

Abstract
Tilletia caries and T. laevis, which are the causal agents of common bunt, as well as T. controversa, which causes dwarf bunt of wheat, threaten especially organic wheat farming. The three closely related fungal species differ in their teliospore morphology and partially in their physiology and infection biology. The gene content as well as intraspecies variation in these species and the genetic basis of their separation is unknown. We sequenced the genome of four T. caries, five T. controversa, and two T. laevis and extended this dataset with five publicly available ones. The genomes of the three species displayed microsynteny with up to 94.3% pairwise aligned regions excluding repetitive regions. The majority of functionally characterized genes involved in pathogenicity, life cycle, and infection of corn smut, Ustilago maydis, were found to be absent or poorly conserved in the draft genomes and the biosynthetic pathway for trimethylamine in Tilletia spp. could be different from bacteria. Overall, 75% of the identified protein-coding genes comprising 84% of the total predicted carbohydrate utilizing enzymes, 72.5% putatively secreted proteins, and 47.4% of effector-like proteins were conserved and shared across all 16 isolates. We predicted nine highly identical secondary metabolite biosynthesis gene clusters comprising in total 62 genes in all species and none were species-specific. Less than 0.1% of the protein-coding genes were species-specific and their function remained mostly unknown. Tilletia controversa had the highest intraspecies genetic variation, followed by T. caries and the lowest in T. laevis. Although the genomes of the three species are very similar, employing 241 single copy genes T. controversa was phylogenetically distinct from T. caries and T. laevis, however these two could not be resolved as individual monophyletic groups. This was in line with the genome-wide number of single nucleotide polymorphisms and small insertions and deletions. Despite the conspicuously different teliospore ornamentation of T. caries and T. laevis, a high degree of genomic identity and scarcity of species-specific genes indicate that the two species could be conspecific.
Supplementary Information
The online version contains supplementary material available at 10.1186/s43008-022-00098-y.
Keywords: Fungal pan-genomes, Functional genomics, Closely related fungal species, Trimethylamine biosynthesis, Basidiomycota
Introduction
The basidiomycete genus Tilletia (Tilletiales, Exobasidiomycetes, Ustilaginomycotina) comprises about 186 described species causing smut disease on Poaceae (Vánky 2012; Denchev and Denchev 2013, 2018a, b; Li et al. 2014; Denchev et al. 2018). Tilletia species are biotrophs that do not develop specialized cellular infection structures, but form so-called local interaction zones in the host tissue (Begerow et al. 2014). The term bunt is used for cereal-infecting species of Tilletia that produce teliospores in the ovary of the host plant (Carris et al. 2006). The infection of cereal crops by bunt species remains asymptomatic up to culm elongation (Purdy et al. 1963). The infected seeds smell like fish due to the production of trimethylamine (Hanna et al. 1932; Nielsen 1963). Contaminated seeds are not suitable for human and animal consumption at a certain infection level and must be treated accordingly for use as seeds in organic and conventional farming.
Three kinds of bunt diseases are known from wheat species (Triticum spp.) namely common, dwarf, and karnal bunt. Only common and dwarf bunt affect wheat production in Central Europe, where they are under phytosanitary regulation for seed certification in organic and conventional farming. Tilletia caries (syn. T. tritici) and T. laevis (syn. T. foetida) cause common bunt of wheat (Woolma and Humphrey 1924; Vánky 2012), a disease that occurs in wheat-growing areas worldwide (Hoffmann 1982; Goates 1996). Dwarf bunt is caused by T. controversa, which is reported to be limited to higher elevations (Goates 1996) or regions with prolonged cooler temperatures (Carris 2010). However, in recent years the disease has also been observed to extend to lowland regions in Germany (Rudloff et al. 2020). Tilletia controversa is economically important for international seed trading because it is a quarantine pathogen in several countries (Mathre 1996; Whitaker et al. 2001; Peterson et al. 2009; Jia et al. 2013).
Tilletia caries, T. controversa, and T. laevis differ in several biological and physiological features. Firstly, the morphology of teliospores varies from smooth in T. laevis to deep and broadened reticulations in T. controversa and an intermediate form in T. caries. Secondly, the teliospores of T. caries and T. laevis germinate within a week at 12 to 15 °C under illumination or in dark, while germination of T. controversa teliospores requires up to eight weeks at the optimum temperature of 3 to 5 °C and light is essential for germination (Purdy et al. 1963). Furthermore, the infection of wheat by common bunt pathogens occurs before the emergence of the coleoptile, whereas T. controversa attacks the same organ after emergence (Carris 2010). Disease symptoms differ moderately between common and dwarf bunt. Substantial wheat stunting and enhanced tillering occur in dwarf bunt and its severity varies among wheat cultivars (Goates 1996; Carris 2010), while stunting in common bunt diseased wheat is not readily distinguishable. Despite the different morphological and physiological features, molecular phylogenetic analysis based on three loci could not resolve the three species unequivocally (Carris et al. 2007). However, a phylogenomic study of five Tilletia species based on 4,896 single-copy orthologous genes but limited specimens sampling (one T. caries, two T. controversa, two T. laevis, three T. indica, and two T. walkeri) suggested that they are distinct (Nguyen et al. 2019).
Despite the growing concern about common and dwarf bunt as major threat to especially organic wheat production due to a limited number of durably resistant cultivars (Ruzgas and Liatukas 2008; Matanguihan et al. 2011; Aydoğdu and Kaya 2020), and the fact that T. controversa is a quarantine pathogen, little is known about the genomic structure and gene contents of the three species.
Here, we report draft genome sequences of four T. caries isolates, five T. controversa isolates, and two isolates of T. laevis, obtained from single teliospore cultures that except for one isolate of T. laevis, originated from recent European populations. The sequenced isolates were selected from a sample collection of common and dwarf bunt characterized by morphological characters and germination behavior of teliospores and later by MALDI-TOF MS (Forster et al. 2022). The genome sequences of these 11 isolates were analyzed together with five additional publicly available common and dwarf bunt genomes (Nguyen et al. 2019) to provide a first insight into the genomic features as well as genomic diversity within and between these three important pathogens.
Methods
Isolates and single teliospore cultures
Isolates of four T. caries, five T. controversa, and two T. laevis were whole-genome sequenced in this study (Table 1). To obtain DNA for genome sequencing, cultures of T. caries, T. controversa, and T. laevis were grown from single teliospores. For the production of single teliospore cultures, teliospores were surface-sterilized as described by Castlebury et al. (2005). Briefly, bunt balls were crushed using a pair of sterile fine-point forceps and the wheat ovary tissue was carefully removed. The teliospores were immersed in 0.26% (v/v) NaClO (Carl Roth, Karlsruhe, Germany) for 30 s, pelleted by centrifugation for 10 s and rinsed twice with sterile, distilled water. For germination, surface-sterilized teliospores were streaked on 1.5% water-agar and incubated either at 5 °C under constant light (T. controversa germination) or at 15 °C in darkness (T. caries and T. laevis germination). A single germinated teliospore of each specimen was then transferred to M-19 agar medium (Trione 1964) using a sterile needle. Fungal cultures on M-19 were maintained at 15 °C in the dark to establish colonies of mononucleate, non-pathogenic, and non teliosporogenic hyphal stage for nucleic acid extraction. The medium was supplemented with penicillin G (240 mg/L) and streptomycin sulfate (200 mg/L). The mycelium was freeze-dried (Christ ALPHA1-4 LSC, Martin Christ Gefriertrocknungsanlagen, Osterode am Harz, Germany) at −40 °C for 48 h and kept at 4 °C until use. Duplicates of the single teliospore cultures obtained in this study were deposited at the CBS-KNAW culture collection of the Westerdijk Fungal Biodiversity Institute (Utrecht, The Netherlands) under the accession numbers provided in Table 1.
Table 1.
List of the Tilletia isolates used for genome comparison
| Taxon | Isolate (Accession numbera) | Collection year | Geographical origin | Host | BioProject accession | Sequencing technology | References |
|---|---|---|---|---|---|---|---|
| T. caries | AA11 (CBS 144825) | 2015 | Austria | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. caries | AI (CBS 145171) | 2015 | Italy | Triticum durum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. caries | AO (CBS 145172) | 2014 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. caries | AZH3 (CBS 145166) | 2015 | Switzerland | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. caries | DAOMC 238032 | 1996 | USA | Triticum sp. | PRJNA317434 | Illumina HiSeq | Nguyen et al. (2019) |
| T. controversa | DAOMC 236426 | 1998 | Canada | Triticum sp. | PRJNA317433 | Illumina HiSeq, MiSeq, PacBio | Nguyen et al. (2019) |
| T. controversa | DAOMC 238052 | 1997 | Canada | Triticum sp. | PRJNA393324 | Illumina MiSeq | Nguyen et al. (2019) |
| T. controversa | OA2 (CBS 145169) | 2015 | Austria | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. controversa | OL14 (CBS 145167) | 2014 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. controversa | OR (CBS 144827) | 2013 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000, PacBio RS II | This study |
| T. controversa | OV (CBS 145170) | 2011 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. controversa | OW (CBS 145168) | 2013 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. laevis | ATCC 42080 | – | USA | Triticum sp. | PRJNA393337 | Illumina MiSeq | Nguyen et al. (2019) |
| T. laevis | DAOMC 238040 | 1997 | Australia | Triricum sp. | PRJNA393335 | Illumina MiSeq | Nguyen et al. (2019) |
| T. laevis | LLFL (CBS 144826) | 2015 | Germany | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
| T. laevis | L-19 (CBS 145173) | – | – | Triticum aestivum | PRJEB40624 | Illumina HiSeq 4000 | This study |
aWesterdijk Fungal Biodiversity Institute (CBS-KNAW; Utrecht, The Netherlands)
High molecular weight nucleic acid extraction and whole-genome sequencing
Total genomic DNA from freeze-dried mycelium was extracted using a modified CTAB-based method (Brandfass and Karlovsky 2008) as follows: 30–50 mg lyophilized mycelium was pulverized in 2 mL reaction tubes with four 4 mm sterile tungsten carbide beads at 22 Hz for 50 s using a tissue lyser (Qiagen Tissuelyser II, Qiagen, Hilden, Germany). The bead-beating step was repeated twice. The reaction tubes were shaken vigorously between the two disruption steps to loosen the mycelium from the bottom of the tubes after bead beating.
The CTAB buffer (600 µL) was supplemented directly before use with 400 µg RNase (Carl Roth, Karlsruhe, Germany), 5 µL β-mercaptoethanol and 0.2 mg proteinase K (Carl Roth, Karlsruhe, Germany) and then added to the ground mycelium. The samples were incubated for 60 min at 65 ºC and 400 rpm in a thermoshaker (Eppendorf, Hamburg, Germany). One volume of phenol/chloroform/isoamylalcohol (25:24:1 v/v/v) was added (Carl Roth, Karlsruhe, Germany) to the mixture followed by 10 min of incubation on ice before centrifugation for 10 min at 13,000×g at room temperature. The upper phase was transferred to a new reaction tube and inverted a few times to mix with chloroform (v/v). The mixture was incubated at room temperature for 10 min before being separated by centrifugation at 13,000×g at room temperature for 10 min. Finally, the DNA was precipitated for 20 min at room temperature using 0.6 volume of isopropanol (Merck, Darmstadt, Germany). The obtained DNA pellet was washed twice with 70% (v/v) ethanol and dried at room temperature, before it was finally dissolved in 500 µL commercially available elution buffer (10 mM Tris–Cl, pH 8.5) (Qiagen, Hilden, Germany) at room temperature.
To digest the remaining RNA, 100 µg RNase (Carl Roth, Karlsruhe, Germany) were added to the extracted gDNA, the mixture was inverted multiple times and incubated for in total 1 h at 42 °C at 100 rpm. After 30 min 7 µg proteinase K (Carl Roth, Karlsruhe, Germany) was also added. RNA and proteins were removed by adding one volume of phenol/chloroform/isoamylalcohol (25:24:1 v/v/v), and DNA was precipitated by isopropanol (1:1 v/v) (Merck, Darmstadt, Germany) as described above. Polar fractions were retrieved through 13,000×g centrifugation. The obtained DNA pellet was washed twice with 70% (v/v) ethanol and resuspended in the elution buffer. DNA from different extraction replicates was pooled. The quality and quantity of the isolated DNA was measured with a Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Darmstadt, Germany) and stored at −20 ºC until shipping.
For whole-genome sequencing, the DNA from single teliospore cultures of eleven isolates was shipped to GATC biotech (Konstanz, Germany) for fragmentation, library preparation, and sequencing on an Illumina HiSeq 4000 platform (125 bp, paired-end reads). Whole-genome shotgun sequencing of one isolate (T. controversa OR) was additionally performed using a Pacific Biosciences (PacBio) RS II instrument, and P6-C4 chemistry. A total of seven Single Molecule Real-Time (SMRT) cells were sequenced for this isolate.
