

Abstract
Two years after its emergence, SARS-CoV-2 still represents a serious and global threat to human health. Antiviral drug development usually takes a long time and, to increase the chances of success, chemical variability of hit compounds represents a valuable source for the discovery of new antivirals. In this work, we applied a platform of variably oriented virtual screening campaigns to seek for novel chemical scaffolds for SARS-CoV-2 main protease (Mpro) inhibitors. The study on the resulting 30 best hits led to the identification of a series of structurally unrelated Mpro inhibitors. Some of them exhibited antiviral activity in the low micromolar range against SARS-CoV-2 and other human coronaviruses (HCoVs) in different cell lines. Time-of-addition experiments demonstrated an antiviral effect during the viral replication cycle at a time frame consistent with the inhibition of SARS-CoV-2 Mpro activity. As a proof-of-concept, to validate the pharmaceutical potential of the selected hits against SARS-CoV-2, we rationally optimized one of the hit compounds and obtained two potent SARS-CoV-2 inhibitors with increased activity against Mpro both in vitro and in a cellular context, as well as against SARS-CoV-2 replication in infected cells. This study significantly contributes to the expansion of the chemical variability of SARS-CoV-2 Mpro inhibitors and provides new scaffolds to be exploited for pan-coronavirus antiviral drug development.
Keywords: SARS-CoV-2, Main protease Mpro, Virtual screening, Antivirals
Graphical abstract

Abbreviations
- CoV
coronavirus
- EC50
effective concentration at half-maximal response
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine;
- FRET
fluorescence resonance energy transfer
- HRMS
High-Resolution Mass Spectroscopy
- Hz
hertz
- LBVS
ligand-based virtual screening
- MIFs
Molecular Interaction Fields
- Mpro
main protease
- PPVS
pocket-pocket virtual screening
- PRA
Plaque reduction assays
- RdRP
RNA-dependent RNA polymerase
- SARS-CoV-2
severe acute respiratory syndrome coronavirus-2
- SBVS
structure-based virtual screening
- SI
selectivity index
- TLC
thin-layer chromatography
1. Introduction
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has caused to date ∼ 521 million infections worldwide and almost 6.3 million related deaths (https://covid19.who.int/, accessed on May 20th, 2022). SARS-CoV-2 belongs to the Coronavirinae subfamily (Coronaviridae family), which comprises four genera, i.e., α-, β-, γ-, and δ-coronavirus. Humans can be infected only by coronavirus (HCoV) belonging to the α (HCoV-229E and HCoV-NL63) and β genera (SARS-CoV and SARS-CoV-2, MERS-CoV, HCoV-OC43, and HCoV-HKU1) (Su et al., 2016).
Anti-SARS-CoV-2 vaccines alone have not completely succeeded in containing the severity of the ongoing pandemic and drug treatments for COVID-19 are still in their infancy. Moreover, vaccine hesitance, non-durable immunity, and emergence of novel variants might further affect their efficacy. Thus, drugs against SARS-CoV-2 could be determinant while waiting for vaccine reformulation and might also be useful in the case of future new emerging coronaviruses.
In drug discovery projects one may aim at hitting either a single or several targets involved in a certain pathway (e.g., viral replication pathways) (Knowles and Gromo, 2003). As a result of extensive research efforts, a few anti-SARS-CoV-2 drugs are starting to be licensed and several others are in advanced clinical trials (Hu et al., 2022). Besides the Merck's viral RNA polymerase inhibitor molnupiravir, which received an emergency use authorization by FDA (www.fda.gov), inhibitors of SARS-CoV-2 main protease (Mpro) are the most promising drug candidates under clinical evaluation (NCT04483973, NCT04484025, NCT04594343, NCT04485130) (Cui et al., 2020). Among them, Pfizer's Paxlovid (PF-07321332, Fig. S1, associated with ritonavir) currently represents the sole Mpro inhibitor approved by FDA for COVID-19 with an emergency use authorization (www.fda.gov). Mpro represents an ideal target since it is an essential enzyme for viral replication very conserved across CoVs and with a high substrate specificity (Shitrit et al., 2020). Moreover, SARS-CoV-2 Mpro recognizes also non-canonical cleavage sites in host proteins linked to SARS-CoV-2 pathogenesis (Pablos et al., 2021; Wenzel et al., 2021). In addition to those already under clinical investigation, other promising SARS-CoV-2 Mpro inhibitors have been reported (Gao et al., 2021).
The availability of novel chemical scaffolds endowed with pharmaceutical potential represents an advantage in antiviral drug discovery. To explore the chemical space and test large numbers of different chemotypes, in silico screenings are usually performed. Both ligand-based and structure-based approaches can be applied, in which an active compound or a given protein cavity is used as a template, respectively (Goracci et al., 2016; Lian et al., 2021; Maia et al., 2020; Muratore et al., 2012a), or alternatively a pharmacophore-based approach can be also applied (Chapy et al., 2015; Pautasso et al., 2014). In addition, combinations of different virtual screening approaches (e.g., ligand-based followed by structure-based screenings) can maximize the use of the available information on ligands and target (Vazquez et al., 2020). More recently, additional computational, compound-repurposing approaches based on pocket similarity search, such as BioGPS (Siragusa et al., 2015), PoSSuM (Ito et al., 2012), or FragVLib (Khashan, 2012) also proved useful to indirectly identify new ligands by comparing databases of druggable protein cavities co-crystallized with a ligand.
In this work, we explored three different virtual screening strategies (Fig. 1 ) with the aim of extending the chemical diversity of Mpro inhibitors, which may serve as starting points for further development of new anti-SARS-CoV-2 drugs. The 30 most promising hits emerging from the virtual screenings were biologically evaluated both in vitro and in infected cells. As a proof-of-concept, one active hit was subjected to optimization by rational design. This approach eventually led to the synthesis of a series of promising SARS-CoV-2 Mpro inhibitors that exhibit antiviral activity in the low micromolar range and represent promising starting platforms for further antiviral drug development.
Fig. 1.

VS campaigns against SARS-CoV-2 Mproperformed in this study. (A) Alignment of the two Mpro covalent inhibitors N3 and 13b. (B) Details of the VS workflows: 1) LBVS on commercial databases followed by SBVS; 2) pocket-pocket comparison VS for ligand repurposing based on the BioGPS approach, followed by SBVS; 3) SBVS on a dataset of compounds synthesized in house as potential anti-influenza compounds targeting PA-PB1 interaction. (C) Superposition of 6lu7 (shape in orange, MIFs as surface) and 6y2g (shape in cyan, MIFs as wireframe) binding sites.
2. Materials and methods
2.1. In silico screenings
2.1.1. FLAP ligand-based virtual screening
Ligand based virtual screening (LBVS) was performed using the FLAP software (implemented in FLAP, developed and licensed by Molecular Discovery Ltd., UK) (Baroni et al., 2007). Three different approaches were employed, differing in the used database and templates. In approaches (1-1) and (1–2) (Fig. 1B), a database of small molecules composed of structures from Specs and ChemDiv vendors (overall 793,212 compounds) was generated by applying the following filters: molecular weight range = 150–500; logP >5; protonation state = most abundant protomer at pH = 7.4; up to 50 conformers with RMSD >0.1. As templates, the two co-crystallized Mpro ligands from PDB ID codes 6lu7 (N3) and 6y2g (13b) were used in (1-1) and (1–2) approaches, respectively. The GRID (Carosati et al., 2004) probes defining shape (H), hydrophobic interactions (DRY), H-bond donor (N1), and H-bond (O) acceptor interactions were used to generate the molecular interaction fields and evaluate the similarity between the screened compounds and the templates. Compound 13b (from PDB ID code 13b) was also used as a template to screen a target library of ligands for protease by Vitas-M containing 2500 small molecules (1–3 approach). Screening was conducted at the -fast accuracy level. At the end of each screening, compounds displaying a similarity score (Glob-Prod) greater than 0.3 were selected and screened through a structure-based approach (Dataset S1).
