

Abstract
This is a review of the current state of molecular profiling in gastrointestinal (GI) cancers and what to expect from this evolving field in the future. Individualized medicine is moving from broad panel testing of numerous genes or gene products in tumor biopsy samples, identifying biomarkers of prognosis and treatment response, to relatively noninvasive liquid biopsy assays, building on what we have learned in our tumor analysis and growing into its own evolving predictive and prognostic subspecialty. Hence, the field of GI precision oncology is exploding, and this review endeavors to summarize where we are now in preparation for the journey ahead.
Keywords: Gastrointestinal, GI; Molecular profiling; Next-generation sequencing, NGS; Liquid biopsy; ctDNA; CTC; Gastroesophageal cancer; Colorectal cancer; Pancreatic cancer; Hepatobiliary cancer; Biomarker
Introduction
Comprehensive molecular profiling has evolved over the last decade. The evolution of molecular profiling changed the face of oncology from standard chemotherapy based on histology to personalized therapy. Using immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), and whole-genome sequencing, oncologists are able to recognize the genomic drivers of tumorigenesis and provide patients with prognostic biomarkers and targeted therapy options (Fig. 1; Table 1).
Fig. 1.

Predictive biomarkers in GI malignancies
Table 1.
Current NCCN predictive markers
| Molecular abnormality | Test | Method | When | Details |
|---|---|---|---|---|
| Colon cancers 2.2021 | ||||
| MLH1, MSH2, MSH6 or PMS2 mutations | MMR protein expression | IHC | Work-up |
Universal testing for MMR or MSI is recommended in all patients newly diagnosed with colorectal cancer. dMMR and MSI-H testing are recommended to predict response to pembrolizumab. Patients with Stage II MSI-H tumors generally have a good prognosis and do not benefit from 5-FU adjuvant therapy. |
| MSI | MSI; changes in short repeated DNA sequences |
PCR NGS |
Work-up | |
| KRAS/NRAS | KRAS (exon 2,3,4) gene; NRAS (exon 2,3,4) gene mutations | NGS | Work-up for metastatic disease: primary tumor and/or metastases |
Patients with any known KRAS mutation (exon 2,3,4) or NRAS mutation (exon 2,3,4) should not be treated with either cetuximab or panitumumab. A BRAF V600E mutation makes response to panitumumab or cetuximab highly unlikely unless given with a BRAF inhibitor (e.g., vemurafenib or encorafenib). If patient tumors have a known RAS/RAF mutation, HER2 testing is not indicated. Anti-HER2 therapy is indicated only in HER2-amplified tumors that are also RAS and BRAF wild type. |
| BRAF | V600E mutation |
IHC NGS |
||
| HER2 (ERBB2) | Gene amplification |
FISH IHC NGS |
||
| NTRK1/2/3 | Gene fusion |
FISH IHC RT-PCR NGS |
Not specified |
TRK inhibitors have activity in patients with NTRK fusions (not mutations). Data support limiting testing for NTRK fusions to tumors that are KRAS, NRAS, BRAF, and HER2 wild-type and (arguably) those that are MMR deficient (dMMR)/MSI-H. |
| Gastric/esophageal/GEJ Cancers 2/2021 | ||||
| HER2 | Amplification |
(F)ISH NGS |
Work-up any time for suspected or documented inoperable locally advanced, recurrent, or metastatic adenocarcinoma | Particularly if trastuzumab therapy is being considered. |
| Protein expression | IHC | |||
| PD-L1 (CD274) and HER2 protein | Protein expression | IHC |
HER2 negative status corresponds with higher PD-L1 expression rates. Together with MMR, HER2 is a potential biomarker for anti-PD-L1 therapy PD-L1 and MSI testing should be considered on locally advanced, recurrent, or metastatic esophageal or GEJ cancers in patients who are candidates for treatment with PD-1 inhibitors. In gastric cancer, universal testing for MSI/MMR is recommended in all newly diagnosed patients. If sufficient tissue is available, broader NGS may be contemplated. At present, three targeted therapeutic agents have been approved by the FDA for use in esophageal and GEJ cancers: trastuzumab (HER2 positivity), ramucirumab, and pembrolizumab (MSI/MMR, PD-L1 expression, or high tumor mutation burden [TMB; by NGS]). NGS offers the opportunity to assess numerous mutations simultaneously. |
|
| MSI | MSI; changes in short repeated DNA sequences |
PCR NGS |
||
| MLH1, MSH2, MSH6 or PMS2 mutations | MMR protein expression | IHC | ||
| NTRK1/2/3 | Gene fusion | NGS | Not specified | The FDA approved the use of select TRK inhibitors (entrectinib, larotrectinib) for NTRK gene fusion-positive solid tumors. |
| Hepatobiliary cancer 1.2021 | ||||
| MSI | MSI; changes in short repeated DNA sequences | PCR | Prior to primary treatment of metastatic or unresectable Gallbladder Cancer or metastatic intrahepatic cholangiocarcinoma | The PD-1 inhibitor, pembrolizumab can be used in patients with MSI-H/dMMR/TMB-H tumors. |
| MLH1, MSH2, MSH6 or PMS2 mutations | MMR protein expression | IHC | ||
| FGFR2 | Gene fusion | NGS | Evaluate for subsequent lines of therapy for unresectable or metastatic bile duct cancer | FGFR2 inhibitors (pemigatinib, infigratinib) are an option for cholangiocarcinoma with FGFR2 fusions or rearrangement. |
| IDH1 | Mutation | NGS | Evaluate for subsequent lines of therapy for unresectable or metastatic bile duct cancer | IDH1 inhibitor (ivosidenib) can be used for cholangiocarcinoma with IDH1 mutations. |
| BRAF | V600E mutation | NGS | Evaluate for subsequent lines of therapy for unresectable or metastatic bile duct cancer | Dabrafenib + trametinib can be used for BRAF V600E mutant tumors. |
| NTRK1/2/3 | Gene fusion |
FISH RT-PCR NGS |
Hepatobiliary cancer | TRK inhibitors (entrectinib, larotrectinib) have activity in patients with NTRK fusions. |
| Pancreatic 2/2021 | ||||
|
BRCA1 & BRCA2 BRAF HER2 KRAS PALB2 |
Mutation (somatic and germline) |
IHC NGS |
Initial work-up Tumor and blood |
Cell-free DNA testing can be considered if tumor tissue testing is not feasible. 9% of pancreatic cancers harbor a germline or somatic BRCA1 or BRCA2 mutation. Olaparib can be used if patient disease has not progressed on first-line platinum-based chemotherapy. Other DDR enzyme inhibitors may be effective. An EGFR inhibitor (e.g., erlotinib) = chemotherapy may benefit KRASwt patients. A BRAF mutation makes this response unlikely. |
|
Fusions: inc ALK, NRG1, NTRK, ROS1 |
IHC PCR NGS |
Fusions are rare but, taking NTRK as an example, TRK inhibitors (e.g., larotrectinib) in these patients very effective. Other fusion inhibitors are experimental in pancreatic cancer but an option. |
||
| MLH1, MSH2, MSH6 or PMS2 mutations | MMR protein expression | IHC | Work-up for locally advanced or metastatic disease | Pembrolizumab is an option in first-line and beyond, particularly for patients with MSI-H and dMMR tumors and no other satisfactory treatment options. |
| MSI | MSI; changes in short repeated DNA sequences |
PCR NGS |
||
Most biomarkers in these tables are classed as “useful in certain circumstances”
Testing in a CLIA-approved laboratory using validated tests or panels is recommended. For NGS, a CLIA-approved high-complexity laboratory is recommended
NGS can pick up rare and actionable mutations and fusions and is recommended for all GI cancers
Molecular profiling using tissue next-generation sequencing (NGS) has become a standard of care practice, and recently, circulating tumor DNA (ctDNA) has emerged as a tool for molecular profiling, a predictor of response to systemic treatment, and a powerful way to measure minimal residual disease (MRD) using less invasive approaches [1–3].
A decade ago, the treatment of colorectal cancer (CRC) was the first of all GI malignancies to be influenced by molecular profiling. It was initially observed that patients with KRAS mutant CRC do not respond to epidermal growth factor rector (EGFR)-targeted agents such as the monoclonal antibodies cetuximab and panitumumab [4, 5]. Later, it was recognized that BRAF, NRAS, and PIK3CA mutations and HER2 mutations and amplifications also confer non-responsiveness to EGFR-targeted agents and carry a generally poorer patient prognosis than wild-type disease [6, 7]. Subsequently, patients with BRAF V600E mutant CRC were shown to benefit from treatment with vemurafenib, a small molecule tyrosine kinase inhibitor [8], and later, encorafenib combined with cetuximab emerged as a standard of care for this subset of patients following chemotherapy [9] (Table 1). With the advent of immune checkpoint inhibitors (ICIs) that target PD-1/PD-L1, microsatellite instability (MSI) was found to be the most significant predictor of CRC treatment response. MSI can be sporadic or driven by germline mutations in one of the MMR genes (MLH1, MSH2, MSH6, or PMS2), as found in hereditary Lynch syndrome [10, 11].
MSI testing is essential for all GI malignancies because localized MSI-high (MSI-H) tumors will have a good prognosis, and advanced disease will likely respond to PD-1/PD-L1 inhibitors [12, 13] (Table 1). Until very recently, advanced gastric and gastroesophageal cancers were treated solely with conventional chemotherapy. Now, advances in molecular profiling and signaling pathway knowledge have provided new treatment options for patients with PD-L1 or HER-2 overexpressing tumors, including PD-1/PD-L1 inhibitor therapy, HER2 targeted treatment, and anti-vascular endothelial growth factor (VEGF) antibody therapy [14–17].
Advances in molecular profiling have led to therapeutic options targeting advanced biliary tract cancers with IDH1/2 mutations, FGFR alterations, HER2 amplifications, and BRAF V600E mutations [18–21] (Table 1).
In pancreatic cancer, understanding the role of germline testing for BRCA1, BRCA2, and PALB2 in homologous recombination repair has allowed the emergence of poly(adenosine diphosphate-ribose) polymerase inhibitors (PARPi) as a treatment option [22].
Biomarkers
PD-L1
PD-1 is an inhibitory receptor expressed on several immune cells, particularly cytotoxic T cells. It interacts with 2 ligands: PD-L1 and PD-L2. PD-L1 is expressed on tumor cells and immune cells, whereas PD-L2 is expressed on macrophages and dendritic cells. The interaction of PD-1 with PD-L1 inhibits T-cell activation and cytokine production, which is vital to maintaining homeostasis of the immune response and preventing autoimmunity [23, 24]. However, PD-1/PD-L1 interactions within the tumor microenvironment provide an immune escape pathway for tumor cells by turning off cytotoxic T cells [25]. Tumor cells upregulate the PD-1 receptor or ligand to evade destruction by the host immune system. Thus, in blocking the PD-1 pathway with antibodies to PD-1 and PD-L1/PD-L2, the adaptive immune response is activated against tumor cells resulting in an anticancer response. Tumor cell PD-L1 expression is associated with response to anti-PD-1/anti-PD-L1 therapy [26].
PD-L1 protein expression in many cancer types, assessed via immunohistochemistry (IHC), is one of the FDA-approved predictive biomarkers for anti-PD-1 and anti-PD-L1 ICI monotherapy [26]. However, PD-L1 expression within tumors and between tumor sites may be heterogeneous [27–29], and assays may give variable results. To the latter point, there are multiple qualitative PD-L1 assays involving different antibodies to assess the expression of PD-L1 by IHC using chromogenic methods [30], and different antibody assays may give different results.
In esophageal/GEJ/gastric cancer, the PD-L1 combined positive score (CPS) has been tested as a predictive biomarker for immunotherapy. CPS is the number of cells staining for PD-L1 cells (tumor cells, lymphocytes, and macrophages) divided by the total number of evaluated tumor cells, multiplied by 100 [31]. Tumors are considered PD-L1 positive if they have a CPS > 1. A positive CPS is associated with improved GI cancer patient outcomes upon ICI therapy. In KEYNOTE 062, KEYNOTE 061, and KEYNOTE 059, the PD-1 inhibitor pembrolizumab demonstrated efficacy against gastric and GEJ cancer as first, second, or third-line treatment based on a CPS of > 1 [32]. On the other hand, the PD-1 inhibitor nivolumab is FDA approved in esophageal/GEJ/gastric cancers regardless of CPS, based on CHECKMATE 648 and CHECKMATE 649 studies [14, 33]. How to use CPS in the selection of upper GI cancer patients for frontline ICI therapy remains a point of debate in the oncology community [34].
