Thiery (Guangzhou Laboratory) and Dr. Qiannan Tang (University of Hong Kong) for proofreading this manuscript. We thank Dr. Jianming Zeng (University of Macau) and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes. This study was supported by the National Key Research and Development Plan (Nos. 2021YFE0202800 to P. L, 2017YFE0131600 to Y. L., and 2019YFA0111500 to X. L.), the National Natural Science Foundation of China (Nos. 81961128003 to P.L., 81972672 to P.L., 81773301 to Z. J., 81870121 to P. L., 81873847 to J. Y, 81872069 to Z.Z, and 32170946 to Z. J.), The Youth Innovation Promotion Association of the Chinese Academy of Sciences (No. 2020351 to Z. J.), the Guangdong Provincial Significant New Drugs Development (Nos. 2019B020202003 to P. L., 2019A1515010062 to Y. Y., and 2020A1515011516 to X. W.), the Guangzhou Science and Technology Plan Project (No. 201907010042 to P. L.), 2020B1212060052, the Frontier Research Program of the Guangzhou Regenerative Medicine and Health Guangdong Laboratory (No. 2018GZR110105003 to P. L.), Science and Technology Planning Project of Guangdong Province, China (2020B1212060052), the Science and Technology Program of Guangzhou, China (No. 202002020083 to X. L.), the Open project of the State Key Laboratory of Respiratory Disease (No. SKLRD‐OP‐202002 to Z. Z.), and the University Grants Committee/Research Grants Council of the Hong Kong (Project No. AoE/M‐401/20), and Innovation and Technology Fund (ITF) from Hong Kong SAR Government.
EMBO reports (2022) 23: e54275.
Contributor Information
Yinxiong Li, Email: li_yinxiong@gibh.ac.cn.
Peng Li, Email: li_peng@gibh.ac.cn.
Data availability
RNA‐Seq data: Gene Expression Omnibus GSE168852 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168852).
References
- Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li LI, Scholler J, Monslow J, Lo A, Han W et al (2019) Targeting cardiac fibrosis with engineered T cells. Nature 573: 430–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma H, Paulk N, Ranade A, Dorrell C, Al‐Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M (2007) Robust expansion of human hepatocytes in Fah‐/‐/Rag2‐/‐/Il2rg‐/‐ mice. Nat Biotechnol 25: 903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B et al (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 173: 371–385 e318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabe F, Kennedy JA, Hope KJ, Dick JE (2007) Modeling the initiation and progression of human acute leukemia in mice. Science 316: 600–604 [DOI] [PubMed] [Google Scholar]
- Bleijs M, van de Wetering M, Clevers H, Drost J (2019) Xenograft and organoid model systems in cancer research. EMBO J 38: e101654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R, Rotter V (2009) When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 9: 701–713 [DOI] [PubMed] [Google Scholar]
- Broutier L, Mastrogiovanni G, Verstegen MMA, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes‐Benito R, Sidorova O, Gaspersz MP et al (2017) Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med 23: 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N (2017) Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169: 1327–1341 e1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Tong JH, Chan SL, Lai PB, To KF (2014) Expression of stemness markers (CD133 and EpCAM) in prognostication of hepatocellular carcinoma. Histopathology 64: 935–950 [DOI] [PubMed] [Google Scholar]
- Dai H, Tong C, Shi D, Chen M, Guo Y, Chen D, Han X, Wang H, Wang Y, Shen P (2020) Efficacy and biomarker analysis of CD133‐directed CAR T cells in advanced hepatocellular carcinoma: a single‐arm, open‐label, phase II trial. OncoImmunology 9: 1846926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H et al (2015) Sequential cancer mutations in cultured human intestinal stem cells. Nature 521: 43–47 [DOI] [PubMed] [Google Scholar]
- Escobar‐Hoyos LF, Penson A, Kannan R, Cho H, Pan C‐H, Singh RK, Apken LH, Hobbs GA, Luo R, Lecomte N et al (2020) Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell 38: 198–211.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, Huang C, Li J, Dong X, Zhou Y et al (2019) Integrated proteogenomic characterization of HBV‐related hepatocellular carcinoma. Cell 179: 561–577 e522 [DOI] [PubMed] [Google Scholar]
- Govaere O, Komuta M, Berkers J, Spee B, Janssen C, de Luca F, Katoonizadeh A, Wouters J, van Kempen LC, Durnez A et al (2014) Keratin 19: a key role player in the invasion of human hepatocellular carcinomas. Gut 63: 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468 [DOI] [PubMed] [Google Scholar]
- Hsu HC, Peng SY, Lai PL, Sheu JC, Chen DS, Lin LI, Slagle BL, Butel JS (1994) Allelotype and loss of heterozygosity of p53 in primary and recurrent hepatocellular carcinomas. A study of 150 patients. Cancer 73: 42–47 [DOI] [PubMed] [Google Scholar]
- Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC (1991) Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350: 427–428 [DOI] [PubMed] [Google Scholar]
- Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, Cen J, Chen X, Liu C, Hu Y et al (2014) Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 14: 370–384 [DOI] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD (2015) Biochemical and structural analysis of common cancer‐associated KRAS mutations. Mol Cancer Res 13: 1325–1335 [DOI] [PubMed] [Google Scholar]
- Hussain SP, Schwank J, Staib F, Wang XW, Harris CC (2007) TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 26: 2166–2176 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, Lin S, Wang S, Wu Q, Liang Q et al (2016) Anti‐GPC3‐CAR T cells suppress the growth of tumor cells in patient‐derived xenografts of hepatocellular carcinoma. Front Immunol 7: 690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel‐Reid S, Letarte M, Sirard C, Doedens M, Grunberger T, Fulop G, Freedman MH, Phillips RA, Dick JE (1989) A model of human acute lymphoblastic leukemia in immune‐deficient SCID mice. Science 246: 1597–1600 [DOI] [PubMed] [Google Scholar]
- Kang T‐W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A et al (2011) Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature 479: 547–551 [DOI] [PubMed] [Google Scholar]
- Kasprzak A, Adamek A (2019) Mucins: the old, the new and the promising factors in hepatobiliary carcinogenesis. Int J Mol Sci 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B (1992) Oncogenic forms of p53 inhibit p53‐regulated gene expression. Science 256: 827–830 [DOI] [PubMed] [Google Scholar]
- Klocke R, Bartels T, Jennings G, Brand K, Halter R, Strauss M, Paul D (2001) Lack of p53 accelerates hepatocarcinogenesis in transgenic mice constitutively overexpressing c‐myc in the liver. FASEB J 15: 1404–1406 [DOI] [PubMed] [Google Scholar]
- Kusano N, Shiraishi K, Kubo K, Oga A, Okita K, Sasaki K (1999) Genetic aberrations detected by comparative genomic hybridization in hepatocellular carcinomas: their relationship to clinicopathological features. Hepatology 29: 1858–1862 [DOI] [PubMed] [Google Scholar]
- Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P (2017) Current status and perspectives of patient‐derived xenograft models in cancer research. J Hematol Oncol 10: 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, Jiang Z, Li B, Lin S, Wang S et al (2018) Toll‐like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia 32: 801–808 [DOI] [PubMed] [Google Scholar]
- Liao P, Zeng SX, Zhou X, Chen T, Zhou F, Cao B, Jung JH, Del Sal G, Luo S, Lu H (2017) Mutant p53 gains its Function via c‐Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol Cell 68: 1134–1146.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, Sun L, Zhan S, Chen L, Cheng C et al (2020) RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3‐mediated autophagy. EMBO J 39: e103181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Xu Y, Xiang J, Long LI, Green S, Yang Z, Zimdahl B, Lu J, Cheng N, Horan LH et al (2017) Targeting alpha‐fetoprotein (AFP)‐MHC complex with CAR T‐cell therapy for liver cancer. Clin Cancer Res 23: 478–488 [DOI] [PubMed] [Google Scholar]
- Llovet JM, Burroughs A, Bruix J (2003) Hepatocellular carcinoma. Lancet 362: 1907–1917 [DOI] [PubMed] [Google Scholar]
- Llovet JM, Montal R, Sia D, Finn RS (2018) Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 15: 599–616 [DOI] [PubMed] [Google Scholar]
- Llovet JM, Villanueva A, Lachenmayer A, Finn RS (2015) Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 12: 408–424 [DOI] [PubMed] [Google Scholar]
- Mehta S, Campbell H, Drummond CJ, Li K, Murray K, Slatter T, Bourdon JC, Braithwaite AW (2021) Adaptive homeostasis and the p53 isoform network. EMBO Rep 22: e53085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menyhart O, Nagy A, Gyorffy B (2018) Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R Soc Open Sci 5: 181006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels BE, Mosa MH, Streibl BI, Zhan T, Menche C, Abou‐El‐Ardat K, Darvishi T, Członka E, Wagner S, Winter J et al (2020) Pooled in vitro and in vivo CRISPR‐Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell 26: 782–792.e787 [DOI] [PubMed] [Google Scholar]
- Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C, Powers RS, Ladanyi M, Shen R (2013) Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci USA 110: 4245–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell MR, Huang JL, Whitney‐Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N et al (2012) Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res 72: 1557–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, MacIsaac KD, Rolfe PA, Conboy CM, Gifford DK, Fraenkel E (2007) Tissue‐specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet 39: 730–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon TC, Wong N, Lai PB, Rattray M, Johnson PJ, Sung JJ (2006) A tumor progression model for hepatocellular carcinoma: bioinformatic analysis of genomic data. Gastroenterology 131: 1262–1270 [DOI] [PubMed] [Google Scholar]
