

Abstract
Coronavirus infection causes endoplasmic reticulum stress inside the cells, which inhibits protein folding. Prolonged endoplasmic reticulum stress causes an apoptotic process of unfolded protein response-induced cell death. Endoplasmic reticulum stress rapidly induces the activation of mTORC1, responsible for the induction of the IRE1-JNK pathway. IRE1-JNK stands out for its dual nature: pro-apoptotic in the first stage of infection, anti-apoptotic in persistently infected cells. Once penetrated the cells, the virus can deflect the mitochondrial function by implementing both waterfalls pro-apoptotic and anti-apoptotic response. The virus prevents, through Open Reading Frame 9b (ORF-9b) interacting with mitochondria, the response of the type I interferon of the cells affected by the infection and is fundamental for generating an antiviral cellular state. ORF-9b has effects on mitochondrial dynamics, inducing fusion and autophagy and promoting cell survival. The recognition of ORF-9b has made it possible to identify it as a molecular target of some existing potentially effective drugs (Midostaurin and Ruxolitinib). Other drugs, with the same target, are currently being tested. Given the great importance of mitochondria in virus-host interaction, in-depth knowledge of the actors and pathways involved is essential to continue developing new therapeutic strategies against SARS CoV2. (www.actabiomedica.it)
Keywords: Coronavirus, endoplasmic reticulum stress, mitochondria, apoptosis, autophagy, mitochondrial fusion, immune system
The origins of the Coronaviruses
Coronaviruses (CoV) comprise a large family of zoonotic viruses, characterized by single-stranded RNA, belonging to the Coronaviridae family, Nidovirales orders (1, 2). In humans, CoV mainly causes respiratory and gastrointestinal symptoms ranging from the common cold to more severe diseases, such as bronchitis, pneumonia, acute respiratory distress syndrome (ARDS), coagulopathy, multiorgan failure, and death (3, 4). The classification was initially based on antigenic relationships and subsequently confirmed by comparing the sequences of the viral genomes (5). Previously, six coronaviruses were identified as human sensitive viruses, including α - CoV HCoV-229E, HCoV-NL63, β - CoV HCoV-HKU1 and HCoV-OC43, responsible for mild respiratory symptoms similar to a common cold. The other two known β - CoV, SARS - CoV, and MERS - CoV are responsible for severe and potentially fatal respiratory tract infections (6, 7). Coronaviruses attracted attention as a highly pathogenic virus for humans with the spread of SARS CoV (severe acute respiratory syndrome) in China in 2002 (8-10). SARS- CoV uses the angiotensin 2 converting enzyme (ACE2) as a receptor and mainly infects ciliated bronchial epithelium and type II pneumocytes (11, 12), the same mechanism used by the more recent SARS-CoV2 responsible for COVID-19 (13, 14). SARS-CoV2 is a non-segmented beta-coronavirus with RNA (+) (positive sense), which shares 79.5% of genomic identity with SARS- CoV (15).
Structure and mechanism of coronavirus infection
Morphologically, coronaviruses have a spherical or pleomorphic shape with an average diameter of 80-120 nm. Strongly glycosylated S proteins on the surface (2) are mainly responsible for the virus›s entry into cells by binding to ACE2. The other structural proteins are: membrane proteins (M), envelope proteins (E) (16), and finally, nucleocapsid proteins (N), linked to genomic RNA. The genome is around 30 kb (17). The Coronavirus S proteins are divided by the host protease into two functional subunits: an S1 domain, which interacts with the host cell receptor, and an S2 domain, responsible for membrane fusion (18, 19, 20).
The viral and cell membrane juxtaposition allows the fusion of the lipid double layers, allowing the transposition of the viral nucleocapsid into the cytoplasm (2). Once penetrated inside the cell, the genomic RNA acts as an mRNA for the translation of the polyprotein replicase. The replicase gene contains two open reading frames, ORF1a and ORF1b, which encode the polyproteins 1a (pp1a) and 1ab (pp1ab) (21). Genomic RNA (+) is then used by the replicase enzyme to synthesize RNA (-) (negative sense), which in turn is used as a template for the synthesis of viral RNA (+). Furthermore, replicase synthesizes a set of subgenomic RNAs (sgRNAs), coding for structural and accessory proteins (22).