2.1.2. FLAP structure-based virtual screening
Selected compounds from LBVS were further screened in a structure-based approach, using the catalytic site of Mpro (PDB ID: 6lu7) as a template. The FlapSite algorithm (implemented in FLAP) (Baroni et al., 2007) was used to define the pocket, which was described in terms of GRID MIFs (Carosati et al., 2004). Compounds to be screened were modelled in their most abundant protomer at pH = 7.4 and considering up to 50 conformers with RMSD >0.1 for each compound. The same GRID probes used in LBVS were used to describe templates and compounds to be screened. Virtual screening was performed using the -normal accuracy level and by using 100 minima point for quadruplets generation. Top ranked compounds (Glob-Prod > 0.6) were visually inspected to finally select 20 hits to be tested (Dataset S1).
2.1.3. Pocket-pocket virtual screening
The FLAPsite algorithm was used for the identification of cavities in two Mpro 3D protein structures (PDB IDs: 6lu7, 6y2g). Since pockets are very similar in the two structures (data not shown), we decided to use the one having the best X-ray resolution (PDB ID: 6lu7). Furthermore, the two binding sites (pockets allocating the two different inhibitors N3 and O6K) in 6lu7 and 6y2g are almost identical (Fig. 1C). Five pockets were detected on the biological unit that is indicated as a dimer (data not shown). The pocket corresponding to the catalytic site of 6lu7 was selected for pocket-pocket virtual screening. We first collected from the PDB the structures of human proteins co-crystallized with a ligand (11,655 structures; October 2018). The protein residues, solvent molecules, co-crystallized ligands, cofactors, water molecules and ions contained in the PDB protein structures were processed by using Fixpdb tool (Siragusa et al., 2015), in order to remove ligands and retain cofactors, structural water molecules and ions. Binding sites were then detected by using the FLAPsite algorithm (Baroni et al., 2007) (19,873 binding sites). The BioGPS technology (Siragusa et al., 2015), a virtual screening algorithm developed and licensed by Molecular Discovery Ltd. (http://www.moldiscovery.com), was used to calculated MIFs for each binding site and compare the Mpro pocket template against the 19,873 MIFs cavities dataset. The BioGPS algorithm compares binding sites by means of their MIF similarity. Initially, the approach uses the GRID force field to evaluate the type, strength, and direction of the interactions that a cavity is capable of making, taking into account shape and hydrophobic, H-bond donor, and H-bond acceptor interactions. Indeed, the GRID probes H, CRY, O, and N1 are used to compute the shape, the hydrophobic interactions, the H-bond donor interactions, and the H-bond acceptor interactions, respectively, for each cavity considered in the analysis. The algorithm overlaps the 3D cavity structures with a specific orientation, according to the matching MIFs, and then calculates the MIFs similarity of the overlapping areas. BioGPS calculates a similarity score representing the degree of volume overlap for all the probes (Global Product score). The Global Product score was set to 0.7 as restrictive threshold for selecting the most similar cavities to the Mpro pocket template. We thus obtained 116 pockets, from which 116 co-crystallized ligands were extracted and used for the following steps.
2.1.4. Rational optimization of compound 7
From a quick retrosynthetic analysis, compound 7 can be obtained by the combination of three main moieties: a (2-aminobenzoyl)valine core, an aniline moiety, and a benzoic acid moiety (coloured in black, blue, and red, respectively, for compound 7 in Fig. S2). Thus, as a first optimization we decided to explore both the end moieties of the molecule (aniline and benzoic acid moieties), while maintaining the (2-aminobenzoyl)valine core. Indeed, with the aim to rationally select the derivatives to be synthesized, a combinatorial virtual library was initially generated entailing all the possible compounds derived from the simultaneous substitution of both ends of the (2-aminobenzoyl)valine central core with different benzoic acids and aniline moieties. In particular, the latter was combined with all the commercial in house available anilines and amines differently substituted (20 units) and all the commercial in house available benzoic acids differently substituted, as well as aliphatic carboxylic acids (28 units) (Fig. S2), thus obtaining a final database of 560 structures (Dataset S2 in Supplementary material). For the generation of the combinatory library, initially the.mol2 files of all the different anilines/amines N-substituted by a dummy atom (Du), as well as the.mol2 files of all the different carboxylic acids as C(O)Du were generated. Then, an in-house developed algorithm was used to generate.mol2 files of all the molecules resulting from the combination of the different fragments, thus obtaining a final database of 560 structures. These structures were successively screened in a structure-based approach, using the catalytic site of Mpro (PDB ID 6lu7) as a template. The FlapSite algorithm (implemented in FLAP) (Baroni et al., 2007) was used to define the pocket, which was described in terms of GRID MIFs (Carosati et al., 2004). Compounds to be screened were modelled in their most abundant protomer at pH = 7.4 and considering up to 50 conformers with RMSD >0.1 for each compound. The same GRID probes used in LBVS were used to describe templates and compounds to be screened. Virtual screening was performed using the -normal accuracy level and by using 100 minima point for quadruplets generation. Considering 7 Glob-Prod value (0.62) as reference, top ranked analogs (Glob-Prod > 0.62) were visually inspected to finally select the derivatives to be synthesized and tested.
2.2. Chemicals
Compounds 1–5 were purchased from ChemDiv, compounds 6–8 from Specs, compounds 9–20 from Vitas-M, compound 21 from Fluorochem, and compounds 22–24 from Sigma Aldrich (more details in Dataset S1). Boceprevir (BOC) and remdesivir (RMV) were purchased from Selleckchem. Synthesis and chemical characterization of compounds 25–30 were already reported (Lepri et al., 2014), while the experimental procedures for the synthesis of compounds 31–37 together with the experimental protocols concerning all the intermediates are reported in the Supplementary material.
2.3. Expression and purification of SARS-CoV-2 Mpro
The plasmid pGEX-6p-1-MPro encoding SARS-CoV-2 Mpro (ORF1ab polyprotein residues 3264–3569, GenBank code: MN908947.3) with Escherichia coli (E. coli) codon usage was kindly provided by Prof. Rolf Hilgenfeld (Institute of Biochemistry, Center for Structural and Cell Biology in Medicine, University of Lübeck, Germany). The protocol for expression and purification of the Mpro was performed as reported by Zhang et al. (2020), with some modifications. The bacterial pellets were resuspended in 15 mL of cold Resuspension buffer [20 mM Tris HCl pH 7.5, 0.5 M NaCl, 10% glycerol, 1 mg/mL lysozyme, 1.15 mg/mL complete EDTA-free protease inhibitor cocktail (Roche)]. Cells were lysed by two freeze/thaw cycles and then sonicated. The lysate was clarified by centrifugation at 13,000 rpm at 4 °C for 45 min. The supernatant was then loaded onto an NiNTA Agarose column (GE Healthcare) equilibrated with 10 mL of Binding buffer (20 mM Tris HCl pH 7.5, 0.5 M NaCl, 10% glycerol). The column was washed with 20 mL Wash buffer (20 mM Tris HCl pH 7.5, 0.5 M NaCl, 10% glycerol, 25 mM imidazole) and elution was performed using 6 mL of elution buffer (20 mM Tris HCl pH 7.5, 0.5 M NaCl, 10% glycerol, 125 mM imidazole). Fractions were dialyzed against 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 30% (vol/vol) glycerol, 5 mM DTT and stored at −80 °C.
2.4. SARS-CoV-2 Mpro activity assay in vitro
To evaluate the SARS-CoV-2 Mpro activity in vitro, we employed a fluorescence resonance energy transfer (FRET)-based cleavage assay with a peptide substrate as previously described (Zhang et al., 2020) with minor modifications. Briefly, a volume of 10 μL of SARS-CoV Mpro (final concentration 0.15 μM) was added to 30 μL of assay buffer (20 mM Tris-HCl pH 7.3, 0.1 M NaCl, 1 mM EDTA, 1 mM DTT) and 10 μL of either 5 × test compounds or 5% DMSO into a 96-well black plate (Nunc) and incubated at 37 °C under agitation for 15 min. Boceprevir was included as a positive control of inhibition. Reaction was then started by the addition of 10 μL of fluorescent substrate (Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2; GL Biochem, final concentration 10 μM) to reach a final reaction volume of 50 μL. The plate was incubated at 37 °C under agitation for 1 h and finally the fluorescence signal deriving from the cleavage of the FRET-peptide substrate by SARS-CoV-2 Mpro was measured with a Victor X2 multiplate reader (PerkinElmer, excitation at 360 nM and emission at 460 nM).
