

Abstract
Understanding the pathogenesis of nigral dopaminergic neurodegeneration is critical for developing mechanism-based treatments for Parkinson's disease (PD). In the nigral dopaminergic neurons of postmortem human PD brains, we found that CREB, a well-recognized pro-survival transcription factor in neurons, was inactivated by dephosphorylation at Ser133. CREB dephosphorylation correlated with decreased expression of NURR1, one of its target genes crucial for dopaminergic neuron survival, confirming that CREB function was impaired in nigral dopaminergic neurons in PD. An MPTP mouse model was used to further elucidate the mechanism underlying CREB dephosphorylation. Protein phosphatase 1γ (PP1γ), which dephosphorylates CREB, was constitutively associated with histone deacetylase 1 (HDAC1). HDAC1 promotes CREB Ser133 dephosphorylation via a stable interaction with PP1γ. We found that CREB interacted with the HDAC1/PP1γ complex during dopaminergic neurodegeneration. Importantly, increased CREB/HDAC1 interaction occurred in the nigral dopaminergic neurons of PD patients as demonstrated using a proximity ligation assay. Disrupting CREB/HDAC1 interaction via either overexpression of GAL4 M1, a CREB mutant, or administration of trichostatin A, a pan-HDAC inhibitor, restored the expression levels of phospho-CREB (Ser133) and NURR1, and protected nigral dopaminergic neurons in the MPTP-treated mouse brain. Collectively, our results demonstrated that HDAC1/PP1γ-mediated CREB inactivation contributed to dopaminergic neuronal degeneration. Disruption of CREB/HDAC1 interaction has the potential to be a new approach for PD therapy.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is the most common movement disorder attributed to the progressive loss of dopaminergic neurons in the substantia nigra. Understanding the pathogenesis of nigral dopaminergic neurodegeneration is critical for developing mechanism-based treatments for PD. We found in nigral dopaminergic neurons of postmortem human PD brains that CREB, a well-recognized pro-survival transcription factor in neurons, was inactivated by dephosphorylation at Ser133. HDAC1, constitutively associated with PP1γ, interacted with CREB to mediate its dephosphorylation during dopaminergic neurodegeneration. Disrupting CREB/HDAC1 interaction restored CREB activity and protected nigral dopaminergic neurons in the MPTP mouse brains. This work suggests that disruption of the CREB/HDAC1 interaction to restore CREB activity may be a potential therapeutic approach in PD.
Keywords: CREB, HDAC, neurodegeneration, Parkinson's disease, PP1
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder worldwide, affecting 1% of the population over the age of 60 (de Lau and Breteler, 2006). The cardinal symptoms of PD are motor dysfunctions, including resting tremors, bradykinesia, and rigidity, which are attributed to the progressive loss of dopaminergic neurons in the substantia nigra (SN). For over 40 years, dopamine replacement therapy has been a first-line treatment, but it cannot halt the progression of nigral dopaminergic neurodegeneration (Armstrong and Okun, 2020). Therefore, there is an urgent need for neuroprotective strategies capable of mitigating these processes. Elucidation of the pathogenic molecular mechanisms underlying parkinsonian dopaminergic neurodegeneration will help identify neuroprotective drug targets for PD.
CREB is a pro-survival transcription factor in neurons (Sakamoto et al., 2011). Reversible phosphorylation of CREB at Ser133 site is the primary mechanism regulating its activity (Mayr and Montminy, 2001; Steven et al., 2020). Once CREB Ser133 is phosphorylated, a binding site for the coactivator CREB-binding protein (CBP)/p300 becomes available, which promotes transcription activation through the recruitment of RNA polymerase II and its intrinsic acetylase activity (Mayr and Montminy, 2001). Phospho-CREB has been shown to increase and accumulate in the cytoplasm of nigral dopaminergic neurons in postmortem tissue from PD patients and cellular models (Chalovich et al., 2006). However, CREB Ser133 dephosphorylation in the SN was demonstrated in animal models of PD in other studies (C. Sun et al., 2019; Zheng et al., 2019; Zhong et al., 2020), inconsistent with the results in humans. Our most recent study also demonstrated in situ CREB Ser133 dephosphorylation in nigral dopaminergic neurons (Xu et al., 2021). In addition, we showed that transcriptional downregulation of NURR1, a nuclear receptor essential for the maintenance of midbrain dopaminergic neurons, is caused by the inactivation of CREB in the MPTP-induced PD mouse model (Xu et al., 2021), implying that CREB inactivation is a critical event during dopaminergic neurodegeneration. Further studies are needed to confirm whether CREB is inactivated by Ser133 dephosphorylation in patients with PD and to elucidate the underlying mechanisms.
CREB Ser133 phosphorylation can be regulated by upstream kinases, such as PKA, ERK, and CaMKIV, and protein phosphatases, such as PP2A and PP1 (Sakamoto et al., 2011). Recent studies have shown the involvement of PP2A (Chung et al., 2007; Lou et al., 2010; Durgadoss et al., 2012) and PP1 (X. Sun et al., 2018) in PD neurodegeneration. Both PP1 and PP2A holoenzymes contain highly conserved catalytic subunits complexed with regulatory subunits, which determine the substrate specificity, cellular localization, and enzymatic activity of the holoenzymes (Bertolotti, 2018; Clark and Ohlmeyer, 2019). Either HDAC1 or HDAC8 can play such a role, assisting in the CREB/PP1 complex formation to promote CREB dephosphorylation (Canettieri et al., 2003; Gao et al., 2009). However, whether and how these phosphatases act on CREB phosphorylation during parkinsonian pathogenesis are still unknown.
In this study, we first identified CREB inactivation via Ser133 dephosphorylation in SN dopaminergic neurons in postmortem specimens from patients with PD. We then examined the effect of CREB inactivation on parkinsonian neurodegeneration in an MPTP mouse model. Finally, we showed that the interaction between CREB and the HDAC1/PP1γ complex was responsible for CREB dephosphorylation and subsequent dopaminergic neurodegeneration, revealing a new mechanism underlying parkinsonian neurodegeneration.
Materials and Methods
Postmortem human brain samples
Human tissue was obtained from the NIH NeuroBioBank at the University of Miami Brain Endowment Bank). Sections from 6 patients (3 males and 3 females) and 5 age- and gender-matched control subjects (2 males and 3 females) were paraffin-embedded and supplied at 5 μm thickness (Table 1). The average age and postmortem intervals for patients and control subjects were not significantly different (two-tailed Student's t test; age: t = 0.2814, df = 9, p = 0.7848; postmortem intervals: t = 1.497, df = 9, p = 0.1687).
Table 1.
Data for Parkinson's disease patients and control subjects
| Tissue code | Type of fixed biospecimens | Brain type | Sex | Age (yr) | Race | Autolysis (h) |
|---|---|---|---|---|---|---|
| 991_HBHV_01_18_SN | Mid-brain SN | PD | F | 79 | White | 5.05 |
| 991_HBEV_01_18_SN | Mid-brain SN | PD | F | 76 | White | 20.40 |
| 991_HBMD_01_18_SN | Mid-brain SN | PD | F | 79 | White | 25.00 |
| 991_HBFS_01_18_SN | Mid-brain SN | PD | M | 91 | White | 16.06 |
| 991_HBJE_01_18_SN | Mid-brain SN | PD | M | 80 | White | 10.75 |
| 991_HBEH_01_18_SN | Mid-brain SN | PD | M | 73 | White | 23.92 |
| 991_HctZA_01_18_SN | Mid-brain SN | Unaffected control | F | 79 | White | 17.80 |
| 991_HctZZC_01_18_SN | Mid-brain SN | Unaffected control | F | 82 | White | 14.20 |
| 991_Hct15HBF_01_18_SN | Mid-brain SN | Unaffected control | F | 83 | White | 6.00 |
| 991_HctYY_01_18_SN | Mid-brain SN | Unaffected control | M | 90 | White | 7.16 |
| 991_Hct15HBM_01_18_SN | Mid-brain SN | Unaffected control | M | 70 | White | 8.67 |
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee, Sun Yat-Sen University (Guangzhou, China). Eight- to 12-week-old male C57BL/6 mice weighing 22-28 g were purchased from the Beijing Vital River Laboratory Animal Technological. All mice were housed in specific pathogen-free facilities, maintained on a 12 h light/dark cycle with controlled temperature and air humidity, and allowed free access to food and water. Mice were randomly assigned into groups. The animal data reporting followed the ARRIVE guidelines.
