

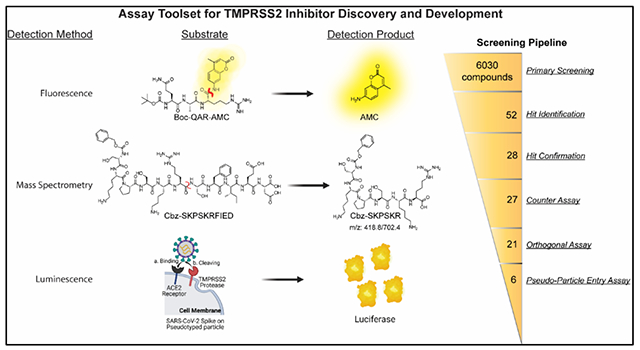
Abstract
SARS-CoV-2 is the causative viral pathogen driving the COVID-19 pandemic that prompted an immediate global response to the development of vaccines and antiviral therapeutics. For antiviral therapeutics, drug repurposing allows for rapid movement of existing clinical candidates and therapies into human clinical trials to be tested as COVID-19 therapies. One effective antiviral treatment strategy used early in symptom onset is to prevent viral entry. SARS-CoV-2 enters ACE2-expressing cells when the receptor-binding domain of the spike protein on the surface of SARS-CoV-2 binds to ACE2 followed by cleavage at two cut sites by TMPRSS2. Therefore, a molecule capable of inhibiting the protease activity of TMPRSS2 could be a valuable antiviral therapy. Initially, we used a fluorogenic high-throughput screening assay for the biochemical screening of 6030 compounds in NCATS annotated libraries. Then, we developed an orthogonal biochemical assay that uses mass spectrometry detection of product formation to ensure that hits from the primary screen are not assay artifacts from the fluorescent detection of product formation. Finally, we assessed the hits from the biochemical screening in a cell-based SARS-CoV-2 pseudotyped particle entry assay. Of the six molecules advanced for further studies, two are approved drugs in Japan (camostat and nafamostat), two have entered clinical trials (PCI-27483 and otamixaban), while the other two molecules are peptidomimetic inhibitors of TMPRSS2 taken from the literature that have not advanced into clinical trials (compounds 92 and 114). This work demonstrates a suite of assays for the discovery and development of new inhibitors of TMPRSS2.
Keywords: COVID-19, TMPRSS2, Antiviral, Drug Repurposing, High-Throughput Screening
Graphical Abstract
The severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) pandemic has driven the need for development of effective therapeutics to be used as prophylactics and as a treatment for infected patients. At the outset of the pandemic, there were no approved therapeutics for treating any coronaviruses, which led to the initiation of several human clinical trials on existing clinical candidates and approved drugs. Drug repurposing allows for drugs to be fast-tracked to the clinic for trials addressing a different disease than it was originally intended. An initial step in drug repurposing is the development of in vitro assays capable of generating efficacy data on the candidates for drug repurposing, which is critical for either supporting or denying their movement into human clinical trials. An example of this was remdesivir (GS-5734, Gilead Sciences Inc.) that had previously been in clinical trials for treating the Ebola virus but was repurposed as a SARS-CoV-2 treatment following its demonstration as a viral polymerase inhibitor within an in vitro experiment using live SARS-CoV-2 virus, leading to FDA approval.1, 2
Host cell targets have also received significant attention, partly because pharmacologic modulation should be invariable against viral variants/mutants.3 Host antiviral targets are often human proteins whose function has been co-opted and are essential to the viral replication cycle.4 Transmembrane protease serine 2 (TMPRSS2) is a human protein within the family of type-II transmembrane serine proteases (TTSP) expressed in epithelial cells of the human lungs, airways, and gastrointestinal tracts.5, 6 TMPRSS2 has become a therapeutic target due to its role in SARS-CoV-2 infection of a cell, and the existence of approved therapeutics able to inhibit its protease activity and be assessed for drug repurposing.7 A mechanism for SARS-CoV-2 entry includes binding of its surface glycoprotein spike (S) with the human cell-surface receptor angiotensin-converting enzyme 2 (ACE2), followed by cleavage at two sequences on the spike protein that can be done by human cell surface proteases, such as TMPRSS2, which causes a conformational change in spike resulting in membrane fusion and release of the viral RNA into the host cell.8–10 TMPRSS2 is considered the primary protease within the lung due to it having the highest co-expression with ACE2.5
We have reported an in vitro enzymatic assay using recombinant TMPRSS2 and a fluorogenic substrate, BOC-QAR-AMC, to demonstrate whether drug repurposing candidates, such as camostat and nafamostat, could inhibit the protease activity.11 Additionally, the enzymatic assay was used to evaluate hits identified from virtual screens.12, 13 Since our initial report on the development of an in vitro assay using recombinant TMPRSS2, several other in vitro assays have been developed. A cell-based in vitro assay using the fluorogenic substrate, BOC-QAR-AMC, in HEK293T cells overexpressing TMPRSS2 allows for quantification of proteolytic activity of TMPRSS2 and assessment of inhibitors within the cellular context compared to recombinant protein.14 Fluorescence resonance energy transfer (FRET)-based peptides mimicking the spike cleavage sites (S1/S2 and S2’) with recombinant TMPRSS2 demonstrated the capability of TMPRSS2 to cleave at both relevant spike cleavage sites.15 A split-luciferase in vitro assay using an engineered full-length human TMPRSS2 expressing a C-terminal HiBiT tag reported on both plasma membrane-associated and intracellular TMPRSS2 expression levels and demonstrated that homoharringtonine and halofuginone reduced total TMPRSS2 expression.16 Cell lines expressing TMPRSS2 (either endogenously or through transfection) have been used with pseudotyped particles or live virus assays to demonstrate the importance of TMPRSS2 in SARS-CoV-2 infection, and assess therapeutic candidates.17, 18 Altogether, assays on TMPRSS2 have been extensively used to assess therapeutic effectiveness.
Here, we report the screening of inhibition of TMPRSS2 from several drug repurposing candidates within 6030 molecules in the NCATS drug libraries and an in-house compiled protease inhibitor library by using the fluorogenic peptide assay. Furthermore, we report the development of a liquid chromatography tandem mass spectrometry (LC-MS/MS)-based quantification assay using recombinant TMPRSS2 and an unlabeled peptide substrate that mimics the SARS-CoV-2 spike cleavage site, S2’. We demonstrate the suitability of the assay in 384-well plates, and its utility as an orthogonal assay capable of detecting false positive hits from the fluorogenic peptide assay. Additionally, the assay showed the ability of TMPRSS2 to cleave the spike cleavage site mimicked by the peptide, which agrees with results derived from use of FRET peptides within another study.15 Hits from the biochemical assays were further tested in a pseudotyped particle cell entry assay. The particles were pseudotyped with either the spike protein of the ‘wildtype’ (WA1 + D614G) or Delta strain (B.1.617.2), in Calu-3 human non-small cell lung cancer cells to demonstrate inhibition and cellular efficacy. Altogether, six molecules demonstrated activity in the biochemical and cell-based assays while remaining inactive in the counter assays. Two of these molecules, camostat and nafamostat, were the most potent inhibitors of TMPRSS2 and are both in human clinical trials as an antiviral against COVID-19.19, 20 Another two drug repurposing candidates, otamixaban and PCI-27483, demonstrated activity but have not entered human clinical trials for treatment of COVID-19. The final two molecules were identified from the literature as peptidomimetic inhibitors of TMPRSS2.21 Several other molecules were found to be weaker inhibitors. This work demonstrates an assay toolset for the discovery and development of new TMPRSS2 inhibitors.
Results and Discussion
Assay design
Following our initial report on the development of a biochemical assay capable of monitoring TMPRSS2 activity and inhibition, we set out to conduct a drug repurposing screen.11 To briefly summarize the biochemical assay, yeast-expressed recombinant TMPRSS2 (aa 106-492, CUSAbio) was used at 175 nM along with a fluorogenic peptide substrate, BOC-QAR-AMC at 10 μM, near its Km of 33 μM, to monitor enzymatic activity via release of the caged 7-amino-4-methylcourmarin (AMC) fluorophore from the peptide at an emission wavelength of 440 nm (Figure 1). The commercial source for TMPRSS2 from our initial report was Creative BioMart.11 When comparing commercial sources of TMPRSS2, CUSAbio allowed for use of a lower TMPRSS2 concentration (0.175 μM) within the assay than Creative BioMart (1 μM). However, for both sources of enzyme, the active concentration of TMPRSS2 was calculated by fitting dose-response data from nafamostat to the Morrison Equation (Supplementary Figure 4G). This was done due to reporting IC50 values lower than the enzyme concentration within the assay. Buffer conditions were tested and optimized to include: 50 mM Tris pH 8, 150 mM NaCl and 0.01% Tween20. We achieved a signal-to-background (S:B) and Z’ of 3.2 and 0.86, respectively in a 1536-well plate. This demonstrated appropriate performance for this assay to be useful for quantitative high-throughput screening (qHTS).22
Figure 1.
