

Abstract
The mechanisms underlying tumor immunosurveillance and their association with the immune-related adverse events (irAEs) associated with checkpoint inhibitor immunotherapies remain poorly understood. We describe a metastatic melanoma patient exhibiting multiple episodes of spontaneous disease regression followed by the development of several irAEs during the course of anti–programmed cell death protein 1 antibody immunotherapy. Whole-exome next-generation sequencing studies revealed this patient to harbor a pyrin inflammasome variant previously described to be associated with an atypical presentation of familial Mediterranean fever. This work highlights a potential role for inflammasomes in the regulation of tumor immunosurveillance and the pathogenesis of irAEs.
Key Words: melanoma, immune-related adverse events, pyrin, inflammasome, spontaneous regression, checkpoint inhibitor immunotherapy, familial Mediterranean fever
As the utilization of checkpoint inhibitor immunotherapy continues to expand in oncology, the diagnosis and management of immune-related adverse events (irAEs) has become an important health care issue.1,2 To improve our ability to manage and predict the development of these irAEs, a more sophisticated understanding of the underlying molecular pathogenesis of these conditions is necessary.3 Given the diversity of the clinical presentations of the irAEs, studying these conditions can be challenging. However, the identification and characterization of single patients with remarkable clinical outcomes can help to overcome these barriers.
Cases of spontaneous melanoma regression are rare, particularly in the metastatic setting. Estimated to occur in 0.2% of cases of metastatic melanoma and with 76 reported cases between 1866 and 2009, this phenomenon is thought to occur due to a spontaneous antitumor response generated by the host immune system.4,5 Despite this longstanding perception, our mechanistic understanding underlying the development of these events remains poor. In addition, whether patients with a prior history of spontaneous disease regression or resolution are at higher risk of developing irAEs during a future course of immunotherapy is unclear.
A spectrum of related autoinflammatory syndromes have been described and include the autosomal recessive disorder, familial Mediterranean fever (FMF).6 FMF is characterized by recurrent fevers, joint and skin inflammation, and a constellation of symptoms associated with serositis and vasculitis.6 Genetic and biochemical studies have demonstrated that the clinical symptomatology of FMF varies widely in correlation with the underlying genotype of the patient.7 FMF is associated with mutations involving the MEFV gene which encodes pyrin, a cytoplasmic pathogen recognition receptor and member of the inflammasome family on chromosome 16p13.3.8 Upon activation, pyrin binds to the apoptosis-associated speck-like protein with a caspase recruitment domain (ASC), which activates caspase-1 and mediates the conversion of pro–interleukin (IL)-1β and pro-IL-18 into mature cytokines. This process further triggers the cleavage and activation of gasdermin D (GSDMD) which subsequently facilitates the release of IL-1β and IL-18 and a proinflammatory form of cell death called pyroptosis.8 Together, these processes drive a proinflammatory cascade that manifests in FMF.
We have previously demonstrated that the tumor-intrinsic NLRP3 inflammasome plays a role in promoting adaptive resistance to anti–programmed cell death protein 1 (PD-1) immunotherapy in melanoma.9,10 However, how germline mutations involving the pyrin inflammasome effect the development of antitumor immunity, as well as the responses and toxicities associated with checkpoint inhibitor immunotherapy, remain unknown. Seemingly innocuous mutations in inflammasome genes that do not generate detectable clinical signs or symptoms at baseline may manifest as a severe autoinflammatory or autoimmune syndrome during the course of immunotherapy. This finding would reveal the existence of clinically relevant gene-environment interactions that influence responses to immunotherapy. Herein, we present the clinical course and germline exome sequencing of a patient with stage IV melanoma that shines new light on the role that inflammasomes may play in affecting immunotherapy outcomes.
CASE PRESENTATION
A woman in her 60s with a reported history of polymyalgia rheumatica (PMR) and giant cell arteritis (GCA) presented in June 2017 with left axillary lymphadenopathy. A mammogram detected a large mass in the left axilla that was confirmed to be an enlarged lymph node (LN) by ultrasound. Biopsy of the LN initially revealed fragments of fibroadipose tissue with hemorrhage and acute inflammation, suggestive of a necrotic neoplasm. While the patient had no prior history of melanoma, concerns were raised by the pathologist for metastatic melanoma given the identification of pigment deposition.