2.5. Cells and viruses
African Green Monkey Vero E6 (ATCC® CRL-1586™), MRC-5 (ATCC® CCL-171™), human embryonic kidney 293T cells (ATCC® CRL-3216™) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Life Technologies), while HCT-8 (ATCC® CCL-244) were cultured in RPMI supplemented with 10% FBS. Calu-3 cells (ATCC® HTB-55™) were cultured in EMEM supplemented with nonessential amino acids and 10% FBS. All cells were cultured in the presence of 100 U/mL penicillin and 100 μg/mL streptomycin (Life Technologies), were maintained at 37 °C in a humidified atmosphere supplemented with 5% CO2, and periodically tested for the absence of mycoplasma contamination.
SARS-CoV-2/NL/2020 strain was obtained from European Virus Archive Global (EVAg) and was propagated and titrated in Vero E6 cells. HCoV-OC43 (VR-1558™) and HCoV-229E (VR-740™) strains were purchased from ATCC and were propagated in HCT-8 or MRC-5 cells, respectively.
2.6. Plaque reduction assays
Plaque reduction assays (PRA) with different CoVs were performed as previously described (Desantis et al., 2021). For SARS-CoV-2, Vero E6 cells were seeded at a density of 1 × 105 cells per well in 24-well plates and the next day, were infected with 80 Plaque Forming Unit (PFU) per well of SARS-CoV-2 (SARS-CoV-2/NL/2020 strain). After incubation, the inoculum was removed, and media containing various concentrations of each compound, 3% FBS, and 0.6% methylcellulose were added. After 3 days of incubation, cell monolayers were fixed and stained, and viral plaques were counted. For infection with HCoV-OC43 and HCoV-229E strains, MRC-5 cells were seeded at a density of 6 × 104 cells per well in 24-well plates. The next day, the cells were infected with 80 Plaque Forming Unit (PFU) per well of the different HCoVs. After incubation, the inoculum was removed, and media containing various concentrations of each compound, 3% FBS, and 0.6% methylcellulose were added. After 6 days of incubation, cell monolayers were fixed and stained, and viral plaques were counted. All work with SARS-CoV-2 virus was performed in a biosafety level 3 (BSL3) laboratory according to the safety practices as approved by the Department of Molecular Medicine (University of Padua, Italy) Committee on Microbiological Safety.
2.7. Virus yield reduction assays
For virus yield reduction assays of SARS-CoV-2 in Calu-3, cells were seeded at a density of 2 × 104 cells per well in 96-well plates and the next day, were infected with SARS-CoV-2 at multiplicity of infection (MOI) of 0.01 PFU/cell. After incubation, the inoculum was removed, and media containing various concentrations of test compounds were added. At 36 h post-infection (p.i.), supernatants were collected and viral titers were determined by titration in fresh Vero E6 cells. Statistical analysis was performed with GraphPad Prism version 8.0.
2.8. Cell viability assays
The effect on cell viability of test compounds was determined in Vero E6 and MRC-5 cells at 72 h, in Calu-3 cells at 48 h, and in 293T at 24 h by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich) method as described previously (Mercorelli et al., 2021).
2.9. Time-of-addition experiments
Vero E6 cells were seeded at a density of 1 × 104 cells per well in 96-well plates and the next day, were infected with SARS-CoV-2 at MOI of 1 PFU/cell for 1 h at 37 °C. Test compounds (50 μM) or BOC (25 μM) as a control for Mpro inhibition were added to the cells at different times during the viral replication cycle, i.e., at −2, 0, +2, +4 h p.i. The supernatants were collected at 9 h p.i., after a single replication cycle, and were then titrated in fresh Vero E6 monolayers.
2.10. Plasmids
The pNLF-nsp4-Mpro plasmid, expressing the NanoLuc® fused at the N-terminus of the nsp4-Mpro of SARS-CoV-2 was obtained by PCR amplification of nsp4-Mpro sequence from pGEX-6p-1-MPro and subsequent cloning into pNLF1-N vector (Promega) at EcoRI/SacI sites. The pNLF-nsp4-Mpro C145A plasmid, which expresses a catalytically inactive form of Mpro, was obtained by site-directed mutagenesis. Primers sequences are listed in Table S1. The correct sequences were verified by Sanger sequencing.
2.11. Cell-based assay to detect SARS-CoV-2 Mpro activity
For transfection experiments, 293T cells were seeded at a density of 2 × 105 cells/well in 24-well plates. The next day, cells were pre-treated for 2 h with test compounds or 0.1% DMSO as a control and then were transfected with 100 ng of different plasmids using Lipofectamine 2000 (ThermoFisher), according to the manufacturer's instructions. Cells were then incubated for 16 h in the presence of test compounds or BOC as a control and at the end were collected for the analysis of intracellular Mpro activity by Western Blot (WB). Whole-cell protein extracts were prepared as previously described (Mercorelli et al., 2016) and then analyzed by WB with antibodies against SARS-CoV-2 Mpro (1: 2,000, Cell Signaling) or against β-actin (1: 10,000, Merck) as a control for protein loading. Immunocomplexes were detected with goat anti-mouse and anti-rabbit immunoglobulin antibodies conjugated to horseradish peroxidase (Life Technologies).
3. Results and discussion
3.1. Virtual screenings against SARS-CoV-2 Mpro
In this study, Mpro was selected as a target to run virtual screenings. At the time of the beginning of this study, only two X-ray structures of Mpro were available, i.e., PDB IDs 6lu7 and 6y2g. Both protein structures were co-crystallized with two covalently bound peptide-like ligands, named N3 and 13b, which share a certain structure similarity, but with N3 larger than 13b, as it emerges from the aligned structures in Fig. 1A. Since the information about Mpro and its ligands was rather limited, we performed multiple explorative virtual screening campaigns, as shown in the scheme of Fig. 1B. First, we performed LBVS campaigns on N3 and 13b using a dataset of commercially available compounds (almost 800,000 small molecules from Specs and ChemDiv) employing the FLAP software (Baroni et al., 2007) (Fig. 1B, approaches 1-1 and 1–2). Compound 13b was also used as a template to screen a target library of ligands for protease by Vitas-M, as we noticed that commercially available databases often include a small number of peptide-like compounds (Fig. 1B, approach 1–3). The best hits, displaying a similarity score (Glob-Prod) greater than 0.3, were subsequently screened through a structure-based virtual screening (SBVS) using the 6lu7 catalytic site of Mpro as a template, since the catalytic sites of 6lu7 and 6y2g share a high similarity in terms of their GRID Molecular Interaction Fields (MIFs) (Carosati et al., 2004), as shown in Fig. 1C, but 6lu7 had the best X-ray resolution compared to 6y2g. SBVS results were further screened (Glob-Prod > 0.6) and resulting compounds were visually inspected to finally select 20 hits to be tested (Compounds 1 to 20, Dataset S1 in Supplementary material).
As a second approach, we used the BioGPS software (Siragusa et al., 2015) to perform a pocket-pocket virtual screening (PPVS, Fig. 1B, approach 2). We used the Mpro catalytic site detected with FlapSite algorithm as a template and compared it against a database of 19,873 human protein pockets containing a known ligand co-crystallized, with the purpose of reusing the ligand as an antiviral drug. Several VS approaches have been successfully performed with BioGPS and are reported in the literature (Duran-Frigola et al., 2017; Lo Piparo et al., 2020; Siragusa et al., 2015, 2016). We thus obtained 116 pockets (with Global Product score major than 0.7), from which 116 co-crystallized ligands were extracted and the reduced dataset was screened on the Mpro cavity using a SBVS approach (Glob-Prod > 0.6) followed by visual inspection to get 4 hits (Compounds 21 to 24, Dataset S1).
Finally, we wished to explore more chemical structures with a peptide-like pattern. In the last years, we have extensively worked on the development of inhibitors of the influenza virus RNA-dependent RNA polymerase (RdRP) (Desantis et al., 2017; Lepri et al., 2014; Massari et al., 2013, 2015, 2021; Muratore et al., 2012b; Nannetti et al., 2019). We identified a series of compounds able to inhibit the interaction between PA and PB1 subunits of viral RdRP complex, which have a chemical structure designed to fit a highly hydrophobic cavity and to mimic protein-protein interactions, therefore possessing a peptidomimetic nature. Thus, we screened by SBVS 24 compounds with various activity profiles towards influenza virus RdRP that were available in house (Fig. 1B, approach 3). Among these, the six most promising compounds were selected as hits (Compounds 25 to 30, Dataset S1).