TMB: tumor mutation burden
TMB, defined as the total number of exon mutations in a tissue sample [35], has emerged as an important biomarker associated with immunotherapy response in multiple tumor types [26]. TMB is a critical driver in the generation of immunogenic neopeptides presented on major histocompatibility complexes on the tumor cell surface [36]. These immunogenic components influence response to ICIs, meaning that TMB impacts ICI efficacy. This TMB effect on ICI efficacy is reflected in many retrospective studies, including the Phase II Keynote-158 trial [37], which led to the FDA approval of pembrolizumab in patients with unresectable or metastatic high TMB (≥ 10 mutations/megabase) solid tumors [38].
Detecting ctDNA in the blood is a noninvasive test called liquid biopsy (see the section on Molecular profiling in the blood). With an increasing interest in ctDNA, studies have been carried out to develop methods, including NGS, that can estimate the tumor fraction in a patient’s plasma and measure the TMB from their blood with high accuracy [39]. Chalmers et al. [40] demonstrated that TMB can be accurately measured in blood by sequencing targeted gene panels, but accuracy is compromised when the sequenced genome region is less than 0.5 MB. The Guardant Health Omni panel (500 genes, 2.1 MB) and Foundation Medicine bTMB panel (394 genes, 1.14 MB) are plasma-based NGS assays containing sufficiently large genome region sizes to measure TMB across a broad range of TMB values. In a study by Qiu et al. [41], Guardant Health and Foundation Medicine tests were evaluated and compared in their ability to evaluate TMB from ctDNA. The investigators ascertained that tissue and plasma TMB correlated well using both assays as long as analyzed samples contained a high TMB; the correlation was compromised if samples contained only low to medium TMB [41].
TMB has been used as a predictor for response to ICI therapy, and early measurements of ctDNA were shown to help detect treatment failure [42]. MRD is another important biomarker of treatment failure. It refers to residual tumor cells present after cancer treatment and is associated with disease recurrence. MRD can be detected in blood using techniques like quantitative PCR and NGS. However, most recently, ctDNA has been used to detect MRD in the blood, serving as a powerful diagnostic and predictive tool (Fig. 2) [43].
Fig. 2.
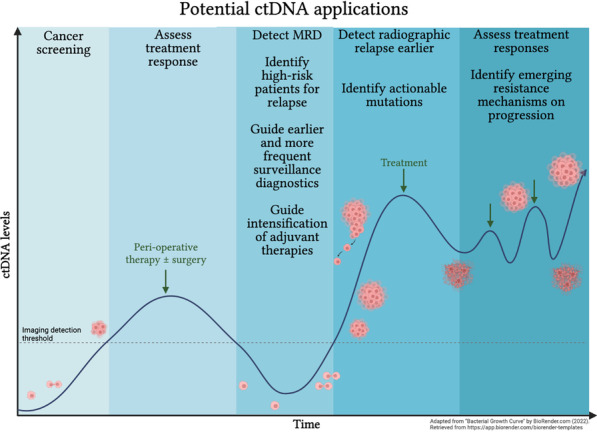
Potential ctDNA applications
MSI
Microsatellite instability (MSI) is a molecular fingerprint for defects in the mismatch repair system (dMMR), which is associated with an increased risk of cancer [44]. The MMR system is composed of heterodimers (MSH2/MSH6 and MSH2/MSH3 complexes) that ensure the specific recognition of mispaired nucleotides generated due to DNA damage [45]. In humans, these complexes initiate DNA repair and recruit MLH1/PMS1, MLH1/PMS2, and MLH1/MLH3 heterodimers to catalyze the excision of the mispaired nucleotides as well as error-free DNA resynthesis. Genetic and epigenetic inactivation of MMR genes cause MMR defects (dMMR) and give rise to spontaneous, genome-wide mutations [45]. This mainly affects the short tandem repeat DNA sequences termed microsatellites, which occur at specific foci throughout the genome.
MSI-H tumors contain many mutation-associated neoantigens, which, it is believed, are recognized as foreign by the immune system. The benefit of ICIs for patients with MSI-H/dMMR tumors was first documented in a Phase II trial, in which patients with metastatic cancer (78% colorectal) with and without dMMR received pembrolizumab. Only the patients with MSI-H/dMMR tumors benefited from the ICI therapy [46]. Results from this trial were confirmed in the larger Phase II KEYNOTE 158 study evaluating pembrolizumab in dMMR metastatic colorectal patients [37]. Later, results from the KEYNOTE 177 trial of pembrolizumab as first-line treatment for patients with MSI-H/dMMR metastatic colorectal cancer showed longer progression-free survival (PFS) compared to standard chemotherapy [47]. Similarly, the CHECKMATE 142 trial suggested durable benefit from combined nivolumab plus ipilimumab (anti-CTLA-4) in patients with MSI-H/dMMR tumors [48].
Conventional methods used for MSI testing include immunohistochemistry (IHC) and PCR-based assays performed on tumor tissue samples. Tumor tissue-based NGS can also determine MSI status.
HER2
HER2 is a member of the human epidermal growth factor receptor (HER) family. This family includes HER1 (ErbB1; EGFR), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) [49–52]. The HER2 receptor regulates normal cell proliferation, survival, and differentiation via different signal transduction pathways. Amplification or overexpression of HER2 is found in 2–6% of patients with metastatic colorectal cancer [53–55]. The frequency of HER2 overexpression in gastric and gastroesophageal cancer ranges from 4.4 to 53.4%, with a mean of 17.9% [56, 57].
Several strategies have been developed to target HER2, including extracellular antibodies like trastuzumab, which targets domain IV of the receptor, and pertuzumab which binds to domain II and inhibits the heterodimerization of HER2 with other ErbB receptors [58]. Additionally, small tyrosine kinase inhibitors like lapatinib, tucatinib, or neratinib inhibit HER2 activity, while antibody–drug conjugates (ADCs), such as trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-Dxd), bind HER2 and introduce a potent cytotoxic agent into cells overexpressing the receptor [16].
The Phase III ToGA (Trastuzumab for Gastric Cancer) trial for patients with gastric or GEJ cancer with overexpression of HER2 or gene amplification was the first study to demonstrate the therapeutic benefit of targeting HER2 in GI cancers [15]. The US FDA subsequently approved trastuzumab for the first-line treatment of patients with metastatic, HER2-positive gastric or gastroesophageal cancer.
Subsequent trials in patients with GI cancers include the Phase 2 HERACLES trial [59] and the ongoing MyPathway basket trial [60]. In the HERACLES trial [59], standard treatment-refractory patients with KRAS wild-type (wt) CRC harboring a HER2 amplification received trastuzumab and lapatinib. In a subset of the ongoing MyPathway basket trial [60], patients with HER2-amplified metastatic CRC received pertuzumab plus trastuzumab. The objective response rates were around 30% in both studies, and several other patients had stable disease, demonstrating that HER2 amplification is an actionable target. More recently, a trial of T-Dxd in previously treated patients with gastric or GEJ cancer found improved overall survival (OS; 12.5 months vs. 8.4 months; P = 0.0097) and ORR (40.5% vs. 11.3%) compared to standard chemotherapy. These results led to the US FDA approval of T-Dxd in the third or later lines of therapy [16].
The development of tumor resistance to HER2 inhibitors is a problem for which there are multiple possible mechanisms, including loss of HER2 expression and HER3 ligand-dependent HER2-HER3 interactions leading to evasion of apoptosis [61]. A TAF/FGF5/FGFR2/c-Src/HER2 axis might act as a HER2-targeted therapy escape pathway, which seems to be reversed by FGFR inhibition [62].
Prior attempts to demonstrate an OS benefit from second-line HER2-targeted vs. standard cytotoxic therapy have failed, possibly due to loss of HER2 expression following trastuzumab-based first-line treatment. MOUNTAINEER-02 trial investigators hope that dual targeting of HER2 with tucatinib and trastuzumab will overcome this resistance. Tucatinib is a small molecule tyrosine kinase inhibitor, which was shown to have “very potent,” selective activity against HER2, with minimal off-target effects [63]. The ongoing Phase II/III MOUNTAINEER-02 trial [NCT04499924] [64] is enrolling patients with advanced or metastatic HER2-positive (overexpression or amplification) gastric or GEJ cancer with disease progression (PD) after frontline therapy, including a HER2-directed antibody. Patients receive second-line treatment with paclitaxel plus ramucirumab, either with tucatinib plus trastuzumab, tucatinib plus trastuzumab-placebo, tucatinib-placebo plus trastuzumab, or two placebos.
Future directions include using liquid biopsy genotyping assays as a viable, real-time alternative to tissue-based genotyping in the identification of HER2 alterations in the metastatic setting. HER2 copy number is typically assessed using surgically-obtained tissue, but necessary information can now be obtained conveniently and noninvasively using ctDNA (See the “Molecular profiling in blood” section).
FGFR 1–4
Fibroblast growth factors (FGFs) and their receptors (FGFR 1, 2, 3, and 4) are vital to many cellular processes. After ligand stimulation, FGFRs undergo dimerization and phosphorylation, prompting intracellular signaling that triggers a number of intracellular survival and proliferative pathways [65–67]. Aberrant FGFR signaling (found in just over 7% of all cancers) has been shown to have an oncogenic role. FGFR alterations (primarily in FGFR2) are found in approximately 13% of intrahepatic cholangiocarcinomas (CCA), 3% of gallbladder cancers, 9% of gastric cancers in a Western population, and 3% of gastric cancers in an Asian population) [68–73].
Of all FGFR2 aberrations, 66% are amplifications, 26% mutations, and 8% rearrangements [74]. Oncogenesis most often occurs through FGFR pathway activation. For example, FGFR amplifications and rearrangements lead to protein overexpression and dependence on FGFR signaling, although conversely, preclinical models suggest that amplifications also predict increased sensitivity to FGFR inhibition [75, 76]. Mutations in FGFRs cause increased downstream phosphorylation [72, 75].
FGFR has become a molecular target of increasing interest in CCA. There are several FGFR-targeted therapies of interest, mostly in the form of tyrosine kinase inhibitors (TKIs). Pemigatinib is a selective oral FGFR1-3 inhibitor investigated in the open-label single-arm FIGHT-202 trial for previously treated advanced CCA [77]. Among patients with FGFR2 alterations, pemigatinib displayed an overall response rate (ORR) of 36%, disease control rate (DCR) of 80%, and median duration of response (DOR) of 7.5 months. Pemigatininb is now FDA approved for previously treated, unresectable, locally advanced, or metastatic CCA with an FGFR2 alteration. Pemigatininb combined with gemcitabine and cisplatin is currently being studied in the first-line, phase III FIGHT-302 trial (NCT03656536). Infigratinib, another selective FGFR 1–3 TKI, obtained accelerated FDA approval in subsequent-line settings for FGFR2-altered CCA. This drug demonstrated an ORR of 23% and mDOR of 5 months [78]. In CCA, other drugs currently under study include derazantinib (FGFR1-3 inhibitor) and erdafinitib (FGFR1-4 inhibitor). Toxicities of FGFR inhibitors are predictable and similar across this class of therapeutics and include hyperphosphatemia (50–80%), nail toxicity (35%), and ophthalmologic toxicity (4–9%) [77, 79–81].
IDH1/2
Isocitrate dehydrogenase (IDH) is a key enzyme in the tricarboxylic acid cycle and comprises 2 subtypes: IDH1, located in the peroxisomes and cytosol, and IDH2, located in the mitochondria [82, 83]. In CCA, IDH1 mutations are found in 15–25% of cases, particularly in intrahepatic CCA. IDH2 mutations are less frequent, found in up to 3% of CCAs [84, 85]. IDH mutations generally lead to a gain-of-function that disrupts normal catalytic activity. The net effect is increased conversion of α-ketoglutarate to D-2-hydroxyglutarate, which leads to downstream cellular proliferation through pathways including DNA methylation and VEGFR [82, 83, 86].