A follow-up positron emission tomography (PET) scan showed rim uptake of an enlarged 4 cm×3 cm left axillary LN prompting a LN dissection procedure which revealed organizing hemorrhage, pigment-laden macrophages, and fat necrosis (Fig. 1). Immunohistochemical (IHC) stains for pancytokeratin (AE1/AE3) and SOX10 were negative, and no features of malignancy were identified in any of the 14 LNs sampled. Notably, during the course of her evaluation, the patient noted a diminishment in the size of the noted left axillary LN.
FIGURE 1.
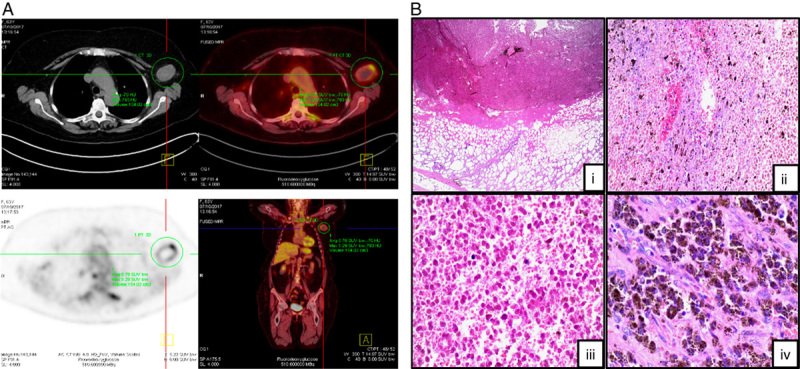
Spontaneous regression of metastatic melanoma involving a left axillary lymph node. A, Transverse and coronal positron emission tomography-computed tomography imaging showing evidence of a necrotic left axillary lymph node. B, Hematoxylin and eosin microscopy of resected left axillary lymph node. (i) Tumoral necrosis, fibrosis, and an inflammatory infiltrate with surrounding fat necrosis (×2). (ii, iii) Coagulative necrosis within tumor tissue with no evidence of viable cells (×10, ×20). (iv) Numerous pigment-laden melanophages identified within the lymph node tumor deposit (×40).
In August 2018, she developed upper abdominal and lower chest discomfort. A computerized tomography (CT) angiogram of the chest to rule out pulmonary embolism incidentally discovered multiple hepatic lesions not present on prior PET imaging. Dedicated CT of the abdomen and pelvis confirmed multiple hypodensities within the right and left hepatic lobes, highly suspicious for metastatic disease (Fig. 2A). A CT-guided needle biopsy of the liver was performed revealing pathology consistent with metastatic melanoma. IHC findings were diffusely positive for SOX10, Mart-1, and HMB45, while S100 staining was weakly positive. IHC was negative for HepAR1, GATA3, and AE1/AE3.
FIGURE 2.
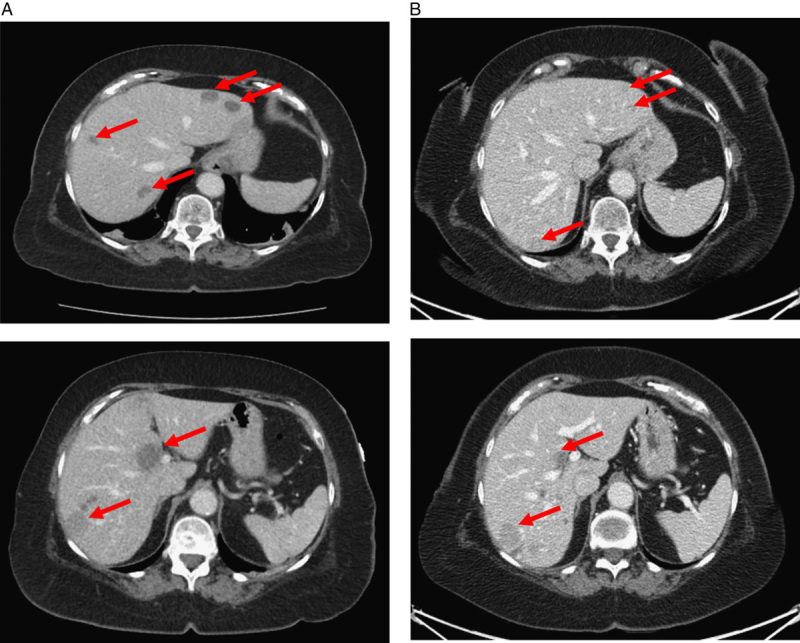
Spontaneous regression of metastatic melanoma in the liver. A, Computed tomography imaging of the abdomen/pelvis in August 2018 demonstrating evidence of multiple hypodense lesions involving the left and right hepatic lobes (red arrows). Computed tomography–guided hepatic tissue biopsy confirmed evidence of metastatic melanoma. B, Computed tomography imaging of the abdomen/pelvis in October 2018 showing decreased size of hepatic lesions without any form of therapy (red arrows). Repeat ultrasound-guided hepatic tissue biopsy also consistent with a diagnosis of metastatic melanoma.