Genome assembly
Trimmomatic v0.36 (Bolger et al. 2014) was used to trim adapters and low quality reads from Illumina data from 11 Tilletia isolates (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:70 AVGQUAL:25). High-quality reads with minimum lengths of 70 bp for both reads and > 25 average quality were retained for further processing. PacBio reads of T. controversa (OR) were corrected using the filtered Illumina reads from the same species using Proovread v2.12 (Hackl et al. 2014). The Prooveread-corrected untrimmed PacBio reads were further corrected and trimmed using the self-correction and trimming method implemented in Canu v1.6 assembler (Koren et al. 2017). The Canu assembler was also used to construct a draft assembly of corrected-trimmed PacBio reads, which was subsequently scaffolded with Illumina paired reads using SSpace-Standard V3.0 (Boetzer et al. 2011) and PacBio reads that were corrected and trimmed by Illumina reads using SSpace-LongRead (Boetzer and Pirovano 2014)). The assembly statistics were generated using assemblathon_stats.pl (Author: Keith Bradnam, Genome Center, UC Davis) and CEGMA v2.5 (Parra et al. 2007) to assess genome completeness. In a separate approach, all the 10 Tilletia isolates were assembled, using the remaining Illumina reads employing Velvet assembler v1.2.10 (Zerbino and Birney 2008) (-scaffolding on). Several assemblies were generated for all the species at different k-mers. Assembly statistics and CEGMA completeness of all the assemblies were tabulated and for individual species the best assembly in terms of statistics and CEGMA completeness was manually chosen.
Additionally, the genome assembly and annotation files of one T. caries, two T. controversa, and two T. laevis isolates sequenced by Nguyen et al. (2019) were retrieved from the National Center for Biotechnology Information (NCBI) repository. In total, 16 genomes comprising five T. caries, seven T. controversa, and four T. laevis isolates were used in this study (Table 1).
Species tree recognition
Genome assemblies used in the phylogentic analyses are listed in Additional file 1: Table 1. For the phylogenetic study, orthologous genes were selected according to a modified approach described by Pizarro et al. (2018). Every assembly was assessed for 303 single-copy genes of the hierarchical catalog of eukaryotic orthologs v9 (Waterhouse et al. 2013) using BUSCO (Benchmarking Universal Single-Copy Orthologs) version 3.0.2 (Simao et al. 2015) in the genomic mode. The putative gene regions identified by BUSCO were extracted. For duplicated genes, we used the sequence that had the higher similarity score to its BUSCO reference. Each BUSCO gene recovered from each of the 27 genomes was aligned using MAFFT V. 7 (Katoh et al. 2017) adopting the iterative refinement algorithms L-INS-i (-localpair -maxiterate 1000—adjustdirectionaccurately). In order to reduce the effect of missing data, alignments with more than 7% of missing data (lacking corresponding sequences in more than two isolates per locus) were removed resulting in 241 genes that could be used for the inferences. Ambiguous regions within each alignment were removed using Gblock v 0.91b (Castresana 2000) with the default parameters (strict).
Species tree inferences based on the multispecies coalescent model (Degnan and Rosenberg 2006) were done because individual phylogenetic analyses based on individual genes can result in different gene trees that differ from the true species tree (Rannala and Yang 2003). First, we constructed approximately-maximum-likelihood phylogenetic trees for every single gene individually using FastTree 2.1.11 (Price et al. 2010) implemented in Geneious version 8.1.2 (Biomatters Limited, Auckland, New Zealand). As nucleotide substitution the Generalized Time-Reversible (GTR) model was used with the optimized gamma20 likelihood distribution. The Accurate Species Tree ALgorithm II (ASTRAL-II) (Mirarab and Warnow 2015) was employed to summarize coalescent inferences resulting from all trees. Clade support was evaluated by computing the local posterior probability (LPP) (Sayyari and Mirarab 2016). The trees were visualized in FigTree v1.4.4 (tree.bio.ed.ac.uk/software/figtree/).
Identification of repetitive regions, simple sequence repeats (SSRs), and transposable elements (TEs)
Draft genome sequences were used to identify SSRs, also known as microsatellites by using the tool MIcroSAtellite identification (MISA) (Beier et al. 2017). The search criteria were at least ten repeat units for mononucleotide, six repeats for dinucleotide, and five for tri-, tetra-, penta-, and hexanucleotide motifs. SSRs with less than 100 bp distance from each other were considered as compound. The relative abundance for each SSR type was calculated by the number of repeats per Mb of genome.
Transposable elements were identified computing TransposonPSI (Haas 2010) with default settings. The program employs PSI-BLAST search (Altschul et al. 1997) against a database of various collections of TE families to identify matching regions in the genome. Additionally, we used RepeatModeler version 1.0.11 (Smit et al. 2008–2019) with the default settings to create a library comprising de novo identified repetitive elements. RepeatModeler employs three de novo repeat finders of RECON (Bao and Eddy 2002), RepeatScout (Price et al. 2005), and Tandem Repeats Finder (Benson 1999). The number of identified TEs and SRRs (see above) was subtracted from the total number of repeats identified by RepeatModeler as unclassified repetitive elements.
The resulting library of RepeatModeler was used to mask respective elements in the target genome sequences using RepeatMasker 4.0.9 (Smit et al. 2013–2015). For different purposes (see below), we generated both soft-masking (i.e. repeats replaced by lowercase letters) and hard-masking (i.e. repeats replaced by N).
Detection of single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels)
For pairwise SNP and indel identification, we used hard-masked genomes generated by RepeatMasker. For each genome pair, SNPs and the total length of indels in the aligned regions were counted using dnadiff wrapper from MUMmer 3.0 package (show-snps-C) (Kurtz et al. 2004). Average nucleotide identities of one-to-one alignments were also obtained from dnadiff output.
Gene model prediction
Genes in the newly sequenced genomes (n = 11) were predicted from the soft-masked assemblies while for the publicly available genomes we used the existing gene annotations. We used a combination of ab initio and homology-based approach for gene model prediction. Gene models were created first by the incorporation of multiple sources of evidence using Gene Model Mapper (GeMoMa pipeline: V1.6.2 beta) (Keilwagen et al. 2016, 2018). GeMoMa is a homology-based gene prediction program and uses RNA-Seq data to incorporate evidence for splice site prediction. Afterwards, BRAKER2 (Brůna et al. 2020), which utilizes the ab initio gene predictor Augustus 3.3.3 (Stanke et al. 2006) and GeneMark-ET 4.33 (Lomsadze et al. 2014) self-training algorithms were applied. To do so, publicly available genome sequences and structural annotations of Acaromyces ingoldii, Ceraceosorus guamensis, Jaminaea rosea, Meira miltonrushii, Pseudomicrostroma glucosiphilum, Tilletiopsis washingtonensis isolates (Kijpornyongpan et al. 2018) and Ustilago maydis (Kamper et al. 2006) were downloaded from GenBank as references (accession numbers are given in Additionalfile 1: Table 2). Additionally, three RNA-Seq datasets were derived from two different T. caries isolates (DAOMC 238032 and WSP 72095/517) and one T. controversa isolate (DAOMC 236426) were downloaded from GenBank (Additionalfile 1: Table 1).
For adapter clipping and read trimming of the RNA-Seq data, the utility program Trim Galore version 0.4.0 (Krueger 2012–2019) was employed (qval > = PHRED 30, minimal read length of 50 bp). Trimmed reads were mapped to the assembled genome sequences using STAR version 2.4.0d-2 (Dobin et al. 2012) with default parameters. The two RNA-Seq datasets of T. caries (SRR2513861 and SRR3337311) were mapped to T. laevis assemblies because no RNA-Seq data was available for T. laevis and the two species are closely related.
Protein-coding exons were extracted from the seven reference genomes by GeMoMa module Extractor (part of GeMoMaPipeline) using the default parameters. The GeMoMa Extract RNA-Seq evidence (ERE) was used to extract intron boundaries of each target genome by utilizing RNA-Seq data (coverage = true). We permitted alternative transcripts. The rest of the parameters were set as follows: maximum intron length = 2500, tBLASTn = false, ambiguity = ambiguous, score = ReAlign, rename = no. Filtered predictions (start = ’M’ and stop = ’*’ sorting = score/AA > = 0.50) file for each genome generated by GeMoMa was used with align2hints command to produce hint file for BRAKER2. The corresponding softmasked genome, the STAR RNA-Seq mapped file, and the GeMoMa hint file were used to run BRAKER2 (UTR = on) for each genome. The generate gene model by BRAKER2 was used for further analysis. To predict the coding regions of transfer RNA (tRNAs), tRNAscan-SE 2.0 (Lowe and Chan 2016) was used with eukaryotic sequence source in the default search mode.
To estimate the completeness of the gene model predictions, BUSCO was used. We used the lineage dataset for Fungi-OrthoDB9 (Zdobnov et al. 2016) in the proteome mode.
Functional annotation of the predicted genes
Genome-wide annotation was done to relate putative biological functions to the predicted genes. To make functional annotation comparable between all draft genomes, we analyzed all 16 genomes used in this study. The putative functions were assigned to the predicted proteins through one-to-one orthology assignments by eggNOG-Mapper 5.0.0 (Huerta-Cepas et al. 2018) (one-to-one ortholog, auto taxonomic adjust mode). Only functional annotations derived from Eukaryote or fungal sequence sources were accepted. Functional descriptions of Gene Ontology (Go) terms (Ashburner et al. 2000; Gene Ontology 2015), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and modules (Kanehisa et al. 2013), and COG/KOG functional categories (Levasseur et al. 2013; Galperin et al. 2014), and SMART/PFAM domains (Letunic and Bork 2018) were obtained using eggNOG-Mapper.
Prediction of encoded carbohydrate-active enzymes (CAZymes), secreted proteins, and secondary metabolites
Carbohydrate-active enzymes derived from the draft genomes were predicted using the HMM-based-dbCAN server (HMMdb v8.0) with a cut-off E-value < 1e−17 (suggested for fungi) and coverage > 0.50 (Zhang et al. 2018). Out of this prediction, the potential plant cell wall degrading enzymes were classified for their substrate according to Kijpornyongpan et al. (2018) and Benevenuto et al. (2018).
Putatively secreted proteins (referred to secretome in their totality) were identified by the presence of a signal peptide and absence of transmembrane domains in the predicted proteomes of each genome according to the suggestions of (Min 2010). Briefly, the proteome of each draft genome was first screened by SignalP 5.0 (Almagro Armenteros et al. 2019). To check whether the prediction belonged to an integral membrane protein, transmembrane α-helix predictor TMHMM v. 2.0 (Krogh et al. 2001) in tandem was employed. Those signal-peptide-like proteins showing any transmembrane helix topology were filtered out. Additionally, the signal peptides were predicted using Phobius (Kall et al. 2007) webserver accessed on Sep. 2019 with default parameters. In the end, only those putative proteins containing signal peptides that had been predicted by both independent approaches were annotated as secretome. To predict the effector repertoire from the predicted secretome of each genome, EffectorP 2.0 (Sperschneider et al. 2018) accessed on Sep. 2019 was used.
The draft genome sequences were searched for secondary metabolites and biosynthetic gene clusters using the fungal version of antiSMASH 5.0 (antibiotics and Secondary Metabolite Analysis Shell) (Medema et al. 2011). Since genes of a cluster may be dispersed on different contigs, the presence, completeness and order of each gene cluster was validated by aligning Illumina reads of each isolate to a reference sequence from each gene cluster group according to a mapping approach described by Weber et al. (2019). Briefly, the reference sequence was selected for each gene cluster group based on either length or high sequence conservation among the different isolates. Illumina reads of each isolate (Additionalfile 1: Table 1) were trimmed using Trimmomatic version 0.36 (Bolger et al. 2014) with a 4:15 sliding window. The trimmed reads were aligned to the different gene cluster references using Bowtie v2.4.1 (Langmead and Salzberg 2012). SAMtools v1.10 (Li et al. 2009) was used for file conversion to bam, validation of read pairing information (fixmate), removal of reading duplicates (rmdup), removal of mapped singleton readings, and calculation of position-specific reading depth (depth). Read depths were plotted with R version 3.5.1 (R Development Core Team 2013).
Orthologous gene identification and clustering
The OrthoMCL pipeline v 2.0 (Li et al. 2003) was used to identify clusters of orthologous genes among all 16 isolates of the three species (inflate = 1.8 and E-value = 1e−10). Input translated protein sequences of all predicted genes contained also alternative transcripts per gene. The OrthoMCL program applies all-against-all BLASTp to estimate similarities between proteins and identifies groups using Markov clustering algorithm. The output of the program was parsed by using a custom-made phyton script to define; (i) orthologous genes that were present in all isolates referred to shared genes at the interspecies level and core genome at intraspecies level; (ii) orthologues genes shared between all isolates of a species and absent in the others (species-specific genes); (iii) the accessory (at intraspecies level) or variable (at interspecies level) genes which were dispensable and not present in all genomes; (iv) singletons presented only in a single isolate (isolate-specific genes). Putative functional prediction of an orthology cluster was reported only when at least 75% of the genes within shared an identical annotation.
Results
Genome assembly and annotation of T. caries, T. controversa, and T. laevis
The assembly of ten draft genomes based only on Illumina reads (four T. caries, four T. controversa, and two T. laevis isolates) resulted in assembled genome sizes of 30.3 to 31.7 Mb (T. caries), 29.4 to 31.9 Mb (T. controversa), and 30.8 Mb (T. laevis) with GC contents between 56.5 to 56.7% (Fig. 1). N50 values varied between 9.3 and 17.8 kb. The hybrid assembly of the T. controversa isolate OR, which was sequenced using both PacBio and Illumina reads, resulted in a draft genome size of 49.3 Mb (scaffold N50 = 137 kb) distributed in 985 scaffolds with a GC content of 55.7% (Fig. 1).