3.2. Activity of hit compounds against Mproin vitro
We first investigated whether the 30 small molecules selected by virtual screening could actually inhibit SARS-CoV-2 Mpro activity in vitro. To this aim, we employed a FRET-based assay to measure the activity of E. coli-expressed and purified Mpro as previously described (Zhang et al., 2020). The 30 hit compounds were initially tested at a fixed concentration of 100 μM and the known SARS-CoV-2 Mpro inhibitor boceprevir (BOC, Fig. S1) was used as a positive control. As reported in Fig. 2 A, only hit compounds 1, 5, 7, 10, 25, 26, and 27 exerted an inhibitory effect on Mpro catalytic activity at 100 μM (cut-off 50% of inhibition). We then repeated the experiments with different concentrations of these active compounds and observed a dose-dependent inhibition for all of them (Fig. 2B and Table 1 ). Under the experimental condition of our assay, for BOC we measured an inhibitory concentration at half-maximal response (IC50) toward Mpro of 18.9 μM.
Fig. 2.

Anti-coronavirus activity of hit compounds. SARS-CoV-2 Mpro activity in vitro in the presence of (A) 100 μM or (B) different doses of selected hits. Graphs represent mean ± SD of n ≥ 3 experiments in duplicate. Data were normalized to the control (Mpro samples containing the same % of DMSO [vol/vol]). (C) Dose-dependent inhibition of the replication of SARS-CoV-2 in infected Vero E6 cells by selected hits. Graphs represent the mean ± SD of n ≥ 3 experiments in duplicate. (D) Antiviral activity of selected hit compounds in human Calu-3 cells infected with SARS-CoV-2 and treated with different concentrations of test compounds or 50 μM boceprevir (BOC) and 10 μM remdesivir (RMV) as controls. At 36 h p.i., supernatants were collected and titrated onto fresh Vero E6 cell monolayers. Graph represents the mean ± SD of n = 3 experiments in duplicate. Data were analyzed by a one-way ANOVA followed by Dunnet's multiple comparison test. ****p < 0.0001 compared to control (infected, DMSO-treated samples). (E) Time-of-addition studies with selected hit compounds. Test compounds at nontoxic concentrations (i.e., 50 μM for hit compounds and 25 μM for BOC) were added to Vero E6 cells prior to (−2 h), at the time of (0 h), or after (+2 h, +4 h) infection with SARS-CoV-2 at MOI of 1 PFU/cell. At 9 h p.i., supernatants were collected and titrated onto fresh Vero E6 cell monolayers. Graph represents the mean ± SD of n = 3 experiments in duplicate. (F) Dose-dependent inhibition of HCoV-OC43 and (G) HCoV-229E replication in MRC-5 cells by selected hit compounds. Graphs represent the mean ± SD of n ≥ 3 experiments in duplicate.
Table 1.
Biological profile of hit compounds
| Compound | VSa | Structure | Antiviral Activity EC50b (μM) | Cytotoxicity CC50c (μM) | SId | Mpro Inhibitory Activity IC50e (μM) |
|---|---|---|---|---|---|---|
| 1 | 1–1 | 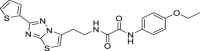 |
>50 | >500 | >10 | 141.5 ± 12.0 |
| 2 | 1–1 | 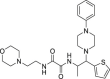 |
>50 | >500 | >10 | ND |
| 3 | 1–1 | 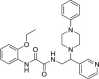 |
12.5 ± 2.1 | >500 | >40 | ND |
| 4 | 1–1 | 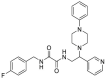 |
20.9 ± 8.5 | >500 | >24 | ND |
| 5 | 1–1 | 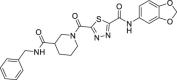 |
7.1 ± 3.5 | 207 ± 14 | 29 | 29.0 ± 3.7 |
| 6 | 1–1 | 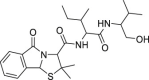 |
3.0 ± 1.6 | >500 | >167 | >100 |
| 7 | 1–1 | 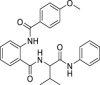 |
>50 | >500 | >10 | 110.7 ± 2.2 |
| 8 | 1–2 | 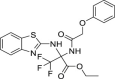 |
23.1 ± 5.5 | 68.8 ± 15.4 | 3 | ND |
| 9 | 1–3 | 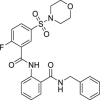 |
>50 | >500 | >10 | ND |
| 10 | 1–3 | 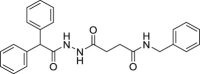 |
>50 | >500 | >10 | 128.1 ± 18.8 |
| 11 | 1–3 | 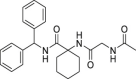 |
>50 | >500 | >10 | ND |
| 12 | 1–3 | 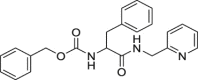 |
20.7 ± 1.1 | 428 ± 11 | 21 | ND |
| 13 | 1–3 | 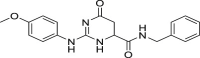 |
>50 | >500 | >10 | ND |
| 14 | 1–3 | 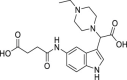 |
>50 | >500 | >10 | ND |
| 15 | 1–3 | 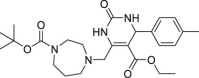 |
15.2 ± 2.5 | 113 ± 3 | 8 | ND |
| 16 | 1–3 | 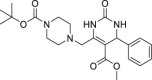 |
36.0 ± 13.4 | 401 ± 52 | 10 | ND |
| 17 | 1–3 | 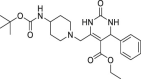 |
>50 | 228 ± 6 | >5 | ND |
| 18 | 1–3 | 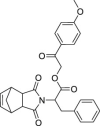 |
>50 | >500 | >10 | ND |
| 19 | 1–3 | 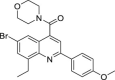 |
7.2 ± 1.0 | 228 ± 69 | 32 | ND |
| 20 | 1–3 | 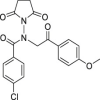 |
>50 | >500 | >10 | ND |
| 21 | 2 | 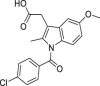 |
>50 | >500 | >10 | ND |
| 22 | 2 | 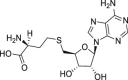 |
>50 | >500 | >10 | ND |
| 23 | 2 | 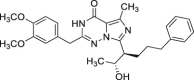 |
16.7 ± 5.9 | 433 ± 54 | 26 | ND |
| 24 | 2 | 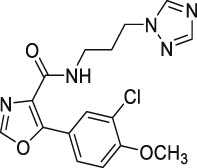 |
11.2 ± 5.5 | >500 | >45 | ND |
| 25 | 3 | 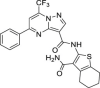 |
8.1 ± 3.4 | >250 | >31 | 26.6 ± 7.4 |
| 26 | 3 | 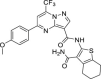 |
9.8 ± 4.5 | 68.9 ± 10.0 | 7 | 57.6 ± 4.1 |
| 27 | 3 | 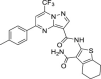 |
14.3 ± 9.6 | 67.9 ± 16.5 | 5 | 46.2 ± 7.7 |
| 28 | 3 | 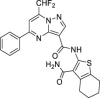 |
>50 | >250 | >5 | ND |
| 29 | 3 | 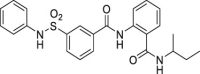 |
9.4 ± 4.5 | >500 | >51 | ND |
| 30 | 3 | >50 | >500 | >10 | ND | |
| BOC | – | 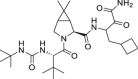 |
22.0 ± 2.6 | >500 | >23 | 18.9 ± 6.5 |
| RMV | – | 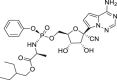 |
0.15 ± 0.04 | >250 | >1667 | ND |
All data were obtained by analysis with nonlinear regression function of GraphPad Prism 8.0.
ND, Not Determined.
Virtual screening methods applied to identify each compound. Numbers refer to the workflows described in Fig. 1B.
50% Effective Concentration at half-maximal response, i.e., the compound concentration that inhibits 50% of plaque formation as determined by PRA against SARS-CoV-2. Reported values represent the means ± SD of data derived from n ≥ 3 independent experiments in duplicate.