Multiple IDH-selective inhibitors are being investigated in vitro and in clinical trials. Ivosidenib, an oral small-molecule inhibitor, was among the first to be studied clinically: A phase I study confirmed tolerability and demonstrated a median PFS (mPFS) of 3.8 months in previously treated patients with IDH1-mutated CCA [87]. The recent ClarIDHy Phase III placebo-controlled trial demonstrated an OS benefit from ivosidenib that trended towards statistical significance (mOS 10.3 vs. 7.5 months, HR 0.79, 95% CI 0.56–1.12; p = 0.093), becoming statistically significant once a mathematical model adjusting for treatment crossover effects was employed (mOS 10.3 vs. 5.1 months, HR = 0.49; 95% CI 0.34–0.70; p < 0.0001) [88]. Ivosidenib was granted FDA approval in August 2021 [89]. Additional promising IDH1-targeting drugs are under early investigation, including another small-molecule inhibitor, olutasidenib (NCT03684811).
BRCA/PALB2
Germline mutations in BRCA1 and BRCA2 are well studied and associated with a high risk of cancer, particularly breast and ovarian cancers, with high hereditary penetrance in an autosomal dominant pattern [90–92]. This associated risk has been established across other cancers, including pancreatic cancer, where BRCA2 mutations pose a relative risk of 3.5–10 compared to non-carriers.
BRCA1 and BRCA2 are suppressor genes of the same family, located on long arms of chromosome 17 and 13, respectively [93, 94]. They play instrumental roles in DNA damage response, particularly in maintaining chromosomal stability in the process of homologous recombination repair [95, 96]. One of the early successes of whole-genome sequencing was the identification of the BRCA2 and its partner and localizer gene PALB2 in 1–4% of familial PDAC [90, 97, 98]. PALB2 colocalizes with BRCA2 at the site of DNA damage to enable DNA repair [99].
Deficiencies in homologous recombination and DNA repair pathways predict sensitivity to platinum-based chemotherapy regimens as well as poly(ADP-ribose) polymerase inhibitors (PARPi). Patients with PDAC and homologous recombination gene mutations had improved PFS and OS when treated with frontline platinum-based therapy compared to patients without such mutations (HR 0.44, 95% CI 0.29–0.67; P < 0.01) [100, 101]. Similarly, PARPi appear to be active in PDAC; the phase III POLO trial demonstrated sensitivity to olaparib of patient tumors with homologous recombination gene mutations. Maintenance olaparib after platinum-based induction therapy showed superior mPFS to placebo (7.4 vs. 3.8 months, HR 0.53, 95% CI 0.35–0.82) [102]. Olaparib is FDA-approved in the maintenance setting for PDAC.
BRAF V600E mutation
The BRAF V600E mutation is found in about 8–10% of CRCs [103] and 3–7% of bile duct cancers [71].
In the NCI-MATCH EAY131-H trial, a combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) produced favorable response rates in a total of 35 pretreated patients with a range of solid tumors, all harboring a BRAF V600E mutation [104]. A confirmed objective response rate (ORR) of 37.9% (90% CI 22.9–54.9%; P < 0.0001 against a null rate of 5%) was reported. The median duration of response was 25.1 months. Four of the 35 patients enrolled on trial had CCA, and 3 of these 4 experienced a partial response (PR) [104].
The same drug combination was tested in patients with bile duct cancer harboring BRAFV600E mutations as part of the ROAR Basket Trial [21]. The ROAR Basket Trial enrolled 9 different cohorts of 178 patients with rare malignancies, all harboring BRAF V600E mutations. The bile-duct cancer cohort included 35 patients treated with the combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib. Most patients (74%) had stage IV disease at enrollment, and all 35 patients with biliary tract cancer had received prior chemotherapy (80% had received at least 2 prior lines). The median duration of treatment exposure was 6 months, with 86% of the patients being treated for more than 3 months. The median follow-up was 8 months. A PR was reported in 42% of the cohort by investigator assessment and 36% by independent review. Stable disease was achieved in 45% and 39%, respectively, and 12% had progressive disease as their best response (by either assessment method). The mPFS by investigator assessment was 9.2 months, and the median OS was 11.7 months. With regard to safety, adverse effects (AEs) were found to be comparable to those previously reported with each agent alone, with no new toxicities observed. Potential study treatment-related toxicities included fatigue, neutropenia, hyponatremia, and hypophosphatemia. No grade 5 events were observed [21].
Molecular profiling in the blood
A liquid biopsy identifies components of a tumor in the blood, such as ctDNA, circulating tumor cells (CTC), circulating tumor RNAs, and circulating tumor exosomes [105]. Of all these entities, ctDNA is the most studied to date and comprises fragments of cell-free DNA (cfDNA) that retain tumor-specific mutations and epigenetic characteristics [106–108]. These fragments are released into the circulation spontaneously or after apoptosis or necrosis. ctDNA makes up a highly variable fraction of total cfDNA in peripheral blood, and this fraction (reportedly ranging from less than 0.1% to almost 90%) is impacted by disease stage, tumor type, and analysis technique, to name a few. The half-life of ctDNA is 16 min to 2 h, allowing for indirect real-time tumor characterization [109–111]. Although the liquid biopsy was described as early as the 1940s, it is only recently, with enhancements in genomic sequencing techniques and identification of novel biomarkers, that we have seen more commercialized applications of liquid biopsy platforms [112–114].
Overview
The advantages of blood-based molecular profiling include the ability to study intra- and inter-tumor genomic heterogeneity, the ability to perform tumor profiling in the absence of available tissue, avoidance of invasive biopsy procedures, the feasibility of frequent longitudinal testing, and a quick turnaround time to inform treatment plans [115]. To date, numerous commercial liquid biopsy profiling assays are available to providers. Methodologies overlap with tissue profiling and involve comprehensive genomic, single-gene, or hotspot gene testing. In hotspot testing, commonly altered regions within select genes are evaluated. Of the comprehensive genomic tests, only two are FDA approved today. Guardant Health’s Guardant360 CDx, targeting 55–74 genes using DNA NGS, was first FDA approved on August 7, 2020. Roche’s FoundationOne Liquid CDx, targeting over 300 genes using DNA NGS, was first approved on August 26, 2020 [116–118]. Although approved as companion diagnostic tests for therapeutics in lung, prostate, ovarian, and breast cancer, these assays are increasingly studied and adopted in GI cancers to identify prognostic and predictive biomarkers, actionable mutations with FDA-approved therapies, responses to treatment, and mechanisms of resistance. These and other comprehensive gene-based assays utilize NGS and can identify single nucleotide variants (SNVs), insertion and deletions, gene rearrangements, copy number variants (CNVs), TMB scoring, and microsatellite status [112, 116, 119, 120].
Numerous studies have recently evaluated ctDNA genomic profiling in GI cancers to identify actionable mutations, monitor disease response, and understand resistance mechanisms (Fig. 2; Table 2). Concordance rates of 85–98% have been reported for ctDNA and tissue genomic profiling, and promising sensitivities and specificities in NGS/PCR-based ctDNA assays, for example, 50–93% and 97–100% respectively for RAS mutations, 63–100%, and 98–100% for BRAF mutations, and 33–98% and 98% for ERBB2 amplifications, have been observed [109, 121–123]. In addition, studies have suggested that ctDNA captures genomic heterogeneity between primary and metastatic sites, which should act to enhance therapy selection [124]. As a noninvasive and convenient test, blood-based genomic profiling has the potential to replace or complement tissue testing, particularly when considering targeted therapies.
Table 2.
Select Examples ctDNA studies in GI cancers
| Gene | Tumor | Study type and details | Sample # | Assay utilized | Findings | Reference |
|---|---|---|---|---|---|---|
| RAS | CRC | Prospective cohort | 98 pts | BEAMing expanded RAS mutation panel |
RAS mut status was evaluable in plasma RAS plasma-tissue concordance was 91.8% ctDNA MAF was associated with clinical stage RAS testing using BEAMing was comparable to tissue testing |
[125] |
| RAS | mCRC | Prospective | 236 pts | OncoBEAM |
RAS plasma-tissue concordance was 92% Plasma false-negatives were more frequent in lung-metastases-only disease |
[126] |
| RAS | mCRC | Prospective | 280 pts | OncoBEAM |
RAS plasma-tissue concordance was 86.4%, positive percent agreement was 82.1%, and negative percent agreement was 90.4% Lung-metastases-only disease was associated with more discordance (concordance rate of 64.5%), but concordance improved with larger tumor burden |
[127] |
| RAS/EGFR/BRAF | mCRC |
Retrospective cohort Plasma samples from RAS/EGFR/BRAF wt mCRC pts analyzed after PD on anti-EGFR therapy |
135 pts 496 pts in the validation cohort |
Guardant 360 NGS |
RAS and EGFR mut clones decrease exponentially from the time of EGFRi discontinuation Confirmed ECD EGFR mutation as a potential driver for EGFRi resistance Identified half-life of RAS/EGFR clones to guide the timing of re-challenge to therapy |
[128] |
| RAS/EGFR/BRAF | mCRC |
Phase II EGFRi therapy rechallenge after prior PD guided by RAS/BRAF/EGFR status in ctDNA |
27 pts | ddPCR and NGS |
69% of screened pts were wt First interventional trial of liquid biopsy—ctDNA molecular selection—driving EGFRi rechallenge in mCRC RAS/BRAF/EGFR wt pts rechallenged to EGFRi demonstrated 30% ORR, 40% SD, 59% DCR with SD > 4 months, mPFS 16 weeks |
[129] CHRONOS |
| RAS/BRAF | mCRC | Prospective | 72 pts | Idylla Biocartis |
RAS/BRAF plasma-tissue concordance was 81.94%, with higher concordance in liver metastases cases Emerging KRAS mutations were identified in 33% of pts treated with EGFRi |
[130] |
| RAS/BRAF | mCRC |
Prospective Plasma mutational testing prospective series |
278 pts | OncoBEAM |
RAS/BRAF plasma-tissue concordance in chemotherapy naïve pts with liver metastases was 91.8% Supports ctDNA as a surrogate marker to tissue testing for RAS and BRAF status |
[131] ColoBEAM |
| RAS/BRAF | mCRC |
Prospective, non-randomized EGFRi therapy rechallenge after prior PD guided by RAS/BRAF status in ctDNA |
22 pts | PyroMark Q24 MDx Workstation, Genetic Analyzer ABI3130, Idylla RT-PCR, QZ200 System ddPCR |
70% of enrolled pts were wt Wt pts who underwent rechallenge experienced 27% ORR, 55% DCR, 7-month mOS, and 3-month mPFS Rechallenge strategy is feasible with molecular selection through ctDNA |
[132] |
| RAS | mCRC |
Prospective, phase II, single-arm EGFRi therapy rechallenge in 3rd line setting after prior PD |
28 pts | ddPCR and Ion Torrent S5 XL ultra-deep NGS |
48% samples at rechallenge baseline were RAS mut wt patients experienced 21% ORR, 32% SD, 54% DCR, mPFS 3.4 mo, and mOS 9.8 mo ctDNA RAS status predicted for responses to EGFRi. RAS wt was associated with a longer mPFS compared to RAS mut (4.0 vs. 1.9 mo, p = 0.03), with a trend toward longer mOS (12.5 vs. 5.2 mo, p = 0.24) |
[133] CRICKET |
| RAS | mCRC | Retrospective post-hoc biomarker study (pts from JACCRO CC-08 and 09) | 16 pts | OncoBEAM Ras CRC |
38% pts at rechallenge were RAS mut DCR was better in RAS wt compared to RAS mut pts (80% vs. 33%, respectively) mPFS in RAS mut vs. RAS wt pts was 2.3 vs. 4.7 mo (p = 0.0042) and mOS 3.7 vs 16 mo (p = 0.0002), respectively ctDNA RAS status was significantly associated with clinical outcomes in pts receiving EGFRi rechallenge |
[134] |
| BRAF | mCRC | Retrospective | 64 pts | MD Anderson/GuardantHealth LB70 NGS |
BRAF V600E plasma-tissue concordance was 80% Lower BRAF V600E VAF was associated with acquired resistance to EGFRi ctDNA to detect BRAF V600E mut is feasible |
[135] |
| Multiple genes | mGEC | Cohort study |
26 pts 28 pts |
Guardant 360 |