Fig. 1.

Bubble plots of descriptive numbers for each genome. The bubble sizes are scaled only within categories. The genome assemblies done in this work are presented in black font. Those in grey font relate to genomes published by Nguyen et al. (2019). Two T. controversa isolates OR and DAOMC 236426 were sequenced both on Illumina and PacBio platforms and marked with asterisks
A combination of ab initio and order-specific gene model data was used after assessing annotation completeness for each annotation approach separately employing a genome (see the results in Additional file 1: Table 3). In total, 9,807 to 9,943 protein-coding genes were annotated in the T. caries genomes, 9,679 to 10,459 in the T. controversa genomes, and 10,160 and 10,203 in the two T. laevis genomes (Table 2). Coding sequences (CDS) consisted of 3.5 exons on average. Alternative splicing forms were predicted for up to 1.4% of the total CDS (Table 2). Genomes contained 110 to 178 genes encoding for tRNAs. The specificity of these tRNAs covered up to 48 of 61 possible anticodons and the codon usage was identical in all three species. To check whether the tRNA genes were clustered, we examined the location of tRNA genes in the most contiguous genome (OR isolate). A total of 178 putative tRNA genes were distributed over 102 scaffolds. The maximum number of 14 tRNA genes plus 9 pseudogenes spanned a 151,829 bp long region on scaffold number OR-9 (accession number CAJHJB010000889). The genome annotation completeness was estimated between 91.1 to 96.9% in the draft genomes (Table 2).
Table 2.
Summary of gene model prediction results
| Annotation statistics | T. caries | T. controversa | T. laevis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA11 | AI | AO | AZH3 | OA2 | OL14 | OR* | OV | OW | LLFL | L-19 | |
| Gene model | 9913 | 9943 | 9807 | 9930 | 9679 | 9956 | 10,459 | 9680 | 9797 | 10,203 | 10,160 |
| tRNA | 119 | 128 | 114 | 123 | 119 | 116 | 178 | 110 | 118 | 116 | 116 |
| Average gene length (bp) | 1790 | 1734 | 1756 | 1766 | 1813 | 1706 | 1931 | 1720 | 1755 | 1671 | 1674 |
| Total CDS length (Mb) | 14.9 | 14.8 | 14.5 | 14.8 | 14.8 | 14.6 | 16.9 | 14.1 | 14.5 | 14.6 | 14.6 |
| Number of exons | 36,009 | 35,788 | 35,117 | 35,601 | 35,266 | 34,951 | 40,922 | 33,870 | 34,789 | 35,521 | 35,439 |
| Exons per gene | 3.63 | 3.6 | 3.58 | 3.58 | 3.64 | 3.51 | 3.91 | 3.49 | 3.55 | 3.48 | 3.48 |
| Number of introns | 26,443 | 26,440 | 25,784 | 26,103 | 25,944 | 25,832 | 30,399 | 24,833 | 25,549 | 26,258 | 26,216 |
| Introns per gene | 2.66 | 2.65 | 2.62 | 2.62 | 2.68 | 2.59 | 2.9 | 2.56 | 2.6 | 2.57 | 2.58 |
| % of genome covered by genes | 56.3 | 54.1 | 56.5 | 55.8 | 54.7 | 55 | 40.5 | 56.3 | 55 | 55.03 | 54.86 |
| Transcripts | 10,052 | 10,069 | 9930 | 10,053 | 9800 | 10,066 | 10,585 | 9801 | 9914 | 10,334 | 10,292 |
| Annotation completeness | 96.6 | 95.8 | 93.8 | 95.1 | 95.5 | 94.5 | 96.9 | 91.1 | 93.8 | 93.1 | 94.1 |
*Reference genome
Although repetitive elements had been generally regarded as junk DNA or remnants of molecular evolution, some repetitive sequences were shown to play diverse roles in environmental adaptation and genome evolution (Wöstemeyer and Kreibich 2002). The genome fraction assigned to repetitive elements in Tilletia species ranged from 7.8 to 13.7% for T. caries, 8.9 to 13.6% for T. controversa, and 9.1 to 11.8% for T. laevis (Additionalfile 1: Table 4) and overall, a higher proportion of repetitive elements was found in the newly sequenced genomes. Exceptionally, roughly a four times higher number of repetitive elements (37%) was revealed in the genome sequence of T. controversa isolate OR and its 49.3 Mb assembled genome size. Transposition is one of the causes of genomic plasticity and plays an important role in pathogenicity and adaptive evolution (Casacuberta and González 2013; Muszewska et al. 2019; Razali et al. 2019). Transposable elements made up to 3.7% of all repetitive elements in the studied genomes. The values were very similar for all three species. Transposable elements can move or copy from one locus to another and are classified based on their mode of dispersion (Levin and Moran 2011). The detected TEs were classified into one of 15 superfamilies (Fig. 2), of which DDE-1, gypsy, hAT, helitronORF, Itr-Roo, Line, mariner-ant1, MuDR-A-B, and TY1-copia were more prevalent in the genomes of European isolates compared to the North American genomes. In all isolates, regardless of the sequencing platform used, the gypsy-like and TY1-copia-like superfamilies were most common, accounting for more than half of the total TEs in each genome (Fig. 2). Additionally, we classified a total of 6,564 to 10,031 repetitive elements as SSRs (Additionalfile 1: Table 5), accounting for 0.53 to 0.63% of the entire genomes. Trinucleotide SSRs (35.2 to 42.8% of all SSRs) were the most abundant.
Fig. 2.

Number of detected transposable elements and their classification in 16 isolates of common and dwarf bunt. Significant differences in the total number of detected transposable elements between sequenced genomes of this study (black font) and publicly available ones are marked with * (Tukey HSD, α = 0.001)
Phylogenetic analysis
From a total of 303 initial BUSCO genes, 241 were used for the construction of individual gene trees on which the multispecies coalescence inferences of 27 strains representing six Tilletia species were based on. In the phylogenetic tree (Fig. 3) the sequenced strains clustered in highly supported groups according to their species circumscription with one exception: The strains of T. caries and T. laevis clustered intermingled in a polyphyletic manner within a joint monophyletic group suggesting their conspecificity.
Fig. 3.
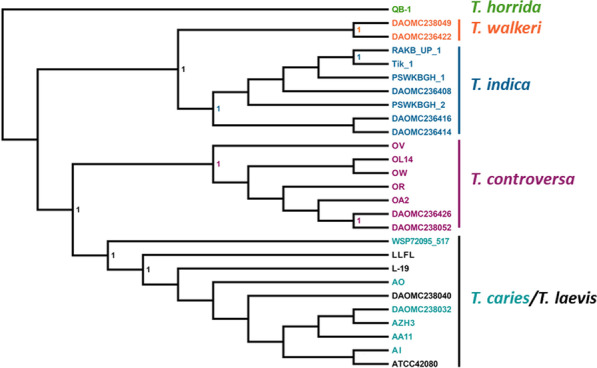
Coalescence-based phylogenetic analysis on a set of 241 genes and 27 strains of six Tilletia species. Individual trees were obtained from approximately maximum likelihood analysis per locus and the coalescence inference was based on all the resulting trees using ASTRAL II. Each isolate is color coded based on species identification. Node support values are given as local posterior probabilities. Only supporting values above 0.95 are indicated at nodes
Genomic synteny and genome-wide diversity
Overall, 82.7 to 94.3% of the assembled genomes could be aligned pairwisely with an average nucleotide identity between 98.7 to 99.6% in one-to-one aligned regions, excluding repetitive sequences (Additionalfile 1: Table 6). Based on the number of SNPs and the total length of indels within species, T. laevis isolates, with max. 0.52 SNPs/kb and 1.09 bp indels/kb, were the most homogeneous, while T. controversa showed the greatest degree of nucleotide diversity (max. 1.47 SNPs/kb and 2.48 indels bp/kb) (Fig. 4). On the interspecies level, low nucleotide variation was observed between T. caries and T. laevis species while all isolates of these two species exhibited a greater distance to the isolates of T. controversa (Fig. 4). No correlation between the sequencing platform and genomic diversity was observed.
Fig. 4.

The number of SNPs per kb (upper triangle) and the total length of indels per kb (lower triangle) in the repeat masked genomes of 16 isolates of common and dwarf bunt using NUCmer. The isolates and species are given as row and column labels where the sequenced genomes in this study are presented in black. Density of the SNPs and indels are represented by two color scales
Functional genomics
Functional information was assigned to gene products based on protein sequence homology. To ensure comparability, functional annotation was performed for the protein-coding genes of all genomes including new functional annotation of published genomes. At least 55.5% of all coding sequences in each genome were functionally annotated (Fig. 5A). In general, the identified biological pathways and functional categories were remarkably similar across the three species.
Fig. 5.

In silico functional genomic prediction. A: Stacked histograms of functional annotation results of each genome. B: Total predicted secretome and effectors and their distribution in different orthology clusters at the interspecies level. Shared fraction refers to the secreted proteins and effectors which are shared between all 16 isolates. Variables are represented in an absent-present manner in different genomes. Secretome and effectors detected in singletons are isolate specific. Values shown are the total numbers of proteins
To overcome plant defense systems for successful colonization, plant pathogens employ plant cell wall-degrading enzymes (PCWDE) that are part of the carbohydrate-active enzymes, effectors which are a subgroup of secreted proteins, and secondary metabolites (Kimura et al. 2001; Chisholm et al. 2006). We searched the predicted proteomes for these important modulators as described below:
Carbohydrate-active enzymes
Carbohydrate-active enzymes are responsible for the biosynthesis, modification, and breakdown of complex carbohydrates and glycoconjugates (Cantarel et al. 2009). Different CAZyme classes comprising of Glycoside Hydrolases (GHs), Polysaccharide Lyases (PLs), Carbohydrate Esterases (CEs), Glycosyltransferases (GTs), Auxiliary Activities (AAs), and Carbohydrate-Binding Modules (CBMs) were searched in each of the predicted proteomes. Similar numbers of CAZymes were predicted in the genomes of T. caries (up to 212), T. controversa (up to 213), and T. laevis (up to 209). The predicted CAZymes belonged to 72 families and 22 subfamilies (Additionalfile 1: Table 7), of which GH followed by GT were most prominent (74 to 96 and 45 to 48 genes, respectively). All three species possessed a limited number of CAZyme genes for starch/glycogen metabolism. Starch can presumably be metabolized by the activities of GH13, GH37, GH65 (Trehalase), and also by GH15, GT3, and CBM21, which are involved in glycogen hydrolases.
Thirteen to 20 enzymes assigned to six families and seven subfamilies were referred to as PCWDEs (Additionalfile 1: Table 7). Although the majority of main CAZymes involved in cellulose breakdowns such as GH6, GH7, GH12, GH44, AA9, and CMB1 were absent in common and dwarf bunt, an expansion of the GH45 family was observed. The GH45 family is composed mainly of β-1,4-endoglucanases which play a role in the hydrolysis of soluble β-1,4 glucans of plant cell wall polysaccharides such as cellulose. More than half of the predicted PCWDEs were assigned to this family. Harboring genes putatively encoding peroxidases of the family AA2 indicates that the three Tilletia species may have a capacity to degrade lignin.
Secreted proteins, effectors, and genes related to pathogenicity, life cycle, and infection
Secreted proteins modulate interactions of plant pathogenic fungi with their hosts and decisively affect the outcome of the interaction (Girard et al. 2013; Delaunois et al. 2014; Kim et al. 2016). The size of the predicted secretomes in T. caries (433 to 510), T. controversa (419 to 516), and T. laevis (459 to 514) (Fig. 5B) was comparable. Effectors are a subset of the secretome that can suppress plant defense against infection by manipulating the plant’s immune system (Kamoun 2006, 2007; Dodds et al. 2009; Hogenhout et al. 2009). Among the secreted proteins, 107 to 144 effector-like proteins were predicted. Effectors can be localized in a cluster (Brefort et al. 2014). A total of 123 putative effectors were distributed across 75 scaffolds in the genome of the T. controversa isolate OR, which was sequenced using both PacBio and Illumina platforms. Maximum five putative effectors were detected on a scaffold spanning a 23,909 bp region, indicating that the genes were not located in clusters.
Functionally characterized proteins involved in pathogenicity, life cycle, and infection in U. maydis and a few other smut fungi had mostly no orthologs in the proteome of common and dwarf bunt (Table 3). Out of 56 sequences of functionally characterized proteins, only six putative orthologous genes were identified by a tBLASTn search (at least 60% coverage and > 20% identity). These genes included (1) Clp1 required for the proliferation of dikaryotic filaments in planta (Scherer et al. 2006); (2) defense-suppressing virulence effector Pep1 (Doehlemann et al. 2009; Hemetsberger et al. 2012, 2015; Sharma et al. 2018); (3) Pep4 involved in dimorphism and pathogenesis (Soberanes-Gutiérrez et al. 2015); (4) precursor of the peptide conferring surface hydrophobicity to hyphae Rep1 (Wösten et al. 1996; Müller et al. 2008; Teertstra et al. 2009); (5) peroxisomal sterol carrier protein Scp2 (Krombach et al. 2018); and (6) high-affinity sucrose transporter required for virulence Srt1 (Wahl et al. 2010).
Table 3.