Adeno-associated virus (AAV) production and infection
AAV2/9-hSyn-NLS-GAL4 M1 (amino acids 1-283, S133A)-WPRE-pA (2.87 × 1012 vg/ml) was generated from the plasmid pCDNA GAL4 CREB 1-341 M1 (a gift from Marc Montminy, Addgene plasmid #22973; https://www.addgene.org/22973/: 22973; RRID:Addgene_22973), and AAV2/9-hSyn-NLS-GAL4-WPRE-pA (2.87 × 1012 vg/ml) was used as a control virus. All viruses were packaged in BrainVTA. Eight-week-old male C57BL/6 mice were anesthetized with 3% isoflurane and then placed into a stereotaxic instrument (Model 940, Kopf Instruments). Anesthesia was maintained using 1.5% isoflurane through a nose tip built into the stereotactic frame. Injections were conducted with a 10 μl syringe connected to a 33-Ga needle (Hamilton) using a microsyringe pump (KDS Legato 130). Two microliters of AAV virus were delivered at 100 nl/min unilaterally into the right SN. The coordinates indicating distance (mm) from bregma were as follows: AP −2.9, ML −1.3, and DV −4.35. After the injection, the needle remained in place for at least 5 min to prevent retrograde flow along the needle track.
Drug administration
MPTP (#M0896, Sigma) treatments were performed as previously described (Li et al., 2020). Briefly, 8- to 12-week-old male mice weighing 22-28 g were injected intraperitoneally with five MPTP doses (30 mg/kg body weight) at 24 h intervals. Control mice received a corresponding volume of saline alone. For investigation of dopaminergic lesions, mice were killed 21 d after the last MPTP treatment. Under anesthesia, mice were injected unilaterally with 2 μl of 250 μm okadaic acid (OA) or vehicle into the SN at a rate of 100 nl/min, using stereotaxic surgery as described for the AAV injection. After the surgical procedure, mice were placed on a heating pad to maintain body temperature at 37.5 ± 0.5°C and kept there until the return of spontaneous movement. The mice received a single 30 mg/kg MPTP injection 1 h after OA injection. Trichostatin A (TSA) (#V900931, Sigma) with a dose of 0.5 mg/kg body weight or vehicle (5% DMSO + 95% PBS) was administered intraperitoneally 1 h before and 1.5 h after intraperitoneal injection of MPTP.
Tissue preparation
For cryosections, mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and intracardially perfused with ice-cold PBS (0.01 m, pH 7.4) for 3 min, followed by cold 4% PFA at a flow rate of 10 ml/min for 8 min. Mouse brains were removed and postfixed overnight in 4% PFA at 4°C and then immersed in 20% and 30% sucrose. Tissues were embedded in embedding media OCT (#4583, Sakura) and cut into 20-μm-thick cryosections for immunofluorescence and 40-μm-thick cryosections for immunohistochemistry. For paraffin sections, tissue was processed in the same way as above except after dissection. Tissue was postfixed for 24 h in 4% PFA with a phosphatase inhibitor cocktail (#B15001, Bimake) at 4°C, then processed and embedded in paraffin, and cut into 5 μm sections for a proximity ligation assay (PLA). For Western blotting and immunoprecipitation, mice were killed by cervical dislocation, and the brains were rapidly removed and washed with ice-cold PBS. The ventral midbrains were rapidly dissected on ice and kept at −80°C for additional experiments.
Western blot analysis
Dissected mouse ventral midbrain tissues were homogenized and lysed in Laemmli buffer (50 μl/mg tissue) containing Tris·Cl (62.5 mm, pH 6.8), SDS (2%, w/v), bromophenol blue (0.005%, w/v), glycerol (10%, v/v), and DTT (8 mg/ml), and then boiled at 95°C-100°C for 5 min. Twenty-five micrograms of protein from each sample were loaded on 10% SDS-PAGE gels followed by transfer to PVDF membranes (#IPVH00010, Millipore). Membranes were blocked with 5% skim milk for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: p-CREB Ser133 (1:1000, #ab32096, Abcam), p-CREB Ser133 (1:1000, #9198, Cell Signaling Technology), CREB (1:500, #9104, Cell Signaling Technology), NURR1 (1:500, #sc-991, Santa Cruz Biotechnology), NURR1 (1:1000, #ab41917, Abcam), tubulin (1:10000, #T4026, Sigma), PP1γ antibody (1:500, #sc-6108, Santa Cruz Biotechnology), PP1α (1:1000, #067070-1, Proteintech), PP1β (1:500, #sc-365678, Santa Cruz Biotechnology), PP2A (1:500, #05-421, Millipore), HDAC1 (1:2000, #YT2145, Immunoway), HDAC2 (1:5000, #ab12169, Abcam), HDAC4 (1:1000, #7628, Cell Signaling Technology), and HDAC8 (1:500, #sc-11405, Santa Cruz Biotechnology). After washing, membranes were incubated with appropriate HRP-conjugated secondary antibodies (1:5000, Jackson ImmunoResearch Laboratories). All blots were visualized using ECL chemiluminescence (#WBKLS0500, Millipore), and the results were analyzed using ImageJ software.
Immunofluorescence staining
Immunofluorescence analysis was performed as previously described (Li et al., 2020). In brief, cryostat-coronal sections (20 μm) encompassing the entire midbrain were serially collected. Free-floating sections were preincubated in blocking solution containing 5% normal donkey serum and 0.3% Triton X-100 in 50 mm TBS, pH 7.4, at room temperature for 1 h. Primary antibodies were diluted in 1% BSA and 0.3% Triton X-100 in TBS, pH 7.4, and incubated overnight at 4°C. The following primary antibodies were used: TH (1:1000, #AB9702, Millipore), p-CREB Ser133 (1:500, #ab32096, Abcam), p-CREB Ser133 (1:100, #9198, Cell signaling Technology), CREB (1:500, #4820, Cell Signaling Technology), NURR1 (1:500, #sc-5568, Santa Cruz Biotechnology), and GAL4 (1:600, #ab14477, Abcam). After three washes, the sections were then incubated with the secondary antibodies (Thermo Fisher Scientific), which were conjugated with Alexa-488, Alexa-555, or Alexa-647, at room temperature for 1 h. Finally, sections were visualized under a confocal laser scanning microscope (LSM 780, Carl Zeiss).
Immunohistochemistry
For mouse samples, the immunohistochemical method was performed as previously described (Li et al., 2020). In brief, cryo-coronal sections (40 μm) encompassing the entire midbrain and striatum were serially collected. Sections were incubated with anti-TH antibody (1:10000, #AB152, Merck Millipore) and then visualized using the Vectastain Elite ABC Kit (#PK-6101, Vector Labs) and the DAB Peroxidase Substrate Kit (#SK-4100, Vector Labs), following the manufacturer's protocol. For Nissl staining, adjacent midbrain sections were mounted on charged slides, dried for 1 h at room temperature, stained with 0.5% cresyl violet after defat, dehydrated, and coverslipped. For human samples, paraffin-embedded human brain tissue slices were deparaffinized in xylene and rehydrated in graded alcohols. Antigens were retrieved by microwave heating with citrate buffer, pH 6.0, for a total of 30 min. Tissue sections were blocked in 3% H2O2 for 30 min at room temperature in the dark to quench endogenous peroxidases and then blocked in 2.5% normal horse blocking serum for 1 h at room temperature. Primary antibodies were incubated with the tissue overnight at 4°C, followed by washing and incubation with secondary antibodies (#MP-7401 or #MP-7405, Vector Labs) for 1 h at room temperature. The following primary antibodies were used: p-CREB Ser133 (1:500, #ab32096, Abcam), CREB (1:500, #AF2989, R&D Systems), and NURR1 (1:2000, #sc-5568, Santa Cruz Biotechnology). Immunostaining was visualized by DAB (#SK-4100, Vector Labs). Tissue sections were then counterstained with hematoxylin and dehydrated in graded alcohols and xylene before mounting with a permanent mounting medium (#H-5000, Vector Labs).