Replication and transcription of the coronavirus genome involve the formation of replication/transcription complexes (RTC), anchored to intracellular membranes through the proteins nsp3, nsp4 and nsp6 (23) and closely associated with double-membrane vesicles (DMV) formed due to the virus (24, 25).
The S, M, and E proteins are synthesized, inserted, and folded into the endoplasmic reticulum (ER) and then transported to the intermediate ER-Golgi compartment (ERGIC). N proteins are instead translated into the cytoplasm, where they encapsulate genomic RNA.
The assembly of virions occurs in ERGIC(2) (2). When assembled, the virions are exported through the vesicles; the mature particles are finally released by budding (26).
In some coronaviruses, a part of the S protein escapes the viral assembly; then it is exposed on the plasma membrane, causing fusion with the cells and causing the formation of a sizeable multinucleated cell known as syncytium: this allows the virus to spread without leaving the cells (2).
In eukaryotic cells, the endoplasmic reticulum (ER) is the leading site for the synthesis and folding of proteins secreted and transmembrane. When protein synthesis exceeds the folding capacity, the explained proteins accumulate in the ER causing ER stress (27).
Cells have developed homeostatic maintenance mechanisms, collectively known as UPR (unfolded protein response) (28), metabolic pathways that aim to restore the normalcy of ER by stopping translation and increasing the folding capacity (29). If, however, despite the activation of these pathways, homeostasis is not restored, the cell is directed toward death by apoptosis (30).
The studies have highlighted how many viruses and pathogens that target mitochondria affect the ways of programmed cell death (31). Therefore, ER stress and UPR activation may contribute significantly to the pathogenesis of Coronavirus (29).
Mitochondrial structure and function
Mitochondria are organelles key in maintaining cellular homeostasis, in the control of metabolism, in cellular aging, in starting some answers and in the activation of innate immunity pathways apoptotic, anti-apoptotic, and numerous other ways of the signal (32). Mitochondria are therefore the ideal target for many viruses that regulate the balance between the proteins of the Bcl-2 family (regulators of cell death), increasing their survival within the host cell on one side and implementing processes of apoptosis on the other hand when cell survival cannot be guaranteed (31, 33, 34).
The inner membrane (IM) is almost impermeable under physiological conditions, thus allowing to maintain the mitochondrial membrane potential (MMP, Δψm), given in turn by the electrochemical gradient created by the respiratory chain. An alteration or transition of MMP (Δψm) leads to the arrest of the normal biosynthetic and bioenergetic functions of the cells, to the release of various pro-apoptotic proteins (35) and finally to the generation of a “crisis” inside the cell. During viral infection, there is a transition of MMP (Δψm), and a prolonged loss of MMP (Δψm) leads to sometimes irreversible damage. Furthermore, the potential of the mitochondrial membrane (∆Ψm) plays an essential role in the process of sorting and segregating defective mitochondria (36, 37). Therefore, the action of a pathogen on mitochondria may impact cell fate, such as to induce or, on the contrary, hinder their death (35).
Physiologically, mitochondria undergo a series of processes: they lengthen by fusion, divide by fission, and undergo a controlled turnover through mitophagy (38). The dynamic processes of fusion, fission, and mitophagy support homeostasis and act as quality control of the mitochondria themselves (39), and play, more generally, an essential role in maintaining cellular health (40). Viruses interfere with these processes by altering mitochondrial dynamics to facilitate their proliferation.
The role of mitochondria in viral infection
Viruses behave as obligate parasites of host cells, modulate metabolism and physiology strategically, causing a dramatic alteration in cell architecture, the subcellular functions (41), and hijack the mitochondria impairs undo the degree of cellular functioning.
The host has different defense mechanisms, some of which depend precisely on the mitochondrial activity which, if on the one hand, it allows undertaking a path of activation of the cell cycle, on the other, it allows to start the programmed cell death (PCD), at the to prevent the spread of viral infection to neighboring cells (42).