Demonstrated genomic heterogeneity within the primary tumor and in disseminated disease Found discordance between primary tumor and metastases in 36% of patients and high concordance between metastases and ctDNA (85%) ctDNA profiling may enhance selection of therapy by identifying heterogeneous mutation profiles |
[124] |
| HER2 | GC |
Prospective Biomarker study in pts treated with neoadjuvant capecitabine + oxaliplatin + lapatinib in HER2 + GC |
32 pts | Guardant 360 |
Plasma ERBB2 amplification predicted for chemotherapy + lapatinib responses Changes in plasma ERBB2 copy number were associated with responses to therapy Plasma genomics at the time of PD revealed emergence of MYC, EGFR, FGFR2, and MET amplification Targeting MET kinase alongside HER2 in PDX model tumor that progressed on afatinib and had MET amplification resulted in tumor regression |
[136] |
| HER2 | mGEC |
Biomarker analysis from phase II (NCT02954536) HER2 positive mGEC treated with trastuzumab and chemotherapy in first-line setting |
25 pts | Guardant 360 NGS | Baseline ctDNA ERBB2 amp and decreasing VAF on treatment were predictive biomarkers for response to HER2-directed therapy | [137] |
| HER2 | mGC |
Biomarker analysis from DESTINY-Gastric01 mGC pts treated with T-DXd |
151 pts | GuardantOMNI |
ORR in pts with baseline plasma ERBB2 amp was 60.6% and in pts without amp was 34.2% ORR in pts with baseline plasma ERBB2 copy number above 6 had ORR 75.8% compared to 40.8% in those below 6.0 |
[138] |
| HER2 | mCRC |
Prospective, phase II Pertuzumab plus trastuzumab in mCRC (refractory/intolerant to chemotherapy and EGFRi) with RAS wt and HER2 amp by tumor or ctDNA analysis |
30 pts | Guardant 360 NGS |
ORR was 30% in tissue HER2 amp pts and 28% in ctDNA HER2 amp pts Pts with tissue + /ctDNA– HER2 amp had significantly lower ctDNA fraction compared to tissue + /ctDNA + HER2 amp pts, likely due to low tumor shedding Baseline alterations in resistance pathways RTK/RAS/PI3K were enriched in non-responders and more frequently identified by ctDNA (67%) compared to tissue (19%) testing Baseline ctDNA profiles predicted those who would benefit from pertuzumab plus trastuzumab Decreasing ctDNA fraction on tx was associated with superior PFS and radiographic response ctDNA identified an actionable new alteration in 62% of pts after PD |
[139] TRIUMPH |
| FGFR2 | mGC | Retrospective | 365 pts | Guardant 360 NGS and Illumina NextSeq 550 |
FGFR2 amp were detected more frequently with ctDNA than with tissue analysis (7.7% vs. 2.6–4.4%, respectively) 2 pts with FGFR2 amp by ctDNA after PD but not on pretreatment tissue analysis responded to FGFR inhibitors |
[140] |
| FGFR2 | CCA | Biomarker analysis from 3 pts enrolled in phase II study with infigratinib in FGFR mut CCA | 3 pts | Guardant 360 NGS |
ctDNA testing at the time of progression on infigratinib revealed new FGFR2 point mutations (resistance mechanisms) ctDNA can reveal heterogeneous concurrent resistance mutations, unlike individual tissue biopsies |
[141] |
| FGFR2 | CCA | Retrospective | 137 pts | Tempus xF liquid biopsy |
ctDNA identified more actionable mutations in liquid biopsies (33.1%) compared to tissue biopsies (23.2%) Prevalence of FGFR2 fusions was higher in liquid biopsies (11.3%) than tissue biopsies (3.4%) ctDNA may be used to guide therapy selection |
[142] |
| Multiple genes | CCA | Prospective | 24 pts | 15-gene and 710 gene oncopanel |
Plasma-tissue concordance was 74% (higher at 94% in intrahepatic CCA and lower at 55% in extrahepatic CCA) Baseline ctDNA VAF correlated with initial tumor loads Baseline ctDNA VAF correlated with PFS in intrahepatic CCA |
[143] |
| IDH1 | CCA | Biomarker analysis from ClarIDHy | 210 samples | BEAMing digital PCR test |
IDH1 mut plasma-tissue concordance was 92% A subset of pts with longer PFS on treatment with ivosidenib had plasma IDH1 mut clearance |
[144] |
| KRAS | PDAC | Prospective | 78 pts | ddPCR | Longitudinal ctDNA KRAS status was prognostic and predictive of responses to chemotherapy | [145] |
| KRAS | PDAC | Prospective | 194 pts | ddPCR |
KRAS plasma-tissue concordance was over 95% ctDNA detection was prognostic for survival |
[146] |
| Multiple genes | PDAC | Prospective | 77 pts | Guardant 360 NGS |
Baseline ctDNA mutations included TP53 (12.6%), KRAS (9.7%), MET (6.8%), ARID1A (4.8%), NF1 (4.8%), and others < 3% ctDNA levels of TP53 and KRAS were associated with radiographic responses New TP53 subclonal variant mutations were the most common resistance mutations in progressions (75%) |
[147] |
| Multiple genes (mostly KRAS) | PDAC | Systematic review and meta-analysis | 2326 pts | various | ctDNA muts and high concentrations of ctDNA are prognostic for survival (PFS and OS) | [148] |
Amp, amplification; BEAM, beads, emulsion, amplification, and magnetics; CCA, cholangiocarcinoma; CRC, colorectal cancer; ctDNA, circulating tumor DNA; DCR, disease control rate; ddPCR, digital droplet polymerase chain reaction; EGFRi, EGFR inhibitor; MAF, mean allele frequency; mCRC, metastatic colorectal cancer; mGC, metastatic gastric cancer; mGEC, metastatic gastric and esophageal cancer; mo, months; mut, mutated; NGS, next-generation sequencing; ORR, overall response rate; (m)OS, (median) overall survival; PDAC, pancreatic ductal adenocarcinoma; PDX, patient-derived xenograft; (m)PFS, (median) progression-free survival; PD, progression of disease; SD, stable disease; T-DXd, trastuzumab deruxtecan-nxki; VAF, variant allele frequency; WT, wild-type
Taking anti-EGFR therapy as an example [125–127, 131], ctDNA studies demonstrate that RAS/EGFR mutant clones emerge during treatment, which might regress upon the withdrawal of anti-EGFR therapy, thereby allowing for rechallenge with the targeted therapy [128]. This regression could not be reasonably assessed using tumor tissue because it would mean the risk of repeated biopsying. Being much less invasive, liquid biopsy and ctDNA analysis may allow for uncomplicated identification of patients suitable for rechallenge based on real-time genomic analysis. This idea was evaluated in the CHRONOS phase II trial, the first prospective interventional study to use liquid biopsy to guide anti-EGFR rechallenge therapy in CRC. Hence, liquid biopsy genotyping differentiated between patients with RAS/BRAF/EGFR mutated versus wild-type tumors, and one-third of wild-type patients had an objective response on rechallenge with an anti-EGFR antibody [129]. Other studies have identified specific EGFR and RAS mutations in the plasma after disease progression on treatment and highlight ctDNA as a tool in clinical practice to inform therapeutic development and tailor treatments based on emerging resistance mutations [128]. The feasibility of BRAF plasma testing and similar potential applications in this targetable gene have also been demonstrated [130, 131, 135].
Another good example of ctDNA application is in HER2 amplified disease. Recent prospective trials have suggested that plasma HER2 amplifications predict response to HER2-directed therapies such as lapatinib, trastuzumab, pertuzumab, and fam-trastuzumab deruxtecan-nxki (T-DXd) [136–139]. For example, better responses to T-DXd in gastric cancers were seen when plasma HER2 and higher copy number amplifications were detected [138]. Also, changes in plasma copy number during HER2-directed therapy were associated with therapeutic response and survival in upper GI cancers and CRCs [122, 136, 137, 139]. Baseline and emerging resistance mutations detected in the plasma at the time of progression, such as MYC, EGFR, FGFR2, and MET amplification, have also been reported along with promising therapeutic strategies to overcome resistance, including combining anti-HER2 and other targeted or immune therapies [136, 139].
ctDNA can detect plasma FGFR2 alterations, sometimes at a higher frequency than tissue testing, identify patients who may benefit from infigratinib, and identify emerging resistance point mutations [140–142]. Recently, plasma-tissue accuracy and survival data have also been described in CCA patients with IDH1 plasma mutations treated with ivosidenib [144]. PDAC is highly KRAS-mutated and harbors rarer targetable mutations. Many studies have evaluated KRAS and TP53 ctDNA detection and kinetics as prognostic and predictive biomarkers in PDAC cases [145, 147, 149, 150].
Early data suggest that plasma MSS and TMB have potential roles as prognostic and predictive biomarkers in GI cancers, although further study is needed to validate these assays in the guidance of immunotherapy [151, 152]. Collectively, these data highlight the evolving potential of blood-based molecular profiling to precisely guide patient therapy and overcome tumor resistance using only minimally invasive procedures. Consequently, clinical trials across GI cancers are increasingly focused on genomic testing of liquid biopsies in place of or in conjunction with tumor tissue biopsies to enroll patients and study their outcomes [64, 153, 154].
Limitations
The liquid biopsy has been rapidly integrated into clinical practice for many solid tumors, but physicians should exercise caution when interpreting results. Tumor samples provide an abundance of tumor DNA compared to a liquid biopsy; therefore, liquid-based NGS may be limited by apparent lower sensitivity due to significantly lower levels of ctDNA [119]. Often, performing NGS on several genes means reduced depth of sequencing and sensitivity, not to mention added cost and effort compared to targeted sequencing of one or a few genes [119, 120, 155].
Studies have demonstrated that ctDNA levels and concordance may vary based on tumor histology, anatomic location, and stage [110, 156, 157]. Limitations arise if the tumor is a poor ctDNA shedder [122, 158]. Additionally, the presence of clonal hematopoiesis may lead to false positives in genotyping [159]. Unique molecular identifiers, dual barcode indexing, methylation assays, matched normal WBC DNA analysis, and cfDNA fragment length analysis can improve the accuracy of ctDNA analysis [109, 160, 161]; however, differences between laboratory testing platforms can contribute to discordant results across many mutations [162]. Despite the intrinsic limitations of assays, blood-based genomic profiling remains a promising tool for patient diagnosis, therapy guidance, and identification of patients for trial enrollment.
ctDNA use in screening and diagnosing cases with insufficient tissue
As molecular profiling technology has advanced, there is increasing interest in using ctDNA for cancer screening (Fig. 2), diagnosis of inaccessible tumors, and management of cancers of unknown primary (CUP). Massive genomic profiling efforts, including The Cancer Genome Atlas’s Pan-Cancer initiative and other massive genomic profiling efforts, have identified tumor DNA, RNA, and protein patterns based on histology, anatomic location, and tissue types. These patterns can, in turn, be used to characterize undifferentiated tumors and identify tissues of origin [163]. For example, the highly-specific CancerSeek test [164] used ctDNA and protein biomarkers coupled with machine-learning to diagnose 8 early-stage cancers, including liver, gastric, pancreatic, esophageal, and colorectal cancer. The Circulating Cell-free Genome Atlas (CCGA) study [165] used a methylation-based assay from Grail to allow deeper sequencing and identified over 50 early-stage cancers, including liver/bile duct, gastric, pancreatic, esophageal, colorectal cancer, and anal cancer. The CancerSeek assay demonstrated sensitivities ranging from 69 to 98% in the detection of 5 cancer types, including liver, stomach, pancreatic, and esophageal [164]. The CCGA assay had 67.3% (CI 60.7–73.3%) sensitivity in 12 cancer types, including anal, colorectal, esophageal, liver/bile duct, pancreatic, and stomach [165]. Both assays had greater than 99% specificity, identifying the tissue of tumor origin with great accuracy. In another active study, Grail’s cfDNA methylation assay predicted the tissue of tumor origin with 92.3% accuracy [166]. Although these platforms are promising, they are still limited by inadequate sensitivity in early-stage diseases. Moreover, questions about disease management after detection, feasibility, ethics of general population testing, and cost–benefit ratios remain [167].