Stereological cell counting
To estimate the number of TH-positive and Nissl-positive neurons in the SN, stereological counting was performed as previously described (Li et al., 2020). In brief, stereological cell counting analyses were performed by the stereological method of optical fractionator with the aid of Stereo Investigator (MBF Bioscience). Every fourth 40-μm-thick section, a total of seven sections from the midbrain (AP −2.7 to −3.8 mm) were collected. The SN was delineated under a 4× objective, and the actual counting was performed under a 100× objective. Stereological counting was performed in a double-blind fashion.
Densitometric analysis
The optical density of p-CREB, CREB, and NURR1 staining on human brain samples was analyzed with the aid of the IHC Profiler plugin of ImageJ software (Varghese et al., 2014). The DAB color spectra were separated, the regions of the nuclei of neuromelanin-positive cells were defined by the experimenter, and the optical density of these regions was measured. The optical density of striatal TH-positive fibers was estimated using ImageJ software. The region of the dorsal striatum was determined as previously described (Li et al., 2020). All analyses were performed blinded to the treatments.
Coimmunoprecipitation
The ventral midbrains were homogenized in lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 10% glycerol, and 1% Triton X-100) supplemented with a protease inhibitor cocktail (#B14002, Bimake). Lysates were centrifuged at 13000 rpm for 15 min at 4°C, and the supernatants were collected. Protein concentration was measured using a Pierce BCA Protein Assay Kit following the manufacturer's instructions (#23225, Thermo Fisher Scientific). Lysates were normalized and cleared. The antibody and control IgG were added to 40 µl protein A/G agarose and incubated at 4°C for 6 h. The following antibodies were used for immunoprecipitation: 2 μg rabbit anti-CREB antibody (#4820, Cell Signaling Technology), 5 μg rabbit anti-HDAC1 antibody (#ab7028, Abcam), and equal amounts of rabbit normal IgG (#12-370, Millipore); 2 μg goat anti-PP1γ antibody (#sc-6108, Santa Cruz Biotechnology) and equal amounts of goat normal IgG (#sc-2028, Santa Cruz Biotechnology, SUPPLECA). Precleared lysates (300 μg) were then incubated with agarose/antibody-coupled beads overnight at 4°C. Immunoprecipitates were washed 5 times with wash buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, and 0.5% Triton X-100) supplemented with a protease inhibitor cocktail, resuspended in 3× SDS-PAGE loading buffer, boiled for 5 min, and analyzed by Western blot.
PLA
For human samples, PLA experiments were performed using the Duolink In Situ Detection Reagents Brightfield Kit (#DUO92012, Sigma). Briefly, paraffin-embedded tissue was prepared for brightfield PLA by deparaffinizing, rehydrating, antigen retrieval, and blocking endogenous peroxidases, as in immunohistochemistry experiments. All samples were incubated in Duolink block solution at 37°C for 1 h and then probed with a specific combination of primary antibodies at 4°C overnight. The following antibodies were used for PLA: HDAC1 (1:1500; #PA1-860, Thermo Fisher Scientific) and CREB (1:1500, #AF2989, R&D Systems). Experiments with only one primary antibody (HDAC1) provided our negative control. For mouse sample, in addition to HDAC1 (1:2000; #PA1-860, Thermo Fisher Scientific) and CREB (1:1 500, #AF2989, R&D Systems) antibodies, TH antibody (1:500, #AB9702, Millipore) was also diluted in Duolink antibody diluent to label mouse dopaminergic neurons by immunofluorescence. The PLA reaction was performed using PLA secondary probes diluted 1:5 in Duolink antibody diluent and incubated at 37°C for 1 h. The PLA secondary probes used were Duolink PLA Anti-Rabbit PLUS (#DUO92002, Sigma) and Anti-Goat MINUS (#DUO92006, Sigma). Subsequent ligation, amplification, detection, and substrate development were performed according to the manufacturer's instructions. The tissue sections were then counterstained with hematoxylin and dehydrated in graded alcohols and xylene before mounting with a permanent mounting medium. Images were acquired using a brightfield microscope (BX61, Olympus). PLA signals in the nucleus were counted.
Ultra performance liquid chromatography (UPLC)
Striatal dopamine concentrations were measured using the Waters ACQUITY UPLC H-Class Chromatographic System coupled with 2465 Electrochemical Detector. Striata samples were weighted and homogenized in ice-cold 0.1 m HClO4 (0.1 g original wet tissue/ml) containing 0.1% L-cysteine and 0.3 ng/μl dihydroxybenzylamine as internal standard. Homogenates were centrifuged at 15000 × g for 20 min at 4°C. Supernatants were collected, and 5 μl was injected onto an ACQUITY UPLC BEH C18 1.7 μm 2.1 × 50 mm column. The mobile phase consisted of 3 mm sodium 1-heptanesulfonate, 100 mm sodium acetate, 85 mm citric acid, and 0.2 mm EDTA, final pH 3.7. The flow rate was set at 0.40 ml/min, and typical retention times were as follows: dopamine, 2.694 min and dihydroxybenzylamine, 1.591 min. Dopamine contents were determined by comparing sample peak areas with those recorded from standards run on the same day.
Behavioral test
Behavioral tests were performed 2 d after the final MPTP injection. The challenge beam test was adapted with minor modifications as described previously (Fleming et al., 2013; Mann and Chesselet, 2015; Bhurtel et al., 2019; Li et al., 2020). In brief, a Plexiglas beam consisting of four equal-length sections (25 cm per section, 100 cm total length) gradually decreasing in width from 3.5 to 0.5 cm in 1 cm increments was used. A mesh grid (1 cm squares) with a width corresponding to the beam size was placed over the beam surface, leaving a 1.5 cm space between the grid and the beam surface. Mice were trained to traverse the beam (from widest to narrowest) directly into their home cage for 5 d (5 times per day) before drug administration without mesh grids. On the day of testing, mesh grids were placed over the beam surface, and mice were videotaped while traversing the beam over five trials. Time to traverse was measured and noted in the test. Video records were analyzed at slow motion to count the number of total steps.
Statistics
GraphPad Prism version 8.0 was used for statistical analysis. All data are presented as mean ± SEM. p < 0.05 was considered statistically significant. Statistical tests used are indicated in the figure legends.
Results
CREB is inactivated by dephosphorylation at Ser133 in nigral dopaminergic neurons of PD patients
To assess whether CREB Ser133 dephosphorylation occurs during parkinsonian pathogenesis, we analyzed phospho-CREB Ser133 (p-CREB) expression levels in postmortem SN tissues from 6 PD cases and 5 age-matched control cases. Immunohistochemical staining revealed that p-CREB expression occurred in both neurons and some of the glial cells and was restricted to the nuclei (Fig. 1A). Next, we used neuromelanin, a catecholamine-derived pigment, as a marker for dopaminergic neurons in humans (Zucca et al., 2017). Densitometric analysis of the immunoreactive signal of p-CREB within the nuclei of neuromelanin-positive cells showed that p-CREB expression was 47.97 ± 12.07% lower in the dopaminergic neurons of PD patients (n = 6) than in those from age-matched controls (n = 5) (Fig. 1A; t(9) = 3.972, p = 0.0032, two-tailed t test). To exclude the possibility that this difference in p-CREB expression resulted from a loss of CREB protein nuclear translocation, we performed total CREB staining in the SN tissue. As shown in Figure 1B, in dopaminergic neurons from both PD patients (n = 3) and age-matched controls (n = 3), total CREB staining was limited to the nuclei, and CREB expression levels were similar in PD and control cases (t(4) = 0.5535, p = 0.6094, two-tailed t test). In parallel, we did not observe overt p-CREB downregulation in glial cells. These results suggested that CREB's function as a transcription factor was repressed in nigral dopaminergic neurons of PD patients.