The patient was seen as an initial consult in September 2018 in the Skin Cancer Clinic at the Duke Cancer Institute. Outside tissue biopsy specimens were reviewed by the Duke dermatopathology team revealing pathologic evidence of spontaneous melanoma regression involving a left axillary LN (Fig. 1B). Additional imaging studies were performed in October 2018, including magnetic resonance imaging of the brain showing no evidence of intracranial metastases. A PET-CT confirmed evidence of multiple hepatic metastases, and 2 of 3 lesions were notably of smaller size relative to the prior CT imaging study in August 2018: hepatic segment VIII/IV lesion 3.2×2.2–1.3×1.2 cm, left hepatic lobe lesion 2.1×1.4–1.1×0.9 cm (Fig. 2B). A repeat liver biopsy in November 2018 confirmed metastatic melanoma with IHC stains positive for S100 and MART45. BRAF mutation analysis was positive and detected a V600E mutation (p.Val600Glu;c.1799T>A). Overall, these findings were consistent with spontaneous regression of melanoma involving both the left axillary LN and the liver.
Discussion with the patient revealed that she had been asymptomatic and untreated for several years regarding her history of PMA. This prompted the initiation of pembrolizumab (anti-PD-1 antibody, 200 mg intravenously every 3 wk), and she underwent 7 cycles of treatment from November 2018 to March 2019. Despite the C-reactive protein level being elevated at baseline before initiating pembrolizumab [5.12 mg/dL (normal: ≤0.60 mg/dL)] and increasing throughout the course of her therapy (up to 13.78 mg/dL), she reported only occasional arthralgias. PET-CT imaging in April 2019 showed an increase in the size of hepatic lesions along with a new left lower lobe lung nodule, a new soft tissue density along the left vaginal cuff, and asymmetric thickening of the left inferior rectus muscle, all consistent with disease progression. The patient was started on dabrafenib/trametinib (100 mg every 12 h/2.0 mg daily) in June of 2019. Repeat PET-CT imaging in July of 2019 showed improvement of hepatic and pulmonary metastases, suggestive of response. The soft tissue nodules adjacent to the vaginal cuff and the thickening of the left inferior rectus muscle had resolved. The patient developed persistent diarrhea and a gastrointestinal bleed on dabrafenib/trametinib. She was subsequently transitioned to encorafenib/binimetinib (450 mg daily/45 mg twice per day) in September of 2019 but continued to have nausea, vomiting, low-grade fevers, and loose stools despite several dose reductions. Follow-up PET-CT imaging in October of 2019 showed continued tumor response with a decreased size in the hepatic lesions.
Given the patient’s persistent inability to tolerate BRAF/MEK inhibitor therapy, she was transitioned to ipilimumab (1 mg/kg) and nivolumab (3 mg/kg) in November of 2019 (intravenously every 3 wk). On follow-up, she was found to have developed grade 3 hepatotoxicity after the first cycle and was treated with high-dose steroids (prednisone 1 mg/kg daily) with improvement in aspartate transaminase from 514 to 46 U/L (normal: 15–41 U/L) and alanine transaminase from 286 to 68 U/L (normal: 14–54 U/L). PET-CT in January of 2020 showed a dramatic increase in the size and number of hepatic metastases including a right hepatic lobe lesion that increased from 2.2×1.8 to 11.8×10.4 cm. In light of these findings, the patient was transitioned back to encorafenib monotherapy (225 mg daily) in February of 2020.