Recent studies have shown that most viruses force cells to undergo apoptosis. However, from a virus point of view, host apoptosis appears to provide no benefit, so this is a topic of current research. Therefore, further studies clarifying the mechanisms underlying virus-induced PCD induction and cell lysis may help identify new drug targets for the treatment of viral infections (31).
Another topic that has attracted attention in recent years is related to the role of mitochondria in generating innate immune responses; this consequently implies an essential role in viral pathogenesis. Innate immunity recognizes pathogenic molecular patterns (PAMPs) and activates a series of defensive pathways, which sometimes lead to apoptosis of infected cells (38, 41). The mitochondria centralize some essential innate immune responses, based on the presence of mitochondrial proteins: mitochondrial antiviral signal (MAVS), gene stimulation signal interferon (STING) and NLRX1. Mitochondrial fusion would serve to increase the interactions of the MAVS with the signaling molecules downstream, generating a consequent synthesis of interferon (IFN). The mitochondrial fission instead would block this antiviral signaling (43).
Therefore, mitochondria are vital as regulators of survival and cell death but also play an important role in controlling cellular response functions following invasion by pathogens. From here, it is easy to understand why they are an ideal target for viruses.
SARS- CoV regulates apoptosis through different pathways
Apoptosis can be activated through intrinsic and extrinsic pathways. The intrinsic pathway is mediated by the Bcl2 family of proteins, in which BAX and BAK are pro-apoptotic, while Bcl2, Bcl-xL, and Mcl-1 are anti-apoptotic. As a result of cell stress (including viral infection), pro-apoptotic ones prevail. Bax increases the permeability of the external mitochondrial membrane (MOMP), which leads to the release of cytochrome C, a signal that directs the cell towards the beginning of apoptosis (caspases 3 and 7).
The extrinsic pathway exploits instead other mediators such as FasL, which leads to the activation of other procaspases, such as procaspase 8 and 10. Once activated, these caspases go to cleave and to activate other caspase initiators and executors, directly responsible for cell death.
The two paths must not be seen as clearly separate, but interconnected. For example, among the targets of caspase 8, there is Bid, a cytoplasm-located pro-apoptotic member of Bcl-2, capable of binding and stabilizing Bax which, by forming oligomeric pores on the external mitochondrial membrane (MOM), causes an increased permeability, thus starting the previously cited intrinsic apoptotic pathway (38, 44).
The apoptosis mediated by SARS- CoV occurs not only as a consequence of a direct action towards pro- and anti-apoptotic proteins but also as a result of ER stress due to the overload of the cells biosynthetic pathways. An endoplasmic reticulum under stress presents an accumulation of misfolded proteins, which leads to a saturation of protein synthesis and processing capacity (44).
There are three pathways in the cell, respectively called PERK, IRE1, and ATF6, to implement the aforementioned unfolded protein response, or UPR, which counteract ER stress, and if the cell homeostasis cannot be resumed, prompt the programmed cell death. In particular, the IRE1 pathway, in addition to compensating for ER stress, induces a cascade of signals, which ends with the activation of JNK (N-terminal kinase c- Jun). The activation of the IRE1-JNK pathway is necessary for the induction of autophagy in the cells with the stress of the endoplasmic reticulum, and it is always this pathway that favors the apoptosis of SARS- CoV infected cells. The JNK in fact, being a kinase phosphorylates other molecules, such as the nuclear transcription factors c- Jun and ATF2, it also dimerizes to form the activator protein 1 (AP-1) and by doing so, induces then the transcription of the Bak and FasL(1) (45-48). JNK finally migrates to the mitochondria, where it induces apoptosis by acting directly on the Bcl2 family (49) (Figures 1).
Figure 1.