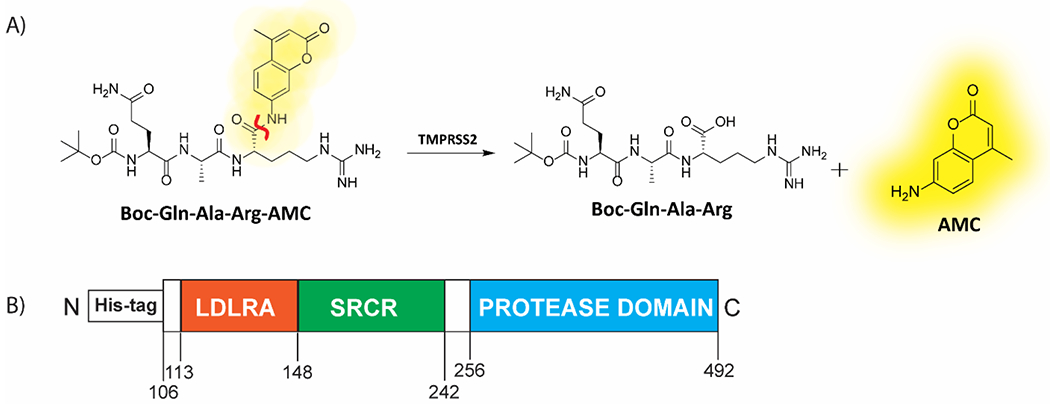
(A) Scheme displaying the enzymatic assay principle for the fluorogenic peptide substrate. The fluorogenic peptide substrate Boc-Gln-Ala-Arg-AMC has low fluorescence compared to the fluorescent 7-amino-4-methylcoumarin (AMC), which is released upon proteolytic cleavage. The scissile bond is indicated in red. (B) Schematic of the truncated yeast-expressed recombinant TMPRSS2 used in the fluorogenic assay, containing the low-density lipoprotein receptor A (LDLRA) domain, scavenger receptor cysteine-rich (SRCR) domain and protease domain.
Drug repurposing qHTS of annotated small molecule libraries
Three compound libraries were selected for qHTS that are ideal for drug repurposing efforts: the NCATS Pharmaceutical Collection (NPC) contains all approved agents from USA, Europe, and Japan as of 2017 along with additional clinical candidates (2,678 compounds), the Mechanism Interrogation Plate (MIPE) is an oncology-focused library with a combination of approved and clinical agents (2,480 compounds), and an in-house compiled protease inhibitor library (PIL, 872 compounds) to screen against TMPRSS2, a total of 6,030 compounds (qHTS assay protocol shown in Table 1). Along with the potential for repurposing, the compounds in these libraries are annotated for published mechanisms of action. Each 1536-well plate contained 32 positive and 64 negative control wells to monitor assay performance. When screening the NPC library, the average plate Z’ was 0.76 with an average S:B of 2.39 (Figure 2A). The MIPE collection had an average plate Z’ of 0.72 with an average S:B of 2.33 (Figure 2B). The PIL collection had an average plate Z’ of 0.87 with an average S:B of 4.86 (Figure 2C).
Table 1.
Detailed TMPRSS2 fluorogenic biochemical assay protocol for qHTS.
| Step # | Process | Notes |
|---|---|---|
| 1 | 20 nL of peptide substrate dispensed into 1536-well plates. | Peptide (250x, dissolved in DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte) into a Corning 1536-well plate (Cat #3724). |
| 2 | Compounds and controls (20 nL) were pre-spotted into 1536-well plates. | Inhibitor (250x) or vehicle control (DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte). Compounds in dose-response were dispensed to columns 5-48 and controls dispensed into columns 1-4. |
| 3 | TMPRSS2 diluted in assay buffer dispensed into 1536-well plates. | TMPRSS2 (reconstituted in 50% glycerol at 8.75 μM, 50x) in assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) was dispensed using a BioRAPTR (Beckman Coulter). Total reaction volume of 5 μL. |
| 4 | Incubated at RT for 1 hr | Final assay conditions were 10 μM peptide and 0.175 μM TMPRSS2 in assay buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.01% Tween20) |
| 5 | Read on PHERAstar FSX (BMG Labtech) | Fastest read settings, Fluorescence Intensity module: 340 nm excitation, 440 nm emission (Cat # 1601A2, BMG Labtech) |
Figure 2.
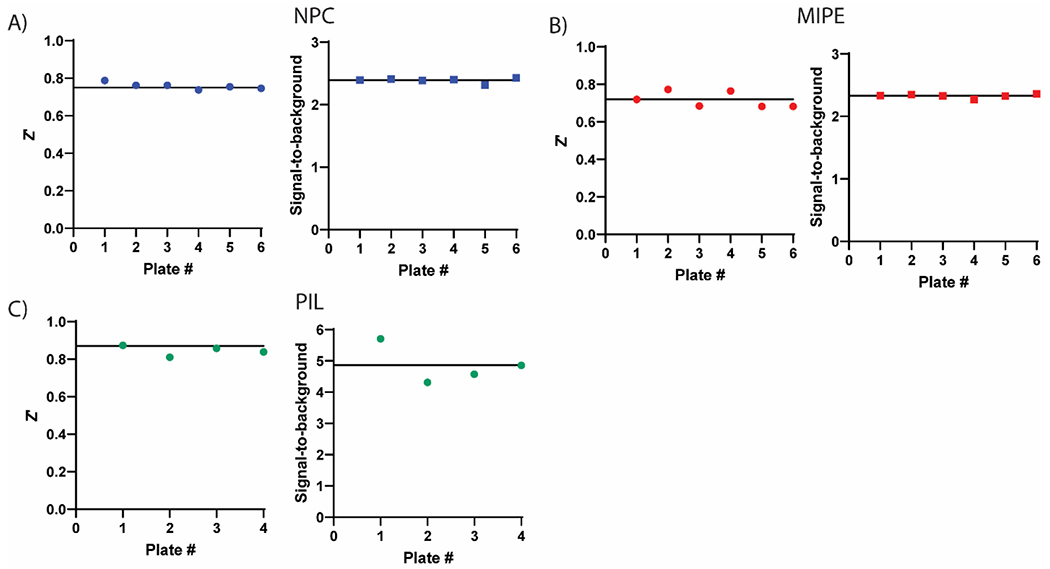
Assay performance for primary screening of the compound libraries in the TMPRSS2 fluorogenic biochemical activity assay. Z’ scores and signal-to-background values are plotted as data points for each plate. There were 32 positive and 64 negative control wells on each 1536-well plate. (A) NCATS Pharmaceutical Collection (NPC), 2,678 compounds, (B) Mechanism Interrogation Plate (MIPE) library, 2,480 compounds and (C) protease inhibitor library (PIL), 872 compounds. Black horizontal lines represent the mean values.
Hit identification
The primary screen for NPC and MIPE were executed using compound concentrations of 40 μM, 8 μM and 1.6 μM, and hit selection criteria was set for those having a maximum response of ≥ −30% inhibition. A total of 14 compounds (0.5% hit rate) were identified as hits from the NPC library and 14 compounds (0.6% hit rate) were identified from the MIPE library. The primary screen for the PIL used compound concentrations of 40 μM, 10 μM, 2.5 μM, and 0.625 μM. Using the same hit selection criterion (max response ≥ −30% inhibition) yielded 37 active compounds (4.2% hit rate). Thus, from a screen of 6,030 compounds a total of 52 unique hits were identified, as there was redundancy from multiple hits presented between the libraries. Within the primary screen, 18,962 data points were generated. All qHTS data from the NPC and MIPE primary screens, and detailed protocol sheets, are available for download at the OpenData Portal website (https://opendata.ncats.nih.gov/covid19/).23
Hit confirmation
To confirm the hits identified from primary screening of the compound libraries, we tested each hit compound in triplicate dose-response in the primary fluorogenic biochemical assay using fresh stock. Compounds were plated in triplicate, 11-point, 1:3 dilution series using an ECHO 655 (LabCyte) acoustic dispenser. Of the 52 hits identified from the primary screening, 28 hits retained a max response inhibition of ≥ −30% when tested in triplicate dose-response. Next, the 28 hits were tested in a fluorescent counterassay, to detect false-positive hit compounds capable of quenching AMC fluorescence and mimicking inhibition, that involved the addition of compounds in dose-response to a buffer containing 1 μM AMC that approximates the estimated 10% enzymatic cleavage in the fluorogenic assay (i.e., starting concentration of substrate is 10 μM, counterassay protocol shown in Table 2). The counter-assay identified one compound (tannic acid) capable of quenching the AMC fluorescent signal ~30%. Altogether, 27 compounds confirmed as inhibitors of TMPRSS2, which had ≥ −30% inhibition and ≤ −10% fluorescence quenching (HTS screening pipeline, Supplementary Figure 1).
Table 2.