Compound concentration that produces 50% of cytotoxicity, as determined by MTT assays at 72 h in Vero E6 cells. Reported values represent the means ± SD of data derived from n = 3 independent experiments in duplicate.
SI, Selectivity Index (determined as the ratio between CC50 and EC50).
50% Inhibitory Concentration at half-maximal response, i.e., the compound concentration that inhibits 50% of SARS-CoV-2 Mpro activity in vitro. Reported values represent the means ± SD of data derived from n ≥ 3 independent experiments in duplicate.
3.3. Antiviral activity of hit compounds against SARS-CoV-2 in infected cells
We then investigated the antiviral effects of the 30 hit compounds selected by VS against SARS-CoV-2 by performing plaque reduction assays (PRA) in Vero E6 cells. Among the 30 compounds evaluated, 15 small molecules were able to inhibit the replication of SARS-CoV-2 in a dose-dependent manner in infected Vero E6 cells with an effective concentration at half-maximal response (EC50) < 50 μM (Table 1). The viral RNA polymerase inhibitor remdesivir (RMV) and BOC were included as controls (Table 1). To exclude that the observed antiviral activity could be due to cytotoxic effects, the cytotoxicity of the compounds was tested in parallel by MTT assays in Vero E6 cells and most of them resulted nontoxic (Table 1). Ten hit compounds (i.e., 3, 4, 5, 6, 12, 19, 23, 24, 25, 29) inhibited SARS-CoV-2 replication in a dose-dependent manner without showing significant cytotoxicity at the tested concentrations, thus having a favorable selectivity index (SI > 20, Table 1 and Fig. 2C). By comparing for each hit compound the anti-SARS-CoV-2 activity in infected cells and the inhibitory activity against Mpro in vitro (Table 1), we noticed that some of the compounds showed activity both in infected cells and against Mpro in vitro (i.e., hit compounds 5, 25, 26, 27). None of the identified Mpro inhibitors contain the scaffolds recently reviewed (Gao et al., 2021). In contrast, some compounds showed activity only in the in vitro Mpro assay (i.e., 1, 7, 10). For these compounds we hypothesized possible cell permeability issues that might explain the lack of antiviral effect in infected cells. On the other hand, for other hit compounds (i.e., 3, 4, 6, 12, 19, 23, 24, 29), under the experimental conditions used in our assays we could observe only an antiviral effect in infected cells but not a significant inhibitory effect against Mpro in vitro. In this case, we could not exclude either a mechanism of inhibitory activity against Mpro that can take place only in the cellular environment due to specific conditions of pH, ionic strength, redox potential, etc., which have been demonstrated to be crucial for SARS-CoV-2 Mpro inhibition (Behnam and Klein, 2021) or a different target. Further investigation will be undertaken in the future to answer to these open questions.
To further characterize the anti-SARS-CoV-2 activity of the most interesting hit compounds, we tested their inhibitory effect also in a human cell line, i.e., lung adenocarcinoma Calu-3 cells. As reported in Fig. 2D, hit compounds 5, 6, and 25 significantly reduced SARS-CoV-2 titers in supernatants of infected and treated Calu-3 cells, comparably to the control compound BOC and without showing toxicity at the tested concentrations (Table S2), thus confirming their antiviral activity.
Next, for target validation, we evaluated hit compounds 5 and 25, along with BOC as a control for Mpro inhibition, in time-of-addition experiments, where the effect of an inhibitor is evaluated at different times during a single virus replication cycle (8 h for SARS-CoV-2). As reported in Fig. 2E, BOC as well as compounds 5 and 25 were able to suppress the production of infectious SARS-CoV-2 when they were added either prior to infection or up to 2 h p.i. The antiviral effect was progressively lost when the compounds were added later, a result in agreement with the inhibition of Mpro during the replication cycle and according to other previously reported SARS-CoV-2 Mpro inhibitors (Bafna et al., 2021).
Noteworthy, regarding the screening of large commercial databases, LBVS followed by SBVS (approaches 1-1 and 1–2 in Fig. 1B) led to the identification of four out of eight compounds with measurable inhibitory activity against SARS-CoV-2 Mpro, with hit compound 5 endowed with good antiviral activity (EC50 = 7.1 μM, SI = 29) and also able to inhibit Mpro in vitro (IC50 = 29.0 μM) (Table 1). On the other hand, compounds that showed activity only in vitro might be the starting platforms for effective hit-to-lead optimization campaigns, as we demonstrated in section 3.5 of this manuscript (see below). By comparing the results of the different VS, the best enrichment was obtained by screening compounds originally designed against influenza virus RNA polymerase (approach 3 in Fig. 1B), with three out of six compounds (i.e., 25, 26, and 27) displaying inhibitory activity against Mpro. Among them, hit compound 25 showed the best antiviral profile (EC50 = 8.1 μM, SI > 31) and also the highest anti-Mpro activity (IC50 = 26.6 μM). The overall good result obtained with approach 3 might be explained by the peptide-like properties of these derivatives that do not usually characterize commercial compound libraries.
3.4. Antiviral activity of hit compounds against other human coronaviruses
Since pan-coronavirus antiviral activity is very important in the development of effective anti-CoV drugs, we further characterized the antiviral properties of the hit compounds with the most potent activity against SARS-CoV-2 coupled to a good SI, i.e., compounds 3, 4, 5, 6, 25, and 29, by testing their activity also against two human endemic CoVs, i.e., another β-coronavirus (HCoV-OC43) and an α-coronavirus (HCoV-229E), in infected MRC-5 cells. Interestingly, compounds 3, 4, 5, 6, 25, and 29 all retained activity towards HCoV-OC43 and most of them (except 25) also towards HCoV-229E, although with higher EC50 values (Fig. 2F–G and Table 2 ). Importantly, all the tested compounds did not show significant cytotoxicity also in human MRC-5 cells, thus excluding that the antiviral activity might be due to cytotoxic effects. We identified a series of structurally unrelated hits with specific antiviral activity against pandemic and endemic CoVs, thus showing a pan-human coronavirus antiviral potential and that could be further developed in the future.
Table 2.
Antiviral activity of selected hit compounds against human coronaviruses
| Compound | HCoV-OC43 |
HCoV-229E |
|||
|---|---|---|---|---|---|
| CC50a (μM) | EC50b (μM) | SIc | EC50b (μM) | SIc | |
| 3 | >500 | 14.5 ± 3.5 | >35 | 34.6 ± 8.5 | >15 |
| 4 | 300 ± 18 | 15.8 ± 1.1 | 19 | 23.8 ± 9.9 | 13 |
| 5 | 100 ± 11.9 | 10.6 ± 5.2 | 9 | 10.4 ± 4.1 | 10 |
| 6 | >500 | 4.3 ± 2.6 | >116 | 22.9 ± 4.9 | >22 |
| 25 | >250 | 10.1 ± 5.8 | >25 | >25 | >10 |
| 29 | >250 | 4.8 ± 0.1 | >52 | 15.7 ± 4.8 | >16 |
| RMV | >125 | 0.10 ± 0.04 | >1250 | 0.09 ± 0.03 | >1389 |
Reported values represent the means ± SD of data derived from n ≥ 3 independent experiments in duplicate. Data were obtained by analysis with nonlinear regression function of GraphPad Prism 8.0.
Compound concentration that produces 50% of cytotoxicity, as determined by MTT assays at 72 h in MRC-5 cells.
50% Effective Concentration at half-maximal response, i.e., the compound concentration that inhibits 50% of plaque formation, as determined by PRA against different CoVs in MRC-5 cells.
SI, Selectivity Index (determined as the ratio between CC50 and EC50).