She then began to report back pain, malaise, and lower extremity weakness, which required hospitalization from February to March of 2020, during which she exhibited worsening dysphonia and dysphagia. She was ultimately diagnosed with progressive polymyositis associated with an elevated aldolase of 12.9 U/L (normal: <7.7 U/L), normal creatine kinase of 107 U/L (normal: 30–220 U/L), a normal estimated sedimentation rate of 13 mm/h (normal: 0–15 mm/h), and an elevated C-reactive protein of 1.47 mg/dL (normal: ≤0.60 mg/dL). Ultimately, her strength gradually improved with high-dose steroids, and a CT scan from that admission showed stable disease.
During the course of her prednisone taper, she experienced worsening dysphagia and dysarthria. She was re-admitted later in March for suspicion of myasthenia gravis with polymyositis, given symptoms of bulbar weakness, gaze difficulty, and nasal dysarthria. Testing for the acetylcholine receptor binding antibody was negative, while assays for acetylcholine receptor modulating antibodies were inconclusive. Additional testing for striational antibody was negative. An electromyogram showed a disfigurative pattern consistent with an inflammatory myositis but could not rule out a neuromuscular disease process. She was ultimately presumed to have a neuromuscular junction disorder overlapping with polymyositis in the setting of checkpoint inhibitor toxicity. She was subsequently treated with 4 days of intravenous immunoglobulin (0.5 g/kg daily).
Unfortunately, her clinical status deteriorated rapidly thereafter with bleeding from hepatic metastases and Gram-negative bacteremia before additional therapeutic measures could be taken. She was transitioned to comfort care and discharged to inpatient hospice in April 2020.
Given the radiologic and pathologic evidence of spontaneous regression of both the left axillary LN and hepatic melanoma metastases, as well as the development of multiple irAEs in the setting of a previous inflammatory diagnosis of PMR, we hypothesized that a germline genetic alteration could provide a unifying pathogenic explanation for each of these immunologic phenomena. As a result, we collected peripheral blood mononuclear cells from this patient and subjected the isolated genomic DNA to whole-exome sequencing analysis.
MATERIALS AND METHODS
The subject of this study signed written formal consent for participation in an ongoing institutional review board–approved clinical protocol to study responses and resistance to immunotherapy in patients with advanced melanoma at Duke University Medical Center and the Duke Cancer Institute (NCT02694965).
Sequencing libraries were generated using Agilent SureSelect Human All Exon Kit (Agilent Technologies, CA) and index codes were added to each sample. Fragmentation was carried out by hydrodynamic shearing (Covaris, MA) to generate 180–280 bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities, and enzymes were removed. After adenylation of 3′ ends of DNA fragments, adapter oligonucleotides were ligated and enriched by polymerase chain reaction. The library was hybridized with biotin-labeled probes, and exons were captured using magnetic beads with streptomycin. Captured libraries were enriched in a polymerase chain reaction to add index tags to prepare for hybridization. Products were purified using AMPure XP system (Beckman Coulter, Beverly, MA) and quantified using the Agilent high sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. Paired-end sequencing was performed using an Illumina NovaSeq. 6000 platform with an average sequencing depth of 133,000,000. The reads were mapped to the human reference genome, GRCh37, using the Burrows-Wheeler Aligner, and haplotype phasing was performed.11 Identified genetic alterations were annotated using the ANNOVAR package.12
Initial germline DNA sequencing identified 1041 insertion-deletion (indel) mutations. This list was narrowed to include only exome mutations totaling 679. Considering the rarity of this clinical phenomenon, specific mutations were then filtered to include only mutations with a prevalence <5% in the general population. In addition, 45 indel mutations without known frequencies were also analyzed, resulting in a total of 156 indel mutations analyzed. A similar process was used to identify 209 single-nucleotide polymorphisms (SNPs) in exome regions with a prevalence of <5% in the general population. Global population mean allele frequencies for each identified SNP was determined based on the 1000 Genomes Project via Ensembl.13
All indel mutations were screened using both the Protein Variation Effect Analyzer (PROVEAN) and the Sorting Intolerant from Tolerant (SIFT) algorithms to determine whether the mutation is predicted to be functionally deleterious.14,15 Of the initial 156 mutations, 19 mutations were predicted to alter protein function based on the PROVEAN algorithm, 8 of which were validated to be likely functionally deleterious using the SIFT tool (Table 1). Each of the identified SNPs were also reviewed utilizing the Polymorphism Phenotyping v2 (PolyPhen-2) tool to predict those SNPs more likely to lead to a functional alteration (Table 2).16
TABLE 1.