- Rao MS, Khan AA, Parveen N, Habeeb MA, Habibullah CM, Pande G (2008) Characterization of hepatic progenitors from human fetal liver during second trimester. World J Gastroenterol 14: 5730–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen C‐C, Ho Y‐J, Sanchez‐Rivera FJ, Feucht J et al (2018) NK cell‐mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362: 1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho Y‐J, Fennell M et al (2020) Senescence‐induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181: 424–441.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales‐Gil R, Kommer DC, de Castro IJ, Amin HA, Vinciotti V, Sisu C, Vagnarelli P (2021) Non‐redundant functions of H2A.Z.1 and H2A.Z.2 in chromosome segregation and cell cycle progression. EMBO Rep 22: e52061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R, Lichter P et al (2008) Etiology‐dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 47: 511–520 [DOI] [PubMed] [Google Scholar]
- Schnalzger TE, de Groot MH, Zhang C, Mosa MH, Michels BE, Röder J, Darvishi T, Wels WS, Farin HF (2019) 3D model for CAR‐mediated cytotoxicity using patient‐derived colorectal cancer organoids. EMBO J 38: e100928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F et al (2015) Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47: 505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, Lu Q, Gao H, Jiang H, Wang H et al (2020) Chimeric antigen receptor‐glypican‐3 T‐cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin Cancer Res 26: 3979–3989 [DOI] [PubMed] [Google Scholar]
- Shultz LD, Ishikawa F, Greiner DL (2007) Humanized mice in translational biomedical research. Nat Rev Immunol 7: 118–130 [DOI] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Szabo TG, Braun VZ, Bongiovanni L, Bos H, Spierings DCJ, Westendorp B, Curinha A, Stojakovic T et al (2020) PIDDosome‐induced p53‐dependent ploidy restriction facilitates hepatocarcinogenesis. EMBO Rep 21: e50893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang Y, Cen J, Ma X, Cui L, Qiu Z, Zhang Z, Li H, Yang R‐Z, Wang C et al (2019) Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat Cell Biol 21: 1015–1026 [DOI] [PubMed] [Google Scholar]
- Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H et al (2004) Near completely humanized liver in mice shows human‐type metabolic responses to drugs. Am J Pathol 165: 901–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H et al (2014) Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46: 1267–1273 [DOI] [PubMed] [Google Scholar]
- Trerotoli P, Fransvea E, Angelotti U, Antonaci G, Lupo L, Mazzocca A, Mangia A, Antonaci S, Giannelli G (2009) Tissue expression of Squamous Cellular Carcinoma Antigen (SCCA) is inversely correlated to tumor size in HCC. Mol Cancer 8: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng H‐C, Xiong W, Badeti S, Yang Y, Ma M, Liu T, Ramos CA, Dotti G, Fritzky L, Jiang J‐G et al (2020) Efficacy of anti‐CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun 11: 4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, de Oliveira RL, Morris B, Gadiot J, Wang W et al (2019) Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574: 268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Lai Y, Li J, Qin LE, Xu Y, Zhao R, Li B, Lin S, Wang S, Wu Q et al (2017) PSCA and MUC1 in non‐small‐cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 6: e1284722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Nishimura T, Nishimura S, Zhang H, Zheng M, Guo YY, Masek M, Michie SA, Glenn J, Peltz G (2014) Fialuridine induces acute liver failure in chimeric TK‐NOG mice: a model for detecting hepatic drug toxicity prior to human testing. PLoS Med 11: e1001628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Jiang Z, Li G‐X, Xiao Y, Lin S, Lai Y, Wang S, Li B, Jia B, Li Y et al (2015) Quantitative evaluation of the immunodeficiency of a mouse strain by tumor engraftments. J Hematol Oncol 8: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhang L, Liu W, Ma X, Cen J, Sun Z, Wang C, Feng S, Zhang Z, Yue L et al (2018) In vitro expansion of primary human hepatocytes with efficient liver repopulation capacity. Cell Stem Cell 23: 806–819 e804 [DOI] [PubMed] [Google Scholar]
- Zhao K, Wang D, Zhao X, Wang C, Gao Y, Liu K, Wang F, Wu X, Wang X, Sun L et al (2020) WDR63 inhibits Arp2/3‐dependent actin polymerization and mediates the function of p53 in suppressing metastasis. EMBO Rep 21: e49269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ding L, Wang D, Ye Z, He Y, Ma L, Zhu R, Pan Y, Wu Q, Pang K et al (2019) EZH2 cooperates with gain‐of‐function p53 mutants to promote cancer growth and metastasis. EMBO J 38: e99599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, Mukherjee P (2019) CAR T cells targeting the tumor MUC1 glycoprotein reduce triple‐negative breast cancer growth. Front Immunol 10: 1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu GN, Zuo L, Zhou Q, Zhang SM, Zhu HQ, Gui SY, Wang Y (2004) Loss of heterozygosity on chromosome 10q22‐10q23 and 22q11.2‐22q12.1 and p53 gene in primary hepatocellular carcinoma. World J Gastroenterol 10: 1975–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou B, Liu X, Zhang B, Gong Y, Cai C, Li P, Chen J, Xing S, Chen J, Peng S et al (2018) The expression of FAP in hepatocellular carcinoma cells is induced by hypoxia and correlates with poor clinical outcomes. J Cancer 9: 3278–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]