3.5. Optimization and synthesis of analogs of hit compound 7
Since virtual screenings in Fig. 1 were aimed at identifying novel chemical scaffolds as starting points for subsequent hit-to-lead optimization, as a proof-of concept we decided to optimize hit compound 7. A fast-track rational design was performed in order to boost the structural optimization aimed at improving both solubility and flexibility as well as virtual affinity with the Mpro catalytic site. It is noteworthy that the commercially available compound 7 was a mixture of the two enantiomers; therefore, we first modelled the most probable binding pose within the Mpro catalytic site for (R)-7 and (S)-7. As shown in Fig. 3 , the binding of the (S)-enantiomer of hit 7 led to a slightly higher similarity score for the Glob-Prod descriptor compared to the (R)-enantiomer and the two isomers also displayed different orientation modes (Fig. 3A–B). The major difference, as expected, relied on the orientation of the aniline moiety, which folds to interact with the p-methoxy-benzoic moiety in the (R)-enantiomer, while it assumes a more elongated pose in the (S)-enantiomer to interact with H163. In addition, the amidic groups nicely fit in the region where C145, S144, and N142 are located, suggesting stabilization by hydrogen bonds. Therefore, we hypothesized that the (S)-enantiomer could be a better ligand for Mpro. Based on this assumption, (S)-7 compound was synthesized (compound 31 in Table 3 ) and the S-valine core was used to design compound 7 derivatives. With the aim to boost the optimization campaign of compound 7, we built a combinatorial virtual library of compound 7 analogs (Dataset S2) that, after screening using the 6lu7 catalytic cavity as a template, helped in the selection of the derivatives to be synthesized (details can be found in Material and Methods Section), such as compounds 32–37 (Table 3).
Fig. 3.

Prediction of the binding poses in the Mpro catalytic site (from PDB ID: 6lu7). The binding poses for the two enantiomers of 7 and for its synthesized derivatives are shown according to the most representative FLAP poses by Glob-Prod descriptor. (A) (R)-enantiomer of 7; (B) (S)-enantiomer of 7 (compound 31); (C) compound 32; (D) compound 33; (E) compound 34; (F) compound 35; (G) compound 36; (H) compound 37. Amino acidic residues more involved in the protein-ligand interactions and discussed in the text are highlighted.
Table 3.
Biological profile of synthesized derivatives of hit 7
| Compound | Structure | Antiviral Activity EC50a (μM) | Cytotoxicity CC50b (μM) | SIc | Mpro Inhibitory Activity IC50d (μM) |
|---|---|---|---|---|---|
| 7 | 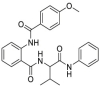 |
>50 | >500 | >10 | 110.7 ± 2.2 |
| 31 | 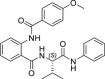 |
>50 | >250 | >5 | 43.7 ± 9.2 |
| 32 | 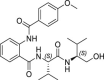 |
30.0 ± 6.9 | >250 | >8 | >200 |
| 33 | 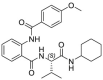 |
35.7 ± 3.8 | >250 | >7 | 26.7 ± 7.3 |
| 34 |  |
>50 | >250 | >5 | 17.1 ± 6.9 |
| 35 | 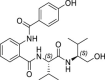 |
25.8 ± 5.7 | >250 | >10 | 19.0 ± 8.4 |
| 36 | 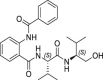 |
1.3 ± 0.5 | >250 | >192 | 42.9 ± 8.3 |
| 37 | 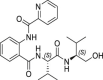 |
2.1 ± 1.1 | >250 | >119 | 41.3 ± 7.6 |
All data were obtained by analysis with nonlinear regression function of GraphPad Prism 8.0.
50% Effective Concentration at half-maximal response, i.e., the compound concentration that inhibits 50% of plaque formation as determined by PRA against SARS-CoV-2. Reported values represent the means ± SD of data derived from n ≥ 3 independent experiments in duplicate.
Compound concentration that produces 50% of cytotoxicity, as determined by MTT assays at 72 h in Vero E6 cells. Reported values represent the means ± SD of data derived from n = 3 independent experiments in duplicate.
SI, Selectivity Index (determined as the ratio between CC50 and EC50).
50% Inhibitory Concentration at half-maximal response, i.e., the compound concentration that inhibits 50% of SARS-CoV-2 Mpro activity in vitro. Reported values represent the means ± SD of data derived from n ≥ 3 independent experiments in duplicate.
Among the synthesized derivatives, except for compounds 33 and 34, in which the aniline moiety was replaced with a cyclohexylamine and ethylamine, respectively, we selected hybrid species bearing the valinol moiety present in compound 6, which emerged as the most active in inhibiting SARS-CoV-2 replication (Table 1). Indeed, compounds 32 and 35–37 displayed a Glob-Prod score similar or even better than compound 31. Since a slightly lower similarity score was related to compound 34, which is the only compound in which the hydroxyl group in the valinol moiety was depleted, we hypothesized that higher scores in the valinol-containing compounds could be due to specific interaction with the hydroxyl group. A closer inspection revealed that hydroxyl group in compound 32 interacted with Q166 while the same group in compounds 35–36 interacted with the polar region generated by C145 and S144 by multiple potential H-bonds (Fig. S3). For compound 37 the interaction with the thiol group of C145 by H-bonds occurred through the amide group between the phenyl group and the leucine moiety, and the binding was also stabilized by the H-bond interaction between Q189 and the amide group linking the pyridine and the phenyl rings. The mentioned residues have been already observed interacting with other known Mpro inhibitors (Jin et al., 2020; Shitrit et al., 2020). Concerning compound 35, the location of the hydroxyl group of the valinol moiety in a region of the Mpro cavity in which multiple possibilities for H-bond establishment are available, could represent the key of its good Mpro inhibitory effect in vitro, and therefore the polar region defined by S144 and H163 deserves further investigation.
3.6. Biological evaluation of derivatives 31–37
From the biological evaluation of derivatives 31–37 (Table 3), it emerged that, compared to parental hit 7, the (S)-enantiomer (compound 31), even though still lacking anti-SARS-CoV-2 activity, resulted >2-fold more effective in inhibiting Mpro, while the replacement of the aniline ring with the L-valinol moiety also allowed to improve compounds’ antiviral activity in infected cells (exhibited by compounds 35, 36 and 37). In particular, 36 and 37 were endowed with low-micromolar range anti-SARS-CoV-2 activity also coupled to >2-fold increased inhibitory potency against Mpro (Table 3 and Fig. 2B–C). Compounds 36 and 37, which exhibited the highest EC50 among the compounds reported in this study (EC50 = 1.3 and 2.1 μM, SI > 192 and > 119, respectively), were confirmed active against SARS-CoV-2 also in the human Calu-3 cell line (Fig. 2D). To further investigate the activity of 36 and 37 against Mpro, we performed the time-of-addition studies and both 36 and 37 demonstrated an antiviral effect consistent with Mpro inhibition (Fig. 2E). Interestingly, their addition at 4 h p.i. still exerted an antiviral effect, thus for these compounds also an additional mechanism might be envisaged. To demonstrate that the hit compounds 36 and 37 are able to inhibit SARS-CoV-2 Mpro also in the cellular context, we set up a cell-based assay that allows detecting the proteolytic cleavage of a NanoLuc-nsp4-Mpro fusion protein expressed in 293T cells and the release of an authentic Mpro protein. As reported in Fig. S4, the expression of NanoLuc-nsp4-Mpro in the absence of an inhibitor resulted in the cleavage of Mpro from the NanoLuc tag (thanks to the cleavage recognition site placed between the two proteins), which was not detected when either the catalytically inactive mutant C145A was transfected or the Mpro inhibitor boceprevir was added to the cells (Fig. S4). As reported in Fig. 4 , treatment of cells transfected with NanoLuc-nsp4-Mpro with different doses of hit compounds 36 and 37 resulted in a dose-dependent inhibition of the proteolytic cleavage and consequent accumulation of uncleaved NanoLuc-nsp4-Mpro fusion protein, indicating that the compounds inhibit Mpro activity also in a cellular context, without showing cytotoxicity (Table S2).
Fig. 4.

Hit compounds 36 and 37 inhibit SARS-CoV-2 Mpro in a cellular context. 293T cells were transfected to express either NanoLuc-nsp4-Mpro fusion protein (NLuc-Mpro) or its catalytically inactive mutant form (C145A). Whole cell lysates obtained from cells collected at 16 h post-transfection were analyzed by Western Blot with an antibody recognizing SARS-CoV-2 Mpro. The expression of NLuc-Mpro in DMSO-treated cells resulted in the cleavage of Mpro from the NanoLuc tag and in the release of an authentic Mpro protein (Mpro, with a molecular mass of ∼33 kDa), which was not detected when the inactive mutant C145A (with a molecular mass of ∼52 kDa) was expressed. The dose-dependent inhibitory effect of hit compounds 36 and 37 on Mpro activity was detected through the accumulation of the 52-kDa, uncleaved NLuc-Mpro fusion protein. Nontransfected 293T cells (NT) and transfected cells treated with 100 μM boceprevir (BOC) were included as controls. β-actin was used as a loading control. Molecular masses in kDa are indicated on the left.