Figure 1.

CREB is inactivated in SN dopaminergic neurons of PD patients. A, B, Sections of human SN obtained from control or PD individuals were immunostained using the p-CREB (A) or CREB antibody (B), respectively, and visualized by DAB staining. Boxed areas are shown at higher magnification in the images below. Scale bars: low magnification, 100 μm; high magnification, 20 μm. C, Adjacent sections of human SN from control or PD individuals were immunostained using the p-CREB or NURR1 antibody, respectively, and visualized by DAB staining. Representative images of p-CREB (top) and NURR1 (bottom) from the same dopaminergic neuron. Scale bar, 20 μm. D, Analysis of the correlation between p-CREB and NURR1 expression in control (n = 3, 133 dopaminergic neurons) and PD (n = 3, 60 dopaminergic neurons) individuals. Pearson correlation coefficient r = 0.4306, p < 0.0001.
If the transcriptional activity of CREB was suppressed, the expression levels of CREB target genes should be downregulated. Indeed, a decrease in NURR1 expression was detected in SN dopaminergic neurons of PD patients compared with age-matched controls (Fig. 1C). Furthermore, a correlation analysis of the expression levels of p-CREB and NURR1, one of CREB's target genes expressed in the ventral midbrain (Volakakis et al., 2010), was conducted. Immunohistochemical staining of p-CREB and NURR1 was performed on adjacent sections from serial brain sections. Correlation analysis showed a positive correlation between the expression levels of p-CREB and NURR1 in the same neurons (Fig. 1D; r = 0.4306, p < 0.0001), confirming that CREB was inactivated by Ser133 dephosphorylation in SN dopaminergic neurons in PD.
Increased interaction of CREB with HDAC1/PP1γ complex in PD
To investigate the mechanism underlying CREB dephosphorylation during dopaminergic neurodegeneration, an MPTP-induced mouse model was used. This model is a well-accepted PD model that recapitulates many of the pathologic and behavioral symptoms of human PD (Przedborski et al., 2001). Specifically, we have previously described the downregulation of CREB dephosphorylation and NURR1 expression in this MPTP mouse model (Xu et al., 2021). Intranigral injection of OA, a commonly used phosphatase inhibitor, restored MPTP-induced dephosphorylation of CREB at Ser133 (Fig. 2A; F(1,8) = 16.00, p = 0.0040). Because OA treatment did not alter the basal levels of p-CREB expression in saline-treated mice as shown in Figure 2A, it is plausible that these phosphatases acted on CREB only after MPTP treatment. To identify the specific phosphatase that interacts with CREB, we performed coimmunoprecipitation experiments. The results showed that PP1γ, one of the isoforms of the catalytic subunit of PP1, was detected in immunoprecipitates of CREB antibody in the SN tissue of mice treated with MPTP, but not in samples from saline-treated mice or control immunoglobulin (Fig. 2B). In parallel, other isoforms of PP1 (PP1α and PP1β) or PP2A were not detected in the immunoprecipitates of CREB antibody (Fig. 2B). These results suggested that PP1γ mediated CREB dephosphorylation during MPTP-induced neurodegeneration.
Figure 2.

CREB is dephosphorylated by HDAC1/PP1γ after MPTP intoxication. A, Western blot analysis for p-CREB in the mouse ventral midbrain by intranigral application of OA before MPTP injection. Quantification of p-CREB normalized to CREB (right). n = 3 per group. Two-way ANOVA (Tukey's): **p < 0.01 versus Saline group; ##p < 0.01 as indicated. B-D, Detection of protein–protein interaction as indicated in the mouse SN by coimmunoprecipitation after MPTP treatment. Representative results from three separate experiments.
The versatility and specificity of PP1 function are largely determined by the binding of its catalytic subunit to different specific regulatory subunits (Bollen et al., 2010). PP1 can be targeted to CREB by binding with HDAC1 (Canettieri et al., 2003). Coimmunoprecipitation experiments in mouse SN tissue showed increased HDAC1 in CREB precipitates derived from MPTP-treated mice (Fig. 2C), while PP1γ constitutively combined with HDAC1 (Fig. 2D). Other HDAC1 homologs (HDAC2, HDAC8, and HDAC4) have also been shown to interact with CREB (Canettieri et al., 2003; Gao et al., 2009; Li et al., 2012). However, our data indicated no interaction between CREB and these HDACs (Fig. 2C).
Since HDAC1 acts as a regulatory protein to join CREB and PP1γ together, a PLA was performed to confirm in situ the presence of the HDAC1/CREB complex in nigral dopaminergic neurons. The average PLA dots signals per nucleus from HDAC1 and CREB interaction were significantly increased in the dopaminergic neurons of MPTP-treated mice (2.01 ± 0.19 in MPTP vs 1.05 ± 0.12 in Saline group), while the PLA signals from HDAC1 antibody alone, used as background signals, were significantly lower than PLA signals derived from HDAC1 and CREB antibody-mediated proximity ligation (Fig. 3A; F(1,12) = 16.88, p = 0.0014).
Figure 3.

Increased interaction between CREB and HDAC1 in SN dopaminergic neurons in PD. A, PLA was performed with HDAC1 and CREB in mouse SN dopaminergic neurons (top); n = 4 per group. Scale bars, 10 μm. Quantification of dopaminergic neuron nuclear PLA signal (bottom). B, PLA was performed with HDAC1 and CREB in the human SN dopaminergic neurons (top); n = 4 per group. Scale bars, 10 μm. Quantification of dopaminergic neuron nuclear PLA signal (bottom). Two-way ANOVA (Tukey's): *p < 0.05, **p < 0.01, ****p < 0.0001 versus Saline/Control group; #p < 0.05, ###p < 0.001, ns, not significant as indicated.
Next, we used PLA to investigate whether increased interaction between CREB and HDAC1 appeared in PD patient brain samples. In nigral neurons containing neuromelanin, PLA signals of CREB/HDAC1 interaction were detected in the nucleus. On average, 2.83 ± 0.35 dots per nucleus were observed in PD patients, significantly higher than those in age-matched controls (1.42 ± 0.27 dots per nucleus), and the PLA signal from PD brains was significantly above the background signal detected with HDAC1 antibody alone (Fig. 3B; F(1,12) = 9.526, p = 0.0094). These results suggested that increased interaction between CREB and HDAC1 occurred in SN neurodegeneration.
Interaction of CREB with HDAC1/PP1γ results in CREB inactivation
To determine whether CREB dephosphorylation was because of the interaction with the HDAC1/PP1γ complex, we interrupted CREB/HDAC1 interaction by overexpressing a CREB polypeptide (amino acids 1-283, S133A; named GAL4 M1) containing the interacting domain with HDAC1 (Canettieri et al., 2003). The sequence for GAL4 M1 was packaged in AAVs, which were injected into the SN to achieve peptide expression in dopaminergic neurons. As expected, GAL4 M1 is competitively bound to HDAC1 in MPTP-treated mice, inhibiting the endogenous interaction of CREB with HDAC1 (Fig. 4A). Immunofluorescence analysis showed that GAL4 M1 and its control protein GAL4 were mainly transduced into nigral TH-positive neurons and that GAL4 M1 restored MPTP-induced downregulation of p-CREB and NURR1 within TH-positive neurons (Fig. 4B). Western blot analyses confirmed that GAL4 M1 expression did not change the basal levels of p-CREB and NURR1 but significantly restored MPTP-induced downregulation of p-CREB and NURR1 (Fig. 4C; p-CREB/CREB: F(1,8) = 6.925, p = 0.0301; NURR1/Tubulin: F(1,12) = 8.904, p = 0.0114).