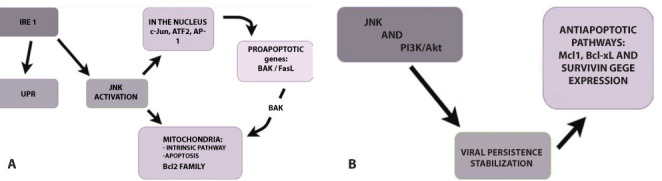
Panel A: Role of the pathway JNK (c- Jun kinase N-terminal) in the first stage of infection with SARS-CoV2: the protein is not explained response (UPR, in the figure); translocates transcription factors such as c- Jun and ATF2 into the nucleus and phosphorylates, dimers to form the activator protein 1 (AP-1) and thus induces the transcription of pro-apoptotic genes such as Bak and FasL; it migrates into the mitochondria where it activates the intrinsic pathway of apoptosis, acting on the Bcl2 family of proteins.
Panel B: Role of the JNK pathway in the advanced stages of infection: emerges through the anti-apoptotic. The switching of this path towards an attitude conducive to cell survival occurs when the infection has become persistent, to use the cell as a source of new viral particles.
A characteristic aspect of coronaviruses is the fact of “playing” with the life of the cells they infect: causing their death, now allowing them to survive. The tendency of JNK to bring the cell through pro-apoptotic cascade is valid only in the first stage of infection with SARS- CoV. It is known that, in persistently infected cells, this pathway tends to promote cell survival.
The pro-survival role of JNK has been discovered through experiments in which the treatments of cultured cells with JNK inhibitors effectively abolishes the persistence of SARS- CoV infection, suggesting the anti-apoptotic aspect of the JNK pathway. It is worth nothing that the transition to a pro-survival attitude would only occur when the infection became persistent, making the cell a reservoir of new viruses (1, 50).
SARS- CoV and antiviral response pathways
Numerous studies found that cells infected with SARS- CoV exhibit dysfunction of the typical response to interferon 1, a fundamental antiviral mechanism (51) (50).
To activate an antiviral response, the virus’ presence must be, first of all, detected. Infected cells have molecular sensors that recognize viral RNA, i.e., RIG-I and MDA5 (retinoic inducible gene I and gene 5 associated with melanoma differentiation), two essential innate immune receptors whose pathways converge in the mitochondrial antiviral signaling protein or MAVS. MAVS also is known as ISP-1 (which stands for interferon Beta promoter stimulator 1, suggesting its fundamental role in the antiviral response), in turn, facilitates the induction of an antiviral cell state by activating specific IRFs (interferon regulatory factors) a group of transcriptional activators/repressors (52, 53).
However, RNA viruses have developed strategies to antagonize these signaling pathways. In particular, the open reading frame 9b (ORF-9b) of SARS-CoV is found in the mitochondria, hindering those mentioned above MAVS-based antiviral signaling path and, at the same time, altering mitochondria’s morphology of the affected cells. The consequences in the whole-cell system of this last-mentioned effect will be explained further in this review.
SARS-CoV infected cells show decreased levels of MAVS protein expression, and consequently lower levels of TRAF3 and TRAF6 (TNF receptor-associated factors), which are two more downstream proteins usually recruited from MAVS.
The virus’ ORF-9b (open reading frame 9b) exploits the physiological role of two proteins: PCBP2 and AIP4 (poly (rC)-binding Protein 2 and atrophin-1 interacting protein 4). They usually account for the inhibition of the MAVS signalosome through ubiquitin-mediated degradation. The conclusion that ORF-9b’s action involves the elements in the MAVS signalosome could be drawn by observing an increased recruiting and enhanced action of PCBP2 and AIP4. The resulting decline in MAVS, TRAF3, and TRAF6 levels is, therefore, a limitation of the cell’s ability to generate a robust defensive interferon response (38, 52, 54).
SARS- CoV promotes the formation of autophagosomes
As previously noted, one of the effects of ORF-9b is a change in mitochondrial dynamics, more specifically by damaging them and inducing their fusion and elongation, as verified by electronic microscopy observations. Mitochondrial damage and elongation are two events strictly related to autophagy; indeed, Coronaviruses have been shown to act as triggers for autophagic processes (55). Mitochondrial elongation is an effect in stark contrast to what usually happens to mitochondria during a non-coronavirus viral infection, i.e., mitochondrial fission/fragmentation. In this case, too, ORF-9b comes into play, by shifting to mitochondria, it induces the formation of autophagosomes. This link between ORF-9b and both autophagy and mitochondrial fusion has been demonstrated as follows:
increased incorporation of a protein called LC3 into the membranes of the autophagosomes has been reported, following the expression of ORF-9b, and LC3 is a well-known index of cellular autophagy (45, 56).