To note, the optimization that led to compounds 36 and 37 enhanced the potency toward SARS-CoV-2, but led to the loss of activity against other CoVs (Table S3). Unfortunately, there is no structural information about HCoV-OC43 protease, but in the case of HCoV-229E this latter experimental evidence appears to be in agreement with the predicted FLAP poses of these compounds in SARS-CoV-2 Mpro cavity (Fig. S3). Indeed, the two residues identified as key points for interaction of compounds 36 and 37 (i.e., S144 and Q189) are not conserved in the HCoV-229E structure (pdb ID: 1P9S), as they are replaced with an alanine and a proline residue, respectively. This variation in the Mpro sequence significantly alters the hydrophilicity and the H-bond capability of the corresponding protein regions likely resulting in a lack of activity (Fig. S3). Intriguingly, compound 5 does not seem to interact with the above-mentioned non-conserved residues S144 and Q189 (Fig. S5). Indeed, its predicted binding pose in the SARS-CoV-2 Mpro protein cavity is stabilized by several interaction including H41 and C145, which are conserved also in HCoV-229E protease catalytic site (Fig. S5). Another key interaction occurs with T25, which corresponds to D24 in HCoV-229E. This different amino acid, however, still allows the H-bond interaction to occur. These observations based on our model may explain why compound 5 retains activity against other CoVs, while analogs of 7 lost pan-coronavirus potential.
4. Conclusions
In summary, we identified new SARS-CoV-2 Mpro inhibitors with undescribed scaffolds and proved that simple optimization strategies guided by rational design and synthesis can lead to analogs with significantly improved potency that represent promising chemotypes to be further developed as candidate anti-CoV drugs.
Concerning the various virtual screening campaigns, our results highlighted that, although the application of a combination of LBVS and SBVS based on the ligand and Mpro co-crystallized in the PDB ID 6lu7 provided the best performance for virtual screenings of commercially available compounds, the large databases used are probably devoid of molecules bearing peptide-like patterns.
The rational optimization of a weak Mpro inhibitor such as hit 7 using the FLAP approach proved useful to increase inhibitory activity against the target, thus validating the computational model used in this study. Moreover, the optimization of hit 7 led to two additional considerations. Firstly, the best Mpro inhibitory activity among compound 7 analogs was observed for compound 35, in which the hydroxyl group of the valinol moiety is located in the polar region defined by S144 and H163 and therefore next studies will be oriented towards the optimization of such H-bond interactions. Secondly, a comparison of the antiviral activity data with the Mpro inhibition data for compounds 36 and 37 (the analogs of compound 7 resulting good inhibitors of Mpro and also displaying the best antiviral activity against SARS-CoV-2) suggests that the generation of hybrid molecules in which the scaffold of compound 7 was modified to contain a terminal valinol moiety as in compound 6 (our best hit for antiviral activity, although not a Mpro inhibitor in vitro itself) may lead to a multiple target mechanism, and therefore compounds 36 and 37 will deserve further investigation.
Finally, the most potent hit compounds identified in the present study by virtual screening (3, 4, 5, 6, 25, and 29) retained activity towards the β-coronavirus HCoV-OC43 and most of them (except 25) also towards the α-coronavirus HCoV-229E, although with higher EC50 values. Conversely, the analogs of compound 7, optimized in the SARS-CoV-2 Mpro structure, did not show this broad antiviral potential. We believe that sharing our results on the exploration of novel chemical structures of CoVs Mpro inhibitors but also on the antiviral properties of L-valinol moiety in hybrid derivatives of compound 7 could contribute to move forward in the development of new antiviral agents against SARS-CoV-2.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by Fondazione Cassa di Risparmio di Padova e Rovigo, Italy - Bando Ricerca Covid-2019 Nr. 55777 2020.0162 - ARREST-COV: AntiviRal PROTAC-Enhanced Small-molecule Therapeutics against COronaViruses” (to A. L.); by Associazione Italiana per la Ricerca sul Cancro, AIRC, Italy (IG 2021 - ID. 25899 project to A.L.); by Ministero dell’Istruzione, dell’Università e della Ricerca, MIUR, Italy (PRIN 2017 n. 2017KM79NN to A.L.), and by University of Padua, Italy (PRID 2021 to B.M). This publication was also supported by the European Virus Archive Global (EVA-GLOBAL) project that has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 871029, which provided free access to the SARS-CoV-2/NL/2020 virus. We thank Prof. Hilgenfeld for kindly providing the pGEX-6P-1-Mpro plasmid and Prof. Richter and Dr. Frasson for providing Calu-3 cell line. The University of Perugia and MIUR are gratefully acknowledged for financial support to the project AMIS, through the program “Dipartimenti di Eccellenza-2018−2022”. We thank Giulia Crescentini and Massimo Baroni for the support in synthesis and in building the library of compound 7 derivatives.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.antiviral.2022.105350.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Bafna K., White K., Harish B., Rosales R., Ramelot T.A., Acton T.B., Moreno E., Kehrer T., Miorin L., Royer C.A., García-Sastre A., Krug R.M., Montelione G.T. Hepatitis C virus drugs that inhibit SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroni M., Cruciani G., Sciabola S., Perruccio F., Mason J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J. Chem. Inf. Model. 2007;47:279–294. doi: 10.1021/ci600253e. [DOI] [PubMed] [Google Scholar]
- Behnam M.A.M., Klein D.C. Inhibitor potency and assay conditions: A case study on SARS-CoV-2 main protease. Proc. Natl. Acad. Sci. U. S. A. 2021;118 doi: 10.1073/pnas.2106095118. e2106095118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carosati E., Sciabola S., Cruciani G. Hydrogen bonding interactions of covalently bonded fluorine atoms: from crystallographic data to a new angular function in the GRID force field. J. Med. Chem. 2004;47:5114–5125. doi: 10.1021/jm0498349. [DOI] [PubMed] [Google Scholar]
- Chapy H., Goracci L., Vayer P., Parmentier Y., Carrupt P.A., Decleves X., Scherrmann J.M., Cisternino S., Cruciani G. Pharmacophore-based discovery of inhibitors of a novel drug/proton antiporter in human brain endothelial hCMEC/D3 cell line. Br. J. Pharmacol. 2015;172:4888–4904. doi: 10.1111/bph.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W., Yang K., Yang H. Recent progress in the drug development targeting SARS-CoV-2 main protease as treatment for COVID-19. Front. Mol. Biosci. 2020;7 doi: 10.3389/fmolb.2020.616341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis J., Mercorelli B., Celegato M., Croci F., Bazzacco A., Baroni M., Siragusa L., Cruciani G., Loregian A., Goracci L. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur. J. Med. Chem. 2021;226 doi: 10.1016/j.ejmech.2021.113814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis J., Nannetti G., Massari S., Barreca M.L., Manfroni G., Cecchetti V., Palù G., Goracci L., Loregian A., Tabarrini O. Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity. Eur. J. Med. Chem. 2017;138:128–139. doi: 10.1016/j.ejmech.2017.06.015. [DOI] [PubMed] [Google Scholar]