Figure 4.
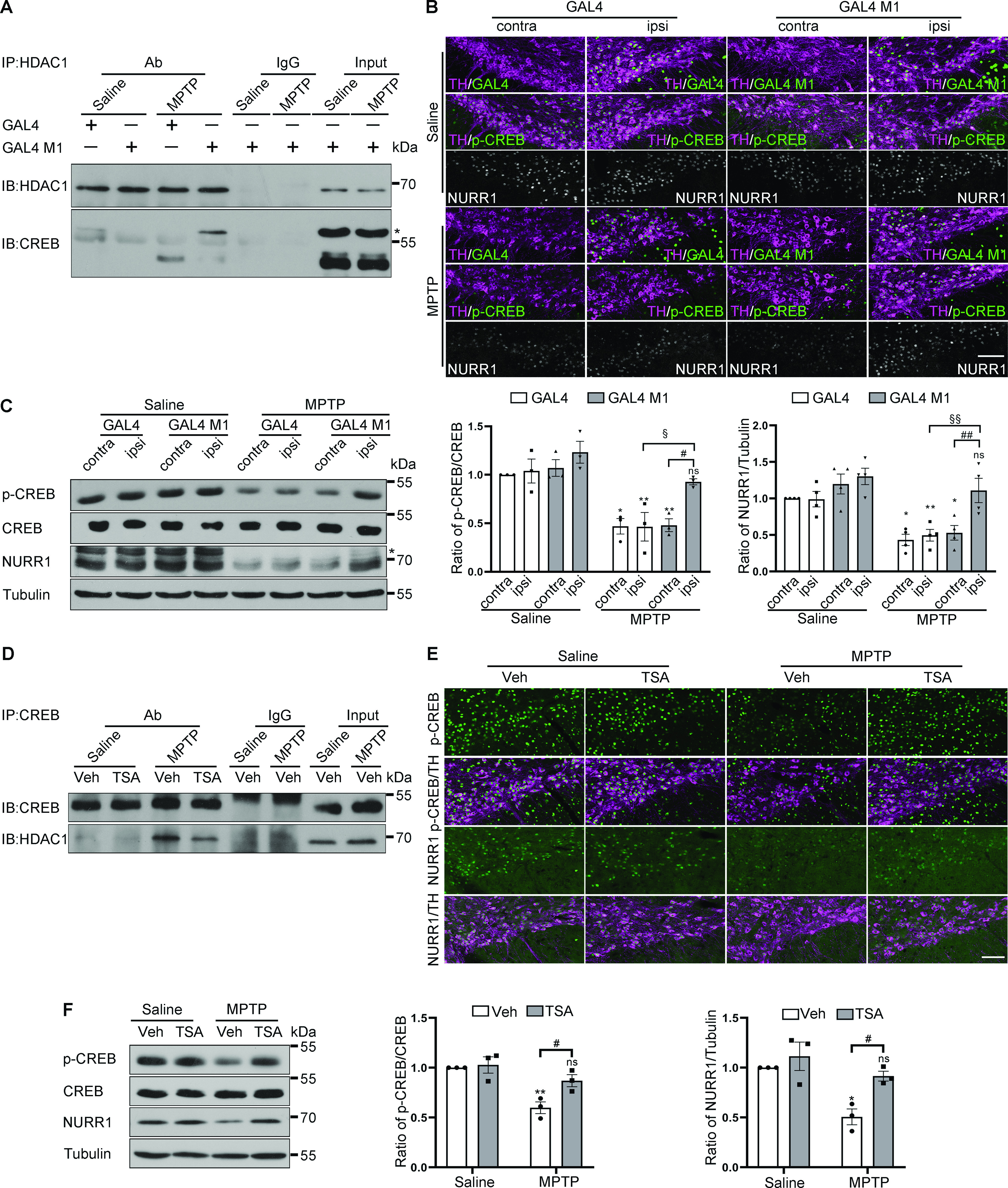
Interruption of CREB/HDAC1 interaction rescues MPTP-induced CREB inactivation. A, Detection of the association between HDAC1 and CREB in mice overexpressing GAL4 or GAL4 M1 by immunoprecipitation from three separate experiments. *The overexpressed GAL4 M1. B, C, Immunofluorescence and Western blot analysis for p-CREB and NURR1 (probed with #ab41917, Abcam) expression in dopaminergic neurons of mice overexpressing GAL4 or GAL4 M1. *A nonspecific band detected by the anti-NURR1 antibody. n = 3 or 4 per group. Scale bars, 100 μm. Quantification of the blots normalized to CREB (middle) or tubulin (right). Three-way ANOVA (Bonferroni's). D, The mouse midbrains from Saline/MPTP mice treated with TSA or its vehicle were subjected to immunoprecipitation. Detection of the association between HDAC1 and CREB. Representative results from three separate experiments. E, F, Immunofluorescence and Western blot analysis for p-CREB and NURR1 (probed with sc-991, Santa Cruz) expression in the indicated groups. Scale bars, 100 μm. Quantification of the blots in F normalized to CREB (middle) or tubulin (right). Two-way ANOVA (Tukey's): *p < 0.05, **p < 0.01 versus Saline group; #p < 0.05, ##p < 0.01, §p< 0.05, §§p< 0.01, ns, not significant as indicated.
To obtain pharmacological evidence for the detrimental role of the interaction between CREB and the HDAC1/PP1γ complex, we examined the effect of a pan-HDAC inhibitor TSA (Hu et al., 2003; Khan et al., 2008), which has been reported to disrupt the association of PP1 with HDAC1 (Brush et al., 2004). TSA treatment significantly blocked the interaction of CREB with HDAC1 (Fig. 4D). Immunofluorescent and Western blot analysis showed that TSA treatment counteracted CREB dephosphorylation and NURR1 downregulation induced by MPTP (Fig. 4E,F; p-CREB/CREB: F(1,8) = 6.478, p = 0.0344; NURR1/Tubulin: F(1,8) = 9.477, p = 0.0152). These data suggested that an increased CREB/HDAC1 interaction led to the blockade of the CREB-NURR1 pathway in SN neurons.
Interruption of CREB/HDAC1 interaction exerts neuroprotection
The effect of CREB/HDAC1 interaction on dopaminergic neuronal degeneration was subsequently determined. Histologic analysis of MPTP mice demonstrated a 39.2% loss of TH-positive neurons in the AAV-GAL4 group, while only 6.3% of the TH-positive neurons degenerated in the AAV-GAL4 M1 group (Fig. 5A,B; F(1,44) = 24.14, p < 0.0001). Nissl staining revealed similar trends (47.3% and 10.5% losses in the AAV-GAL4 and AAV-GAL4 M1 groups, respectively, Fig. 5C; F(1,44) = 48.63, p < 0.0001), confirming a real nigral neuronal loss instead of a loss of TH expression. Consistently, AAV-GAL4 M1 mice retained striatal TH-positive fiber density (42.2% and 8.8% reductions in AAV-GAL4 and AAV-GAL4 M1 groups, respectively, Fig. 5D,E; F(1,44) = 23.97, p < 0.0001). To assess the functional integrity of dopaminergic neurons, we measured striatal dopamine concentrations by UPLC. The results showed that GAL4 M1 gene delivery restored dopamine levels in the MPTP model (Fig. 5F; F(1,32) = 24.43, p < 0.0001).
Figure 5.