Identified Deleterious Indels
| Gene | Protein Role | Type of Mutation | PROVEAN Score* | SIFT Confidence Value† |
|---|---|---|---|---|
| AHDC1 (AT-hook DNA binding motif containing 1) | DNA binding | Deletion | −2.77 | 0.667 |
| DCAF8 (DDB1 and CUL4-associated factor 8) | WD repeat-containing protein that interacts with ligase macromolecular complex | Deletion | −5.44 | 0.667 |
| EVX1 (even skipped homeobox 1) | Transcriptional repressor during embryogenesis | Insertion | −6.63 | 0.667 |
| OR2W3 (olfactory receptor family 2 subfamily W member 3) | G-protein–coupled receptor responsible for transduction of odorant signals | Deletion | −11.58 | 0.894 |
| RBM19 (RNA binding motif protein 19) | Regulates ribosome biogenesis | Deletion | −8.88 | 0.943 |
| SEC. 16B (SEC. 16 homolog B, endoplasmic reticulum export factor) | Organizes transitional endoplasmic reticulum sites and protein export | Insertion | −6.32 | 0.826 |
| VSIG10 (V-set and immunoglobulin domain containing 10) | Cell adhesion molecule binding | Deletion | −5.82 | 0.894 |
| ZNF83 (zinc finger protein 83) | Herpes simplex virus type 1 infection pathway | Deletion | −7.57 | 0.894 |
Protein Variation Effect Analyzer (PROVEAN) score threshold=−2.5.
Sorting Intolerant from Tolerant (SIFT) confidence value threshold >0.6.
TABLE 2.
Identified Deleterious Single-nucleotide Polymorphisms
| Gene | rsID | Position | Transcript ID | Coding Sequence Change | Protein Change | Global Allele Frequency |
|---|---|---|---|---|---|---|
| MEFV | rs11466023 | chr16:3249586 | NM_000243.3 | c.1105G>A | P369S | 0.020 |
| MEFV | rs11466024 | chr16:3249468 | NM_000243.2 | c.1223C>T | R408Q | 0.017 |
| MC1R | rs1805009 | chr16:89920138 | NM_002386.3 | c.880G>C | D294H | 0.003 |
RESULTS AND DISCUSSION
All identified mutations predicted to be deleterious were investigated using multiple different databases (including PubMed, Ensembl, the HUGO Gene Nomenclature Committee, UniProt Knowledgebase, and ClinVar) to examine an association between the mutation and possible effect on immunologic function. Using this approach, we were unable to identify a convincing indel mutation capable of explaining the unusual clinical presentation of this patient (Table 1). However, the SNP analysis revealed 3 SNPs predicted to exhibit deleterious function in genes relevant to this patient’s clinical course (Table 2). One of these SNPs involved the melanocortin-1 receptor (MC1R; D294H; rs1805009), previously linked with an elevated risk of cutaneous melanoma.17 The second SNP was associated with the Mediterranean fever gene (MEFV), otherwise known as pyrin and encoded by MEFV (P369S; rs11466023).18 A previous study identified this MEFV SNP to be associated with a highly variable phenotype and occasionally with FMF symptoms when found in combination with another MEFV (R408Q; rs11466024) mutation.19 Of 44 patients found to exhibit this MEFV (P369S/R408Q) variant, 40 were noted to be symptomatic with a wide spectrum of symptoms. However, only 5 of these patients met Tel-Hashomer criteria for the clinical diagnosis of FMF. Indeed, an additional search of our patient’s sequencing data revealed a MEFV-associated SNP encoding the mutation, MEFV (R408Q) (Table 2). Prior studies suggest that the P369S and R408Q alleles are likely to be in linkage disequilibrium (LD) and to cosegregate in cis.19,20 Consistent with this, the P369S and R408Q mutations are noted to be in LD based on the 1000 Genomes Project with R 2=0.85 in examining combined populations in LDlink (P<0.001).21,22 Therefore, this identified combination mutation variant of pyrin, MEFV (P369S/R408Q), is potentially an important contributor to this patient’s overall presentation and clinical course. This could include her presumed previous diagnosis of PMR/GCA, 2 episodes of spontaneous regression of metastatic melanoma lesions, as well the development of several irAEs associated with anti-PD-1 immunotherapy. Based on these data, we propose that mutations in inflammasome genes that generate only mild clinical signs or symptoms at baseline may manifest as a severe autoinflammatory or autoimmune syndrome during the course of immunotherapy.