Functionally characterized genes involved in pathogenicity, life cycle, and infection
| Gene ID | Gene name | Source species | Putative orthologs | References |
|---|---|---|---|---|
| smut_2965 | Tilletia horrida | NA | Wang et al. (2020) | |
| smut_5844 | T. horrida | NA | Wang et al. (2020) | |
| sr10077 | SAD1 | Sporisorium reilianum | NA | Ghareeb et al. (2015) |
| UHOR_10022 | UhAvr1 | Ustilago hordei | NA | Ali et al. (2014) |
| UMAG_00715 | stp3 | U. maydis | NA | Lanver et al. (2018) |
| UMAG_01235 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_01236 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_01238 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_01240 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_01241 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_01375 | Pit2 | U. maydis | NA | Doehlemann et al. (2011) |
| UMAG_01829 | afu1 | U. maydis | NA | Lanver et al. (2018) |
| UMAG_01987 | Pep1 | U. maydis | YES | Doehlemann et al. (2009) |
| UMAG_02012 | ApB73 | U. maydis | NA | Stirnberg and Djamei (2016) |
| UMAG_02135 | eff1-5 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02136 | eff1-6 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02137 | eff1-7 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02138 | eff1-8 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02140 | eff1-10 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02141 | eff1-11 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_02239 | See1 | U. maydis | NA | Redkar et al. (2015a), Redkar et al. (2015b) |
| UMAG_02374 | Srt1 | U. maydis | YES | Wahl et al. (2010) |
| UMAG_02438 | Clp1 | U. maydis | YES | Scherer et al. (2006) |
| UMAG_02473 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_02474 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_02475 | Stp1 | U. maydis | NA | Schipper (2009) |
| UMAG_03172 | Rbf1 | U. maydis | NA | Heimel et al. (2010) |
| UMAG_03223 | Mig1 | U. maydis | NA | Basse et al. (2000) |
| UMAG_03274 | rsp3 | U. maydis | NA | Ma et al. (2018) |
| UMAG_03313 | eff1-3 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_03314 | eff1-4 | U. maydis | NA | Khrunyk et al. (2010) |
| UMAG_03924 | rep1 | U. maydis | YES | Teertstra et al. (2009) |
| UMAG_04926 | pep4 | U. maydis | YES | Soberanes-Gutiérrez et al. (2015) |
| UMAG_05295 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05297 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05299 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05300 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05301 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05302 | Tin2 | U. maydis | NA | Skibbe et al. (2010), Brefort et al. (2014) |
| UMAG_05304 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05305 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05306 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05307 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05308 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05311 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05312 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05313 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05314 | U. maydis | NA | Skibbe et al. (2010) | |
| UMAG_05318 | Tin4 | U. maydis | NA | Skibbe et al. (2010), Brefort et al. (2014) |
| UMAG_05319 | Tin5 | U. maydis | NA | Brefort et al. (2014) |
| UMAG_05731 | Cmu1 | U. maydis | NA | Djamei et al. (2011) |
| UMAG_06098 | UmFly1 | U. maydis | NA | Ökmen et al. (2018) |
| UMAG_10067 | stp2 | U. maydis | NA | Lanver et al. (2018) |
| UMAG_10556 | Tin3 | U. maydis | NA | Brefort et al. (2014) |
| UMAG_11938 | Scp2 | U. maydis | YES | Krombach et al. (2018) |
| UMAG_12197 | cce1 | U. maydis | NA | Seitner et al. (2018) |
Biosynthesis of secondary metabolites
Secondary metabolites (syn. specialized metabolites) are compounds of low-molecular weight, which are typically species-specific and are believed to possess ecological functions (Pusztahelyi et al. 2015; Macheleidt et al. 2016). Nine putative secondary metabolite biosynthesis clusters comprising 62 genes were identified, which were highly conserved (> 95% identity) across all three species (Fig. 6 and Additionalfile 1: Table 8). Eight out of these nine secondary metabolite gene clusters were recovered almost completely in the different draft genomes by aligning Illumina reads to the respective cluster sequences (Additionalfile 2: Fig. 1). Of those clusters two different clusters encoded for terpene synthases, one cluster was putatively responsible for the synthesis of indole alkaloids, four clusters contained non-ribosomal peptide like synthetases (NRPS-like) genes, and one cluster comprised an NRPS gene. The last secondary metabolite gene cluster was a hybrid cluster encoding for a type 1 polyketide synthases (T1PKS)/NRPS-like and was only partially found in most of the tested isolates. Two highly similar NRPS-like gene clusters were exclusively identified in the T. controversa isolate OR (cluster V in Additionalfile 1: Table 8).
Fig. 6.

The gene organization of the nine putative secondary metabolite gene clusters found in all 16 T. caries, T. controversa, and T. laevis isolates
The translated sequences of each of the core NRPS and NRPS-like genes were searched for the adenylation domains according to NRPSpredictor2 (Röttig et al. 2011) which predicts the amino acid substrate based on the Stachelhaus codes (Stachelhaus et al. 1999). The results are presented in Additionalfile 1: Table 9. The only predicted NRPS gene cluster, which potentially encodes three modules, that are possibly used twice to synthesize a hexapeptide, may participate in siderophores production because the adenylation domains showed 45 to 53% identity to the U. maydis ferrichrome siderophore peptide synthetase.
Genomic insight into trimethylamine synthesis in Tilletia spp.
Fishy smell of grains infected with smut is caused by trimethylamine (TMA), which was isolated already in 1887 from ergot (Claviceps purpurea) and Ustilago sp. (Diehl 1887) and in 1932 from T. laevis-infected wheat (Hanna et al. 1932). It is not known how fungi synthesize TMA while the biosynthesis of TMA in bacteria was unraveled (Craciun and Balskus 2012). The precursor of TMA in bacteria is choline and the reaction is catalyzed by choline trimethylamine-lyase CutC, which is activated by activating protein CutD. The sequences of both proteins are highly conserved (Martínez-del Campo et al. 2015). Based on the report that TMA in ergot also originates from choline (Brieger 1887), we assumed that TMA biosynthesis in bacteria and fungi follow a convergent path or the pathway was transferred from bacteria to fungi, as was reported for other genes (Jaramillo et al. 2015; Navarro-Muñoz and Collemare 2020). We therefore searched for homologs of cutC in Tilletia spp. No such protein was found in the proteome of common and dwarf bunt, indicating that Tilletia spp. do not possess choline trimethylamine-lyase. Searches for proteins similar to activating protein CutD failed, too. Both genes were also missing from the genomes of Ustilago and Claviceps, indicating that the biosynthesis of TMA in smut and ascomyceteous fungi is different from bacteria.
Ethanolamine is a structural analog of choline. In bacteria, the degradation of ethanolamine to ammonia is catalyzed by vitamin B12-dependent ethanolamine ammonia-lyase EutBC (Garsin 2010). This enzyme inspired the search for choline degradation pathway that eventually led to the discovery of CutC/CutD (Craciun and Balskus 2012). We searched Tilletia genomes for genes similar to eutBC, too, but no such gene was found, indicating that the synthesis of TMA in fungi does proceed by the removal of the hydroxyethyl group from choline by an enzyme related to ethanolamine ammonia-lyase.
Inter- and intraspecies variation of protein-coding genes
To compare protein-coding genes within and among the three species, all 159,834 predicted CDS were grouped into orthology clusters based on the sequence homology of their products. From the total of the CDS, 97.6% were grouped into 11,463 orthology clusters; the remaining 2.4% genes were singletons that did not group to any orthology cluster (Fig. 7, Additionalfile 1: Table 10). A total of 5,919 orthology clusters (comprising 75.4% of total CDS) were shared by all 16 isolates. Many of them (4,167) were single-copy genes that did not have any paralog in any isolate. Additional 3,203 orthology clusters were shared among all species by at least one but not all isolates per species, indicating that these genes were neither essential nor species-specific (Additionalfile 1: Table 10). Interestingly, 84% of the total predicted CAZymes (Fig. 8), 72.5% of the total secretome (Fig. 5B), and 47.4% of the genes encoding effectors (Fig. 5B) were among the 5,919 orthology clusters shared and conserved across all species.
Fig. 7.

Distribution of orthologues clusters and CDS among 16 isolates of T. caries, T. controversa, and T. laevis species. Out of 159,834 total CDS, the majority (120,600) which clustered in 5,919 orthology clusters were shared between all the 16 isolates
Fig. 8.

Distribution of putative CAZymes in the shared and variable orthology clusters, and singleton genes between five isolates of T. caries, seven isolates of T. controversa, and four isolates of T. laevis. The majority of the CAZymes are conserved and shared between the three species. Glycoside Hydrolases (GHs), Polysaccharide Lyases (PLs), Carbohydrate Esterases (CEs), Glycosyltransferases (GTs), Auxiliary Activities (AAs), and Carbohydrate-Binding Modules (CBMs)
The number of species-specific orthology clusters defined by CDS that were present in all isolates of the target species only, but not in any isolate of the other species varied between 1 (T. caries), 21 (T. controversa), and 3 (T. laevis) (Additionalfile 1: Table 11). With a more relaxed definition, allowing the gene of an orthology cluster to be missing at most in one isolate of the target species, the numbers increased to 7, 39, and 10 in T. caries, T. controversa, and T. laevis, respectively. We were also interested in the genes that were present in both causal agents of common bunt (T. caries and T. laevis) only. Using the strict definition, we found 19 common bunt-specific orthology clusters. Under relaxed criteria, allowing an orthology cluster to be missing in at most two isolates, we found 40 common bunt-specific orthology clusters. Putative functions were assigned to only 16 out of 96 total common and dwarf bunt-specific orthology clusters (relaxed and strict) (Additionalfile 1: Table 11). In total, species-specific orthology clusters comprised only 0.09% of the total CDS (Fig. 7). Not a single effector nor CAZyme was detected among the species-specific genes. However, two orthology clusters comprised genes that putatively encode secreted proteins. One of them was specific for T. controversa and the other for the common bunt species T. caries and T. laevis. A more specific functional prediction of these interesting genes was not possible.
Based on the orthology clusters, we assigned CDS to core genomes (present in all isolates of a species) and pan-genomes (all CDS present in at least one isolate of a species) of each species individually. The largest core genome belonged to T. laevis (95.4% of the pan-genome), while T. controversa had the largest accessory genome (6.1% of the pan-genome) (Table 4).
Table 4.
Pan-genomes of T. caries, T. controversa, and T. laevis
| Species | Isolates | Pan-genome | Core genome | Accessory genome | Singleton | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CDS | Orthology cluster | CDS | Orthology cluster | % | CDS | Orthology cluster | % | CDS | % | ||
| T. caries | 5 | 50,056 | 9187 | 46,492 | 7020 | 92.8 | 2238 | 2167 | 4.5 | 1284 | 2.5 |
| T. controversa | 7 | 69,702 | 9732 | 63,728 | 6238 | 91.4 | 4297 | 3494 | 6.1 | 1677 | 2.4 |
| T. laevis | 4 | 40,076 | 9153 | 38,264 | 7200 | 95.4 | 1187 | 1953 | 2.9 | 625 | 1.5 |
Discussion
Insights into the genomic features of common and dwarf bunt
In this study, whole-genome sequencing, assembly, as well as structural and functional annotation of 11 isolates of T. caries, T. controversa, and T. laevis were performed. We additionally included the assembled genomes of five isolates of these species, which were previously sequenced by Nguyen et al. (2019) to assess the inter- and intraspecific genetic variation of these important wheat pathogens. Additionally, phylogenetic analyses were performed to obtain an evolutionary framework for interpretation of the results.
The size of the assembled genomes of ten Tilletia isolates based on Illumina reads ranged from 29.4 to 31.9 Mb. These numbers were similar to those obtained by the genome analyses done by Nguyen et al. (2019) who reported genome sizes of 28.1 to 29.9 Mb. A significantly broader range of genome sizes was reported by Russell and Mills (1993) applying an electrophoretic karyotype analysis, where genome sizes were estimated in the range of 28–39 Mb for T. caries and 34–40 Mb for T. controversa, distributed on 14–20 chromosomes in T. caries and 19–20 chromosomes in T. controversa, respectively. The genome of one isolate of T. controversa (OR) was assembled using a combination of PacBio and Illumina reads. This approach resulted in the fewest scaffolds (985 scaffolds) and an increase of N50 to 137 kb. In addition, as more repetitive DNA could be resolved using the long PacBio reads, the assembled genome size increased significantly (49.3 Mb) This higher sequencing depth also resulted in almost 50% more tRNA identification compared to the other isolates.
It is known that the proportion of repetitive regions can differ significantly between closely related species as is exemplified by Fusarium oxysporum and F. graminearum where repetitive elements account for 16.83 Mb and 0.24 Mb respectively (Ma et al. 2010). In our study, the percentage of repetitive regions was variable among the 15 isolates (7.8 to 13.7%) without resulting in a difference between the three species. Moreover, the true proportion of repetitive regions can be expected to be higher as exemplified by the number of 37% found in the isolate OR. This observation suggests that a variable number of repeat elements might have collapsed in the assemblies based on short reads and demonstrates the importance of long reads. The transposon types Gypsy followed by Copia, both belonging to the long terminal direct repeats (LTR) class of retrotransposons, were the most abundant in all three bunt species. Similarly, Gypsys were the most frequent TE reported in T. indica (Gurjar et al. 2019; Mishra et al. 2019) as well as in T. horrida (Wang et al. 2018). Gypsys are the most successful group of TEs in fungi (Gorinsek et al. 2004) and plants (Sabot and Schulman 2006) and are capable to increase their number by autonomous transposition (Elliott and Gregory 2015).