Detailed fluorescence counterassay protocol for qHTS hits.
| Step # | Process | Notes |
|---|---|---|
| 1 | 20 nL of 7-amino-4-methylcoumarin dispensed into 1536-well plates. | AMC (250x, dissolved in DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte) into a Corning 1536-well plate (Cat #3724). |
| 2 | Compounds and controls (20 nL) were pre-spotted into 1536-well plates. | Inhibitor (250x) or vehicle control (DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte). Compounds in dose-response were dispensed to columns 5-48 and controls dispensed into columns 1-4. |
| 3 | Assay buffer dispensed into 1536-well plates. | Assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) was dispensed using a BioRAPTR (Beckman Coulter). Total reaction volume of 5 μL. |
| 4 | Incubated at RT for 5 min | Final assay conditions were 1 μM AMC with inhibitor or control (DMSO) in assay buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.01% Tween20). Columns 3 and 4 had 0.1 μM AMC with control (DMOS) in assay buffer. |
| 5 | Read on PHERAstar FSX (BMG Labtech) | Fastest read settings, Fluorescence Intensity module: 340 nm excitation, 440 nm emission (Cat # 1601A2, BMG Labtech) |
Orthogonal mass spectrometry assay development
We next developed a biochemical assay with an unlabeled peptide substrate and mass spectrometric detection. The marker peptide sequence contained the S2’ cleavage site on the spike protein of SARS-CoV-2 (Figure 3A). The peptide substrate sequence selected was Cbz-SKPSKRSFIED (scissile amide bond bolded) that yielded cleavage product peptides of Cbz-SKPSKR and SFIED by TMPRSS2 (Figure 3B). The LC-MS/MS assay showed the ability of TMPRSS2 to cut the surrogate peptide at the spike cleavage site, which agrees with results derived from use of FRET peptides in a separate study.15 Directly measuring an unlabeled peptide product, rather than a fluorescent product, removes the possibility of false positives from compound fluorescence interference.24, 25
Figure 3.
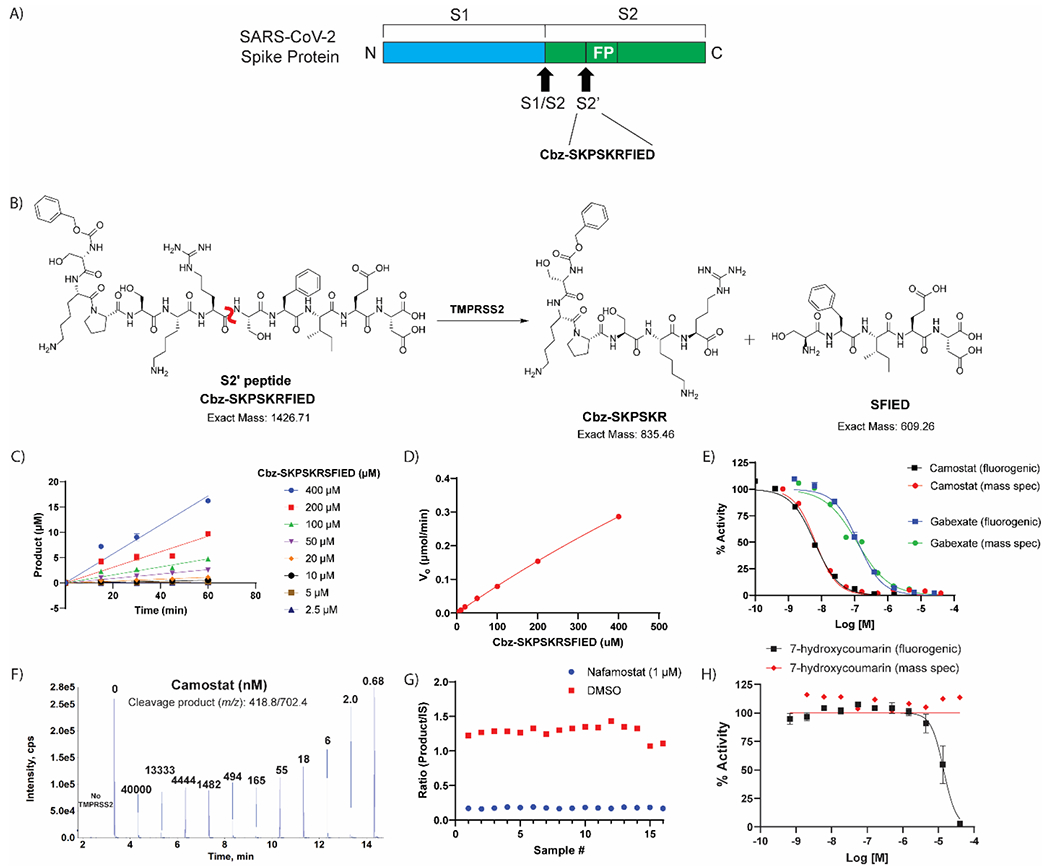
Label-free mass spectrometry biochemical assay. (A) Peptide derived from the known S2’ cleavage site of SARS-CoV-2 Spike (S) protein. (B) Scheme displaying the enzymatic assay principle for the unlabeled peptide substrate. The unlabeled peptide substrate, Cbz-SKPSKRFIED, is cleaved by TMPRSS2 to create two cleavage products, Cbz-SKPSKR and SFIED. The scissile bond is indicated in red. (C) Initial velocity (V0) was calculated for each substrate concentration by plotting product formation vs time. (D) The V0 for each concentration were plotted against the various substrate concentrations to obtain the Vmax and Km. Vmax: 1.95 μmol/min and Km: estimated 2320 μM. (E) Comparison of TMPRSS2 fluorogenic detection and mass spectrometry detection assays by assessing dose-response inhibition by camostat and gabexate. (F) Mass spectrometry traces of the cleavage product (m/z: 418.8/702.4) showing that addition of camostat prevents product formation in dose-response. Each peak is labeled with the concentration of camostat (nM) for that condition. (G) Assay performance from the mass spectrometry detection assay when screening the hits identified from the primary screening. Z’ of 0.71 and S:B of 7.37. (H) Dose-response inhibition of 7-hydroxycoumarin against TMPRSS2 in both fluorogenic and mass spectrometry detection assays showing the fluorescent molecule as a false-positive hit in the fluorogenic assay, but not interfering with the mass spectrometry detection assay.
The optimum charge state, declustering potential, collision energy and fragment ion intensity were determined via automated flow injection analysis using an LS1 autosampler and LeadScape software from Sound Analytics (Supplementary Figure 2). The +6 amu isotopically labeled version of the cleavage product peptide (Cbz-SK-{13C5 15N Pro}-SKR) was used to determine conditions for the unlabeled cleavage product peptide (Cbz-SKPSKR) since a discrete standard of the unlabeled version was not available (Supplementary Figure 3). The linear range of the LC-MS/MS method was assessed using Cbz-SK-{13C5 15N Pro}-SKR, m/z 421.6/708.2. The standard curve was linear in the concentration range from 1 nM to 2000 nM. To determine the Km of the substrate peptide, Cbz-SKPSKRSFIED, the TMPRSS2 concentration was held constant at 400 nM while substrate concentration was varied from 2.5 μM to 400.26 The enzymatic reaction was quenched using an equal volume of Acetonitrile:Water (90:10, 0.1% formic acid) containing 100 nM of Cbz-SK-{13C5 15N Pro}-SKR as an internal standard (assay protocol shown in Table 3). Quantification of the TMPRSS2 protease activity was determined by calculating peak area ratio of the cleavage product peptide (Cbz-SKPSKR, m/z 418.8/702.4) and internal standard peptide (Cbz-SK-{13C5 15N Pro}-SKR, m/z 421.6/708.2). From the substrate titration experiment, the initial velocity was determined at each substrate concentration by plotting the amount of product formed against time (Figure 3C). Initial velocities were plotted against substrate concentrations (Figure 3D), indicating that the Km was indeterminate as it yielded a value of 2320 μM, which is higher than the maximum substrate concentration in the assay. Due to the indeterminate Km, the assay conditions were set using an identical concentration of peptide substrate as the fluorogenic assay (10 μM). Under these conditions, the IC50 obtained from the mass spectrometry detection assay for camostat and gabexate were 6.6 nM (95% CI: 5.4, 8.2) and 119 nM (95% CI: 76.8, 181.3), respectively, which is statistically equivalent to that obtained from the fluorogenic assay, 6.2 nM (95% CI: 5.5, 7.0) and 130 nM (95% CI: 107.2, 147.5) (Figure 3E, F). Positive and negative control conditions were assessed in a 384-well plate format to demonstrate appropriate performance, which showed a S:B and Z’ of 7.37 and 0.71, respectively (Figure 3G).
Table 3.
Detailed TMPRSS2 mass spectrometry detection biochemical assay.