The human pyrin inflammasome is an 86 kDa protein that is expressed predominantly in myeloid cell populations and is composed of 4 functional domains including the pyrin domain, a zinc finger domain (B-box) and a coiled coil domain which are thought to contribute to pyrin oligomerization, and a B30.2/SPRY domain that serves to interact with the pyrin domain and block its interaction with ASC (Fig. 3A).6 While the majority of mutations associated with FMF are located within this B30.2 domain towards the C-terminal region of pyrin, P369S and R408Q reside within the B-box domain (Fig. 3A).8 Previous studies have shown that the B-box domain interacts with proline serine-threonine phosphatase interacting protein (PSTPIP1); however, in vitro coimmunoprecipitation studies with overexpressed proteins showed no effect of P369S/R408Q on pyrin-PSTPIP1 binding.19 Although it seems reasonable to conclude that these mutations are enhancing pyrin oligomerization, the underlying mechanism explaining the relationship between these select mutations and the resulting autoinflammatory phenotype remains unclear (Fig. 3B).
FIGURE 3.
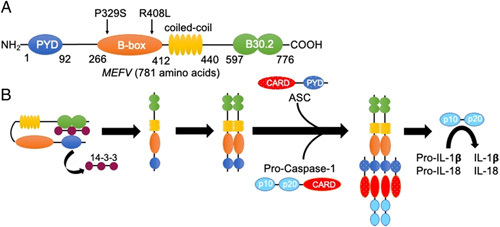
Structure and activation of the pyrin inflammasome. A, Basic domain structure of pyrin. MEFV, encoding gene. The P369S/R408L mutations in this patient are located within the B-box domain which is presumed to play a role in pyrin oligomerization. Most mutations associated with familial Mediterranean fever reside in the B30.2 domain which inhibits pyrin domain (PYD)—ASC interactions. B, Basic mechanism of pyrin inflammasome activation. 14-3-3 proteins bind to pyrin to maintain an inactivated state. The dissociation of 14-3-3 proteins allow for pyrin oligomerization and downstream activation of procaspase-1 followed by activation and release of interleukin (IL)-1β and IL-18.
Examples of spontaneous regression of metastatic melanoma in the literature are rare and have been attributed to interactions between the tumor and the host immune system.23–27 Although the trigger for these events is unknown, they have been conjectured to be potentially associated with surgical trauma or infection.28 In this patient’s case, neither site of spontaneous regression could be related to these events. These findings suggest that spontaneous melanoma regression can occur in the setting of a systemic inflammatory syndrome. Indeed, this observation is consistent with previous retrospective studies demonstrating a significantly lower incidence of cancer diagnoses in Isreali and Turkish FMF patients.29,30 To our knowledge, this represents the first report of spontaneous melanoma regression characterized by whole-exome sequencing.
In addition to spontaneous regression, this case report demonstrates the development of a series of irAEs including hepatitis and polymyositis with a neuromuscular junction syndrome in association with a gain-of-function genotype in the pyrin inflammasome and implicates a potential role for inflammasomes in the pathogenesis of inflammatory toxicities associated with checkpoint inhibitor immunotherapy. Notably, the frequency of the MEFV (P369S/R408Q) genotype in the white population has been estimated to be ~1.3%, while it exceeds 5% in the Japanese population, suggesting that the incidence of certain irAEs during the course of immunotherapy may vary significantly between different ethnic populations.20,31 Thus, future studies to examine the genetic testing for inflammasome mutations as predictive biomarkers for irAE are needed.