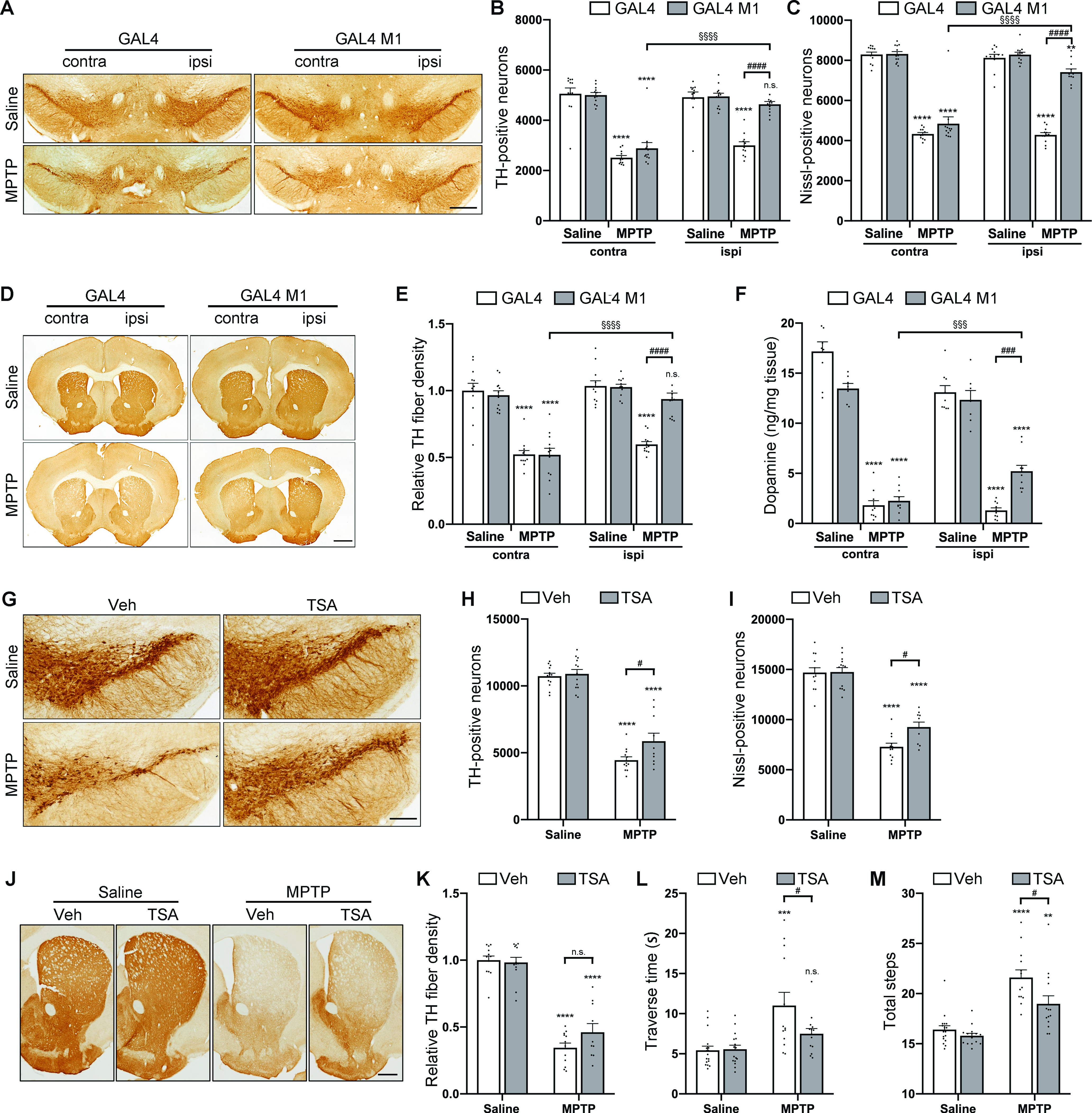
Interruption of CREB/HDAC1 interaction alleviates MPTP-induced dopaminergic neurodegeneration. A-E, Immunohistochemical analysis of MPTP-induced mouse model injected with GAL4/GAL4 M1. A, D, Immunohistochemical TH staining on midbrain (A) sections and striatal (D) sections from saline- and MPTP-treated mice overexpressing GAL4 or GAL4 M1 driven by an AAV vector. Scale bars: A, 500 μm; D, 1000 μm. B, C, TH-positive (B) cell and Nissl-positive (C) cell counts in the SN assessed by unbiased stereology. E, Quantitation of the optical densities of the striatal TH immunostaining. n = 12, 12, 12, and 12 for Saline + GAL4, Saline + GAL4 M1, MPTP + GAL4, and MPTP + GAL4 M1 groups, respectively. F, The striatal dopamine concentrations were detected by UPLC assay 21 d after the final MPTP injection. n = 9, 7, 10, and 10 for Saline + GAL4, Saline + GAL4 M1, MPTP + GAL4, and MPTP + GAL4 M1 groups, respectively. Three-way ANOVA (Bonferroni's). G-K, Immunohistochemical analysis of MPTP-induced mouse model treated with vehicle/TSA. G, J, TH detection in midbrain (G) sections and striatal (J) sections by immunohistochemistry in the indicated groups. Scale bars: G, 200 μm; J, 500 μm. H, I, TH-positive (H) cell and Nissl-positive (I) cell counts in the SN assessed by unbiased stereology. K, Quantitation of optical densities of striatal TH immunostaining. n = 13, 13, 12, and 10 for Saline + Vehicle, Saline + TSA, MPTP + Vehicle, and MPTP + TSA groups, respectively. Two-way ANOVA (Tukey's). L, M, Bar graphs showing the results of challenging beam tests: traverse time (L), numbers of total steps (M), respectively. n = 17, 16, 13, and 14 for Saline + Vehicle, Saline + TSA, MPTP + Vehicle, and MPTP + TSA groups, respectively. Two-way ANOVA (Tukey's). **p < 0.01, ***p < 0.001, ****p < 0.0001 ns, not significant versus Saline group; #p < 0.05, ###p < 0.001, ####p < 0.0001, §§§p < 0.001, §§§§p < 0.0001 ns, not significant as indicated.
We then investigated whether TSA could rescue dopaminergic neurodegeneration provoked by MPTP. TSA per se did not produce obvious neurotoxicity in the saline group (Fig. 5G,H). In the MPTP group, treatment with TSA significantly alleviated TH-positive neuron loss in the MPTP group (the number of TH-positive neurons in the TSA+MPTP group was 53.8% of that in the TSA+saline group), while the survival rate of TH-positive neurons in the vehicle+MPTP group was only 41.5% of that in the vehicle+saline group (Fig. 5G,H; F(1,44) = 5.14, p = 0.0283). Nissl-positive cell counts confirmed TSA neuroprotection rather than a rescue of TH expression (Fig. 5I; F(1,44) = 5.12, p = 0.0286). The decrease in striatal TH-positive fiber density showed similar trends to the reduction in the numbers of nigral neurons (the relative optical density in vehicle+MPTP and TSA+MPTP groups was 34.5% and 46.1% of that in the vehicle+saline group, respectively, Fig. 5J,K; F(1,44) = 1.44, p = 0.2366). Along with defects in the nigrostriatal dopaminergic system, mice treated with MPTP developed motor symptoms, including a longer time to reach the home cage and increased total steps monitored by the challenging beam test. However, TSA treatment shortened the traverse time with a strong trend (Fig. 5L; F(1,56) = 3.77, p = 0.0574) and significantly decreased the number of total steps (Fig. 5M; F(1,55) = 8.33, p = 0.0056), indicating an improvement in motor performance. Collectively, these findings demonstrate that an increased interaction of CREB/HDAC1 leads to nigrostriatal degeneration in PD.
Discussion
The present study represents the first demonstration that CREB is inactivated by dephosphorylation at the Ser133 site in the degenerating nigral neurons of PD patients. Mechanistically, HDAC1/PP1γ complex mediates CREB inactivation to promote dopaminergic neurodegeneration in PD.