| Step # | Process | Notes |
|---|---|---|
| 1 | 200 nL of peptide substrate dispensed into 384-well plates. | Peptide (Cbz-SKPSKRSFIED, 250x, dissolved in DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte) into a Greiner 384-well plate (Cat #781201). |
| 2 | Compounds and controls (200 nL) were pre-spotted into 384-well plates. | Inhibitor (250x) or vehicle control (DMSO) was dispensed using an ECHO 655 acoustic dispenser (LabCyte). Compounds in dose-response were dispensed to columns 5-24 and controls dispensed into columns 1-4. |
| 3 | TMPRSS2 diluted in assay buffer dispensed into 384-well plates. | TMPRSS2 (reconstituted in 50% glycerol at 8.75 μM, 22x) in assay buffer (50 mM Tris pH 8, 150 mM NaCl) was dispensed using a BioRAPTR (Beckman Coulter). Total reaction volume of 50 μL. |
| 4 | Incubated at RT for 1 hr | Final assay conditions are 10 μM peptide and 0.4 μM TMPRSS2 in assay buffer (50 mM Tris-HCl pH 8, 150 mM NaCl) |
| 5 | Addition of quench solution (50 μL) | Quench solution (90:10 ACN:H2O + 0.1% formic acid + 100 nM of IS (Cbz-SK-{13C5 15N Pro}-SKR. Total volume of 100 μL. |
| 6 | Dilution of reaction (10-fold) | Dilution of reaction 10-fold using 20 mM ammonium formate + 0.1% formic acid. |
| 7 | Measure mass detection on SciEx 6500 | The LC mobile phases (MP) were A: 0.1% difluoroacetic acid in water and B: acetonitrile, and a gradient elution was used that went from 5% MPB to 90% MPB in 1.0 min with a flow rate of 0.70 mL/min. The product cleavage fragment (Cbz-SKPSKR): 418.8/702.4 m/z. The IS cleavage fragment: 421.64/708.24 |
The 27 confirmed hits from the fluorogenic assay were tested in the mass spectrometry detection assay. One qHTS hit that failed to inhibit in the mass spectrometry detection assay was 7-hydroxycoumarin, a fluorescent molecule similar to 7-amino-4-methylcoumarin that is released upon cleavage of the fluorogenic substrate. Based on the fluorogenic assay, 7-hydroxycoumarin showed nearly full inhibition of TMPRSS2 at 40 μM; yet there was no inhibition detected within the mass spectrometry detection assay (Figure 3H). The cause for this discrepancy likely comes from 7-hydroxycoumarin being fluorescent; thereby, the endpoint fluorescent detection is overwhelmed by the amount of inhibitor fluorescence compared to the added cleaved product fluorescence. Since there is no increased fluorescence from the cleaved product at T60 (when quench solution is added) compared to T0, it appears that TMPRSS2 was fully inhibited with no cleaved product formation. This is an example of an assay interference false positive due to compound auto-fluorescence.24 The benefits of the mass spectrometry detection assay are twofold: 1) lower number of interference compounds identified, as demonstrated by 7-hydroxycoumarin and 2) capability of using a more physiologically relevant substrate that is unlabeled. Overall, 21 of the 27 compounds assessed for inhibition of TMPRSS2 showed activity. Unlike the assay interference compound, 7-hydroxycoumarin, the other five compounds that did not inhibit TMPRSS2 within the mass spectrometry detection assay was likely due to their weak activity within the fluorogenic assay.
SARS-CoV-2-S pseudotyped particle entry assay
Finally, we demonstrated the effectiveness of these small molecules at preventing TMPRSS2-dependent viral entry into cells using a SARS-CoV-2 spike pseudotyped particle (PP) entry assay performed with the human airway epithelium cell line Calu-3, as previously described (Figure 4A,B).18 In addition to being the airway epithelium cell model, Calu-3 cells rely on serine proteases, like TMPRSS2, for viral entry when compared to other cell line models, which makes this cell line ideal for testing inhibitors of TMPRSS2.17 Two different pseudotyped particles with spike from different variants were used to assess TMPRSS2 inhibitors: one containing the Delta variant (B.1.617.2) spike protein that bears the mutations T19R, G142D, del156-157, R158G, L452R, T478K, D614G, P681R, D950N, and one containing the WA1 + D614G spike sequence. Notably, the Delta variant carries a P681R mutation in the S1/S2 cut site of the spike protein that may affect the cleavage activity by TMPRSS2; however, there are no mutations within the S2’ cut site. It’s unclear how the various mutations may affect the cleavage activity by TMPRSS2. A cytotoxicity counter assay was performed to assess whether hits were cytotoxic towards Calu-3 cells, which would produce a false-positive indicator of activity in the PP assay.
Figure 4.
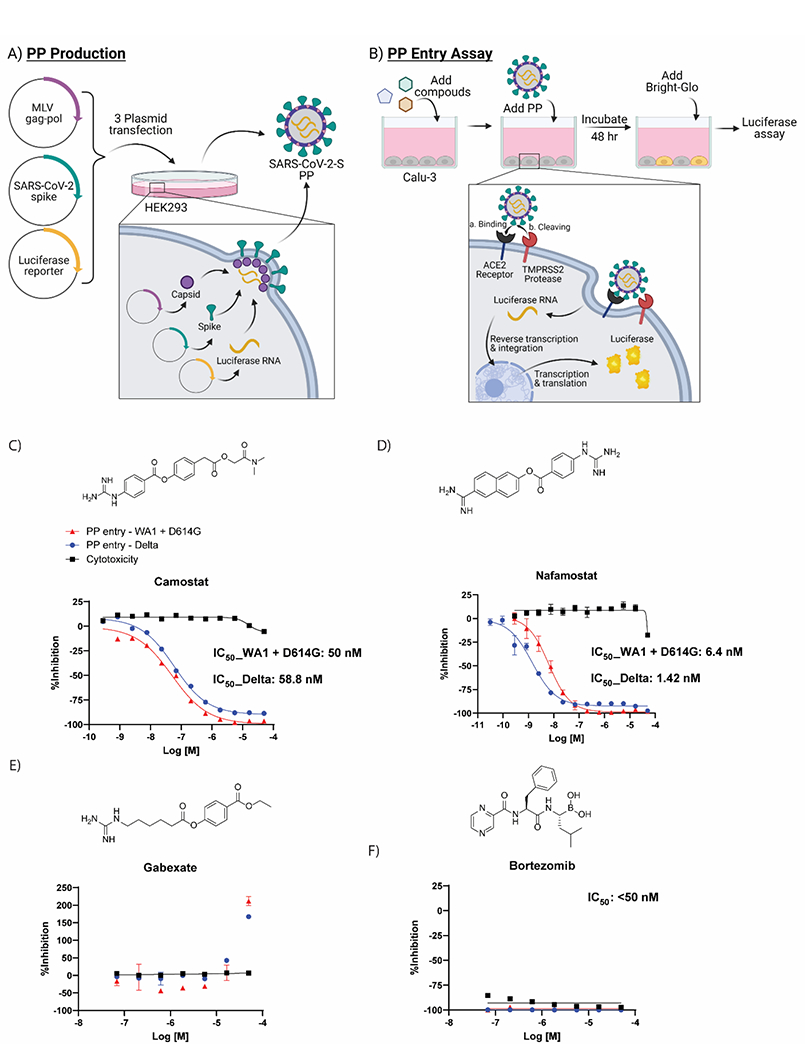
Cell-based pseudotyped particle assay. (A) Pseudotyped particle production. Three plasmids (pCMV-MLVgag-pol, pcDNA-SARS-CoV-2 spike, and pTG-Luciferase) are co-transfected into HEK-293T/17 cells. The plasmids express MLV core gag-pol polyprotein, SARS-CoV-2 spike glycoproteins, and luciferase RNAs, which together assemble into pseudotyped particles. (B) Pseudotyped particle assay scheme. Order of addition of reagents to Calu-3 cells is 1. compounds, 2. pseudotyped particles and 3. the Promega reagent Bright-Glo. Once cell entry of pseudotyped particle occurs, RNAs of pseudotyped particles are released into the cell, where they are reverse transcribed into DNAs, integrated into the genome and express luciferase reporter enzyme. The amount of luminescence following addition of Bright-Glo is proportional to the amount of pseudotyped particle entry into cells. Illustration was made with BioRender. Activity of clinically approved inhibitors in prevention of pseudotyped particle entry using the Spike sequence from the Delta variant (blue), WA1 + D614G variant (red) and cytotoxicity counter-assay (black). The molecular structures and dose-response inhibition of particle entry by (C) camostat, (D) nafamostat, (E) gabexate, (F) bortezomib. The calculated concentrations required for 50% inhibition (IC50) are displayed in nM.
Compounds having confirmed inhibition of TMPRSS2 in both biochemical assays and in the cellular assay are listed in Table 4 (dose-response curves shown in Supplementary Figures 4 and 5). Of the three most potent inhibitors of TMPRSS2 from the biochemical screening, camostat and nafamostat also demonstrated the greatest potency within the PP entry assay against the Delta pseudotyped particle with IC50 of 59 nM and 1.4 nM, respectively (Figure 4C,D). However, gabexate was inactive at inhibiting viral particle entry (Figure 4E). This result is in agreement with other studies reporting gabexate is inactive in cell-based assays testing TMPRSS2 inhibition.27, 28 Notably, bortezomib appeared to demonstrate a highly potent inhibition of viral entry (IC50 <50 nM); however, the cytotoxicity counterassay revealed it was due to high cytotoxicity of the Calu-3 cells, which again demonstrates the importance and necessity of appropriate counterassays in designing an assay pipeline (Figure 4F). Additional evidence that 7-hydroxycoumarin is a false-positive identified from the fluorogenic assay is its lack of activity within the cell-based assay evaluating viral entry inhibition. The rest of the HTS hits had weak to no inhibition in the cell-based PP assay, most likely due to their already weak activity against TMPRSS2 from the biochemical assay (Supplementary Table 1).
Table 4.