As it currently stands, GI cancer-specific but not yet multicancer screening tests have been FDA approved. Epigenomics Epi proColon® detects methylated SEPT9 DNA in the blood and was FDA approved on April 13, 2016, as the first blood-based CRC screening tool [168, 169]. Guardant’s LUNAR-2 ctDNA screening test for CRC is also currently under investigation, among others [170]. Blood-based hydroxymethylation and protein glycosylation signatures are promising biomarkers for the early detection of PDAC [171, 172]. Distinct circulating miRNAs signatures in blood and bile might act as biomarkers to differentiate between biliary cancers and other benign hepatobiliary diseases [173, 174]. However, miRNAs are nonspecific, and the best source of miRNA collection and the translation of miRNA assays into clinical practice are as yet undefined [175, 176].
Finally, the feasibility and utility of analyzing ctDNA from blood to characterize CUPs and identify targetable mutations have been described in multiple studies over the past decade [177–180]. Historically, patients with CUP had limited treatment options and poor prognoses because many standard-of-care therapies are tumor-specific. As more biomarkers are identified, broad blood-based NGS can uncover targetable mutations and identify more previously non-indicated therapies for these patients.
ctDNA in MRD
Efforts are underway to identify patients at high risk of early relapse and develop interventions to lengthen patient survival. The strategy of MRD monitoring (Fig. 2) and eradication is already established in hematologic malignancies and is stimulating interest in GI cancers [181–183].
MRD ctDNA assays are often characterized as tumor-agnostic or tumor-informed. Tumor-agnostic approaches do not require pre-existing knowledge of a tumor’s genomic profile and often employ broad-based NGS, narrower PCR, or methylation assays to identify common cancer markers circulating in the blood. Numerous studies in localized or oligometastatic CRC patients undergoing curative surgery have demonstrated that ctDNA detection post-surgery or post-adjuvant therapy is a strong independent prognostic marker for survival [184–186]. Others suggest that ctDNA levels correlate with tumor burden and response to treatment [187]. Promising results in PDAC (often targeting KRAS) and gastroesophageal cancer have also been reported [188–191]. While limited by decreased sensitivity, tumor-agnostic methods are advantageous in their quick turnaround time, low cost, and simultaneous broad-based genomic profiling and resistance mechanism identification potential.
Although slower and more expensive, a tumor-informed approach offers higher sensitivity and is particularly attractive in assessing MRD when the ctDNA level is low, as in early-stage disease. Here, a patient’s tumor tissue is tested using genomic sequencing, and tumor-specific mutations are identified. These mutations are subsequently targeted in the blood using a personalized assay. Examples include SafeSeqS, CAPP-Seq, Archer DX, Radar, and Signatera. To date, most GI cancer studies have been in CRC and demonstrate sensitivities ranging from 48 to 100%, specificities ranging from 90 to 100%, positive predictive values (PPV) over 98%, and median lead times to radiographic relapse of about 8–9 months [192–203]. For example, using the Signatera test, a ctDNA positive status after adjuvant therapy and on postoperative longitudinal testing was found to confer 18 times and 30 to 40 times, respectively, higher risk of relapse compared to a ctDNA negative status. Moreover, ctDNA was found to outperform CEA in predicting relapse [192, 193, 198]. Studies characterizing ctDNA in the provision of early prognostic data are also emerging in gastroesophageal cancer, PDAC, and CCA [188, 189, 204, 205]. Some studies in the adjuvant and metastatic setting also suggest that ctDNA clearance or kinetics may predict treatment response and survival [198, 206].
We know that ctDNA positivity is highly prognostic but still lack knowledge on optimal disease management strategies for patients with MRD. While a positive test may theoretically hasten surveillance diagnostics, the role of local or systemic therapy escalation, in this case, is unclear, especially in the setting of serial, low-level ctDNA without evidence of radiographic relapse. Also, despite the achievable 0.01% level of ctDNA detection using tumor-informed assays, false negatives due to ctDNA levels being below the limit of detection should be considered when interpreting negative results. This is likely to be the case for low-shedding tumors, for example [207]. Therefore, in the absence of prospective, randomized data to support de-escalation strategies, a patient with a negative test should receive standard adjuvant therapy if not otherwise contraindicated. It is also important to realize that each company’s tests are uniquely constructed, using different error-correcting techniques that are frequently updated.
Circulating tumor cells (CTCs)
It has been shown that CTCs are an intermediate stage of cancer metastasis. Like ctDNA, CTCs are obtained from peripheral blood; however, CTCs may have a greater clinical impact as they can be grown, propagated, and extensively studied in vitro and in vivo under optimal conditions [208, 209]. However, it is unclear if a single cell assay accurately reflects entire tumor heterogeneity.
Data supporting CTC enumeration as a predictor of clinical outcome dates back as early as 2004, when it was shown that patients with metastatic breast cancer had shorter mPFS and mOS if they had higher CTC levels [210]. Since then, this finding has been further validated across a wide range of tumor types, including GI cancers [208]. CTC enumeration shows promise in clinical management guidance; for example, as shown in breast cancer cases, the discovery of discordant driver mutations between an individual’s CTCs and their primary tumor may inform targeted treatment decisions [211–213].
Several commercial systems and clinical services (Epic, RareCyte™, CytoTrack, SRI FASTcell™) exist [214]. Current methods of CTC detection rely on one of three basic principles. The CellSearch® system was the first and is the only FDA-approved device for CTC enumeration [215]. The CellSearch® platform relies on antibody detection of CTC markers [208, 216]. Cohen et al. used the CellSearch™ system to estimate CTCs in their prospective multicenter mCRC study [217]. Their results showed that patients with ≥ 3 CTCs/7.5 mL blood had shorter mOS than patients with < 3 CTCs (P < 0.0001), and these differences persisted at follow-up time points after therapy. It was concluded that the number of CTCs was an independent predictor of disease-free survival (DFS) and OS in metastatic cancer [217]. An alternative CTC enumeration approach relies on isolating CTCs according to prespecified cancer-specific gene products (RNAs and proteins) [208, 218]. However, this method involves the lysing of captured CTCs, limiting their use in downstream analyses. A third technique isolates CTCs according to their physical characteristics; CTCs are generally much larger than blood cells (30 µm vs. 7–9 µm), allowing their isolation and enumeration [219].
Future directions
CTCs and organoids
CTC study has already added to our understanding of cancer metastases. For example, it has been described that CTCs often carry genetic variations in driver mutations that are different from the primary tumor; these differences would likely help explain the propensity for primary tumors to metastasize and seed into other organs [220].
As the next frontier in precision medicine, the ability to grow and expand CTCs ex vivo is an invaluable, noninvasive tool in the study of cancer biology and metastasis [208, 209, 216]. One step further, the ability to create CTC-derived xenografts by injecting CTCs directly into immunocompromised mouse hosts holds vast implications in both research and clinical settings. These organoids maintain tumor heterogeneity and allow investigation of therapeutic elements on the xenograft that mirror patient response to the same treatment [221, 222]. Such clinical applications have already been used to perform in vivo drug screens with high success and hold implications for new drug discovery [221]. For research purposes, organoids can be used for disease modeling to understand the process of carcinogenesis. They can be manipulated easily using retroviruses and inhibitors, for example, and can be used to identify key driver mutations, as shown already in some GI cancer organoid studies [223, 224]. The clinical role of CTCs is currently limited but is expected to expand on the heels of technology improvements, including CTC-isolation and organoid-development techniques.
What can we learn from the blood, and how can we use biomarker testing in the future?
Tissue molecular profiling provides clinically significant subtyping of all GI cancers. The liquid biopsy promises dynamic tumor characterization through various platforms, and we believe these capabilities will be increasingly incorporated into clinical cancer management. The liquid biopsy is already integrated into the standard of care for gastric, esophageal, and GEJ cancers, for which NCCN guidelines recommend plasma ctDNA profiling by NGS to detect targetable alterations or clones with altered treatment sensitivity when patients are not candidates for tumor-tissue biopsy and NGS [207].
In the future, molecular profiling of the liquid biopsy will likely complement or replace the GI tumor-tissue biopsy in select scenarios. Future therapeutic studies should include ctDNA analyses to identify prognostic and predictive liquid biopsy biomarkers. Serial testing should also be further assessed as a way to quickly and noninvasively characterize disease response or mechanisms of resistance. Finally, “MRD with no evidence of radiographic disease” might become a theoretical “new stage,” warranting novel treatment strategies. As liquid biopsy techniques improve, blood-based testing will hopefully better identify MRD and screen early-stage GI tumors with hopes of curing more patients and improving outcomes.
Summary
Molecular profiling for patients with GI malignancies is clearly making an impact and has become the standard of care in many situations. In fact, in 2021, ASCO chose molecular profiling in GI cancers as its Advance[ment] of the Year [225]. An increasing number of actionable biomarkers are being identified, and drugs that act on these biomarkers are continually being developed, providing patients with better treatment options, improved quality of life, and increased survival compared to standard therapy alone. Likewise, analytical methods using tumor tissue and, more recently, blood are constantly being developed and improved, promoting the identification of biomarkers and gene signatures that help diagnose disease and predict therapy success in this oftentimes refractory group of malignancies. Together with machine learning, our evolving biomarker technology is promising to help us fight an even smarter war against GI cancers.
Acknowledgements
Not applicable.