Carbon utilization and establishment of fungal biotrophy
As other biotrophic pathogens, T. caries, T. controversa, and T. laevis encode for a relatively low number of CAZymes (Zhao et al. 2013; Lyu et al. 2015). Moreover, the three species were quite similar in the diversity and abundance of identified CAZyme families. Plant-parasitic fungi secrete a variety of CAZymes, which may play a role in pathogenicity and virulence and are needed to successfully degrade plant cell walls and to complete host invasion (Annis and Goodwin 1997; Gibson et al. 2011; Kubicek et al. 2014). Based on the in silico analyses, all 16 isolates had two putatively secreted CAZymes in common; one belonged to one chitin deacetylases of the family CE4 and one to the family GH152. CE4 is suggested to play a role in the modification of fungal cell walls for masking hyphae to escape from enzymatic hydrolysis by host chitinases through de‐N‐acetylation of chitin (El Gueddari et al. 2002; Boneca et al. 2007). The enzyme β-1,3-glucanase (GH152) is suggested to play a role in cell wall softening during morphogenesis (Mouyna et al. 2013). Common and dwarf bunt, similar to T. indica (Gurjar et al. 2019), have genes encoding for GH8 (broad activity hydrolase), which was suggested to be present in all Ustilaginomycotina (Kijpornyongpan et al. 2018). Interestingly, Tilletia spp. harbored families coding for PL14 and AA2 enzymes that are involved in lignin decomposition, which were completely absent in other studied Ustilaginomycotina, but present in Agaricomycotina (Kijpornyongpan et al. 2018) where the majority of lignin decomposing white-rot fungi belong to. Putative genes encoding for PL14 and AA2 are also reported from T. indica CAZyme analyses (Gurjar et al. 2019). The role of these genes in Tilletia is puzzling.
Almost half of the putative genes encoding for effectors in the multi-species comparison of T. caries, T. controversa, and T. laevis were among the variable genes (dispensable and not present in all genomes), which is in agreement with their ability to undergo rapid diversification including duplication, deletions, and point mutations (Oliver and Solomon 2010; Rouxel et al. 2011). Interestingly, most of the genes that have been functionally characterized in the model fungi were either absent or poorly conserved in the genomes of the common and dwarf bunt agents. Those which had orthologoues in the genome of the three species were suggested to be either essential for establishing biotrophy in smuts such as Pep1 or important virulence factors such as Srt1 (Hemetsberger et al. 2015; Kijpornyongpan et al. 2018; Lanver et al. 2018). The absence of other such genes in Tilletia species correlates with the fact that Tilletiales are unique within Exobasidiomycetes and Ustilaginomycotina in several aspects of the life cycle and the ultrastructure of the host-parasite interaction.
Secondary metabolites pathways and trimethylamine synthesis
Putative secondary metabolite gene clusters were predicted for T. caries, T. controversa, and T. laevis in this study. Secondary metabolites are known as virulence factors (Oide et al. 2006), toxins, inhibitors (Shwab and Keller 2008), and antifeedants or deterrents (Tanaka et al. 2005; Xu et al. 2019). Non-ribosomal peptide synthetase gene clusters are often repetitive in their internal structures and the identification of a higher number of them in the PacBio sequenced genome can be explained by a higher coverage of repetitive regions. The in silico analyses of putative secondary metabolites gene clusters revealed that they were nearly identical across the three species.
Production of TMA gave the stinking smut its name. The biological function of TMA in Tilletia spp. (Ettel and Halbsguth 1963; Singh and Trione 1969) as well as in Geotrichum candidum (Robinson et al. 1989) is the autoinhibition of spore germination. Other metabolites of Tilletia were shown to inhibit spore germination in vitro (Trione and Ross 1988), but they are not volatile and therefore cannot fulfill the function of autoinhibitors. The precursor of TMA in smut fungi and bacteria is choline, but the lack of homologous genes in smut fungi indicates that the biosynthetic pathway is different. The biosynthesis of TMA in smut fungi also appears unrelated to ethylamine degradation by bacteria. We therefore hypothesize that smut fungi possess a different biosynthetic pathway to TMA, which has yet to be discovered.
Inter- and intraspecies genomic comparisons
Genomic synteny, genome-wide diversity, and phylogenetic inferences
The genomes of the three Tilletia species appeared to be largely syntenic as up to 94% of non-repetitive DNA regions could be aligned in a pairwise manner. Furthermore, we detected more than 98.7% average nucleotide identity in one-to-one aligned DNA regions, which was in general agreement with Nguyen et al. (2019) and Sedaghatjoo et al. (2021). The high genomic synteny observed between the three species can be explained by their close phylogenetic relationship (Carris et al. 2007). High genomic identity among closely related species has also been reported for some Fusarium species (De Vos et al. 2014) and within Dothideomycetes (Ohm et al. 2012).
Genome-wide average diversity was least between T. caries and T. laevis with 0.51 SNPs/kb and 1.04 indels bp/kb. This observation is in concordance with the results of the phylogenetic analysis based on 241 genes, where representatives of T. caries and T. laevis did not cluster according to their species affiliation but formed a joint monophyletic group. Independent support for these observations was also found in a study establishing the use of MALDI-TOF MS for differentiation of the three species of interest (Forster et al. 2022). Also in that study, using a broader sample collection, T. caries and T. laevis could not be resolved as distinct entities. Altogether, this may suggest that the two species could in fact only represent two morphotypes of one species, a so-called pseudomorphospecies (Vánky 2008). It is interesting to note that T. laevis is the only species in the genus lacking teliospore ornamentation. However, Vánky (2012) reports that isotypes of T. laevis he studied microscopically contained not only smooth teliospores, but also reticulate spores resembling T. caries within the same sorus and therefore speculated about a hybridogen origin of the species. In fact, experimental approaches on hybrids of T. caries and T. laevis point into a similar direction: the hybrid progenies of the crosses between T. caries and T. laevis displayed teliospores surfaces varying from completely smooth to various degrees of reticulated (Holton 1942). Although the genetic basis for the teliospores reticulation in Tilletia species is unknown, in some species of the smut genus Ustilago it has been suggested that only two dominant and complementary genes encode for the teliospore wall ornamentation (Nielsen 1968; Huang and Nielsen 1984). If this is also true for Tilletia bunts, the different teliospore ornamentation between T. caries and T. laevis would be caused by different alleles resulting from mutation, because there is no evidence that one phenotype has advantages over the other. This would explain, that overall the genomes can be nearly identical, while a few genes would account for the significant morphological features defining the two species in its current circumscription. Whether this is sufficient to maintain the two species as separate entities – given their basically identical (infection) biology and genome similarity will need to be discussed based on broader specimens samplings in the near future.
At the same time, T. caries and T. laevis showed almost equal distance to T. controversa correlating with the fact that both are identical in germination requirements and infection biology, but different in these from T. controversa. This genomic similarity and distances are especially striking, because T. laevis, with smooth teliospores, can be relatively easily identified among these three species. In contrast, both T. caries and T. controversa have ornamented spores differing only slightly and can be problematic to be differentiated morphologically as is required e.g. in seed health testing. However, these two species can be easier distinguished using germination tests requiring different germination time and temperatures, while T. caries and T. laevis do not differ in this respect.
At species level, up to three times higher nucleotide polymorphisms were observed among the seven isolates of T. controversa (max. 1.47 SNPs/kb) compared to the five isolates of T. caries (max. 0.53 SNPs/kb) and four isolates of T. laevis (max. 0.52 SNPs/kb). This is especially remarkable because the common bunts isolates’ geographic origins were more distant to each other (Austria, Italy, Germany, Switzerland, and USA) compared to those of the dwarf bunt isolates, which were mainly collected from Germany. All in all, the number of SNPs were generally low for all three bunt species compared to reports for other species of Basidiomycota suggesting that the causal agents of common bunt and dwarf bunt might have only diverged recently. For instance, different genotypes of Heterobasidion irregulare had 4 SNPs/kb (Sillo et al. 2015) and Melamspora larici-populina 6 SNPs/kb (Persoons et al. 2014), respectively. The especially low genetic diversity observed within the common bunt species compared to the slightly higher genetic diversity of T. controversa, could be the consequence of different mating systems. All causal agents of common and dwarf bunt display bipolar mating behaviors meaning that selfing by direct mating of compatible basidiospores while still attached to the basidium is the dominant reproduction form (Goates 1996) and is thus limiting the chances of outcrossing. However, the mating system is biallelic in T. caries and T. laevis (Holton 1951; Holton and Kendrick 1957), while it is multiallelic in T. controversa (Hoffmann and Kendrick 1969) leading to a somewhat higher chance of occasional outcrossing and consequently a higher degree of diversity in T. controversa as suggested by Pimentel et al. 2000.
Inter- and intraspecies diversification
We compared all the predicted protein-coding genes of the three species by clustering orthologues to define variation across the 16 studied isolates and to find genes, which may be involved in different biological features of the three species. The shared fraction of genes between the three species of T. caries, T. controversa, and T. laevis together was 75.4% of the total predicted proteomes. McCarthy and Fitzpatrick (2019) reported that at species level 83.2% and 85% of the total genes represented the core genomes of Aspergillus fumigatus and Saccharomyces cerevisiae, respectively. Similarly, the size of the core genome of six Aspergillus niger isolates was also 80% of the pan-genome (Vesth et al. 2018) while they mostly have asexual reproduction form. The three bunt species together had only 5% lower than the minimum core genome size reported for single fungal species. The high degree of gene conservation between the three Tilletia species is in line with the phylogenetic analyses suggesting that T. caries, T. controversa, and T. laevis share a recent common ancestor (Carris et al. 2007).
We surprisingly found very few species-specific genes in our study and their functions could mostly not be predicted in silico. Wet lab analysis is required to determine whether these few genes are involved in the different physiological and morphological features of the three species. It is however noteworthy that the species-specific orthology clusters found were higher in T. controversa (21) compared to either T. caries (1) or T. laevis (3). But if T. caries and T. laevis were treated as if they were one specific entity, 19 specific orthology clusters could be found corresponding very well with the numbers of specific orthology clusters in T. controversa suggesting that they might be hierarchically comparable genetic entities.
Conclusions
In conclusion, the genomic analyses presented in the present paper show a broad overall similarity in the genomic structure and features of the studied wheat bunt fungi. However, several lines of evidence obtained by the genomic analyses including phylogenetics suggest that the low amount of genomic differences that could reliably be observed reveals a pattern that correlates better with the definition of the diseases ‘common bunt’ versus ‘dwarf bunt’ instead of the three currently defined species.
Supplementary Information
Additional file 1: Table 1. List of the genome assemblies, raw reads, and RNA-Seq data used in this study. Table 2. List of the additional Basidiomycetes genomes and their accession numbers used for gene model prediction. Table 3. Assessment of the genome annotation completeness when different approaches (homology-based, ab initio and a combination) were used. Table 4. Percentage of repetitive regions in five isolates of T. caries, seven isolates of T. controversa, and four isolates of T. laevis. Table 5. Number and relative abundance of total SSRs identified in the T. caries, T. controversa, and T. laevis genomes. Table 6. Percentage of the maximum aligned bases in non-repetitive regions. Table 7. The number of putative enzymes in each CAZyme family in different isolates and their substrate. Table 8. Prediction of the secondary metabolite biosynthesis gene clusters. Table 9. Analysis of adenylation domains of putative NRPS genes in Tilletia spp. Table 10. List of the identified orthology clusters. Table 11. In silico functional prediction of the species-specific orthology clusters.
Additional file 2: Figure 1. Coverage of putative secondary metabolites and biosynthetic gene clusters. Trimmed Illumina reads of each isolate were aligned to the selected reference gene cluster sequence using default mapping parameters. The plot shows the read depth up to 500 (blue line) and lowest regression (filter: 1/25; orange line). The median coverage is indicated on the right y-axis in red and the x-axes shows length of the predicted secondary metabolite gene cluster in bp. Regions which were annotated as core genes by antiSMASH are shaded in grey. The reference secondary metabolite gene clusters used for mappings are as follow: The predicted gene cluster of T. controversa strain OR for, Indole, NRPS-like I, NRPS-like II, T1PKS/NRPS-like, T. caries isolate AA11 for NRPS, Terpene I, Terpene II, T. laevis isolate ATCC 42080 for NRPS-like IV, and T. controversa isolate DAOMC 236426 for NRPS-like III. The region marked with arrow represent non-covered regions by reads. While the present and the order of nearly all gene clusters were also supported by Illumina reads, the observed drop in reads coverage in T1PKS/NRPS-like synthases was probably due to the present of long stretches of Ts and Ns nucleotides within this gene cluster in our selected reference. Part of the NRPS-like I gene cluster in the T. caries isolate DOAMC 238032 could not be recovered by mapping.
Acknowledgements
The authors acknowledge the critical reviews and helpful comments by Stephan Fuchs. Anne Fiebig’s support for the submission of genomes is acknowledged. Many thanks are extended to A. E. Müllner, Veronica Weyermann, Robert Bauer, Blair Goates, and David Hole who freely shared their samples with us. Anke Brißke-Rode’s and Thomas Berner’s excellent technical assistance are greatly appreciated.