Compounds having confirmed inhibition of TMPRSS2 in both biochemical assays and in the cellular assay.
| Biochemical (fluorogenic) | Biochemical (fluorogenic counterassay) | Biochemical (mass spectrometry) | SARS-CoV-2 PP (Delta) | SARS-CoV-2 PP (WA1 + D614G) | Calu-3 (cytotoxicity) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound Name | Structure | AC50 (μM) | Max Response (%) | AC50 (μM) | AC50 (μM) | Max Response (%) | AC50 (μM) | Max Response (%) | AC50 (μM) | Max Response (%) | Status for Human Use |
| Nafamostat mesilate |
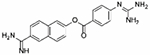
|
0.00011 | −1.9 | 0.0002 | 0.0014 | −100% | 0.0064 | −100% | N/D | 0% | Approved |
| Camostat |

|
0.0062 | −5 | 0.0055 | 0.059 | −92% | 0.05 | −95% | N/D | 0% | Approved |
| PCI-27433 |
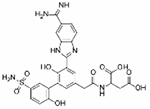
|
1.8 | 61.3 | 1.4 | 15.85 | −66% | 16.5 | −66% | N/D | 0% | Clinical Trials |
| Cmpd 92 (Meyer) |
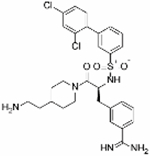
|
0.41 | −5.6 | 0.28 | 3.98 | 92% | 2.05 | −70% | N/D | 0% | N/T |
| Cmpd 114 (Meyer) |
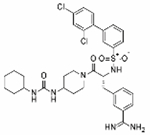
|
0.57 | −6.2 | 0.26 | 15.27 | 99% | 5.35 | −88% | N/D | 0% | N/T |
| Otamixaban |
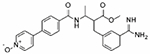
|
1.64 | −1 | 1.23 | 18.01 | 54% | 20 | −50% | N/D | 0% | Clinical Trials |
N/D = Not Detected
N/T = Not Tested
Here we developed the first reported mass spectrometry-based detection assay for TMPRSS2 activity, and demonstrate a TMPRSS2 early drug discovery pipeline. To demonstrate proof-of-concept, we performed a HTS using our drug repurposing libraries.
The advantages to developing a mass spectrometry-based detection assay are twofold: 1) the unlabeled substrate can be more physiologically relevant leading to higher quality inhibitors being identified, 2) the unlabeled substrate avoids identification of a false positive that may arise in a fluorescence-based detection assay from inhibitor fluorescent quenching or compound fluorescence.24 For example, 7-hydroxycoumarin was identified as a hit within the fluorogenic assay. However, at the highest concentration tested of 40 μM, this molecule increases fluorescence to 1300x above the baseline level of fluorescence coming from the fluorogenic peptide substrate. Consequently, there is no increased fluorescence upon TMPRSS2 cleavage of the AMC from the fluorogenic peptide substrate; therefore, it instead appears that the enzyme was fully inhibited. The 7-hydroxycoumarin molecule did not show inhibition of TMPRSS2 in the mass spectrometry-based detection assay nor any inhibition of pseudotyped particle entry into Calu-3 cells.
Drug repurposing screening on in vitro assays yields initial proof-of-concept data that can be used to fast-track a drug to the clinic for trials addressing a different disease than it was originally intended. Our primary drug repurposing screen used recombinant TMPRSS2 with a fluorogenic peptide substrate with fluorescence detection. The screen included compounds from three in-house libraries totaling 6,030 compounds with many being drug repurposing candidates. Each compound was tested at multiple concentrations leading to generation of 18,962 data points. The primary screen yielded 52 hits. Upon assessing each of these compounds in an 11-point dose-response and in a fluorescence counter assay, only 27 compounds remained as confirmed hits. Each of these compounds were tested in dose-response for inhibition of TMPRSS2 in the mass spectrometry detection assay and in dose-response for inhibition of pseudotyped particle entry into Calu-3 cells.
Notably, potency of the molecules was not significantly shifted between the two different types of pseudotyped particles from the two variants (WA1 + D614G and Delta). This demonstrates the validity of drugging host targets such as TMPRSS2 to avoid viral escape of the antiviral therapy. The current variant of concern (VOC), omicron, has quickly become the dominant variant in the US and many parts of the world. The omicron spike protein sequence bears numerous mutations, and data suggests that these lead to an increased preference for cathepsin proteases over TMPRSS2 cleavage entry pathway compared to earlier variants, such as WA1 + D614G and delta.29, 30 This correlates with a shift in tropism of omicron SARS-CoV-2 virus and PP towards cells of the respiratory tract, compared with that of the lungs, which show higher TMPRSS2 expression.29, 30 In addition, omicron virus and PP are less sensitive to the TMPRSS2 inhibitor camostat, and more sensitive to the cathepsin inhibitor E64d, than previous variants. However, it should be noted that omicron spike can still utilize TMPRSS2 entry pathway despite the lower potency than previous variants.29, 30 This, combined with the fact that the TMPRSS2 vs cathepsin usage of future variants remains to be seen, places TMPRSS2 as an important target for antiviral therapeutic development.
Altogether, six compounds demonstrated inhibition of TMPRSS2 in biochemical and cellular assays. Those six compounds include two approved drugs (nafamostat and camostat, both approved in Japan to treat pancreatitis), two clinical candidates (PCI-27483 and otamixaban) and two literature compounds (compound 92 and compound 114).21
Camostat is an oral drug used for the treatment of chronic pancreatitis, with a typical regimen of 200 mg taken three times a day (TID), and for the treatment of postoperative reflux esophagitis (100 mg, TID).31 It is a pro-drug with a terminal ester which is rapidly and efficiently cleaved to give 4-(4-guanidinobenzoyloxy)phenylacetic acid (FOY-251). The parent drug is not detected in plasma and FOY-251 has a short, 1 hour terminal half-life.32 This presents a problem for the maintenance of efficacious plasma levels of drug if the inhibition is rapidly reversible. However, camostat forms a stable, covalent, acyl-enzyme intermediate. The half-life of this intermediate, which is identical to the intermediate formed from nafamostat, was determined to be 14.7 hours.33 The slow enzyme off-rate, if it were to translate in vivo, would help to compensate for the rapid clearance of FOY-251 enabling a test of the hypothesis that camostat could be a useful oral treatment for COVID-19.
The first evidence of efficacy of camostat in a randomized, double-blind, placebo-controlled trial, was in the early treatment of mild to moderate severity COVID-19. This trial showed that camostat (200 mg) taken three times a day (TID) for 7 days resulted in a shorter illness course, with attenuation of loss of smell and taste. The trial did not however achieve the primary endpoint of reducing the viral load by PCR in the upper respiratory tract.34
A trial in mild to moderate patients (ACTIV-2) with camostat (200 mg) given four times a day for seven days, did not show an improvement in symptoms, although smell and taste was not evaluated, and there was no effect on viral load by PCR.35 Another trial testing a higher dose of camostat (600 mg) given four times daily for up to 14 days in a similar patient population, did not show an improved time to negative SARS-CoV-2 test.36 Finally, treatment of hospitalized, patients with camostat (200 mg TID for 5 days) did not affect time to clinical improvement or disease progression.37 The efficacy of camostat in the one outpatient trial is consistent with the accumulated experience that antivirals are best used early in the course of COVID-19, soon after diagnosis and prior to the onset of the inflammatory phase of the disease.
Nafamostat is marketed as an IV treatment for acute symptoms of pancreatitis and for disseminated intravascular coagulopathy (DIC).38 This derives from it actions as an inhibitor of trypsin-like serine proteases involved in hemostasis. This raises the possibility that it could be useful in the care of moderate to severe COVID-19 as advanced disease is associated with acute respiratory distress syndrome, including coagulopathy, and particularly lung microvascular thrombosis.11 Its antiviral action is a bonus, although as noted with camostat, the benefit of that is likely to be attenuated in advanced disease. Currently nafamostat is being studied in the hospital setting where it can be titrated against coagulation markers to tests its utility for the management of acute respiratory distress syndrome. A randomized, double blind, placebo controlled clinical trial is underway to test the hypothesis that nafamostat can lower lung function deterioration and the need for ICU admission of hospitalized patients.39
PCI-27483 was originally developed as a highly potent and selective inhibitor of the serine protease Factor VIIa (FVIIa) when in complex with tissue factor (TF) that has a normal physiological role in the initiation of the blood clotting cascade in response to an injured blood vessel wall.40 However, TF has been shown to be overexpressed in numerous primary tumors and is associated with tumor progression and worsened survival in cancer patients. This complex may contribute to tumor invasiveness by promoting cell migration and angiogenesis.41 Consequently, PCI-27483 entered clinical trials for patients with advanced pancreatic cancer in combination with gemcitabine. This trial has completed and though the combination was well tolerated, there was no demonstrated efficacy.42 PCI-27483 was first identified as a drug repurposing candidate as an inhibitor of TMPRSS2 by using a structure-based phylogenetic computational tool called 3DPhyloFold to identify structurally similar serine proteases, such as FVIIa, with known inhibitors that docked well to the TMPRSS2 structural model.43 Later, PCI-27483 was identified and confirmed to be an inhibitor of TMPRSS2 by NCATS scientists using a structural modeling and binding-site analysis of TMPRSS2 followed by a structure-based virtual screening and molecular docking approach.12 Though it has been demonstrated within in vitro studies as an inhibitor of TMPRSS2, it has yet to enter clinical trials as an antiviral treatment for COVID-19. The other clinical candidate, otamixaban, is an inhibitor of factor Xa (fXa), a critical serine protease in the blood coagulation cascade that catalyzes the conversion of prothrombin to thrombin.44, 45 Otamixaban entered clinical trials as an anticoagulant for patients with non-ST-segment elevation acute coronary syndromes (NSTE-ACS) against a control arm using unfractionated heparin plus eptifibatide. Otamixaban was shown to be safe but lack efficacy relative to the control arm.46 Otamixaban was first identified as a drug repurposing candidate as an inhibitor of TMPRSS2 within a virtual screen.47 Corroborating data came from the Noe lab and our group that included computational, biochemical and cellular data to demonstrate its inhibition of TMPRSS2.12, 13 It has yet to enter clinical trials as an antiviral treatment for COVID-19.