Author contributions
All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
CY, None; RM, None; RH, None; AZA, None; MK, None; ARH, Grant or research support-Merck (Merck Sharp & Dohme a subsidiary of Merck & Co, Inc.), grant or research support-Genentech, Inc., Speakers Bureau-Eisai, Speakers Bureau-BMS; BAW, Personal fees from Taiho, Lilly, Sirtex, Bayer, and Daiichi Sankyo/AstraZeneca, and grants from Ipsen outside the submitted work; JLM, Honoraria—Pfizer, Inc., and Daiichi Sankyo, Inc. Advisor—Bayer Corporation, and Taiho Oncology; MLH, None; MSN: Support from Ipsen outside the submitted work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shu Y, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7:583. doi: 10.1038/s41598-017-00520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dasari A, et al. ctDNA applications and integration in colorectal cancer: an NCI Colon and Rectal-Anal Task Forces whitepaper. Nat Rev Clin Oncol. 2020;17:757–770. doi: 10.1038/s41571-020-0392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parikh AR, et al. Serial ctDNA monitoring to predict response to systemic therapy in metastatic gastrointestinal cancers. Clin Cancer Res. 2020;26:1877–1885. doi: 10.1158/1078-0432.CCR-19-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moroni M, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–286. doi: 10.1016/S1470-2045(05)70102-9. [DOI] [PubMed] [Google Scholar]
- 5.Lievre A, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 6.De Roock W, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 7.Sartore-Bianchi A, et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist. 2019;24:1395–1402. doi: 10.1634/theoncologist.2018-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopetz S, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406) J Clin Oncol. 2017;35:3505–3505. doi: 10.1200/JCO.2017.35.15_suppl.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabernero J, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol. 2021;39:273–284. doi: 10.1200/JCO.20.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roudko V, et al. Lynch Syndrome and MSI-H cancers: from mechanisms to "off-the-shelf" cancer vaccines. Front Immunol. 2021;12:757804. doi: 10.3389/fimmu.2021.757804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 12.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–756. doi: 10.1002/cam4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonneville R, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017:1–15. doi: 10.1200/PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janjigian YY, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40. doi: 10.1016/S0140-6736(21)00797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bang YJ, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 16.Shitara K, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med. 2020;382:2419–2430. doi: 10.1056/NEJMoa2004413. [DOI] [PubMed] [Google Scholar]
- 17.Wilke H, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–1235. doi: 10.1016/S1470-2045(14)70420-6. [DOI] [PubMed] [Google Scholar]
- 18.Zhu AX, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical ClarIDHy trial. JAMA Oncol. 2021;7:1669–1677. doi: 10.1001/jamaoncol.2021.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saborowski A, Lehmann U, Vogel A. FGFR inhibitors in cholangiocarcinoma: what's now and what's next? Ther Adv Med Oncol. 2020;12:1758835920953293. doi: 10.1177/1758835920953293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Javle M, et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021;22:1290–1300. doi: 10.1016/S1470-2045(21)00336-3. [DOI] [PubMed] [Google Scholar]
- 21.Subbiah V, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020;21:1234–1243. doi: 10.1016/S1470-2045(20)30321-1. [DOI] [PubMed] [Google Scholar]
- 22.Reiss KA, et al. Phase II study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin Oncol. 2021;39:2497–2505. doi: 10.1200/JCO.21.00003. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 24.Reiss KA, Forde PM, Brahmer JR. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: a promising new anticancer strategy. Immunotherapy. 2014;6:459–475. doi: 10.2217/imt.14.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinberg BA, Hameed R, Marshall JL. Biomarkers for immune therapy in gastrointestinal cancers. Clin Adv Hematol Oncol. 2019;17:109–119. [PubMed] [Google Scholar]
- 26.Suda K, Mitsudomi T. Inter-tumor heterogeneity of PD-L1 status: is it important in clinical decision making? J Thorac Dis. 2020;12:1770–1775. doi: 10.21037/jtd-20-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haragan A, et al. Heterogeneity of PD-L1 expression in non-small cell lung cancer: implications for specimen sampling in predicting treatment response. Lung Cancer. 2019;134:79–84. doi: 10.1016/j.lungcan.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manson QF, Schrijver W, Ter Hoeve ND, Moelans CB, van Diest PJ. Frequent discordance in PD-1 and PD-L1 expression between primary breast tumors and their matched distant metastases. Clin Exp Metastasis. 2019;36:29–37. doi: 10.1007/s10585-018-9950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stovgaard ES, et al. PD-L1 diagnostics in the neoadjuvant setting: implications of intratumoral heterogeneity of PD-L1 expression in triple negative breast cancer for assessment in small biopsies. Breast Cancer Res Treat. 2020;181:553–560. doi: 10.1007/s10549-020-05655-w. [DOI] [PubMed] [Google Scholar]
- 30.Mino-Kenudson M. Programmed cell death ligand-1 (PD-L1) expression by immunohistochemistry: could it be predictive and/or prognostic in non-small cell lung cancer? Cancer Biol Med. 2016;13:157–170. doi: 10.20892/j.issn.2095-3941.2016.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs CS, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4:e180013. doi: 10.1001/jamaoncol.2018.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wainberg ZA, et al. Efficacy of pembrolizumab monotherapy for advanced gastric/gastroesophageal junction cancer with programmed death ligand 1 combined positive score ≥10. Clin Cancer Res. 2021;27:1923–1931. doi: 10.1158/1078-0432.CCR-20-2980. [DOI] [PubMed] [Google Scholar]
- 33.Chau I, et al. Nivolumab (NIVO) plus ipilimumab (IPI) or NIVO plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced esophageal squamous cell carcinoma (ESCC): first results of the CheckMate 648 study. J Clin Oncol. 2021;39:LBA4001. doi: 10.1200/JCO.2021.39.15_suppl.LBA4001. [DOI] [Google Scholar]
- 34.Zhao JJ, et al. Low programmed death-ligand 1-expressing subgroup outcomes of first-line immune checkpoint inhibitors in gastric or esophageal adenocarcinoma. J Clin Oncol. 2022;40:392–402. doi: 10.1200/JCO.21.01862. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Perez E, Molina-Vila MA, Marino-Buslje C. Panels and models for accurate prediction of tumor mutation burden in tumor samples. NPJ Precis Oncol. 2021;5:31. doi: 10.1038/s41698-021-00169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sha D, et al. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 2020;10:1808–1825. doi: 10.1158/2159-8290.CD-20-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marabelle A, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–1365. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 38.Marcus L, et al. FDA approval summary: pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin Cancer Res. 2021;27:4685–4689. doi: 10.1158/1078-0432.CCR-21-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang T, et al. Accurate measurement of tumor mutation burden in liquid biopsy (bTMB) using a 500 gene panel. Ann Oncol. 2018;29:viii51. doi: 10.1093/annonc/mdy269.161. [DOI] [Google Scholar]
- 40.Chalmers ZR, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu P, Poehlein CH, Marton MJ, Laterza OF, Levitan D. Measuring tumor mutational burden (TMB) in plasma from mCRPC patients using two commercial NGS assays. Sci Rep. 2019;9:114. doi: 10.1038/s41598-018-37128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forschner A, et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—results of a prospective biomarker study. J Immunother Cancer. 2019;7:180. doi: 10.1186/s40425-019-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng Y, Mei W, Ma K, Zeng C. Circulating tumor DNA and minimal residual disease (MRD) in solid tumors: current horizons and future perspectives. Front Oncol. 2021;11:763790. doi: 10.3389/fonc.2021.763790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilson P, Merlin JL, Harle A. Detection of microsatellite instability: state of the art and future applications in circulating tumour DNA (ctDNA) Cancers (Basel) 2021;13:1491. doi: 10.3390/cancers13071491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reyes GX, Schmidt TT, Kolodner RD, Hombauer H. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124:443–462. doi: 10.1007/s00412-015-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andre T, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J Clin Oncol. 2020;38:LBA4. doi: 10.1200/JCO.2020.38.18_suppl.LBA4. [DOI] [Google Scholar]
- 48.Overman MJ, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 49.Gordon MA, et al. Assessment of HER2 gene amplification in adenocarcinomas of the stomach or gastroesophageal junction in the INT-0116/SWOG9008 clinical trial. Ann Oncol. 2013;24:1754–1761. doi: 10.1093/annonc/mdt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Press MF, et al. HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J Clin Oncol. 1997;15:2894–2904. doi: 10.1200/JCO.1997.15.8.2894. [DOI] [PubMed] [Google Scholar]
- 51.Press MF, et al. Amplification and overexpression of HER-2/neu in carcinomas of the salivary gland: correlation with poor prognosis. Cancer Res. 1994;54:5675–5682. [PubMed] [Google Scholar]
- 52.Saffari B, et al. Amplification and overexpression of HER-2/neu (c-erbB2) in endometrial cancers: correlation with overall survival. Cancer Res. 1995;55:5693–5698. [PubMed] [Google Scholar]
- 53.Ingold Heppner B, et al. HER2/neu testing in primary colorectal carcinoma. Br J Cancer. 2014;111:1977–1984. doi: 10.1038/bjc.2014.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross JS, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. 2018;124:1358–1373. doi: 10.1002/cncr.31125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo AN, et al. HER2 status in colorectal cancer: its clinical significance and the relationship between HER2 gene amplification and expression. PLoS ONE. 2014;9:e98528. doi: 10.1371/journal.pone.0098528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He C, et al. Correlation of human epidermal growth factor receptor 2 expression with clinicopathological characteristics and prognosis in gastric cancer. World J Gastroenterol. 2013;19:2171–2178. doi: 10.3748/wjg.v19.i14.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jørgensen JT, Hersom M. HER2 as a prognostic marker in gastric cancer—a systematic analysis of data from the literature. J Cancer. 2012;3:137–144. doi: 10.7150/jca.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esparís-Ogando A, Montero JC, Arribas J, Ocaña A, Pandiella A. Targeting the EGF/HER ligand-receptor system in cancer. Curr Pharm Des. 2016;22:5887–5898. doi: 10.2174/1381612822666160715132233. [DOI] [PubMed] [Google Scholar]
- 59.Sartore-Bianchi A, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–746. doi: 10.1016/S1470-2045(16)00150-9. [DOI] [PubMed] [Google Scholar]
- 60.Meric-Bernstam F, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20:518–530. doi: 10.1016/S1470-2045(18)30904-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe S, et al. Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Med. 2019;8:1258–1268. doi: 10.1002/cam4.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernández-Nogueira P, et al. Tumor-associated fibroblasts promote HER2-targeted therapy resistance through FGFR2 activation. Clin Cancer Res. 2020;26:1432–1448. doi: 10.1158/1078-0432.CCR-19-0353. [DOI] [PubMed] [Google Scholar]
- 63.Kulukian A, et al. Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol Cancer Ther. 2020;19:976–987. doi: 10.1158/1535-7163.MCT-19-0873. [DOI] [PubMed] [Google Scholar]
- 64.Strickler JH, et al. MOUNTAINEER-02: phase II/III study of tucatinib, trastuzumab, ramucirumab, and paclitaxel in previously treated HER2+ gastric or gastroesophageal junction adenocarcinoma—trial in progress. J Clin Oncol. 2021;39:TPS252. doi: 10.1200/JCO.2021.39.3_suppl.TPS252. [DOI] [Google Scholar]
- 65.Kong-Beltran M, Donoghue DJ. Signaling from FGF receptors in development and disease. In: Bradshaw RA, Dennis EA, editors. Handbook of cell signaling. Burlington: Academic Press; 2003. pp. 861–865. [Google Scholar]
- 66.Presta M, Chiodelli P, Giacomini A, Rusnati M, Ronca R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol Ther. 2017;179:171–187. doi: 10.1016/j.pharmthera.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 67.Dienstmann R, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25:552–563. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goyal L, Kongpetch S, Crolley VE, Bridgewater J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat Rev. 2021;95:102170. doi: 10.1016/j.ctrv.2021.102170. [DOI] [PubMed] [Google Scholar]
- 69.Ross JS, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–242. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Graham RP, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45:1630–1638. doi: 10.1016/j.humpath.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 71.Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 2017;7:943–962. doi: 10.1158/2159-8290.CD-17-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cristescu R, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–456. doi: 10.1038/nm.3850. [DOI] [PubMed] [Google Scholar]
- 73.Network TCGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. [DOI] [PMC free article] [PubMed]
- 74.Helsten T, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22:259–267. doi: 10.1158/1078-0432.CCR-14-3212. [DOI] [PubMed] [Google Scholar]
- 75.Weiss J, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dutt A, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS ONE. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abou-Alfa GK, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21:671–684. doi: 10.1016/S1470-2045(20)30109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Javle MM, et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. J Clin Oncol. 2021;39:265–265. doi: 10.1200/JCO.2021.39.3_suppl.265. [DOI] [Google Scholar]
- 79.Tabernero J, et al. Phase I Dose-Escalation Study Of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401–3408. doi: 10.1200/JCO.2014.60.7341. [DOI] [PubMed] [Google Scholar]
- 80.Kommalapati A, Tella SH, Borad M, Javle M, Mahipal A. FGFR Inhibitors in oncology: insight on the management of toxicities in clinical practice. Cancers (Basel) 2021;13:2968. doi: 10.3390/cancers13122968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goyal L, et al. FOENIX-CCA2: A phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J Clin Oncol. 2020;38:108–108. doi: 10.1200/JCO.2020.38.15_suppl.108. [DOI] [Google Scholar]
- 82.Salati M, et al. IDH signalling pathway in cholangiocarcinoma: from biological rationale to therapeutic targeting. Cancers (Basel) 2020;12:3310. doi: 10.3390/cancers12113310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao S, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Borger DR, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grassian AR, Pagliarini R, Chiang DY. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2014;30:295–302. doi: 10.1097/MOG.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 86.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lowery MA, et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a phase 1 study. Lancet Gastroenterol Hepatol. 2019;4:711–720. doi: 10.1016/S2468-1253(19)30189-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu AX, et al. Final results from ClarIDHy, a global, phase III, randomized, double-blind study of ivosidenib (IVO) versus placebo (PBO) in patients (pts) with previously treated cholangiocarcinoma (CCA) and an isocitrate dehydrogenase 1 (IDH1) mutation. J Clin Oncol. 2021;39:266–266. doi: 10.1200/JCO.2021.39.3_suppl.266. [DOI] [Google Scholar]
- 89.Administration, U.S.F. & Drug. FDA approves ivosidenib for advanced or metastatic cholangiocarcinoma (2021).