- Duran-Frigola M., Siragusa L., Ruppin E., Barril X., Cruciani G., Aloy P. Detecting similar binding pockets to enable systems polypharmacology. PLoS Comput. Biol. 2017;13 doi: 10.1371/journal.pcbi.1005522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K., Wang R., Chen J., Tepe J.J., Huang F., Wei G.W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021;64:16922–16955. doi: 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goracci L., Deschamps N., Randazzo G.M., Petit C., Dos Santos Passos C., Carrupt P.A., Simoes-Pires C., Nurisso A. A rational approach for the identification of non-hydroxamate HDAC6-selective inhibitors. Sci. Rep. 2016;6 doi: 10.1038/srep29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S., Jiang S., Qi X., Bai R., Ye X.Y., Xie T. Races of small molecule clinical trials for the treatment of COVID-19: an up-to-date comprehensive review. Drug Dev. Res. 2022;83:16–54. doi: 10.1002/ddr.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito J., Tabei Y., Shimizu K., Tsuda K., Tomii K. PoSSuM: a database of similar protein-ligand binding and putative pockets. Nucleic Acids Res. 2012;40:D541–D548. doi: 10.1093/nar/gkr1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Khashan R. FragVLib a free database mining software for generating "Fragment-based Virtual Library" using pocket similarity search of ligand-receptor complexes. J. Cheminf. 2012;4:18. doi: 10.1186/1758-2946-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles J., Gromo G. A guide to drug discovery: target selection in drug discovery. Nat. Rev. Drug Discov. 2003;2:63–69. doi: 10.1038/nrd986. [DOI] [PubMed] [Google Scholar]
- Lepri S., Nannetti G., Muratore G., Cruciani G., Ruzziconi R., Mercorelli B., Palù G., Loregian A., Goracci L. Optimization of small-molecule inhibitors of influenza virus polymerase: from thiophene-3-carboxamide to polyamido scaffolds. J. Med. Chem. 2014;57:4337–4350. doi: 10.1021/jm500300r. [DOI] [PubMed] [Google Scholar]
- Lian X., Xia Z., Li X., Karpov P., Jin H., Tetko I.V., Xia J., Wu S. Anti-MRSA drug discovery by ligand-based virtual screening and biological evaluation. Bioorg. Chem. 2021;114 doi: 10.1016/j.bioorg.2021.105042. [DOI] [PubMed] [Google Scholar]
- Lo Piparo E., Siragusa L., Raymond F., Passeri G.I., Cruciani G., Schilter B. Bisphenol A binding promiscuity: a virtual journey through the universe of proteins. ALTEX. 2020;37:85–94. doi: 10.14573/altex.1906141. [DOI] [PubMed] [Google Scholar]
- Maia E.H.B., Assis L.C., de Oliveira T.A., da Silva A.M., Taranto A.G. Structure-based virtual screening: from classical to artificial intelligence. Front. Chem. 2020;8:343. doi: 10.3389/fchem.2020.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari S., Bertagnin C., Pismataro M.C., Donnadio A., Nannetti G., Felicetti T., Di Bona S., Nizi M.G., Tensi L., Manfroni G., Loza M.I., Sabatini S., Cecchetti V., Brea J., Goracci L., Loregian A., Tabarrini O. Synthesis and characterization of 1,2,4-triazolo[1,5-a]pyrimidine-2-carboxamide-based compounds targeting the PA-PB1 interface of influenza A virus polymerase. Eur. J. Med. Chem. 2021;209 doi: 10.1016/j.ejmech.2020.112944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari S., Nannetti G., Desantis J., Muratore G., Sabatini S., Manfroni G., Mercorelli B., Cecchetti V., Palù G., Cruciani G., Loregian A., Goracci L., Tabarrini O. A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein-basic protein 1 (PA-PB1) subunits. J. Med. Chem. 2015;58:3830–3842. doi: 10.1021/acs.jmedchem.5b00012. [DOI] [PubMed] [Google Scholar]
- Massari S., Nannetti G., Goracci L., Sancineto L., Muratore G., Sabatini S., Manfroni G., Mercorelli B., Cecchetti V., Facchini M., Palù G., Cruciani G., Loregian A., Tabarrini O. Structural investigation of cycloheptathiophene-3-carboxamide derivatives targeting influenza virus polymerase assembly. J. Med. Chem. 2013;56:10118–10131. doi: 10.1021/jm401560v. [DOI] [PubMed] [Google Scholar]
- Mercorelli B., Celegato M., Luganini A., Gribaudo G., Lepesheva G.I., Loregian A. The antifungal drug isavuconazole inhibits the replication of human cytomegalovirus (HCMV) and acts synergistically with anti-HCMV drugs. Antivir. Res. 2021;189 doi: 10.1016/j.antiviral.2021.105062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercorelli B., Luganini A., Nannetti G., Tabarrini O., Palù G., Gribaudo G., Loregian A. Drug repurposing approach identifies inhibitors of the prototypic viral transcription factor IE2 that block human cytomegalovirus replication. Cell Chem. Biol. 2016;23:340–351. doi: 10.1016/j.chembiol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Muratore G., Goracci L., Mercorelli B., Foeglein A., Digard P., Cruciani G., Palù G., Loregian A. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc. Natl. Acad. Sci. U. S. A. 2012;109:6247–6252. doi: 10.1073/pnas.1119817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore G., Mercorelli B., Goracci L., Cruciani G., Digard P., Palù G., Loregian A. Human cytomegalovirus inhibitor AL18 also possesses activity against influenza A and B viruses. Antimicrob. Agents Chemother. 2012;56:6009–6013. doi: 10.1128/AAC.01219-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannetti G., Massari S., Mercorelli B., Bertagnin C., Desantis J., Palù G., Tabarrini O., Loregian A. Potent and broad-spectrum cycloheptathiophene-3-carboxamide compounds that target the PA-PB1 interaction of influenza virus RNA polymerase and possess a high barrier to drug resistance. Antivir. Res. 2019;165:55–64. doi: 10.1016/j.antiviral.2019.03.003. [DOI] [PubMed] [Google Scholar]
- Pablos I., Machado Y., de Jesus H.C.R., Mohamud Y., Kappelhoff R., Lindskog C., Vlok M., Bell P.A., Butler G.S., Grin P.M., Cao Q.T., Nguyen J.P., Solis N., Abbina S., Rut W., Vederas J.C., Szekely L., Szakos A., Drag M., Kizhakkedathu J.N., Mossman K., Hirota J.A., Jan E., Luo H., Banerjee A., Overall C.M. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CL(pro) substrate degradome. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautasso C., Troia R., Genuardi M., Palumbo A. Pharmacophore modeling technique applied for the discovery of proteasome inhibitors. Expert Opin. Drug Discov. 2014;9:931–943. doi: 10.1517/17460441.2014.923838. [DOI] [PubMed] [Google Scholar]
- Shitrit A., Zaidman D., Kalid O., Bloch I., Doron D., Yarnizky T., Buch I., Segev I., Ben-Zeev E., Segev E., Kobiler O. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Sci. Rep. 2020;10 doi: 10.1038/s41598-020-77794-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa L., Cross S., Baroni M., Goracci L., Cruciani G. BioGPS: navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins. 2015;83:517–532. doi: 10.1002/prot.24753. [DOI] [PubMed] [Google Scholar]
- Siragusa L., Luciani R., Borsari C., Ferrari S., Costi M.P., Cruciani G., Spyrakis F. Comparing drug images and repurposing drugs with BioGPS and FLAPdock: the thymidylate synthase case. ChemMedChem. 2016;11:1653–1666. doi: 10.1002/cmdc.201600121. [DOI] [PubMed] [Google Scholar]
- Su S., Wong G., Shi W., Liu J., Lai A.C.K., Zhou J., Liu W., Bi Y., Gao G.F. Epidemiology, genetic recombination, and pathogenesis of Coronaviruses. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez J., Lopez M., Gibert E., Herrero E., Luque F.J. Merging ligand-based and structure-based methods in drug discovery: an overview of combined virtual screening approaches. Molecules. 2020;25:4723. doi: 10.3390/molecules25204723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J., Lampe J., Muller-Fielitz H., Schuster R., Zille M., Muller K., Krohn M., Korbelin J., Zhang L., Ozorhan U., Neve V., Wagner J.U.G., Bojkova D., Shumliakivska M., Jiang Y., Fahnrich A., Ott F., Sencio V., Robil C., Pfefferle S., Sauve F., Coelho C.F.F., Franz J., Spiecker F., Lembrich B., Binder S., Feller N., Konig P., Busch H., Collin L., Villasenor R., Johren O., Altmeppen H.C., Pasparakis M., Dimmeler S., Cinatl J., Puschel K., Zelic M., Ofengeim D., Stadelmann C., Trottein F., Nogueiras R., Hilgenfeld R., Glatzel M., Prevot V., Schwaninger M. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021;24:1522–1533. doi: 10.1038/s41593-021-00926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.