The two literature compounds, 92 and 114, have sulfonylated 3-amindinophenylalanylamide core structures that were among the initial synthetic inhibitors of TMPRSS2.21 The Steinmetzer lab, developed a fluorescence based assay using a fluorogenic peptide substrate used with recombinant TMPRSS2 to identify 92 and 114 as potent inhibitors with a Ki of 0.9 nM and 5 nM, respectively. Both were shown to be not cytotoxic within Calu-3 cells. Additionally, 92 significantly reduced the replication of H1N1 and H3N2 influenza virus strains in Calu-3 cells. However, no further studies had been done with respect to SARS-CoV-2 with these compounds. We demonstrate inhibition of TMPRSS2 with an IC50 of ~300 nM for both compounds and blocking of entry into Calu-3 cells using pseudotyped particles with SARS-CoV-2 spike protein with an IC50 of 4 μM and 15 μM for 92 and 114, respectively.
Methods
Reagents.
Recombinant Human TMPRSS2 protein expressed from yeast (human TMPRSS2 residues 106–492, N-terminal 6x His-tag) (cat.# CSB-YP023924HU) was acquired from Cusabio. The fluorogenic peptide substrate, Boc-QAR-AMC. HCl was obtained from Bachem (cat.# I-1550). The unlabeled peptide substrate, Cbz-SKPSKRSFIED was custom ordered from LifeTein.
Fluorogenic Peptide Screening Protocol 1536-Well Plate.
To a 1536-well black plate was added Boc-QAR-AMC substrate (2.5 mM, 20 nL, 250x) and inhibitor (20 nL, 250x) using an ECHO 655 acoustic dispenser (LabCyte). To that was dispensed TMPRSS2 (reconstituted in 50% glycerol at 8.75 μM, 50x) in assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) using a BioRAPTR (Beckman Coulter) to give a total reaction volume of 5 μM. Following 1 h of incubation at RT, detection was done using the PHERAstar with 340 nm excitation and 440 nm emission (Table 1).
Fluorescence Counter Assay 1536-Well Plate.
To a 1536-well black plate was added 7-amino-4-methylcoumarin (0.25 mM, 20 nL, 250x) and inhibitor (20 nL, 250x) to columns 5-48 using an ECHO 655 acoustic dispenser (LabCyte). To the control columns 1-2 was added 7-amino-4-methylcoumarin (0.25 mM, 20 nL, 250x) and DMSO (20 nL), and to the control columns 3-4 was added 7-amino-4-methylcoumarin (0.025 mM, 20 nL, 250x) and DMSO (20 nL). Assay buffer (50 mM Tris pH 8, 150 mM NaCl, 0.01% Tween20) was added to the full plate to give a total reaction volume of 5 μL. Detection was done using the PHERAstar with 340 nm excitation and 440 nm emission. Fluorescence was normalized relative to a negative control containing 7-amino-4-methylcoumarin at 0.1 μM (−100% activity, low fluorescence) and a positive control containing 7-amino-4-methylcoumarin at 1 μM (0% activity, high fluorescence). An inhibitor causing fluorescence quenching would be identified as having a concentration-dependent decrease on AMC fluorescence resulting in a <0% activity (Table 2).
Mass Spectrometry-based Assay 384-well plate.
To a 384-well plate (Greiner 781201) was added Cbz-SKPSKRSFIED substrate (2.5 mM, 200 nL, 250x) and inhibitor (200 nL, 250x) using an ECHO 655 acoustic dispenser (LabCyte). To that was dispensed TMPRSS2 (reconstituted in 50% glycerol at 8.75 μM, 22x) in assay buffer (50 mM Tris pH 8, 150 mM NaCl) using a BioRAPTR (Beckman Coulter) to give a total reaction volume of 50 μL. Following 1 h of incubation at RT, the reaction was quenched by adding 90:10 ACN:H2O + 0.1% formic acid + 100 nM of internal standard (Cbz-SK-{13C5 15N Pro}-SKR, 50 μL) to give a total volume of 100 μL. Samples were further diluted 10-fold using 20 mM ammonium formate + 0.1% formic acid prior to injection into a 6500 mass spectrometer (Sciex) equipped with an LS1 autosampler, 1290 Binary Gradient pumps (Agilent) and LeadScape software (Sound Analytics, Niantic, CT). A 2x20mm Halo C18 (Mac-Mod) column was used for LC separation and quantification. The mobile phases (MP) were MPA: 0.1% Difluoroacetic acid in water and MPB: Acetonitrile. A linear gradient elution was used running from 5% MPB to 90% MPB in 1.0 min at a flow rate of 0.70 mL/min. Quantification of the TMPRSS2 protease activity was determined by calculating peak area ratio of the cleavage product peptide (Cbz-SKPSKR, m/z 418.8/702.4) and internal standard peptide (Cbz-SK-{13C5 15N Pro}-SKR, m/z 421.6/708.2) (Table 3).
Data Process and Analysis.
To determine compound activity in the assay, the concentration–response data for each sample was plotted and modeled by a four-parameter logistic fit yielding IC50 and maximal response values. Raw plate reads for each titration point were first normalized relative to a positive control containing nafamostat at 1 μM (0% activity, full inhibition) and a negative control containing DMSO-only wells (100% activity, basal activity). Data normalization, visualization, and curve fitting were performed using Prism (GraphPad, San Diego, CA).
SARS-CoV-2-S Pseudotyped Particle Entry Assays.
A SARS-CoV-2 Spike pseudotyped particle (PP) entry assay was performed as previously described (Chen et. al.) In short, 40,000 cells/well Calu3 cells (catalog HTB-55, ATCC) were seeded in white, solid bottom 96-well microplates (Greiner BioOne) in 100 μL/well media and incubated at 37 °C with 5% CO2 overnight (~16 h). Then, the supernatant was removed, and compounds were added as 50 μL/well, 2X solutions in media. Cells were incubated with compounds for 1 h at 37 °C with 5% CO2, before 50 μL/well of SARS-CoV-2-S pseudotyped particles (PP) was added. We used the PP pseudotyped SARS-CoV-2 spike from the Delta variant B.1.617.2 (catalog 21H13, Codex Biosolutions) with a C-terminal 19 amino acid deletion using the murine leukemia virus system as previously described (Millet et. al.). The plates were then spinoculated by centrifugation at 1500 rpm (453g) for 45 min and incubated for 48 h at 37 °C with 5% CO2 to allow the cell entry of PP and the expression of the luciferase reporter. After the incubation, the supernatant was removed, and 20μL/well of the Bright-Glo Luciferase detection reagent (catalog E2620, Promega) was added to assay plates and incubated for 5 min at room temperature. The luminescence signal was measured using a PHERAStar plate reader (BMG Labtech). The data were normalized with wells containing SARS-CoV-2-S PPs as 0% and wells containing control bald PP as −100%. A cytotoxicity counter screen was performed using the same protocol without the addition of PP, with an adenosine triphosphate (ATP) content assay kit (catalog 6016949, Promega).
Supplementary Material
Figure S1. Screening pipeline for inhibitors of TMPRSS2S2
Figure S2. Scheme displaying the enzymatic assay principle and MS/MS molecular fragmentation
Figure S3. Mass spectrometry trace of the +6 amu isotopically labeled version of the cleavage product peptide (Cbz-SK-{13C5 15N Pro}-SKR, m/z 421.6/708.2)
Figure S4. Dose-response data from the fluorescence biochemical assay and fluorescence counterassay for the six actives
Figure S5. Dose-response data from the SARS-CoV-2-S pseudotyped particle entry assays and cytotoxicity counterassay for the six actives
Supplementary Table 1. List of 27 hits identified following confirmation and counterassay testing
Synopsis:
The left half of the TOC describes the substrates, products and detection methods used within the three assays used to identify an inhibitor of TMPRSS2. Those assays are apart of the assay toolset. The right half of the TOC is a summary of the screening results from all assays involved.
Acknowledgments
The authors acknowledge James Inglese for helpful discussions on enzyme kinetics. This work was supported by the National Center for Advancing Translational Sciences, Division of Preclinical Innovation.
Footnotes
Supporting Information
The Supporting Information is available free of charge at _.