ORF-9b increases the ubiquitination and degradation of DRP1 (dynamin-related protein 1), a protein that has a crucial role in mediating mitochondrial fission and elongation.
ORF-9b would, therefore, serve to promote cell survival during viral replication, because mitochondrial fusion would give rise to a process of safeguarding mitochondria from degradation and more generally of the cell from programmed cell death (an enhancement of autophagy and mitochondrial fusion correspond to suppression of mitophagy and apoptotic death (38, 52, 57).
Apoptosis, antiviral response, autophagy: roads that meet in mitochondria
SARS-CoV infection controls the intrinsic pathway of apoptosis, and the signals that determine programmed cell death or cell survival depend on whether or not MOMP is lost. The UPR IRE1-JNK path mediates this ambivalent control. JNK acts both directly at the mitochondrial and nuclear levels. In the JNK nucleus, it influences the expression of the genes involved in apoptosis, whose protein products are localized at the mitochondrial level, while in the mitochondria, it directly influences several members of the Bcl2 family.
The SARS- CoV ORF-9b acts in the mitochondria to induce the degradation of the signal system supported by MAVS / TRAF3 / TRAF6 and to reduce the antiviral signaling. The SARS- CoV ORF-9b causes mitochondrial stress and induces their fusion and elongation, while promoting autophagy and hindering mitophagy at the same time, thus allowing the infected cells to survive (56, 52).
What emerges from the previous paragraphs is that mitochondria exert quite an interesting role during Coronavirus infection, even more, if we take into account the fact that these observations apply to the responsible of the actual pandemics, SARS-CoV2. The IRE1 unfolded protein response pathway is involved in viral cell infection by any coronavirus; for what concerns ORF-9b, it is included in the previously mentioned 79,5% of shared genomic identity with SARS-CoV, and it acts in the same way. The last observation is especially useful if we consider that ORF-9b can act as a molecular target, as explained in the next section.
Orf 9b as a molecular target: current therapeutic applications
Currently, there is no therapy or specific antiviral drugs with proven efficacy to counteract the infection caused by SARS CoV 2; nevertheless, since the pandemic of COVID-19 has started, clinical trials went ongoing based on the knowledge we had from the first SARS CoV with which SARS CoV 2 shares most of the genomic identity.
Through the study of the viral genome, Nevan Krogan’s laboratory at the University of California in San Francisco took the first step towards identifying new drug targets against SARS CoV 2 infection (58). At the basis of this identification is a complete analysis of the interaction between viral proteins and human proteins (59), the study of transcription factors called “main regulators” (60), followed by a chemoinformatic identification of existing compounds which affect proteins in previously studied pathways. Sixty-six human proteins or host factors have been identified as the potential target of 69 American FDA approved drugs or compounds, currently under study (59).
For the moment and particularly for what concerns the molecular targets related to the mitochondrial role during coronavirus infection, two molecules already used in the clinic are known, both protein kinase inhibitors and which have the molecular target ORF-9b: Midostaurin, used in acute myeloid leukemia, in myelodysplastic syndromes, and systemic mastocytosis; Ruxolitinib, used in myelofibrosis. The other molecules, with the same molecular target, are instead tested and act on the phosphatase of the triphosphate nucleotides, a protector of the genome from the incorporation of genotoxic nucleotides.
Conclusions
Many host responses seem to contribute to the pathogenesis of viral infections and recent cellular and molecular studies have shown how viruses specifically affect mitochondria, leading to the loss of MMP, the implementation of pro-apoptotic and anti-apoptotic processes in favor of viral survival with the implementation of autophagy and the reduction of antiviral signaling, reducing the possibility of defense of the host. Mitochondria, therefore, play an essential role in the survival and cell death, so a better understanding of how viruses use mitochondrial responses to control cells can provide a basis for the development of new treatments.