Abbreviations
- AA
Auxiliary Activity
- CAZymes
Carbohydrate-active enzymes
- CBM
Carbohydrate-Binding Module
- CDS
Coding sequences
- CE
Carbohydrate Esterase
- GH
Glycoside Hydrolase
- GT
Glycosyltransferases
- indel
Small insertions and deletion
- LTR
Long terminal direct repeats
- NRPS
Non-ribosomal peptide like synthases
- PCWDE
Plant cell wall-degrading enzyme
- SNP
Single nucleotide polymorphism
- SSR
Simple sequence repeat
- PL
Polysaccharide Lyases
- TE
Transposable element
- TMA
Trimethylamine
- tRNA
Transfer RNA
- T1PKS
Type 1 polyketide synthases
Author contributions
SS conceptualized the study, designed, and performed the experiments, analyzed the data; BM assembled the genomes; MKF acquired samples, assisted in the fungal identification, and sample preparation for PacBio sequencing; YB revised the manuscript; JK assisted in genomes annotations; BK administrated the project and acquired funding and samples; MT assembled the genomes; PK conceptualized the study and contributed to the data analysis; WM administrated the project, acquired funding, contributed to the methodology; SS, WM, and PK wrote the manuscript with the input of other coauthors; All co-authors approved the final version.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by two Grants (Numbers 2812NA128 and 2812NA017) from the German Federal Ministry of Food and Agriculture (BMEL) based on a decision of the German Federal Parliament.
Availability of data and materials
The datasets generated during the current study (raw reads, assemblies, and structural annotations) are available in the European Nucleotide Archive database repository (http://www.ebi.ac.uk/ena) and can be accessed under the project accession number of PRJEB40624.
Declarations
Adherence to national and international regulations
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Petr Karlovsky and Wolfgang Maier: Joint senior authors
References
- Ali S, Laurie JD, Linning R, Cervantes-Chavez JA, Gaudet D, Bakkeren G. An immunity-triggering effector from the Barley smut fungus Ustilago hordei resides in an Ustilaginaceae-specific cluster bearing signs of transposable element-assisted evolution. PLoS Pathog. 2014;10:e1004223. doi: 10.1371/journal.ppat.1004223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37:420–423. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis SL, Goodwin PH. Recent advances in the molecular genetics of plant cell wall-degrading enzymes produced by plant pathogenic fungi. Eur J Plant Pathol. 1997;103:1–14. doi: 10.1023/a:1008656013255. [DOI] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydoğdu M, Kaya Y. Reactions of spring wheat varieties to common bunt (Tilletia laevis) in Turkey. Cereal Research Communications. 2020 doi: 10.1007/s42976-020-00040-1. [DOI] [Google Scholar]
- Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse CW, Stumpferl S, Kahmann R. Characterization of a Ustilago maydis gene specifically induced during the biotrophic phase: evidence for negative as well as positive regulation. Mol Cell Biol. 2000;20:329–339. doi: 10.1128/mcb.20.1.329-339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begerow D, Schäfer AM, Kellner R, Yurkov A, Kemler M, Oberwinkler F, Bauer R. Ustilaginomycotina. In: McLaughlin DJ, Spatafora JW, editors. Systematics and evolution: Part A. Heidelberg: Springer Berlin Heidelberg; 2014. pp. 295–329. [Google Scholar]
- Beier S, Thiel T, Münch T, Scholz U, Mascher M. MISA-web: a web server for microsatellite prediction. Bioinformatics. 2017;33:2583–2585. doi: 10.1093/bioinformatics/btx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benevenuto J, Teixeira-Silva NS, Kuramae EE, Croll D, Monteiro-Vitorello CB. Comparative genomics of smut pathogens: insights from orphans and positively selected genes into host specialization. Front Microbiol. 2018;9:660. doi: 10.3389/fmicb.2018.00660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014;15:211. doi: 10.1186/1471-2105-15-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneca IG, Dussurget O, Cabanes D, Nahori M-A, Sousa S, Lecuit M, Psylinakis E, Bouriotis V, Hugot J-P, Giovannini M, Coyle A, Bertin J, Namane A, Rousselle J-C, Cayet N, Prévost M-C, Balloy V, Chignard M, Philpott DJ, Cossart P, Girardin SE. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci. 2007;104:997–1002. doi: 10.1073/pnas.0609672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandfass C, Karlovsky P. Upscaled CTAB-based DNA extraction and real-time PCR assays for Fusarium culmorum and F. graminearum DNA in plant material with reduced sampling error. Int J Mol Sci. 2008;9:2306–2321. doi: 10.3390/ijms9112306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brefort T, Tanaka S, Neidig N, Doehlemann G, Vincon V, Kahmann R. Characterization of the largest effector gene cluster of Ustilago maydis. PLoS Pathog. 2014;10:e1003866–e1003866. doi: 10.1371/journal.ppat.1003866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieger L. Die Quelle des Trimethylamins im Mutterkorn. Z Physiol Chem. 1887;11:2. [Google Scholar]
- Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. bioRxiv. 2020 doi: 10.1101/2020.08.10.245134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carris LM, et al. smuts. In: Bockus WW, et al., editors. Compendium of wheat diseases and pests. United States: American Phytopathological Society; 2010. pp. 60–65. [Google Scholar]
- Carris LM, Castlebury LA, Goates BJ. Nonsystemic bunt fungi - Tilletia indica and T. horrida: A review of history, systematics, and biology. Annu Rev Phytopathol. 2006;44:113–133. doi: 10.1146/annurev.phyto.44.070505.143402. [DOI] [PubMed] [Google Scholar]
- Carris LM, Castlebury LA, Huang G, Alderman SC, Luo J, Bao X. Tilletia vankyi, a new species of reticulate-spored bunt fungus with non-conjugating basidiospores infecting species of Festuca and Lolium. Mycol Res. 2007;111:1386–1398. doi: 10.1016/j.mycres.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22:1503–1517. doi: 10.1111/mec.12170. [DOI] [PubMed] [Google Scholar]
- Castlebury LA, Carris LM, Vánky K. Phylogenetic analysis of Tilletia and allied genera in order Tilletiales (Ustilaginomycetes; Exobasidiomycetidae) based on large subunit nuclear rDNA sequences. Mycologia. 2005;97:888–900. doi: 10.3852/mycologia.97.4.888. [DOI] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos L, Steenkamp ET, Martin SH, Santana QC, Fourie G, van der Merwe NA, Wingfield MJ, Wingfield BD. Genome-wide macrosynteny among Fusarium species in the Gibberella fujikuroi complex revealed by amplified fragment length polymorphisms. PLoS ONE. 2014;9:e114682. doi: 10.1371/journal.pone.0114682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan JH, Rosenberg NA. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006;2:e68. doi: 10.1371/journal.pgen.0020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunois B, Jeandet P, Clement C, Baillieul F, Dorey S, Cordelier S. Uncovering plant-pathogen crosstalk through apoplastic proteomic studies. Front Plant Sci. 2014;5:249. doi: 10.3389/fpls.2014.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchev CM, Denchev TT. Erratomycetaceae, fam. nov., and validation of some names of smut fungi recently described from India. MYCOBIOTA. 2013;1:9. doi: 10.12664/mycobiota.2013.01.07. [DOI] [Google Scholar]
- Denchev TT, Denchev CM. Tilletia tripogonellae (Tilletiaceae), A New Smut Fungus on Tripogonella spicata (Poaceae) from Argentina. Ann Bot Fenn. 2018;55(273–277):5. [Google Scholar]
- Denchev TT, Van der Zon APM, Denchev CM. Tilletia triraphidis (Tilletiaceae), a new smut fungus on Triraphis purpurea (Poaceae) from Namibia. Phytotaxa. 2018;375:182–186. doi: 10.11646/phytotaxa.375.2.5. [DOI] [Google Scholar]
- Denchev TT, Denchev CM (2018b) Two new smut fungi on Ventenata Poaceae: Tilletia elizabethae from Slovakia and T. ventenatae from Turkey BIOONE
- Diehl L. Report on the progress of pharmacy. Proc Am Pharm Assoc. 1887;35:372. [Google Scholar]
- Djamei A, Schipper K, Rabe F, Ghosh A, Vincon V, Kahnt J, Osorio S, Tohge T, Fernie AR, Feussner I, Feussner K, Meinicke P, Stierhof Y-D, Schwarz H, Macek B, Mann M, Kahmann R. Metabolic priming by a secreted fungal effector. Nature. 2011;478:395–398. doi: 10.1038/nature10454. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds PN, Rafiqi M, Gan PHP, Hardham AR, Jones DA, Ellis JG. Effectors of biotrophic fungi and oomycetes: pathogenicity factors and triggers of host resistance. New Phytol. 2009;183:993–1000. doi: 10.1111/j.1469-8137.2009.02922.x. [DOI] [PubMed] [Google Scholar]
- Doehlemann G, van der Linde K, Aßmann D, Schwammbach D, Hof A, Mohanty A, Jackson D, Kahmann R. Pep1, a secreted effector protein of Ustilago maydis, is required for successful invasion of plant cells. PLoS Pathog. 2009;5:e1000290. doi: 10.1371/journal.ppat.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehlemann G, Reissmann S, Aßmann D, Fleckenstein M, Kahmann R. Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol Microbiol. 2011;81:751–766. doi: 10.1111/j.1365-2958.2011.07728.x. [DOI] [PubMed] [Google Scholar]
- El Gueddari NE, Rauchhaus U, Moerschbacher BM, Deising HB. Developmentally regulated conversion of surface-exposed chitin to chitosan in cell walls of plant pathogenic fungi. New Phytol. 2002;156:103–112. doi: 10.1046/j.1469-8137.2002.00487.x. [DOI] [Google Scholar]
- Elliott TA, Gregory TR. Do larger genomes contain more diverse transposable elements? BMC Evol Biol. 2015;15:69. doi: 10.1186/s12862-015-0339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettel G, Halbsguth W. The effect of trimethylamine, calcium nitrate and light on the germination of bunt spores of Tilletia tritici. Beiträge Zur Biologie Der Pflanzen. 1963;39:37. [Google Scholar]
- Forster MK, Sedaghatjoo S, Maier W, Killermann B, Niessen L. Discrimination of Tilletia controversa from the T. caries/T. laevis complex by MALDI-TOF MS analysis of teliospores. Appl Microbiol Biotechnol. 2022;106:1257–1278. doi: 10.1007/s00253-021-11757-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2014;43:D261–D269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol. 2010;8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghareeb H, Drechsler F, Löfke C, Teichmann T, Schirawski J. SUPPRESSOR OF APICAL DOMINANCE1 of Sporisorium reilianum modulates inflorescence branching architecture in maize and Arabidopsis. Plant Physiol. 2015;169:2789–2804. doi: 10.1104/pp.15.01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DM, King BC, Hayes ML, Bergstrom GC. Plant pathogens as a source of diverse enzymes for lignocellulose digestion. Curr Opin Microbiol. 2011;14:264–270. doi: 10.1016/j.mib.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Girard V, Dieryckx C, Job C, Job D. Secretomes: the fungal strike force. Proteomics. 2013;13:597–608. doi: 10.1002/pmic.201200282. [DOI] [PubMed] [Google Scholar]
- Goates BJ. Common bunt and dwarf bunt. In: Wilcoxson RD, Saari EE, editors. Bunt and smut diseases of wheat; concepts and methods of disease management. Mexico: CIMMYT; 1996. [Google Scholar]
- Gorinsek B, Gubensek F, Kordis D. Evolutionary genomics of chromoviruses in eukaryotes. Mol Biol Evol. 2004;21:781–798. doi: 10.1093/molbev/msh057. [DOI] [PubMed] [Google Scholar]
- Gurjar MS, Aggarwal R, Jogawat A, et al. De novo genome sequencing and secretome analysis of Tilletia indica inciting Karnal bunt of wheat provides pathogenesis-related genes. 3. Biotech. 2019;9:219. doi: 10.1007/s13205-019-1743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B (2010) TransposonPSI: an application of PSI-blast to mine (retro-)transposon ORF homologies. http://transposonpsi.sourceforge.net/. Accessed 20 Sep 2019
- Hackl T, Hedrich R, Schultz J, Förster F. proovread : large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics. 2014;30:3004–3011. doi: 10.1093/bioinformatics/btu392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna WF, Vickery HB, Pucher GW. The isolation of trimethylamine from spores of Tilletia levis, the stinking smut of wheat. J Biol Chem. 1932;97:351–358. doi: 10.1016/S0021-9258(18)76191-7. [DOI] [Google Scholar]
- Heimel K, Scherer M, Vranes M, Wahl R, Pothiratana C, Schuler D, Vincon V, Finkernagel F, Flor-Parra I, Kämper J. The transcription factor Rbf1 is the master regulator for b-mating type controlled pathogenic development in Ustilago maydis. PLoS Pathog. 2010;6:e1001035. doi: 10.1371/journal.ppat.1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemetsberger C, Herrberger C, Zechmann B, Hillmer M, Doehlemann G. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog. 2012;8:e1002684. doi: 10.1371/journal.ppat.1002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemetsberger C, Mueller AN, Matei A, Herrberger C, Hensel G, Kumlehn J, Mishra B, Sharma R, Thines M, Huckelhoven R, Doehlemann G. The fungal core effector Pep1 is conserved across smuts of dicots and monocots. New Phytol. 2015;206:1116–1126. doi: 10.1111/nph.13304. [DOI] [PubMed] [Google Scholar]
- Hoffmann JA. Bunt of wheat. Plant Dis. 1982;66:979. doi: 10.1094/pd-66-979. [DOI] [Google Scholar]