- 90.Hofstatter EW, et al. PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer. 2011;10:225–231. doi: 10.1007/s10689-011-9426-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seeber A, et al. Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open. 2020;5:e000942. doi: 10.1136/esmoopen-2020-000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu G, et al. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics. 2012;13:1523–1535. doi: 10.2217/pgs.12.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Connor F, et al. Cloning, chromosomal mapping and expression pattern of the mouse Brca2 gene. Hum Mol Genet. 1997;6:291–300. doi: 10.1093/hmg/6.2.291. [DOI] [PubMed] [Google Scholar]
- 94.Miki Y, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 95.Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci. 2001;114:3591–3598. doi: 10.1242/jcs.114.20.3591. [DOI] [PubMed] [Google Scholar]
- 96.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/S1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 97.Tischkowitz MD, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137:1183–1186. doi: 10.1053/j.gastro.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Slater EP, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–494. doi: 10.1111/j.1399-0004.2010.01425.x. [DOI] [PubMed] [Google Scholar]
- 99.Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2013;13:66–74. doi: 10.1038/nrc3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park W, et al. Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin Cancer Res. 2020;26:3239–3247. doi: 10.1158/1078-0432.CCR-20-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O'Reilly EM, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol. 2020;38:1378–1388. doi: 10.1200/JCO.19.02931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Golan T, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 104.Salama AKS, et al. Dabrafenib and trametinib in patients with tumors with BRAF(V600E) Mutations: Results Of The NCI-MATCH trial subprotocol H. J Clin Oncol. 2020;38:3895–3904. doi: 10.1200/JCO.20.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mazouji O, Ouhajjou A, Incitti R, Mansour H. Updates on clinical use of liquid biopsy in colorectal cancer screening, diagnosis, follow-up, and treatment guidance. Front Cell Dev Biol. 2021;9:660924. doi: 10.3389/fcell.2021.660924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA. Clin Chim Acta. 2001;313:139–142. doi: 10.1016/S0009-8981(01)00665-9. [DOI] [PubMed] [Google Scholar]
- 107.Bi F, et al. Circulating tumor DNA in colorectal cancer: opportunities and challenges. Am J Transl Res. 2020;12:1044–1055. [PMC free article] [PubMed] [Google Scholar]
- 108.van der Vaart M, Pretorius PJ. The origin of circulating free DNA. Clin Chem. 2007;53(12):2215. doi: 10.1373/clinchem.2007.092734. [DOI] [PubMed] [Google Scholar]
- 109.Bachet JB, et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: the AGEO RASANC prospective multicenter study. Ann Oncol. 2018;29:1211–1219. doi: 10.1093/annonc/mdy061. [DOI] [PubMed] [Google Scholar]
- 110.Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Diehl F, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mandel P, Metais P. Nuclear acids in human blood plasma. C R Seances Soc Biol Fil. 1948;142:241–243. [PubMed] [Google Scholar]
- 113.Domínguez-Vigil IG, Moreno-Martínez AK, Wang JY, Roehrl MHA, Barrera-Saldaña HA. The dawn of the liquid biopsy in the fight against cancer. Oncotarget. 2018;9:2912–2922. doi: 10.18632/oncotarget.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wan JCM, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 115.Shohdy KS, West HJ. Circulating tumor DNA testing-liquid biopsy of a cancer. JAMA Oncol. 2020;6:792. doi: 10.1001/jamaoncol.2020.0346. [DOI] [PubMed] [Google Scholar]
- 116.Finkle JD, et al. Validation of a liquid biopsy assay with molecular and clinical profiling of circulating tumor DNA. NPJ Precis Oncol. 2021;5(1):1–12. doi: 10.1038/s41698-020-00139-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.US Food and Drug Administration. FDA approves first liquid biopsy next-generation sequencing companion diagnostic test. August 07, 2020. Accessed 29 Oct 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-first-liquidbiopsy-next-generation-sequencing-companion-diagnostic-test.
- 118.US Food and Drug Administration. FDA approves liquid biopsy next-generation sequencing companion diagnostic test. September 15, 2020. Accessed 29 Oct 2021. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approvesliquid-biopsy-next-generation-sequencing-companion-diagnostic-test.
- 119.Odegaard JI, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24(15):3539–3549. doi: 10.1158/1078-0432.CCR-17-3831. [DOI] [PubMed] [Google Scholar]
- 120.Woodhouse R, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE. 2020;15(9):e0237802. doi: 10.1371/journal.pone.0237802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li G, et al. Genomic profiling of cell-free circulating tumor DNA in patients with colorectal cancer and its fidelity to the genomics of the tumor biopsy. J Gastrointest Oncol. 2019;10:831–840. doi: 10.21037/jgo.2019.05.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Siravegna G, et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin Cancer Res. 2019;25:3046–3053. doi: 10.1158/1078-0432.CCR-18-3389. [DOI] [PubMed] [Google Scholar]
- 123.Thierry AR, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–435. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- 124.Pectasides E, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 2018;8:37–48. doi: 10.1158/2159-8290.CD-17-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schmiegel W, et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol Oncol. 2017;11:208–219. doi: 10.1002/1878-0261.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.García-Foncillas J, et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br J Cancer. 2018;119:1464–1470. doi: 10.1038/s41416-018-0293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bando H, et al. A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br J Cancer. 2019;120:982–986. doi: 10.1038/s41416-019-0457-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Parseghian CM, et al. Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol. 2019;30:243–249. doi: 10.1093/annonc/mdy509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sartore-Bianchi A, et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: the CHRONOS trial. J Clin Oncol. 2021;39:3506–3506. doi: 10.1200/JCO.2021.39.15_suppl.3506. [DOI] [Google Scholar]
- 130.Vitiello PP, et al. Clinical practice use of liquid biopsy to identify RAS/BRAF mutations in patients with metastatic colorectal cancer (mCRC): a single institution experience. Cancers (Basel) 2019;11:1504. doi: 10.3390/cancers11101504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Harle A, et al. cfDNA for accurate determination of RAS and BRAF mutations using OncoBEAM liquid biopsy in metastatic colorectal cancer patients: results of the real-world multicentric ColoBEAM study. J Clin Oncol. 2019;37:3542–3542. doi: 10.1200/JCO.2019.37.15_suppl.3542. [DOI] [Google Scholar]
- 132.Mariani S, et al. Liquid biopsy-driven anti-EGFR rechallenge in patients with metastatic colorectal cancer. J Clin Oncol. 2021;39:3577–3577. doi: 10.1200/JCO.2021.39.15_suppl.3577. [DOI] [Google Scholar]
- 133.Cremolini C, et al. Rechallenge for patients With RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 2019;5:343–350. doi: 10.1001/jamaoncol.2018.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sunakawa Y, et al. RAS mutations in circulating tumor DNA and clinical outcomes of rechallenge treatment with anti-EGFR antibodies in patients with metastatic colorectal cancer. JCO Precis Oncol. 2020;4:898–911. doi: 10.1200/PO.20.00109. [DOI] [PubMed] [Google Scholar]
- 135.Morris VK, et al. Utility of circulating tumor DNA in the clinical management of patients with BRAFV600E metastatic colorectal cancer. J Clin Oncol. 2021;39:119–119. doi: 10.1200/JCO.2021.39.3_suppl.119. [DOI] [Google Scholar]
- 136.Kim ST, et al. Impact of genomic alterations on lapatinib treatment outcome and cell-free genomic landscape during HER2 therapy in HER2+ gastric cancer patients. Ann Oncol. 2018;29(4):1037–1048. doi: 10.1093/annonc/mdy034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maron SB, et al. Early predictors of benefit to dual anti-PD1/HER2 inhibition: Biomarker analysis from phase 2 trial of pembrolizumab/trastuzumab in HER2-positive metastatic esophagogastric (mEG) cancer. J Clin Oncol. 2021;39:4058–4058. doi: 10.1200/JCO.2021.39.15_suppl.4058. [DOI] [Google Scholar]
- 138.Shitara K, et al. O-14 Exploratory biomarker analysis of trastuzumab deruxtecan in DESTINY-Gastric01, a randomized, phase 2, multicenter, open-label study in patients with HER2-positive or -low advanced gastric or gastroesophageal junction adenocarcinoma. Ann Oncol. 2021;32:S224. doi: 10.1016/j.annonc.2021.05.018. [DOI] [Google Scholar]
- 139.Nakamura Y, et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: a phase 2 trial. Nat Med. 2021;27:1899–1903. doi: 10.1038/s41591-021-01553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jogo T, et al. Circulating tumor DNA analysis detects FGFR2 amplification and concurrent genomic alterations associated with FGFR inhibitor efficacy in advanced gastric cancer. Clin Cancer Res. 2021;27:5619–5627. doi: 10.1158/1078-0432.CCR-21-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Goyal L, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7:252–263. doi: 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kasi PM, Le AD, Barrett A. Comparative landscape of actionable somatic alterations in advanced cholangiocarcinoma from circulating tumor and tissue-based DNA profiling. J Clin Oncol. 2021;39(suppl 3):abstr 342. doi: 10.1200/JCO.2021.39.3_suppl.342. [DOI] [Google Scholar]
- 143.Ettrich TJ, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep. 2019;9:13261. doi: 10.1038/s41598-019-49860-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aguado E, et al. IDH1 mutation detection in plasma circulating tumor DNA (ctDNA) and association with clinical response in patients with advanced intrahepatic cholangiocarcinoma (IHC) from the phase III ClarIDHy study. J Clin Oncol. 2020;38:4576–4576. doi: 10.1200/JCO.2020.38.15_suppl.4576. [DOI] [Google Scholar]
- 145.Watanabe F, et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS ONE. 2019;14:e0227366. doi: 10.1371/journal.pone.0227366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bernard V, et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic cancer. Gastroenterology. 2019;156:108–118.e104. doi: 10.1053/j.gastro.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Banerjee M, et al. Analysis of pancreatobiliary adenocarcinoma (PBC) treatment response and resistance utilizing circulating tumor DNA (ctDNA) J Clin Oncol. 2020;38:758–758. doi: 10.1200/JCO.2020.38.4_suppl.758. [DOI] [Google Scholar]
- 148.Fang Z, et al. Prognostic value of circulating tumor DNA in pancreatic cancer: a systematic review and meta-analysis. Aging (Albany NY) 2020;13:2031–2048. doi: 10.18632/aging.202199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jaworski JJ, Morgan RD, Sivakumar S. Circulating cell-free tumour DNA for early detection of pancreatic cancer. Cancers (Basel) 2020;12:3704. doi: 10.3390/cancers12123704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Waters AM, Der CJ. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen EX, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: The Canadian Cancer Trials Group CO.26 Study. JAMA Oncol. 2020;6:831–838. doi: 10.1001/jamaoncol.2020.0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang Z, et al. Plasma-based microsatellite instability detection strategy to guide immune checkpoint blockade treatment. J Immunother Cancer. 2020;8:e001297. doi: 10.1136/jitc-2020-001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nakamura Y, et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med. 2020;26:1859–1864. doi: 10.1038/s41591-020-1063-5. [DOI] [PubMed] [Google Scholar]
- 154.Wainberg ZA, et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT) J Clin Oncol. 2021;39:160–160. doi: 10.1200/JCO.2021.39.3_suppl.160. [DOI] [Google Scholar]
- 155.Singh RR. Next-generation sequencing in high-sensitive detection of mutations in tumors: challenges, advances, and applications. J Mol Diagn. 2020;22:994–1007. doi: 10.1016/j.jmoldx.2020.04.213. [DOI] [PubMed] [Google Scholar]
- 156.Bando H, et al. Impact of a metastatic site on circulating tumor DNA (ctDNA) analysis in patients (pts) with metastatic colorectal cancer (mCRC) J Clin Oncol. 2021;39:3554–3554. doi: 10.1200/JCO.2021.39.15_suppl.3554. [DOI] [Google Scholar]
- 157.Erve IVT, et al. Abstract 708: limited release of circulating tumor DNA into the systemic circulation by peritoneal metastases from colorectal cancer. Cancer Res. 2020;80:708–708. [Google Scholar]
- 158.Raghav KPS, et al. Assessment of HER2 (ERBB2) amplification (HER2amp) using blood-based circulating tumor DNA (ctDNA) next generation sequencing (NGS) and correlation with tissue-based testing in metastatic colorectal cancer (mCRC) J Clin Oncol. 2021;39:3589–3589. doi: 10.1200/JCO.2021.39.15_suppl.3589. [DOI] [Google Scholar]
- 159.Hu Y, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437–4443. doi: 10.1158/1078-0432.CCR-18-0143. [DOI] [PubMed] [Google Scholar]
- 160.Newman AM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rose Brannon A, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun. 2021;12:3770. doi: 10.1038/s41467-021-24109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Larson K, et al. A comparative analysis of tumors and plasma circulating tumor DNA in 145 advanced cancer patients annotated by 3 core cellular processes. Cancers (Basel) 2020;12:701. doi: 10.3390/cancers12030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hoadley KA, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304 e296. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cohen JD, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu MC, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31:745–759. doi: 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Beer TM, et al. A prespecified interim analysis of the PATHFINDER study: performance of a multicancer early detection test in support of clinical implementation. J Clin Oncol. 2021;39:3070–3070. doi: 10.1200/JCO.2021.39.15_suppl.3070. [DOI] [Google Scholar]
- 167.The Lancet Oncology Editorial Cancer detection: the quest for a single liquid biopsy for all. Lancet Oncol. 2020;21:733. doi: 10.1016/S1470-2045(20)30033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Parikh RB, Prasad V. Blood-based screening for colon cancer: a disruptive innovation or simply a disruption? JAMA. 2016;315:2519–2520. doi: 10.1001/jama.2016.7914. [DOI] [PubMed] [Google Scholar]
- 169.Cancer Network. FDA Approves Blood-Based Colorectal Cancer Test. August 15, 2016. https://www.cancernetwork.com/view/fda-approves-blood-based-colorectal-cancer-test. Accessed 27 Oct 2021.