References
- 1.Wang M; Cao R; Zhang L; Yang X; Liu J; Xu M; Shi Z; Hu Z; Zhong W; Xiao G, Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research 2020, 30 (3), 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eastman RT; Roth JS; Brimacombe KR; Simeonov A; Shen M; Patnaik S; Hall MD, Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Central Science 2020, 6 (5), 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baggen J; Vanstreels E; Jansen S; Daelemans D, Cellular host factors for SARS-CoV-2 infection. Nature Microbiology 2021, 6 (10), 1219–1232. [DOI] [PubMed] [Google Scholar]
- 4.Hall MD; Anderson JM; Anderson A; Baker D; Bradner J; Brimacombe KR; Campbell EA; Corbett KS; Carter K; Cherry S; Chiang L; Cihlar T; de Wit E; Denison M; Disney M; Fletcher CV; Ford-Scheimer SL; Götte M; Grossman AC; Hayden FG; Hazuda DJ; Lanteri CA; Marston H; Mesecar AD; Moore S; Nwankwo JO; O’Rear J; Painter G; Singh Saikatendu K; Schiffer CA; Sheahan TP; Shi P-Y; Smyth HD; Sofia MJ; Weetall M; Weller SK; Whitley R; Fauci AS; Austin CP; Collins FS; Conley AJ; Davis MI, Report of the National Institutes of Health SARS-CoV-2 Antiviral Therapeutics Summit. The Journal of Infectious Diseases 2021, 224 (Supplement_1), S1–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann M; Hofmann-Winkler H; Smith JC; Krüger N; Arora P; Sørensen LK; Søgaard OS; Hasselstrøm JB; Winkler M; Hempel T; Raich L; Olsson S; Danov O; Jonigk D; Yamazoe T; Yamatsuta K; Mizuno H; Ludwig S; Noé F; Kjolby M; Braun A; Sheltzer JM; Pöhlmann S, Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertram S; Heurich A; Lavender H; Gierer S; Danisch S; Perin P; Lucas JM; Nelson PS; Pöhlmann S; Soilleux EJ, Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLOS ONE 2012, 7 (4), e35876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jankun J, COVID-19 pandemic; transmembrane protease serine 2 (TMPRSS2) inhibitors as potential drugs. Translation: The University of Toledo Journal of Medical Sciences 2020, 7, 1–5. [Google Scholar]
- 8.Bestle D; Heindl MR; Limburg H; Van Lam van T; Pilgram O; Moulton H; Stein DA; Hardes K; Eickmann M; Dolnik O; Rohde C; Klenk H-D; Garten W; Steinmetzer T; Böttcher-Friebertshäuser E, TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Science Alliance 2020, 3 (9), e202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peacock TP; Goldhill DH; Zhou J; Baillon L; Frise R; Swann OC; Kugathasan R; Penn R; Brown JC; Sanchez-David RY; Braga L; Williamson MK; Hassard JA; Staller E; Hanley B; Osborn M; Giacca M; Davidson AD; Matthews DA; Barclay WS, The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nature Microbiology 2021, 6 (7), 899–909. [DOI] [PubMed] [Google Scholar]
- 10.Zang R; Castro MFG; McCune BT; Zeng Q; Rothlauf PW; Sonnek NM; Liu Z; Brulois KF; Wang X; Greenberg HB; Diamond MS; Ciorba MA; Whelan SPJ; Ding S, TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Science Immunology 2020, 5 (47), eabc3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shrimp JH; Kales SC; Sanderson PE; Simeonov A; Shen M; Hall MD, An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacology & Translational Science 2020, 3 (5), 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X; Shrimp JH; Guo H; Xu M; Chen CZ; Zhu W; Zakharov AV; Jain S; Shinn P; Simeonov A; Hall MD; Shen M, Discovery of TMPRSS2 Inhibitors from Virtual Screening as a Potential Treatment of COVID-19. ACS Pharmacology & Translational Science 2021, 4 (3), 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hempel T; Elez K; Krüger N; Raich L; Shrimp JH; Danov O; Jonigk D; Braun A; Shen M; Hall MD; Pöhlmann S; Hoffmann M; Noé F, Synergistic inhibition of SARS-CoV-2 cell entry by otamixaban and covalent protease inhibitors: pre-clinical assessment of pharmacological and molecular properties. Chemical Science 2021, 12 (38), 12600–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azouz Nurit P, K. AM, Callahan Victoria, Akhrymuk Ivan V, Elez Katarina, Raich Uuis, Henry Brandon M, Benoit Justin L, Benoit Stefanie W, Noe Frank, Kehn-Hall Kylene, Rothenberg Marc E, Alpha 1 Antitrypsin is an Inhibitor of the SARS-CoV-2–Priming Protease TMPRSS2. Pathogens And Immunity 2021, 6(1), 55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kastenhuber ER; Jaimes JA; Johnson JL; Mercadante M; Muecksch F; Weisblum Y; Bram Y; Schwartz RE; Whittaker GR; Cantley LC , Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. bioRxiv 2021, 2021.03.31.437960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y; Lear TB; Evankovich JW; Larsen MB; Lin B; Alfaras I; Kennerdell JR; Salminen L; Camarco DP; Lockwood KC; Tuncer F; Liu J; Myerburg MM; McDyer JF; Liu Y; Finkel T; Chen BB, A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nature Communications 2021, 12 (1), 3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann M; Kleine-Weber H; Schroeder S; Krüger N; Herrler T; Erichsen S; Schiergens TS; Herrler G; Wu N-H; Nitsche A; Müller MA; Drosten C; Pöhlmann S, SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181 (2), 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CZ; Xu M; Pradhan M; Gorshkov K; Petersen JD; Straus MR; Zhu W; Shinn P; Guo H; Shen M; Klumpp-Thomas C; Michael SG; Zimmerberg J; Zheng W; Whittaker GR, Identifying SARS-CoV-2 Entry Inhibitors through Drug Repurposing Screens of SARS-S and MERS-S Pseudotyped Particles. ACS Pharmacology & Translational Science 2020, 3 (6), 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NIH Clinical Trials of Camostat for COVID-19. https://clinicaltrials.gov/ct2/results?cond=&term=camostat&cntry=&state=&city=&dist=.
- 20.NIH Clinical Trials of Nafamostat for COVID-19. https://clinicaltrials.gov/ct2/results?cond=COVID-19&term=nafamostat&cntry=&state=&city=&dist=
- 21.Meyer D; Sielaff F; Hammami M; Böttcher-Friebertshäuser E; Garten W; Steinmetzer T, Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochemical Journal 2013, 452 (2), 331–343. [DOI] [PubMed] [Google Scholar]
- 22.Inglese J; Auld DS; Jadhav A; Johnson RL; Simeonov A; Yasgar A; Zheng W; Austin CP, Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proceedings of the National Academy of Sciences 2006, 103 (31), 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brimacombe KR; Zhao T; Eastman RT; Hu X; Wang K; Backus M; Baljinnyam B; Chen CZ; Chen L; Eicher T; Ferrer M; Fu Y; Gorshkov K; Guo H; Hanson QM; Itkin Z; Kales SC; Klumpp-Thomas C; Lee EM; Michael S; Mierzwa T; Patt A; Pradhan M; Renn A; Shinn P; Shrimp JH; Viraktamath A; Wilson KM; Xu M; Zakharov AV; Zhu W; Zheng W; Simeonov A; Mathé EA; Lo DC; Hall MD; Shen M, An OpenData portal to share COVID-19 drug repurposing data in real time. bioRxiv 2020. [Google Scholar]
- 24.Thorne N; Auld DS; Inglese J, Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Current Opinion in Chemical Biology 2010, 14 (3), 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adam GC; Meng J; Rizzo JM; Amoss A; Lusen JW; Patel A; Riley D; Hunt R; Zuck P; Johnson EN; Uebele VN; Hermes JD, Use of High-Throughput Mass Spectrometry to Reduce False Positives in Protease uHTS Screens. Journal of Biomolecular Screening 2014, 20 (2), 212–222. [DOI] [PubMed] [Google Scholar]
- 26.Acker MG; Auld DS, Considerations for the design and reporting of enzyme assays in high-throughput screening applications. Perspectives in Science 2014, 1 (1), 56–73. [Google Scholar]
- 27.Yamamoto M; Kiso M; Sakai-Tagawa Y; Iwatsuki-Horimoto K; Imai M; Takeda M; Kinoshita N; Ohmagari N; Gohda J; Semba K; Matsuda Z; Kawaguchi Y; Kawaoka Y; Inoue J.-i., The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12 (6), 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann M; Schroeder S; Kleine-Weber H; Müller MA; Drosten C; Pöhlmann S, Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrobial Agents and Chemotherapy 2020, 64 (6), e00754–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng B; Abdullahi A; Ferreira IATM; Goonawardane N; Saito A; Kimura I; Yamasoba D; Gerber PP; Fatihi S; Rathore S; Zepeda SK; Papa G; Kemp SA; Ikeda T; Toyoda M; Tan TS; Kuramochi J; Mitsunaga S; Ueno T; Shirakawa K; Takaori-Kondo A; Brevini T; Mallery DL; Charles OJ; Baker S; Dougan G; Hess C; Kingston N; Lehner PJ; Lyons PA; Matheson NJ; Ouwehand WH; Saunders C; Summers C; Thaventhiran JED; Toshner M; Weekes MP; Maxwell P; Shaw A; Bucke A; Calder J; Canna L; Domingo J; Elmer A; Fuller S; Harris J; Hewitt S; Kennet J; Jose S; Kourampa J; Meadows A; O’Brien C; Price J; Publico C; Rastall R; Ribeiro C; Rowlands J; Ruffolo V; Tordesillas H; Bullman B; Dunmore BJ; Gräf S; Hodgson J; Huang C; Hunter K; Jones E; Legchenko E; Matara C; Martin J; Mescia F; O’Donnell C; Pointon L; Shih J; Sutcliffe R; Tilly T; Treacy C; Tong Z; Wood J; Wylot M; Betancourt A; Bower G; Cossetti C; De Sa A; Epping M; Fawke S; Gleadall N; Grenfell R; Hinch A; Jackson S; Jarvis I; Krishna B; Nice F; Omarjee O; Perera M; Potts M; Richoz N; Romashova V; Stefanucci L; Strezlecki M; Turner L; De Bie EMDD; Bunclark K; Josipovic M; Mackay M; Butcher H; Caputo D; Chandler M; Chinnery P; Clapham-Riley D; Dewhurst E; Fernandez C; Furlong A; Graves B; Gray J; Hein S; Ivers T; Le Gresley E; Linger R; Kasanicki M; King R; Kingston N; Meloy S; Moulton A; Muldoon F; Ovington N; Papadia S; Penkett CJ; Phelan I; Ranganath V; Paraschiv R; Sage A; Sambrook J; Scholtes I; Schon K; Stark H; Stirrups KE; Townsend P; Walker N; Webster J; Butlertanaka EP; Tanaka YL; Ito J; Uriu K; Kosugi Y; Suganami M; Oide A; Yokoyama M; Chiba M; Motozono C; Nasser H; Shimizu R; Kitazato K; Hasebe H; Irie T; Nakagawa S; Wu J; Takahashi M; Fukuhara T; Shimizu K; Tsushima K; Kubo H; Kazuma Y; Nomura R; Horisawa Y; Nagata K; Kawai Y; Yanagida Y; Tashiro Y; Tokunaga K; Ozono S; Kawabata R; Morizako N; Sadamasu K; Asakura H; Nagashima M; Yoshimura K; Cárdenas P; Muñoz E; Barragan V; Márquez S; Prado-Vivar B; Becerra-Wong M; Caravajal M; Trueba G; Rojas-Silva P; Grunauer M; Gutierrez B; Guadalupe JJ; Fernández-Cadena JC; Andrade-Molina D; Baldeon M; Pinos A; Bowen JE; Joshi A; Walls AC; Jackson L; Martin D; Smith KGC; Bradley J; Briggs JAG; Choi J; Madissoon E; Meyer KB; Mlcochova P; Ceron-Gutierrez L; Doffinger R; Teichmann SA; Fisher AJ; Pizzuto MS; de Marco A; Corti D; Hosmillo M; Lee JH; James LC; Thukral L; Veesler D; Sigal A; Sampaziotis F; Goodfellow IG; Matheson NJ Sato K; Gupta RK; The, C.-N. B. C.-C.; The Genotype to Phenotype Japan, C.; Ecuador, C. C., Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603 (7902), 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hui KPY; Ho JCW; Cheung M.-c.; Ng K.-c.; Ching RHH; Lai K-I; Kam TT; Gu H; Sit K-Y; Hsin MKY; Au TWK; Poon LLM; Peiris M; Nicholls JM; Chan MCW, SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603 (7902), 715–720. [DOI] [PubMed] [Google Scholar]
- 31.NCATS/NIH, Inxight Drugs: Camostat.