- Hoffmann JA, Kendrick E. Genetic control of compatibility in Tilletia controversa. Phytopathology. 1969;59:4. [Google Scholar]
- Hogenhout SA, Van der Hoorn RA, Terauchi R, Kamoun S. Emerging concepts in effector biology of plant-associated organisms. Mol Plant Microbe Interact. 2009;22:115–122. doi: 10.1094/MPMI-22-2-0115. [DOI] [PubMed] [Google Scholar]
- Holton CS. Inheritance of chlamydospore and sorus characters in species and race hybrids of Tilletia caries and T. foetida. Phytopathology. 1942;34:586–592. [Google Scholar]
- Holton CS. Methods and results of studies on heterothallism and hybridization in Tilletia caries and T. foetida. Phytopathology. 1951;41:511–521. [Google Scholar]
- Holton CS, Kendrick EL. Fusion between secondary sporidia in culture as a valid index of sex compatibility in Tilletia caries. Phytopathology. 1957;47:2. [Google Scholar]
- Huang HQ, Nielsen J. Hybridization of the seedling-infecting Ustilago spp. pathogenic on barley and oats, and a study of the genotypes conditioning the morphology of their spore walls. Can J Bot. 1984;62(3):603–608. doi: 10.1139/b84-091. [DOI] [Google Scholar]
- Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen Lars J, von Mering C, Bork P. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo VDA, Sukno SA, Thon MR. Identification of horizontally transferred genes in the genus Colletotrichum reveals a steady tempo of bacterial to fungal gene transfer. BMC Genomics. 2015;16:2. doi: 10.1186/1471-2164-16-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia WM, Zhou YL, Duan XY, Luo Y, Ding SL, Cao XR, Fitt BDL. Assessment of risk of establishment of wheat dwarf bunt (Tilletia controversa) in China. J Integr Agric. 2013;12:87–94. doi: 10.1016/S2095-3119(13)60208-7. [DOI] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 2007;35:W429–W432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol. 2006;44:41–60. doi: 10.1146/annurev.phyto.44.070505.143436. [DOI] [PubMed] [Google Scholar]
- Kamoun S. Groovy times: filamentous pathogen effectors revealed. Curr Opin Plant Biol. 2007;10:358–365. doi: 10.1016/j.pbi.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Kamper J, Kahmann R, Bolker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Muller O, Perlin MH, Wosten HA, de Vries R, Ruiz-Herrera J, Reynaga-Pena CG, Snetselaar K, McCann M, Perez-Martin J, Feldbrugge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, Gonzalez-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Munch K, Rossel N, Scherer M, Vranes M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho EC, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Hauser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schluter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Guldener U, Munsterkotter M, Haase D, Oesterheld M, Mewes HW, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101. doi: 10.1038/nature05248. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2013;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017;20(4):1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, Hartung F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44:e89. doi: 10.1093/nar/gkw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Hartung F, Paulini M, Twardziok SO, Grau J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018;19:189. doi: 10.1186/s12859-018-2203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrunyk Y, Münch K, Schipper K, Lupas AN, Kahmann R. The use of FLP-mediated recombination for the functional analysis of an effector gene family in the biotrophic smut fungus Ustilago maydis. New Phytol. 2010;187:957–968. doi: 10.1111/j.1469-8137.2010.03413.x. [DOI] [PubMed] [Google Scholar]
- Kijpornyongpan T, Mondo SJ, Barry K, Sandor L, Lee J, Lipzen A, Pangilinan J, LaButti K, Hainaut M, Henrissat B, Grigoriev IV, Spatafora JW, Aime MC. Broad genomic sampling reveals a smut pathogenic ancestry of the fungal clade ustilaginomycotina. Mol Biol Evol. 2018;35:1840–1854. doi: 10.1093/molbev/msy072. [DOI] [PubMed] [Google Scholar]
- Kim K-T, Jeon J, Choi J, Cheong K, Song H, Choi G, Kang S, Lee Y-H. Kingdom-wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front Plant Sci. 2016;7:186–186. doi: 10.3389/fpls.2016.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Anzai H, Yamaguchi I. Microbial toxins in plant-pathogen interactions: biosynthesis, resistance mechanisms, and significance. J Gen Appl Microbiol. 2001;47:149–160. doi: 10.2323/jgam.47.149. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Krombach S, Reissmann S, Kreibich S, Bochen F, Kahmann R. Virulence function of the Ustilago maydis sterol carrier protein 2. New Phytol. 2018;220:553–566. doi: 10.1111/nph.15268. [DOI] [PubMed] [Google Scholar]
- Krueger F (2012–2019) Babraham Bioinformatics - Trim Galore! https://www.bioinformatics.babraham.ac.uk/projects/trim_galore. Accessed 20 Jun 2019
- Kubicek CP, Starr TL, Glass NL. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu Rev Phytopathol. 2014;52(52):427–451. doi: 10.1146/annurev-phyto-102313-045831. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanver D, Müller AN, Happel P, Schweizer G, Haas FB, Franitza M, Pellegrin C, Reissmann S, Altmüller J, Rensing SA, Kahmann R. The biotrophic development of Ustilago maydis studied by RNA-Seq analysis. Plant Cell. 2018;30:300–323. doi: 10.1105/tpc.17.00764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46:D493–d496. doi: 10.1093/nar/gkx922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6:41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12:615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Shivas RG, Cai L. Three new species of Tilletia on Eriachne from north-western Australia. Mycoscience. 2014;55:361–366. doi: 10.1016/j.myc.2013.12.003. [DOI] [Google Scholar]
- Lomsadze A, Burns PD, Borodovsky M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014;42:e119–e119. doi: 10.1093/nar/gku557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu X, Shen C, Fu Y, Xie J, Jiang D, Li G, Cheng J. Comparative genomic and transcriptional analyses of the carbohydrate-active enzymes and secretomes of phytopathogenic fungi reveal their significant roles during infection and development. Sci Rep. 2015;5:15565. doi: 10.1038/srep15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-J, van der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim W-B, Woloshuk C, Xie X, Xu J-R, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RAE, Chapman S, Coulson R, Coutinho PM, Danchin EGJ, Diener A, Gale LR, Gardiner DM, Goff S, Hammond-Kosack KE, Hilburn K, Hua-Van A, Jonkers W, Kazan K, Kodira CD, Koehrsen M, Kumar L, Lee Y-H, Li L, Manners JM, Miranda-Saavedra D, Mukherjee M, Park G, Park J, Park S-Y, Proctor RH, Regev A, Ruiz-Roldan MC, Sain D, Sakthikumar S, Sykes S, Schwartz DC, Turgeon BG, Wapinski I, Yoder O, Young S, Zeng Q, Zhou S, Galagan J, Cuomo CA, Kistler HC, Rep M. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L-S, Wang L, Trippel C, Mendoza-Mendoza A, Ullmann S, Moretti M, Carsten A, Kahnt J, Reissmann S, Zechmann B, Bange G, Kahmann R. The Ustilago maydis repetitive effector Rsp3 blocks the antifungal activity of mannose-binding maize proteins. Nat Commun. 2018;9:1711. doi: 10.1038/s41467-018-04149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheleidt J, Mattern DJ, Fischer J, Netzker T, Weber J, Schroeckh V, Valiante V, Brakhage AA. Regulation and role of fungal secondary metabolites. Annu Rev Genet. 2016;50:371–392. doi: 10.1146/annurev-genet-120215-035203. [DOI] [PubMed] [Google Scholar]
- Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. Mbio. 2015 doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matanguihan JB, Murphy KM, Jones SS. Control of common bunt in organic wheat. Plant Dis. 2011;95:92–103. doi: 10.1094/PDIS-09-10-0620. [DOI] [PubMed] [Google Scholar]
- Mathre DE. DWARF BUNT: politics, identification, and biology. Annu Rev Phytopathol. 1996;34:67–85. doi: 10.1146/annurev.phyto.34.1.67. [DOI] [PubMed] [Google Scholar]
- McCarthy CGP, Fitzpatrick DA. Pan-genome analyses of model fungal species. Microbial Genomics. 2019;5:e000243. doi: 10.1099/mgen.0.000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min XJ. Evaluation of computational methods for secreted protein prediction in different eukaryotes. J Proteom Bioinform. 2010;3:143–147. doi: 10.4172/jpb.1000133. [DOI] [Google Scholar]
- Mirarab S, Warnow T. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 2015;31:i44–i52. doi: 10.1093/bioinformatics/btv234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, Maurya R, Gupta VK, Ramteke PW, Marla SS, Kumar A. Comparative genomic analysis of monosporidial and monoteliosporic cultures for unraveling the complexity of molecular pathogenesis of Tilletia indica pathogen of wheat. Sci Rep. 2019;9:8185. doi: 10.1038/s41598-019-44464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouyna I, Hartl L, Latge JP. beta-1,3-glucan modifying enzymes in Aspergillus fumigatus. Front Microbiol. 2013;4:81. doi: 10.3389/fmicb.2013.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller O, Schreier PH, Uhrig JF. Identification and characterization of secreted and pathogenesis-related proteins in Ustilago maydis. Mol Genet Genomics. 2008;279:27–39. doi: 10.1007/s00438-007-0291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszewska A, Steczkiewicz K, Stepniewska-Dziubinska M, Ginalski K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci Rep. 2019;9:4307. doi: 10.1038/s41598-019-40965-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Muñoz JC, Collemare J. Evolutionary histories of type III polyketide synthases in fungi. Front Microbiol. 2020 doi: 10.3389/fmicb.2019.03018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HDT, Sultana T, Kesanakurti P, Hambleton S. Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus. 2019 doi: 10.1186/s43008-019-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. Trimethylammonium compounds in Tilletia Spp. Can J Bot. 1963;41:335–339. doi: 10.1139/b63-032. [DOI] [Google Scholar]
- Nielsen J. The inheritance of spore wall characteristics in Ustilago avenae and U. kolleri. Can J Bot. 1968;46:497–500. doi: 10.1139/b68-074. [DOI] [Google Scholar]
- Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, LaButti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GHJ, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJGM, Zhong S, Goodwin SB, Grigoriev IV. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLoS Pathog. 2012;8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oide S, Moeder W, Krasnoff S, Gibson D, Haas H, Yoshioka K, Turgeon BG. NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell. 2006;18:2836–2853. doi: 10.1105/tpc.106.045633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ökmen B, Kemmerich B, Hilbig D, Wemhöner R, Aschenbroich J, Perrar A, Huesgen PF, Schipper K, Doehlemann G. Dual function of a secreted fungalysin metalloprotease in Ustilago maydis. New Phytol. 2018;220:249–261. doi: 10.1111/nph.15265. [DOI] [PubMed] [Google Scholar]
- Oliver RP, Solomon PS. New developments in pathogenicity and virulence of necrotrophs. Curr Opin Plant Biol. 2010;13:415–419. doi: 10.1016/j.pbi.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Persoons A, Morin E, Delaruelle C, Payen T, Halkett F, Frey P, De Mita S, Duplessis S. Patterns of genomic variation in the poplar rust fungus Melampsora larici-populina identify pathogenesis-related factors. Front Plant Sci. 2014 doi: 10.3389/fpls.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson GL, Whitaker TB, Stefanski RJ, Podleckis EV, Phillips JG, Wu JS, Martinez WH. A risk assessment model for importation of United States milling wheat containing Tilletia contraversa. Plant Dis. 2009;93:560–573. doi: 10.1094/PDIS-93-6-0560. [DOI] [PubMed] [Google Scholar]
- Pimentel G, Peever TL, Carris LM. Genetic variation among natural populations of Tilletia controversa and T. bromi. Phytopathology. 2000;90:376–383. doi: 10.1094/PHYTO.2000.90.4.376. [DOI] [PubMed] [Google Scholar]
- Pizarro D, Divakar PK, Grewe F, Leavitt SD, Huang JP, Dal Grande F, Schmitt I, Wedin M, Crespo A, Lumbsch HT. Phylogenomic analysis of 2556 single-copy protein-coding genes resolves most evolutionary relationships for the major clades in the most diverse group of lichen-forming fungi. Fungal Diversity. 2018;92:31–41. doi: 10.1007/s13225-018-0407-7. [DOI] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy LH, Kendrick EL, Hoffmann JA, Holton CS. Dwarf bunt of wheat. Annu Rev Microbiol. 1963;17:199–222. doi: 10.1146/annurev.mi.17.100163.001215. [DOI] [Google Scholar]
- Pusztahelyi T, Holb I, Pócsi I. Secondary metabolites in fungus-plant interactions. Front Plant Sci. 2015 doi: 10.3389/fpls.2015.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2013) R: A Language and Environment for Statistical Computing, R foundation for Statistical Computing, Vienna, Austria