To counteract SARS CoV 2 infection, it is crucial to understand the host’s hijacking in the way to develop specific drug therapies. According to a medical consensus and the most recent studies, it is crucial to systematically explore the host dependencies of SARS CoV 2. However, the recent emergence of this virus and our limited knowledge preclude the identification of molecule candidates for host-directed therapies (59). This gap may be filled up by a systematic map of the interaction between SARS CoV 2 proteins and human proteins.
Conflict of interest:
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article.
References
- Fung TS, Liu DX. Human Coronavirus: Host-Pathogen Interaction. Annu Rev Microbiol. 2019 Sep 8;73(1):529–57. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- Masters PS. The Molecular Biology of Coronaviruses. Advances in Virus Research. Adv Virus Res. 2006;65:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Curtis N. Pediatric Infectious Disease Journal. Vol. 39. Lippincott Williams and Wilkins; 2020. Coronavirus infections in children including COVID-19: An overview of the epidemiology, clinical features, diagnosis, treatment and prevention options in children; pp. 355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staico MF, Zaffanello M, Di Pietro G, Fanos V, Marcialis MA. The kidney in COVID-19: protagonist or figurant? Panminerva Med [Internet] 2020 May 20 [cited 2020 Jun 10] doi: 10.23736/S0031-0808.20.03965-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32432445 . [DOI] [PubMed] [Google Scholar]
- Gorbalenya AE, Snijder EJ, Spaan WJM. Severe Acute Respiratory Syndrome Coronavirus Phylogeny: toward Consensus. J Virol. 2004 Aug 1;78(15):7863–6. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Wunderink RG. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology [Internet] 2018 Feb 1 [cited 2020 Apr 25];23(2):130–7. doi: 10.1111/resp.13196. Available from: http://doi.wiley.com/10.1111/resp.13196 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. StatPearls. StatPearls Publishing; 2020 [cited 2020 Apr 25]. Features, Evaluation and Treatment Coronavirus (COVID-19) [Internet] Available from: http://www.ncbi.nlm.nih.gov/pubmed/32150360 . [PubMed] [Google Scholar]
- Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003 Oct 25;362(9393):1353–8. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li F, Shi ZL. Nature Reviews Microbiology. Vol. 17. Nature Publishing Group; 2019. Origin and evolution of pathogenic coronaviruses; pp. 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N Engl J Med [Internet] 2012 Nov 8 [cited 2020 Apr 25];367(19):1814–20. doi: 10.1056/NEJMoa1211721. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa1211721 . [DOI] [PubMed] [Google Scholar]
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature [Internet] 2003 Nov 27 [cited 2020 Apr 25];426(6965):450–4. doi: 10.1038/nature02145. Available from: http://www.nature.com/articles/nature02145 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Travanty EA, Oko L, Edeen K, Berglund A, Wang J, et al. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am J Respir Cell Mol Biol. 2013 Jun;48;(6):742–8. doi: 10.1165/rcmb.2012-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Yang X Lou, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 Mar 12;579(7798):270–3. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature [Internet] 2013 Aug 7 [cited 2020 Apr 25];500(7461):227–31. doi: 10.1038/nature12328. Available from: http://www.nature.com/articles/nature12328 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Military Medical Research. NLM (Medline) 2020;7:11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturman LS, Holmes K V, Behnke J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J Virol. 1980;33(1):449–62. doi: 10.1128/jvi.33.1.449-462.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TS, Huang M, Liu DX. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014 Dec 19;194:110–23. doi: 10.1016/j.virusres.