In light of their heterogeneity, it is likely that various pathogenic mechanisms contribute to the development of immunotherapy-associated toxicities. Given the role of inflammasomes in the regulation of neutrophils, it is possible that the pathogenesis of certain irAEs involve the innate immune system.32 Indeed, prior studies have linked neutrophil infiltration with Th17 differentiation, a process associated with a variety of autoimmune conditions.33–35 An improved understanding of how the genetics of the host and environmental influences interact to generate these toxicities will ultimately be necessary to effectively predict and manage patients that have developed irAEs. Given the complexity of these conditions and the growing use of immunotherapeutics with greater potency in the clinic, it is critical that tissue specimen collection protocols be initiated to more thoroughly investigate the underlying mechanisms associated with these irAEs.
CONCLUSIONS
We describe a patient with metastatic melanoma and a reported history of PMR with GCA that experienced 2 episodes of spontaneous distant disease regression before developing several irAEs including inflammatory hepatitis and polymyositis with myasthenic features while undergoing anti-PD-1 antibody immunotherapy. Whole-exome sequencing studies determined this patient to harbor a germline P369S/R408Q variant of the pyrin inflammasome, consistent with a previously reported genotype associated with atypical symptoms of FMF.19 Other authors have also indicated that these mutations may be sufficient to drive inflammatory toxicities in response to foreign stimuli.19 Altogether, these results suggest that an underlying autoinflammatory syndrome associated with inflammasome hyperactivation may influence the development of antitumor immunity as well as the generation of checkpoint inhibitor immunotherapy-associated toxicities.9 While case reports have several limitations, they can be hypothesis-generating. This observation indicates that larger clinical studies and additional preclinical mechanistic experiments are needed to test whether a relationship exists between the state of the inflammasome and both adaptive immune responses to the tumor as well as the evolution of irAEs in patients undergoing anti-PD-1 immunotherapy. We are therefore pursuing studies to investigate potential associations between inflammasome genetic alterations and the development of irAEs. Finally, the current standard-of-care management of irAEs often involves the use of high-dose steroids that are accompanied by their own set of adverse effects. These findings further suggest that studies are warranted to examine pharmacologic inflammasome inhibitors in the management of select irAEs that occur during the course of immunotherapy.
Acknowledgments
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
B.T. is supported by a NIH/NCI 1R37CA249085-01S1. N.C.D. is supported by a Damon Runyon Physician Scientist Award. B.A.H. is supported by a NIH/NCI 1R37CA249085-01, a NIH/NCI 1R01CA251136-01, a DoD ME200215, an ASCO/CCF ACRA, a Duke Health Scholars Award, a Duke Strong Start Award, and funding from Merck & Co. The remaining authors have declared that there are no financial conflicts of interest with regard to this work.
Contributor Information
Cameron J. Oswalt, Email: cameron.oswalt@duke.edu.
Rami N. Al-Rohil, Email: rami.alrohil@duke.edu.
Bala Theivanthiran, Email: bala.theivanthiran@duke.edu.
Tarek Haykal, Email: tarek.haykal@duke.edu.
April K.S. Salama, Email: april.salama@duke.edu.
Nicholas C. DeVito, Email: nicholas.devito@duke.edu.
Alisha Holtzhausen, Email: alishaah@email.unc.edu.
Dennis C. Ko, Email: dennis.ko@duke.edu.
Brent A. Hanks, Email: brent.hanks@duke.edu.