- Rannala B, Yang Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razali NM, Cheah BH, Nadarajah K. Transposable elements adaptive role in genome plasticity, pathogenicity and evolution in fungal phytopathogens. Int J Mol Sci. 2019 doi: 10.3390/ijms20143597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redkar A, Hoser R, Schilling L, Zechmann B, Krzymowska M, Walbot V, Doehlemann G. A secreted effector protein of Ustilago maydis guides maize leaf cells to form tumors. Plant Cell. 2015;27:1332–1351. doi: 10.1105/tpc.114.131086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redkar A, Villajuana-Bonequi M, Doehlemann G. Conservation of the Ustilago maydis effector See1 in related smuts. Plant Signal Behav. 2015;10:e1086855–e1086855. doi: 10.1080/15592324.2015.1086855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PM, McKee ND, Thompson LAA, Harper DB, Hamilton JTG. Autoinhibition of germination and growth in Geotrichum candidum. Mycol Res. 1989;93:214–222. doi: 10.1016/s0953-7562(89)80120-0. [DOI] [Google Scholar]
- Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–W367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, Cozijnsen AJ, Ciuffetti LM, Degrave A, Dilmaghani A, Duret L, Fudal I, Goodwin SB, Gout L, Glaser N, Linglin J, Kema GHJ, Lapalu N, Lawrence CB, May K, Meyer M, Ollivier B, Poulain J, Schoch CL, Simon A, Spatafora JW, Stachowiak A, Turgeon BG, Tyler BM, Vincent D, Weissenbach J, Amselem J, Quesneville H, Oliver RP, Wincker P, Balesdent M-H, Howlett BJ. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun. 2011;2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudloff JE, Bauer R, Büttner P, Sedaghtjoo S, Kirsch N, Maier W. Monitoring zum Vorkommen von Tilletia controversa (Zwergsteinbrand) an konventionell erzeugtem Winterweizen in den Bundesländern Brandenburg, Mecklenburg-Vorpommern, Niedersachsen, Nordrhein-Westfalen, Sachsen, Sachsen-Anhalt Und Schleswig-Holstein. Kulturpflanzen. 2020;72:7. doi: 10.5073/JfK.2020.08.16. [DOI] [Google Scholar]
- Russell BW, Mills D. Electrophoretic karyotypes of Tilletia caries, T. controversa, and their F1 progeny: further evidence for conspecific status. Mol Plant Microbe Interact. 1993;6:66–74. doi: 10.1094/mpmi-6-066. [DOI] [PubMed] [Google Scholar]
- Ruzgas V, Liatukas Ž. Resistance genes and sources for the control of wheat common bunt (Tilletia tritici (DC.) Tul.) Biologija. 2008;54:274–278. doi: 10.2478/v10054-008-0056-y. [DOI] [Google Scholar]
- Sabot F, Schulman AH. Parasitism and the retrotransposon life cycle in plants: a hitchhiker's guide to the genome. Heredity (edinb) 2006;97:381–388. doi: 10.1038/sj.hdy.6800903. [DOI] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol Biol Evol. 2016;33:1654–1668. doi: 10.1093/molbev/msw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Heimel K, Starke V, Kämper J. The Clp1 protein is required for clamp formation and pathogenic development of Ustilago maydis. Plant Cell. 2006;18:2388–2401. doi: 10.1105/tpc.106.043521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper K (2009) Charakterisierung eines Ustilago maydis Genclusters, das für drei neuartige sekretierte Effektoren kodiert Biologie Philipps-Universität Marburg Marburg
- Sedaghatjoo S, Forster MK, Niessen L, Karlovsky P, Killermann B, Maier W. Development of a loop-mediated isothermal amplification assay for the detection of Tilletia controversa based on genome comparison. Sci Rep. 2021;11:11611. doi: 10.1038/s41598-021-91098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitner D, Uhse S, Gallei M, Djamei A. The core effector Cce1 is required for early infection of maize by Ustilago maydis. Mol Plant Pathol. 2018;19:2277–2287. doi: 10.1111/mpp.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Ökmen B, Doehlemann G, Thines M. Pseudozyma saprotrophic yeasts have retained a large effector arsenal, including functional Pep1 orthologs. bioRxiv. 2018 doi: 10.1101/489690. [DOI] [Google Scholar]
- Shwab EK, Keller NP. Regulation of secondary metabolite production in filamentous ascomycetes. Mycol Res. 2008;112:225–230. doi: 10.1016/j.mycres.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Sillo F, Garbelotto M, Friedman M, Gonthier P. Comparative genomics of sibling fungal pathogenic taxa identifies adaptive evolution without divergence in pathogenicity genes or genomic structure. Genome Biol Evol. 2015;7:3190–3206. doi: 10.1093/gbe/evv209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Singh J, Trione E. Self inhibition of the germination of bunt teliospores of Tilletia caries and of Tilletia controversa. Phytopathology. 1969;59:15. [Google Scholar]
- Skibbe DS, Doehlemann G, Fernandes J, Walbot V. Maize tumors caused by Ustilago maydis; require organ-specific genes in host and pathogen. Science. 2010;328:89. doi: 10.1126/science.1185775. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P (2008–2019) RepeatModeler. http://www.repeatmasker.org/RepeatModeler/. Accessed 05 Sep 2019
- Smit A, Hubley R, Green P (2013–2015) RepeatMasker Open-4.0.9. http://www.repeatmasker.org/. Accessed 05 Sep 2019b
- Soberanes-Gutiérrez CV, Juárez-Montiel M, Olguín-Rodríguez O, Hernández-Rodríguez C, Ruiz-Herrera J, Villa-Tanaca L. The pep4 gene encoding proteinase A is involved in dimorphism and pathogenesis of Ustilago maydis. Mol Plant Pathol. 2015;16:837–846. doi: 10.1111/mpp.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider J, Dodds PN, Gardiner DM, Singh KB, Taylor JM. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol Plant Pathol. 2018;19:2094–2110. doi: 10.1111/mpp.12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/s1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnberg A, Djamei A. Characterization of ApB73, a virulence factor important for colonization of Zea mays by the smut Ustilago maydis. Mol Plant Pathol. 2016;17:1467–1479. doi: 10.1111/mpp.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Tapper BA, Popay A, Parker EJ, Scott B. A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol Microbiol. 2005;57:1036–1050. doi: 10.1111/j.1365-2958.2005.04747.x. [DOI] [PubMed] [Google Scholar]
- Teertstra WR, van der Velden GJ, de Jong JF, Kruijtzer JAW, Liskamp RMJ, Kroon-Batenburg LMJ, Müller WH, Gebbink MFBG, Wösten HAB. The filament-specific Rep1-1 repellent of the phytopathogen Ustilago maydis forms functional surface-active amyloid-like fibrils. J Biol Chem. 2009;284:9153–9159. doi: 10.1074/jbc.M900095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trione EJ. Isolation and in Vitro culture of the wheat bunt fungi Tilletia caries and Tilletia controversa. Phytopathology. 1964;54:592–596. [Google Scholar]
- Trione EJ, Ross WD. Lipids as bioregulators of teliospore germination and sporidial formation in the wheat bunt fungi, Tilletia Species. Mycologia. 1988;80:38–45. doi: 10.1080/00275514.1988.12025495. [DOI] [Google Scholar]
- Vánky K. Smut fungi (Basidiomycota p.p., Ascomycota p.p.) of the world. novelties, selected examples, trends. Acta Microbiol Immunol Hung. 2008;55:91–109. doi: 10.1556/AMicr.55.2008.2.2. [DOI] [PubMed] [Google Scholar]
- Vánky K. Smut fungi of the world. Minnesota: American Phytopathological Society; 2012. [Google Scholar]
- Vesth TC, Nybo JL, Theobald S, Frisvad JC, Larsen TO, Nielsen KF, Hoof JB, Brandl J, Salamov A, Riley R, Gladden JM, Phatale P, Nielsen MT, Lyhne EK, Kogle ME, Strasser K, McDonnell E, Barry K, Clum A, Chen C, LaButti K, Haridas S, Nolan M, Sandor L, Kuo A, Lipzen A, Hainaut M, Drula E, Tsang A, Magnuson JK, Henrissat B, Wiebenga A, Simmons BA, Mäkelä MR, de Vries RP, Grigoriev IV, Mortensen UH, Baker SE, Andersen MR. Investigation of inter- and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat Genet. 2018;50:1688–1695. doi: 10.1038/s41588-018-0246-1. [DOI] [PubMed] [Google Scholar]
- Wahl R, Wippel K, Goos S, Kämper J, Sauer N. A novel high-affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis. PLoS Biol. 2010;8:e1000303. doi: 10.1371/journal.pbio.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Pang L, Wang N, et al. The pathogenic mechanisms of Tilletia horrida as revealed by comparative and functional genomics. Sci Rep. 2018;8:15413. doi: 10.1038/s41598-018-33752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Shu X, Niu X, Yi X, Zheng A. Transcriptome analysis and whole genome re-sequencing provide insights on rice kernel smut (Tilletia horrida) pathogenicity. J Plant Pathol. 2020;102(1):155–67. doi: 10.1007/s42161-019-00401-8. [DOI] [Google Scholar]
- Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–D365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber RE, Pietsch M, Frühauf A, Pfeifer Y, Martin M, Luft D, Gatermann S, Pfennigwerth N, Kaase M, Werner G, Fuchs S. IS26-mediated transfer of blaNDM–1 as the main route of resistance transmission during a polyclonal, multispecies outbreak in a German Hospital. Front Microbiol. 2019 doi: 10.3389/fmicb.2019.02817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker TB, Wu J, Peterson GL, Giesbrecht FG, Johansson AS. Variability associated with the official USDA sampling plan used to inspect export wheat shipments for Tilletia controversa spores. Plant Pathol. 2001;50:755–760. doi: 10.1046/j.1365-3059.2001.00640.x. [DOI] [Google Scholar]
- Woolma NHM, Humphrey HB (1924) Summary of literature on bunt or stinking smut of Wheat. United States Department of Agriculture
- Wöstemeyer J, Kreibich A. Repetitive DNA elements in fungi (Mycota): impact on genomic architecture and evolution. Curr Genet. 2002;41:189–198. doi: 10.1007/s00294-002-0306-y. [DOI] [PubMed] [Google Scholar]
- Wösten HA, Bohlmann R, Eckerskorn C, Lottspeich F, Bölker M, Kahmann R. A novel class of small amphipathic peptides affect aerial hyphal growth and surface hydrophobicity in Ustilago maydis. EMBO J. 1996;15:4274–4281. doi: 10.1002/j.1460-2075.1996.tb00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Vinas M, Alsarrag A, Su L, Pfohl K, Rohlfs M, Schäfer W, Chen W, Karlovsky P. Bis-naphthopyrone pigments protect filamentous ascomycetes from a wide range of predators. Nat Commun. 2019;10:3579. doi: 10.1038/s41467-019-11377-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simão FA, Ioannidis P, Seppey M, Loetscher A, Kriventseva EV. OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2016;45:D744–D749. doi: 10.1093/nar/gkw1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu H, Wang C, Xu J-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table 1. List of the genome assemblies, raw reads, and RNA-Seq data used in this study. Table 2. List of the additional Basidiomycetes genomes and their accession numbers used for gene model prediction. Table 3. Assessment of the genome annotation completeness when different approaches (homology-based, ab initio and a combination) were used. Table 4. Percentage of repetitive regions in five isolates of T. caries, seven isolates of T. controversa, and four isolates of T. laevis. Table 5. Number and relative abundance of total SSRs identified in the T. caries, T. controversa, and T. laevis genomes. Table 6. Percentage of the maximum aligned bases in non-repetitive regions. Table 7. The number of putative enzymes in each CAZyme family in different isolates and their substrate. Table 8. Prediction of the secondary metabolite biosynthesis gene clusters. Table 9. Analysis of adenylation domains of putative NRPS genes in Tilletia spp. Table 10. List of the identified orthology clusters. Table 11. In silico functional prediction of the species-specific orthology clusters.
Additional file 2: Figure 1. Coverage of putative secondary metabolites and biosynthetic gene clusters. Trimmed Illumina reads of each isolate were aligned to the selected reference gene cluster sequence using default mapping parameters. The plot shows the read depth up to 500 (blue line) and lowest regression (filter: 1/25; orange line). The median coverage is indicated on the right y-axis in red and the x-axes shows length of the predicted secondary metabolite gene cluster in bp. Regions which were annotated as core genes by antiSMASH are shaded in grey. The reference secondary metabolite gene clusters used for mappings are as follow: The predicted gene cluster of T. controversa strain OR for, Indole, NRPS-like I, NRPS-like II, T1PKS/NRPS-like, T. caries isolate AA11 for NRPS, Terpene I, Terpene II, T. laevis isolate ATCC 42080 for NRPS-like IV, and T. controversa isolate DAOMC 236426 for NRPS-like III. The region marked with arrow represent non-covered regions by reads. While the present and the order of nearly all gene clusters were also supported by Illumina reads, the observed drop in reads coverage in T1PKS/NRPS-like synthases was probably due to the present of long stretches of Ts and Ns nucleotides within this gene cluster in our selected reference. Part of the NRPS-like I gene cluster in the T. caries isolate DOAMC 238032 could not be recovered by mapping.
Data Availability Statement
The datasets generated during the current study (raw reads, assemblies, and structural annotations) are available in the European Nucleotide Archive database repository (http://www.ebi.ac.uk/ena) and can be accessed under the project accession number of PRJEB40624.