- 32.Midgley I; Hood AJ; Proctor P; Chasseaud LF; Irons SR; Cheng KN; Brindley CJ; Bonn R, Metabolic fate of 14C-camostat mesylate in man, rat and dog after intravenous administration Xenobiotica 1994, 24 (1), 79–92. [DOI] [PubMed] [Google Scholar]
- 33.Fraser BJ; Beldar S; Seitova A; Hutchinson A; Mannar D; Li Y; Kwon D; Tan R; Wilson RP; Leopold K; Subramaniam S; Halabelian L; Arrowsmith CH; Bénard F, Structure, activity and inhibition of human TMPRSS2, a protease implicated in SARS-CoV-2 activation. bioRxiv 2021, 2021.06.23.449282. [DOI] [PubMed] [Google Scholar]
- 34.Chupp G; Spichler-Moffarah A; Søgaard OS; Esserman D; Dziura J; Danzig L; Chaurasia R; Patra KP; Salovey A; Nunez A; May J; Astorino L; Patel A; Halene S; Wang J; Hui P; Patel P; Lu J; Li F; Gan G; Parziale S; Katsovich L; Desir GV; Vinetz JM, A Phase 2 Randomized, Double-Blind, Placebo-controlled Trial of Oral Camostat Mesylate for Early Treatment of COVID-19 Outpatients Showed Shorter Illness Course and Attenuation of Loss of Smell and Taste. medRxiv 2022, 2022.01.28.22270035. [Google Scholar]
- 35.ACTG announces Camostat will not advance to phase 3 in outpatient treatment study for COVID-19. https://actgnetwork.org/2021/06/30/actg-announces-camostat-will-not-advance-to-phase-3-in-outpatient-treatment-study-for-covid-19/.
- 36.Ono Pharmaceutical Co., L. Phase III Study Result of FOIPAN® Tablets, a Protease Enzyme Inhibitor, in Japan in Patients with Novel Coronavirus Infection (COVID-19). https://www.ono-pharma.com/news/20210611.html.
- 37.Gunst JD; Staerke NB; Pahus MH; Kristensen LH; Bodilsen J; Lohse N; Dalgaard LS; Brønnum D; Fröbert O; Hønge B; Johansen IS; Monrad I; Erikstrup C; Rosendal R; Vilstrup E; Mariager T; Bove DG; Offersen R; Shakar S; Cajander S; Jørgensen NP; Sritharan SS; Breining P; Jespersen S; Mortensen KL; Jensen ML; Kolte L; Frattari GS; Larsen CS; Storgaard M; Nielsen LP; Tolstrup M; Sædder EA; Østergaard LJ; Ngo HTT; Jensen MH; Højen JF; Kjolby M; Søgaard OS, Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine 2021, 35, 100849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.NCATS/NIH Inxight Drugs: Nafamostat. https://drugs.ncats.io/substance/Y25LQ0H97D (accessed March 21, 2022).
- 39.Padova UH NCT04352400. https://clinicaltrials.gov/ct2/show/NCT04352400. [Google Scholar]
- 40.Morrissey JH; Neuenschwander PF; Huang Q; McCallum CD; Su B; Johnson AE, Factor VIIa-tissue factor: functional importance of protein-membrane interactions. Thromb Haemost 1997, 78 (1), 112–6. [PubMed] [Google Scholar]
- 41.Hembrough TA; Swartz GM; Papathanassiu A; Vlasuk GP; Rote WE; Green SJ; Pribluda VS, Tissue Factor/Factor VIIa Inhibitors Block Angiogenesis and Tumor Growth Through a Nonhemostatic Mechanism. Cancer Research 2003, 63 (11), 2997. [PubMed] [Google Scholar]
- 42.Ramanathan RK; Thomas GW; Khorana AA; Shah S; Zhou C; Wong S; Cole G Jr; James D; Gabrail NY, A Phase 2 Study of PCI-27483, a Factor VIIa Inhibitor in Combination with Gemcitabine for Advanced Pancreatic Cancer. Oncology 2019, 96 (4), 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun YJ; Velez G; Parsons DE; Li K; Ortiz ME; Sharma S; McCray PB Jr.; Bassuk AG; Mahajan VB, Structure-based phylogeny identifies avoralstat as a TMPRSS2 inhibitor that prevents SARS-CoV-2 infection in mice. The Journal of Clinical Investigation 2021, 131 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guertin KR; Choi YM, The discovery of the Factor Xa inhibitor otamixaban: from lead identification to clinical development. Curr Med Chem 2007, 14 (23), 2471–81. [DOI] [PubMed] [Google Scholar]
- 45.Guertin KR; Gardner CJ; Klein SI; Zulli AL; Czekaj M; Gong Y; Spada AP; Cheney DL; Maignan S; Guilloteau J-P; Brown KD; Colussi DJ; Chu V; Heran CL; Morgan SR; Bentley RG; Dunwiddie CT; Leadley RJ; Pauls HW, Optimization of the β-Aminoester class of factor Xa inhibitors, part 2: Identification of FXV673 as a potent and selective inhibitor with excellent In vivo anticoagulant activity. Bioorganic & Medicinal Chemistry Letters 2002, 12 (12), 1671–1674. [DOI] [PubMed] [Google Scholar]
- 46.Steg PG; Mehta SR; Pollack CV Jr.; Bode C; Cohen M; French WJ; Hoekstra J; Rao SV; Ruzyllo W; Ruiz-Nodar JM; Sabaté M; Widimsky P; Kiss RG; Navarro Estrada JL; Hod H; Kerkar P; Guneri S; Sezer M; Ruda M; Nicolau JC; Cavallini C; Ebrahim I; Petrov I; Kim JH; Jeong MH; Ramos Lopez GA; Laanmets P; Kovar F; Gaudin C; Fanouillere KC; Minini P; Hoffman EB; Moryusef A; Wiviott SD; Sabatine MS, Anticoagulation with otamixaban and ischemic events in non-ST-segment elevation acute coronary syndromes: the TAO randomized clinical trial. Jama 2013, 310 (11), 1145–55. [DOI] [PubMed] [Google Scholar]
- 47.Rensi S KA, Lo Y-C, Derry A, McInnes G, Liu T, Altman R , Homology Modeling of TMPRSS2 Yields Candidate Drugs That May Inhibit Entry of SARS-CoV-2 into Human Cells. ChemRxiv 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Screening pipeline for inhibitors of TMPRSS2S2
Figure S2. Scheme displaying the enzymatic assay principle and MS/MS molecular fragmentation
Figure S3. Mass spectrometry trace of the +6 amu isotopically labeled version of the cleavage product peptide (Cbz-SK-{13C5 15N Pro}-SKR, m/z 421.6/708.2)
Figure S4. Dose-response data from the fluorescence biochemical assay and fluorescence counterassay for the six actives
Figure S5. Dose-response data from the SARS-CoV-2-S pseudotyped particle entry assays and cytotoxicity counterassay for the six actives
Supplementary Table 1. List of 27 hits identified following confirmation and counterassay testing