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang IC, Bosch BJ, Li F, Li W, Kyoung HL, Ghiran S, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem. 2006 Feb 10;281(6):3198–203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Liu XB, Fang SG, Tay FPL, Liu DX. Acquisition of cell-cell fusion activity by amino acid substitutions in spike protein determines the infectivity of a coronavirus in cultured cells. PLoS One. 2009 Jul 2;4(7) doi: 10.1371/journal.pone.0006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L, Godeke G-J, Raamsman MJB, Masters PS, Rottier PJM. Retargeting of Coronavirus by Substitution of the Spike Glycoprotein Ectodomain: Crossing the Host Cell Species Barrier. J Virol. 2000 Feb 1;74(3):1393–406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use [Internet] [cited 2020 May 14] doi: 10.1093/nar/gkw530. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5009743/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki SG, Sawicki DL, Siddell SG. A Contemporary View of Coronavirus Transcription. J Virol. 2007 Jan 1;81(1):20–9. doi: 10.1128/JVI.01358-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra M, te Lintelo EG, Deijs M, Verheije MH, Rottier PJM, de Haan CAM. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J Virol. 2007 Nov 15;81(22):12323–36. doi: 10.1128/JVI.01506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K, Kikkert M, Van Den Worm SHE, Zevenhoven-Dobbe JC, Van Der Meer Y, Koster AJ, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008 Sep;6;(9):1957–74. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, Onderwater JJM, van der Meulen J, Koerten HK, et al. Ultrastructure and Origin of Membrane Vesicles Associated with the Severe Acute Respiratory Syndrome Coronavirus Replication Complex. J Virol. 2006 Jun 15;80(12):5927–40. doi: 10.1128/JVI.02501-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse-Locker J, Ericsson M, Rottier PJM, Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: Evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol. 1994;124(1–2):55–70. doi: 10.1083/jcb.124.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud J-M, et al. Lipid-Induced ER Stress: Synergistic Effects of Sterols and Saturated Fatty Acids. Traffic [Internet] 2009 Jun 1 [cited 2020 Apr 25];10(6):673–90. doi: 10.1111/j.1600-0854.2009.00903.x. Available from: http://doi.wiley.com/10.1111/j.1600-0854.2009.00903.x . [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Nature Reviews Molecular Cell Biology. Vol. 8. Nature Publishing Group; 2007. Signal integration in the endoplasmic reticulum unfolded protein response; pp. 519–29. [DOI] [PubMed] [Google Scholar]
- Fung TS, Liu DX. Frontiers in Microbiology. Vol. 5. Frontiers Research Foundation; 2014. Coronavirus infection, ER stress, apoptosis and innate immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Nature Cell Biology. Vol. 13. NIH Public Access; 2011. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress; pp. 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshi L, Wang H-V, Hong J-R. In: Mitochondrial Diseases. InTech; 2018. Modulation of Mitochondria During Viral Infections. [Google Scholar]
- Bardanzellu F, Pintus MC, Fanos V, Marcialis MA. Journal of Pediatric and Neonatal Individualized Medicine. Vol. 8. Hygeia Press di Corridori Marinella; 2019. Once we were bacteria mitochondria to infinity and beyond; p. e080106. [Google Scholar]
- Viruses as Modulators of Mitochondrial Functions [Internet] [cited 2020 May 16] Available from: https://www.hindawi.com/journals/av/2013/738794/ [Google Scholar]
- Wallace DC. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu Rev Genet. 2005 Dec;39;(1):359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshi ML, Su Y-C, Hong J-R. RNA Viruses: ROS-Mediated Cell Death. Int J Cell Biol. 2014;2014 doi: 10.1155/2014/467452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glingston RS, Deb R, Kumar S, Nagotu S. Microbes and Infection. Vol. 21. Elsevier Masson SAS; 2019. Organelle dynamics and viral infections: at cross roads; pp. 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward CL, Prakobwanakit S, Mosessian S, Chow SA. Integrase Interacts with Nucleoporin NUP153 To Mediate the Nuclear Import of Human Immunodeficiency Virus Type 1. J Virol. 2009 Jul 1;83(13):6522–33. doi: 10.1128/JVI.02061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Syed GH, Kim SJ, Siddiqui A. Biochimica et Biophysica Acta - Molecular Cell Research. Vol. 1853. Elsevier; 2015. Mitochondrial dynamics and viral infections: A close nexus; pp. 2822–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Cell. Vol. 125. Elsevier; 2006. Mitochondria: Dynamic Organelles in Disease, Aging, and Development; pp. 1241–52. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet [Internet] 2009 Oct 15;18(R2):R169–76. doi: 10.1093/hmg/ddp326. Available from: https://pubmed.ncbi.nlm.nih.gov/19808793 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [Recognition of Pathogens by Innate Immunity] - PubMed [Internet] [cited 2020 May 17] Available from: https://pubmed.ncbi.nlm.nih.gov/19188883/ [Google Scholar]