Our findings that the levels of p-CREB in the SN dopaminergic neurons in both PD patient and MPTP-treated mouse brain were reduced contradict with a previous report showing increased cytoplasmic expression of p-CREB in the nigral dopaminergic neurons of PD patients and a 6-OHDA-treated cellular model of PD (Chalovich et al., 2006). This discrepancy could be attributed to the different sources of brain samples, antibodies used for immunostaining, or antibody batches. Our results confirmed a recently published study showing CREB dephosphorylation in situ in an MPTP model (Zhong et al., 2020) and were supported by three additional lines of evidence. First, p-CREB downregulation induced by MPTP administration could be restored by OA treatment, indicating the specificity of the p-CREB signal. Second, CREB protein distribution was limited to the nucleus and in line with CREB's function as a transcription factor. Third, our recently published study revealed that CREB occupancy at the CRE site of the promoter region of NURR1 did not differ between saline- and MPTP-treated mice, consistent with CREB's nuclear localization (Xu et al., 2021).
It should be noted that OA is a commonly used phosphatase inhibitor, but it lacks selectivity. It inhibits not only PP1 and PP2A, but also PP4 and PP5 (Swingle et al., 2007). The results of OA administration could provide clues for the role of phosphatases in CREB dephosphorylation. It has been reported that CREB can be dephosphorylated by PP1 and PP2A (Sakamoto et al., 2011). We therefore performed coimmunoprecipitation to investigate whether and which phosphatase was responsible for CREB dephosphorylation in the MPTP model. Only PP1γ was detected in the CREB precipitates (Fig. 2B). It has been shown that PP1γ expression is diffuse in the cytoplasm and nucleoplasm, whereas PP1α and PP1β are expressed primarily in the cytoplasm (Shioda et al., 2015). In addition, PP2A has been reported to have a cytoplasmic distribution of its catalytic subunit (Huang et al., 2020). The subcellular distribution of these phosphatases may explain our observation that only PP1γ was recovered in CREB immunoprecipitates, which is the main isoform expressed in the nucleus.
Of note, the localization of PP1 isoforms is determined mainly by their targeting regulatory subunits, which incorporate them into different signaling complexes (Moorhead et al., 2007). Our data showed that PP1γ constitutively interacted with the targeting subunit HDAC1 (Fig. 2D), which is also shown in the mouse hippocampus (Koshibu et al., 2009). It has been suggested that an HDAC/phosphatase complex could function as a transcriptional repressor (de Castro et al., 2017), implicating that the HDAC1/PP1γ complex could play a role in fundamental cellular processes in the SN.
The HDAC1/PP1γ complex was targeted to CREB in the MPTP mouse SN tissue, demonstrated by coimmunoprecipitation analysis (Fig. 2). These results were further verified by the PLA experiment, confirming that CREB/HDAC1 interaction did occur in dopaminergic neurons after MPTP treatment (Fig. 3A). Notably, a PLA experiment on tissue sections of postmortem human brains provided solid evidence that CREB/HDAC1 interaction increased in patients with PD (Fig. 3B), in line with the results of CREB Ser133 dephosphorylation (Fig. 1A). By disrupting CREB/HDAC1 interaction via overexpression of GAL4 M1 or administration of TSA, we further demonstrated that this interaction was a pathogenic mechanism in dopaminergic neuron death in PD.
Although we have demonstrated that TSA interrupted CREB/HDAC1 interaction to restore CREB phosphorylation, it remains to be clarified whether this effect depends on the inhibition of the deacetylase activity of HDAC1. A catalytically inactive mutant of HDAC1 (H141A) associates with CREB and PP1 at levels comparable to WT HDAC1, while another catalytically inactive mutant of HDAC1 (D174/6N) has no effect (Canettieri et al., 2003), implying that CREB dephosphorylation should be independent of HDAC1 activity. However, another study demonstrates that the HDAC1 H141A mutant is a partially active enzyme, while the D174/6N shows a near-complete inhibition (Hassig et al., 1998). It is worth noting that acetylation modification is shown to be involved in the regulation of CREB phosphorylation. CREB can be acetylated by CBP (Lu et al., 2003) and deacetylated by SIRT1 (Qiang et al., 2011). CBP overexpression is sufficient to promote CREB phosphorylation following increased CREB acetylation (Lu et al., 2003), while SIRT1 overexpression promoted CREB dephosphorylation following decreased CREB acetylation (Qiang et al., 2011), suggesting that acetylation may play a permissive role in CREB phosphorylation. These data imply that the deacetylase activity of HDAC1 would be required for CREB/HDAC1 interaction.
We found that TSA inhibited HDAC1/CREB interaction and rescued CREB dephosphorylation in the MPTP mouse model. These observations are in agreement with the neuroprotective effect shown in the present study and in others (Kidd and Schneider, 2011; Suo et al., 2015). In contrast, a few studies have reported that TSA promotes dopaminergic cell death (Wang et al., 2009; Park et al., 2016). Since HDAC inhibitors induce cell cycle arrest (Eckschlager et al., 2017), conclusions should be drawn with caution when immortalized cells are used to study the effect of HDAC inhibitors.
A limitation of this study is that only male mice were used for this study, which was because male mice are more sensitive to MPTP and exhibit more stability in the extent of neuropathology (Przedborski et al., 2001; Jackson-Lewis and Przedborski, 2007). Since PD is not a disease that only occurs in men, other animal models, including animals of both sexes, should be used to verify this mechanism in the future.
In conclusion, we found CREB inactivation by dephosphorylation in nigral dopaminergic neurons of PD patients. Our study demonstrated that CREB dephosphorylation by the HDAC1/PP1γ complex resulted in NURR1 downregulation and subsequent dopaminergic neuron loss. Importantly, increased CREB/HDAC1 interaction was observed in nigral dopaminergic neurons of PD patients. Our findings further the understanding of the molecular pathogenesis of PD and suggest that disruption of the CREB/HDAC1 interaction to restore CREB activity may be a potential therapeutic approach in PD.
Footnotes
This work was supported in part by National Natural Science Foundation of China Grants U1801681, 32070959, 31871019, and 81771368; Key Realm R&D Program of Guangdong Province 2018B030337001; Guangdong Provincial Key Laboratory of Brain Function and Disease 2020B1212060024; Guangzhou Municipal Science and Technology Project 201804020008; National Key R&D Program of China 2018YFA0108300; and Fundamental Research Funds for the Central Universities 2021qntd46. Human tissue was obtained from the University of Miami Brain Endowment Bank, a National Institutes of Health NeuroBioBank. We thank Dr. Jiayi Li and Dr. Shengtao Hou for professional advice on the manuscript.
The authors declare no competing financial interests.