- 170.Raymond VM, Higashi L, Marino E, Lang K. Evaluation of the ctDNA LUNAR-2 test in an average patient screening episode (ECLIPSE) J Clin Oncol. 2021;39:TPS142. doi: 10.1200/JCO.2021.39.3_suppl.TPS142. [DOI] [Google Scholar]
- 171.Guler GD, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun. 2020;11:5270. doi: 10.1038/s41467-020-18965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kasi PM, et al. Serum glycoproteomic-based liquid biopsy for the detection of pancreatic ductal adenocarcinoma. J Clin Oncol. 2020;38:763–763. doi: 10.1200/JCO.2020.38.4_suppl.763. [DOI] [Google Scholar]
- 173.Loosen SH, et al. Serum levels of miR-29, miR-122, miR-155 and miR-192 are elevated in patients with cholangiocarcinoma. PLoS ONE. 2019;14:e0210944. doi: 10.1371/journal.pone.0210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Voigtlander T, et al. MicroRNAs in serum and bile of patients with primary sclerosing cholangitis and/or cholangiocarcinoma. PLoS ONE. 2015;10:e0139305. doi: 10.1371/journal.pone.0139305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Afonso MB, Rodrigues PM, Simao AL, Castro RE. Circulating microRNAs as potential biomarkers in non-alcoholic fatty liver disease and hepatocellular carcinoma. J Clin Med. 2016;5:E30. doi: 10.3390/jcm5030030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rompianesi G, Di Martino M, Gordon-Weeks A, Montalti R, Troisi R. Liquid biopsy in cholangiocarcinoma: current status and future perspectives. World J Gastrointest Oncol. 2021;13:332–350. doi: 10.4251/wjgo.v13.i5.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Conway AM, et al. Molecular characterisation and liquid biomarkers in Carcinoma of Unknown Primary (CUP): taking the 'U' out of 'CUP'. Br J Cancer. 2019;120:141–153. doi: 10.1038/s41416-018-0332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Laprovitera N, et al. Genetic characterization of cancer of unknown primary using liquid biopsy approaches. Front Cell Dev Biol. 2021;9:666156. doi: 10.3389/fcell.2021.666156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kato S, et al. Utility of genomic analysis in circulating tumor DNA from patients with carcinoma of unknown primary. Cancer Res. 2017;77:4238–4246. doi: 10.1158/0008-5472.CAN-17-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kato S, et al. Therapeutic actionability of circulating cell-free DNA alterations in carcinoma of unknown primary. JCO Precis Oncol. 2021;5:1687–1698. doi: 10.1200/PO.21.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gokbuget N, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131:1522–1531. doi: 10.1182/blood-2017-08-798322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schuurhuis GJ, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–1291. doi: 10.1182/blood-2017-09-801498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Venditti A, et al. GIMEMA AML1310 trial of risk-adapted, MRD-directed therapy for young adults with newly diagnosed acute myeloid leukemia. Blood. 2019;134:935–945. doi: 10.1182/blood.2018886960. [DOI] [PubMed] [Google Scholar]
- 184.Overman MJ, et al. Circulating tumor DNA (ctDNA) utilizing a high-sensitivity panel to detect minimal residual disease post liver hepatectomy and predict disease recurrence. J Clin Oncol. 2017;35:3522–3522. doi: 10.1200/JCO.2017.35.15_suppl.3522. [DOI] [Google Scholar]
- 185.Parikh AR, et al. A plasma-only integrated genomic and epigenomic circulating tumor DNA (ctDNA) assay to inform recurrence risk in colorectal cancer (CRC) J Clin Oncol. 2019;37:3602–3602. doi: 10.1200/JCO.2019.37.15_suppl.3602. [DOI] [Google Scholar]
- 186.Taieb J, et al. Prognostic value and relation with adjuvant treatment duration of ctDNA in stage III colon cancer: a post hoc analysis of the PRODIGE-GERCOR IDEA-France trial. Clin Cancer Res. 2021;27:5638–5646. doi: 10.1158/1078-0432.CCR-21-0271. [DOI] [PubMed] [Google Scholar]
- 187.Symonds EL, et al. Use of circulating tumor DNA in colorectal cancer patients to assess tumor burden and response to therapy: an observational study. J Clin Oncol. 2021;39:3528–3528. doi: 10.1200/JCO.2021.39.15_suppl.3528. [DOI] [Google Scholar]
- 188.Lee JS, et al. Circulating tumor DNA as a prognostic indicator in resectable pancreatic ductal adenocarcinoma: a systematic review and meta-analysis. Sci Rep. 2019;9:16971. doi: 10.1038/s41598-019-53271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ococks E, et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann Oncol. 2021;32:522–532. doi: 10.1016/j.annonc.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 190.Yang J, et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis. 2020;11:346. doi: 10.1038/s41419-020-2531-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yang L, Jiang J, Ye S, Xu Y, Huang D. Circulating tumor DNA analysis to detect minimal residual disease and predict recurrence in patients with resectable pancreatic cancer. J Clin Oncol. 2020;38(suppl 15):abstr e16799. doi: 10.1200/JCO.2020.38.15_suppl.e16799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Henriksen TV, et al. Circulating tumor DNA analysis for assessment of recurrence risk, benefit of adjuvant therapy, and early relapse detection after treatment in colorectal cancer patients. J Clin Oncol. 2021;39:11–11. doi: 10.1200/JCO.2021.39.3_suppl.11. [DOI] [Google Scholar]
- 193.Reinert T, et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol. 2019;5:1124–1131. doi: 10.1001/jamaoncol.2019.0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tie J, et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for Stage III COLON CANCer. JAMA Oncol. 2019;5:1710–1717. doi: 10.1001/jamaoncol.2019.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tie J, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra392. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Diehn M, et al. Early prediction of clinical outcomes in resected stage II and III colorectal cancer (CRC) through deep sequencing of circulating tumor DNA (ctDNA) J Clin Oncol. 2017;35:3591–3591. doi: 10.1200/JCO.2017.35.15_suppl.3591. [DOI] [Google Scholar]
- 197.Khakoo S, et al. MRI tumor regression grade and circulating tumor DNA as complementary tools to assess response and guide therapy adaptation in rectal cancer. Clin Cancer Res. 2020;26:183–192. doi: 10.1158/1078-0432.CCR-19-1996. [DOI] [PubMed] [Google Scholar]
- 198.Loupakis F, et al. Detection of molecular residual disease using personalized circulating tumor DNA assay in patients with colorectal cancer undergoing resection of metastases. JCO Precis Oncol. 2021;5:1166–1177. doi: 10.1200/PO.21.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Scholer LV, et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res. 2017;23:5437–5445. doi: 10.1158/1078-0432.CCR-17-0510. [DOI] [PubMed] [Google Scholar]
- 200.Tarazona N, et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann Oncol. 2019;30:1804–1812. doi: 10.1093/annonc/mdz390. [DOI] [PubMed] [Google Scholar]
- 201.Tie J, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut. 2019;68:663–671. doi: 10.1136/gutjnl-2017-315852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang Y, et al. Prognostic potential of circulating tumor DNA measurement in postoperative surveillance of nonmetastatic colorectal cancer. JAMA Oncol. 2019;5:1118–1123. doi: 10.1001/jamaoncol.2019.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yukami H, et al. Minimal residual disease by circulating tumor DNA analysis for colorectal cancer patients receiving radical surgery: an initial report from CIRCULATE-Japan. J Clin Oncol. 2021;39:3608–3608. doi: 10.1200/JCO.2021.39.15_suppl.3608. [DOI] [Google Scholar]
- 204.Kasi PM, et al. Tumor-informed assessment of circulating tumor DNA and its incorporation into practice for patients with hepatobiliary cancers. J Clin Oncol. 2021;39:4103–4103. doi: 10.1200/JCO.2021.39.15_suppl.4103. [DOI] [Google Scholar]
- 205.Huffman B. Building Together: Performance of a tumor-informed circulating tumor DNA assay from over 250 patients with over 600 plasma time points in esophageal and gastric cancer. Poster presented at: ESMO Congress 2021; Sep 16, 2021; Paris, France.
- 206.Bratman SV, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat Cancer. 2020;1:873–881. doi: 10.1038/s43018-020-0096-5. [DOI] [PubMed] [Google Scholar]
- 207.Cheng ML, et al. Circulating tumor DNA in advanced solid tumors: clinical relevance and future directions. CA A Cancer J Clinicians. 2021;71:176–190. doi: 10.3322/caac.21650. [DOI] [PubMed] [Google Scholar]
- 208.Xiao J, et al. Circulating tumor cells: technologies and their clinical potential in cancer metastasis. Biomedicines. 2021;9:1111. doi: 10.3390/biomedicines9091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Xiao J, et al. Efficient propagation of circulating tumor cells: a first step for probing tumor metastasis. Cancers (Basel) 2020;12:2784. doi: 10.3390/cancers12102784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cristofanilli M, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 211.Fehm T, et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Res Treat. 2010;124:403–412. doi: 10.1007/s10549-010-1163-x. [DOI] [PubMed] [Google Scholar]
- 212.Ignatiadis M, et al. Liquid biopsy-based clinical research in early breast cancer: the EORTC 90091–10093 Treat CTC trial. Eur J Cancer. 2016;63:97–104. doi: 10.1016/j.ejca.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 213.Riethdorf S, et al. Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin Cancer Res. 2010;16:2634–2645. doi: 10.1158/1078-0432.CCR-09-2042. [DOI] [PubMed] [Google Scholar]
- 214.Hardingham JE, et al. Detection and clinical significance of circulating tumor cells in colorectal cancer–20 years of progress. Mol Med. 2015;21(Suppl 1):S25–31. doi: 10.2119/molmed.2015.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rushton AJ, Nteliopoulos G, Shaw JA, Coombes RC. A review of circulating tumour cell enrichment technologies. Cancers (Basel) 2021;13:970. doi: 10.3390/cancers13050970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Riethdorf S, O'Flaherty L, Hille C, Pantel K. Clinical applications of the cell search platform in cancer patients. Adv Drug Deliv Rev. 2018;125:102–121. doi: 10.1016/j.addr.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 217.Cohen SJ, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:3213–3221. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 218.Rossi E, et al. M30 neoepitope expression in epithelial cancer: quantification of apoptosis in circulating tumor cells by cell search analysis. Clin Cancer Res. 2010;16:5233–5243. doi: 10.1158/1078-0432.CCR-10-1449. [DOI] [PubMed] [Google Scholar]
- 219.Zhou J, et al. Isolation of circulating tumor cells in non-small-cell-lung-cancer patients using a multi-flow microfluidic channel. Microsyst Nanoeng. 2019;5:8. doi: 10.1038/s41378-019-0045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Heitzer E, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73:2965–2975. doi: 10.1158/0008-5472.CAN-12-4140. [DOI] [PubMed] [Google Scholar]
- 221.Girotti MR, et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016;6:286–299. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- 222.Praharaj PP, Bhutia SK, Nagrath S, Bitting RL, Deep G. Circulating tumor cell-derived organoids: current challenges and promises in medical research and precision medicine. Biochim Biophys Acta Rev Cancer. 2018;1869:117–127. doi: 10.1016/j.bbcan.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fumagalli A, et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci USA. 2017;114:E2357–e2364. doi: 10.1073/pnas.1701219114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Smith SM, et al. Clinical cancer advances 2021: ASCO's report on progress against cancer. J Clin Oncol. 2021;39:1165–1184. doi: 10.1200/JCO.20.03420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.