- Mitochondria: More than Just “Power Plants” in Stem Cells [Internet] [cited 2020 May 17] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5660791/ [Google Scholar]
- Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010 Feb;11;(2):133–8. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushi M, Yoshinaka Y, Matsuoka Y, Hatakeyama S, Ishizaka Y, Kirikae T, et al. Monitoring of S Protein Maturation in the Endoplasmic Reticulum by Calnexin Is Important for the Infectivity of Severe Acute Respiratory Syndrome Coronavirus. J Virol. 2012 Nov 1;86(21):11745–53. doi: 10.1128/JVI.01250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Nature Reviews Molecular Cell Biology. Vol. 9. Nature Publishing Group; 2008. Apoptosis: Controlled demolition at the cellular level; pp. 231–41. [DOI] [PubMed] [Google Scholar]
- Ogata M, Hino S-i, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol Cell Biol. 2006 Dec 15;26(24):9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang XZ, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (80- ) 2000 Jan 28;287(5453):664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354(6353):494–6. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 Is Phosphorylated and Inactivated by an ASK1/Jun N-Terminal Protein Kinase Pathway Normally Activated at G 2 /M. Mol Cell Biol. 1999 Dec 1;19(12):8469–78. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T, Fukushi S, Saijo M, Kurane I, Morikawa S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim Biophys Acta - Mol Basis Dis. 2005 Jun 30;1741(1–2):4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M, Pichlmair A, Martinez-Sobrido L, Cros J, Garcia-Sastre A, Haller O, et al. Inhibition of Beta Interferon Induction by Severe Acute Respiratory Syndrome Coronavirus Suggests a Two-Step Model for Activation of Interferon Regulatory Factor 3. J Virol. 2005 Feb 15;79(4):2079–86. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C-S, Qi H-Y, Boularan C, Huang N-N, Abu-Asab M, Shelhamer JH, et al. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J Immunol. 2014 Sep 15;193(6):3080–9. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Antiviral Signaling Through Pattern Recognition Receptors. J Biochem [Internet] 2006 Dec 26;141(2):137–45. doi: 10.1093/jb/mvm032. Available from: https://doi.org/10.1093/jb/mvm032 . [DOI] [PubMed] [Google Scholar]
- You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009 Nov 1;10(12):1300–8. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- Maier HJ, Britton P. Viruses. Vol. 4. Multidisciplinary Digital Publishing Institute (MDPI); 2012. Involvement of autophagy in coronavirus replication; pp. 3440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Van Der Bliek AM. Science. Vol. 337. American Association for the Advancement of Science; 2012. Mitochondrial fission, fusion, and stress; pp. 1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011 Jun 21;108(25):10190–5. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathways in COVID-19 replication and pre-clinical drug target identification [Internet] [cited 2020 Apr 25] Available from: https://www.drugtargetreview.com/article/58628/host-pathways-in-coronavirus-replication-and-covid-19-pre-clinical-drug-target-identification-using-proteomic-and-chemoinformatic-analysis/ [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’Meara MJ, et al. A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. bioRxiv [Internet] 2020 Mar 27 [cited 2020 Apr 25] 2020.03.22.002386. Available from: https://www.biorxiv.org/content/10.1101/2020.03.22.002386v1.full . [Google Scholar]
- Guzzi PH, Mercatelli D, Ceraolo C, Giorgi FM. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. J Clin Med [Internet] 2020 Apr 1 [cited 2020 Apr 25];9(4):982. doi: 10.3390/jcm9040982. Available from: https://www.mdpi.com/2077-0383/9/4/982 . [DOI] [PMC free article] [PubMed] [Google Scholar]