REFERENCES
- 1. Puzanov I, Diab A, Abdallah K, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30:2691–2697. [DOI] [PubMed] [Google Scholar]
- 3. Young A, Quandt Z, Bluestone JA. The balancing act between cancer immunity and autoimmunity in response to immunotherapy. Cancer Immunol Res. 2018;6:1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gray A, Grushchak S, Mudaliar K, et al. The microenvironment in primary cutaneous melanoma with associated spontaneous tumor regression: evaluation for T-regulatory cells and the presence of an immunosuppressive microenvironment. Melanoma Res. 2017;27:104–109. [DOI] [PubMed] [Google Scholar]
- 5. Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res. 2009;19:275–282. [DOI] [PubMed] [Google Scholar]
- 6. Schnappauf O, Chae JJ, Kastner DL, et al. The pyrin inflammasome in health and disease. Front Immunol. 2019;10:1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stoler I, Freytag J, Orak B, et al. Gene-dose effect of MEFV gain-of-function mutations determines ex vivo neutrophil activation in familial Mediterranean fever. Front Immunol. 2020;11:716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Eur J Immunol. 2018;48:230–238. [DOI] [PubMed] [Google Scholar]
- 9. Theivanthiran B, Evans KS, DeVito NC, et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J Clin Invest. 2020;130:2570–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Theivanthiran B, Haykal T, Cao L, et al. Overcoming immunotherapy resistance by targeting the tumor-intrinsic NLRP3-HSP70 signaling axis. Cancers (Basel). 2021;13:4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howe KL, Achuthan P, Allen J, et al. Ensembl 2021. Nucleic Acids Res. 2021;49(D1):D884–D891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Choi Y, Sims GE, Murphy S, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- 16. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wolf Horrell EM, Boulanger MC, D’Orazio JA. Melanocortin 1 receptor: structure, function, and regulation. Front Genet. 2016;7:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Touitou I, Lesage S, McDermott M, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24:194–198. [DOI] [PubMed] [Google Scholar]
- 19. Ryan JG, Masters SL, Booty MG, et al. Clinical features and functional significance of the P369S/R408Q variant in pyrin, the familial Mediterranean fever protein. Ann Rheum Dis. 2010;69:1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sugiura T, Kawaguchi Y, Fujikawa S, et al. Familial Mediterranean fever in three Japanese patients, and a comparison of the frequency of MEFV gene mutations in Japanese and Mediterranean populations. Mod Rheumatol. 2008;18:57–59. [DOI] [PubMed] [Google Scholar]
- 21. Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cervinkova M, Kucerova P, Cizkova J. Spontaneous regression of malignant melanoma—is it based on the interplay between host immune system and melanoma antigens? Anticancer Drugs. 2017;28:819–830. [DOI] [PubMed] [Google Scholar]
- 24. Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178–181. [DOI] [PubMed] [Google Scholar]
- 25. Khosravi H, Akabane AL, Alloo A, et al. Metastatic melanoma with spontaneous complete regression of a thick primary lesion. JAAD Case Rep. 2016;2:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ong SF, Harden M, Irandoust S, et al. Spontaneous regression of pulmonary metastatic melanoma. Respirol Case Rep. 2016;4:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schlabe J, Shah KA, Sheerin F, et al. Complete spontaneous regression of a metastatic melanoma of the mandible: a case report and follow-up recommendations. Int J Oral Maxillofac Surg. 2018;47:1519–1522. [DOI] [PubMed] [Google Scholar]
- 28. Tran T, Burt D, Eapen L, et al. Spontaneous regression of metastatic melanoma after inoculation with tetanus-diphtheria-pertussis vaccine. Curr Oncol. 2013;20:e270–e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brenner R, Ben-Zvi I, Shinar Y, et al. Familial Mediterranean fever and incidence of cancer: an analysis of 8534 israeli patients with 258,803 person-years. Arthritis Rheumatol. 2018;70:127–133. [DOI] [PubMed] [Google Scholar]
- 30. Bilgin E, Dizdar O, Guven DC, et al. Cancer incidence in familial Mediterranean fever patients: a retrospective analysis from central Anatolia. Rheumatol Int. 2019;39:1045–1051. [DOI] [PubMed] [Google Scholar]
- 31. Fujikura K. Global epidemiology of familial Mediterranean fever mutations using population exome sequences. Mol Genet Genomic Med. 2015;3:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siwicki M, Gort-Freitas NA, Messemaker M, et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol. 2021;6:eabi7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bedoya SK, Lam B, Lau K, et al. Th17 cells in immunity and autoimmunity. Clin Dev Immunol. 2013;2013:986789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weaver CT, Elson CO, Fouser LA, et al. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol. 2013;8:477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meng G, Zhang F, Fuss I, et al. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. [DOI] [PMC free article] [PubMed] [Google Scholar]