References
- Armstrong MJ, Okun MS (2020) Diagnosis and treatment of Parkinson disease: a review. JAMA 323:548–560. 10.1001/jama.2019.22360 [DOI] [PubMed] [Google Scholar]
- Bertolotti A (2018) The split protein phosphatase system. Biochem J 475:3707–3723. 10.1042/BCJ20170726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhurtel S, Katila N, Srivastav S, Neupane S, Choi DY (2019) Mechanistic comparison between MPTP and rotenone neurotoxicity in mice. Neurotoxicology 71:113–121. 10.1016/j.neuro.2018.12.009 [DOI] [PubMed] [Google Scholar]
- Bollen M, Peti W, Ragusa MJ, Beullens M (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci 35:450–458. 10.1016/j.tibs.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush MH, Guardiola A, Connor JH, Yao TP, Shenolikar S (2004) Deactylase inhibitors disrupt cellular complexes containing protein phosphatases and deacetylases. J Biol Chem 279:7685–7691. 10.1074/jbc.M310997200 [DOI] [PubMed] [Google Scholar]
- Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson SD, Yates JR 3rd, Montminy M (2003) Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol 10:175–181. 10.1038/nsb895 [DOI] [PubMed] [Google Scholar]
- Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT (2006) Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem 281:17870–17881. 10.1074/jbc.M602632200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Endo S, Isacson O (2007) An endogenous serine/threonine protein phosphatase inhibitor, G-substrate, reduces vulnerability in models of Parkinson's disease. J Neurosci 27:8314–8323. 10.1523/JNEUROSCI.1972-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Ohlmeyer M (2019) Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol Ther 201:181–201. 10.1016/j.pharmthera.2019.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro IJ, Amin HA, Vinciotti V, Vagnarelli P (2017) Network of phosphatases and HDAC complexes at repressed chromatin. Cell Cycle 16:2011–2017. 10.1080/15384101.2017.1371883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM (2006) Epidemiology of Parkinson's disease. Lancet Neurol 5:525–535. 10.1016/S1474-4422(06)70471-9 [DOI] [PubMed] [Google Scholar]
- Durgadoss L, Nidadavolu P, Valli RK, Saeed U, Mishra M, Seth P, Ravindranath V (2012) Redox modification of Akt mediated by the dopaminergic neurotoxin MPTP, in mouse midbrain, leads to down-regulation of pAkt. FASEB J 26:1473–1483. 10.1096/fj.11-194100 [DOI] [PubMed] [Google Scholar]
- Eckschlager T, Plch J, Stiborova M, Hrabeta J (2017) Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci 18:1414. 10.3390/ijms18071414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Ekhator OR, Ghisays V (2013) Assessment of sensorimotor function in mouse models of Parkinson's disease. J Vis Exp 17:50303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Siddoway B, Huang Q, Xia H (2009) Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem Biophys Res Commun 379:1–5. 10.1016/j.bbrc.2008.11.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, Schreiber SL (1998) A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci USA 95:3519–3524. 10.1073/pnas.95.7.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M (2003) Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307:720–728. 10.1124/jpet.103.055541 [DOI] [PubMed] [Google Scholar]
- Huang C, Liu T, Wang Q, Hou W, Zhou C, Song Z, Shi YS, Gao X, Chen G, Yin Z, Hu Y (2020) Loss of PP2A disrupts the retention of radial glial progenitors in the telencephalic niche to impair the generation for late-born neurons during cortical development. Cereb Cortex 30:4183–4196. 10.1093/cercor/bhaa042 [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc 2:141–151. 10.1038/nprot.2006.342 [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409:581–589. 10.1042/BJ20070779 [DOI] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS (2011) Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience 194:189–194. 10.1016/j.neuroscience.2011.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshibu K, Graff J, Beullens M, Heitz FD, Berchtold D, Russig H, Farinelli M, Bollen M, Mansuy IM (2009) Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci 29:13079–13089. 10.1523/JNEUROSCI.3610-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, Herrup K (2012) Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med 18:783–790. 10.1038/nm.2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ma S, Chen J, Hu K, Li Y, Zhang Z, Su Z, Woodgett JR, Li M, Huang Q (2020) GSK-3β contributes to parkinsonian dopaminergic neuron death: evidence from conditional knockout mice and tideglusib. Front Mol Neurosci 13:81. 10.3389/fnmol.2020.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Montoya SE, Alerte TN, Wang J, Wu J, Peng X, Hong CS, Friedrich EE, Mader SA, Pedersen CJ, Marcus BS, McCormack AL, Di Monte DA, Daubner SC, Perez RG (2010) Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem 285:17648–17661. 10.1074/jbc.M110.100867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Hutchins AE, Doyle CM, Lundblad JR, Kwok RP (2003) Acetylation of cAMP-responsive element-binding protein (CREB) by CREB-binding protein enhances CREB-dependent transcription. J Biol Chem 278:15727–15734. 10.1074/jbc.M300546200 [DOI] [PubMed] [Google Scholar]
- Mann A, Chesselet MF (2015) Techniques for motor assessment in rodents. In: Movement disorders, Ed 2 (LeDoux MS, ed), pp 139–157. Boston: Academic. [Google Scholar]
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2:599–609. 10.1038/35085068 [DOI] [PubMed] [Google Scholar]
- Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A (2007) Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol 8:234–244. 10.1038/nrm2126 [DOI] [PubMed] [Google Scholar]
- Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z (2016) Regulation of histone acetylation by autophagy in Parkinson disease. J Biol Chem 291:3531–3540. 10.1074/jbc.M115.675488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M (2001) The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem 76:1265–1274. 10.1046/j.1471-4159.2001.00183.x [DOI] [PubMed] [Google Scholar]
- Qiang L, Lin HV, Kim-Muller JY, Welch CL, Gu W, Accili D (2011) Proatherogenic abnormalities of lipid metabolism in SirT1 transgenic mice are mediated through Creb deacetylation. Cell Metab 14:758–767. 10.1016/j.cmet.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K, Obrietan K (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem 116:1–9. 10.1111/j.1471-4159.2010.07080.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Sawai M, Ishizuka Y, Shirao T, Fukunaga K (2015) Nuclear translocation of calcium/calmodulin-dependent protein kinase IIδ3 promoted by protein phosphatase-1 enhances brain-derived neurotrophic factor expression in dopaminergic neurons. J Biol Chem 290:21663–21675. 10.1074/jbc.M115.664920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven A, Friedrich M, Jank P, Heimer N, Budczies J, Denkert C, Seliger B (2020) What turns CREB on? And off? And why does it matter? Cell Mol Life Sci 77:4049–4067. 10.1007/s00018-020-03525-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang Y, Mo M, Song C, Wang X, Chen S, Liu Y (2019) Minocycline protects against rotenone-induced neurotoxicity correlating with upregulation of Nurr1 in a Parkinson's disease rat model. Biomed Res Int 2019:6843265. 10.1155/2019/6843265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Aime P, Dai D, Ramalingam N, Crary JF, Burke RE, Greene LA, Levy OA (2018) Guanabenz promotes neuronal survival via enhancement of ATF4 and parkin expression in models of Parkinson disease. Exp Neurol 303:95–107. 10.1016/j.expneurol.2018.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo H, Wang P, Tong J, Cai L, Liu J, Huang D, Huang L, Wang Z, Huang Y, Xu J, Ma Y, Yu M, Fei J, Huang F (2015) NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson's disease. Neuropharmacology 99:67–78. 10.1016/j.neuropharm.2015.07.015 [DOI] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE (2007) Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol 365:23–38. 10.1385/1-59745-267-X:23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese F, Bukhari AB, Malhotra R, De A (2014) IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One 9:e96801. 10.1371/journal.pone.0096801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, Spiegelman BM, Perlmann T (2010) NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc Natl Acad Sci USA 107:12317–12322. 10.1073/pnas.1007088107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang X, Liu L, Wang X (2009) HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci Lett 467:212–216. 10.1016/j.neulet.2009.10.037 [DOI] [PubMed] [Google Scholar]
- Xu X, He X, Ma S, Li M, Huang Q (2021) Nurr1 downregulation is caused by CREB inactivation in a Parkinson's disease mouse model. Neurosci Lett 759:136045. 10.1016/j.neulet.2021.136045 [DOI] [PubMed] [Google Scholar]
- Zheng M, Liu C, Fan Y, Shi D, Jian W (2019) Total glucosides of paeony (TGP) extracted from Radix Paeoniae Alba exerts neuroprotective effects in MPTP-induced experimental parkinsonism by regulating the cAMP/PKA/CREB signaling pathway. J Ethnopharmacol 245:112182. 10.1016/j.jep.2019.112182 [DOI] [PubMed] [Google Scholar]
- Zhong J, Dong W, Qin Y, Xie J, Xiao J, Xu J, Wang H (2020) Roflupram exerts neuroprotection via activation of CREB/PGC-1alpha signalling in experimental models of Parkinson's disease. Br J Pharmacol 177:2333–2350. 10.1111/bph.14983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2017) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog Neurobiol 155:96–119. 10.1016/j.pneurobio.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]