

Abstract
The nucleocapsid (N) protein is one of the four structural proteins of the SARS-CoV-2 virus and plays a crucial role in viral genome organization and, hence, replication and pathogenicity. The N-terminal domain (NNTD) binds to the genomic RNA and thus comprises a potential target for inhibitor and vaccine development. We determined the atomic-resolution structure of crystalline NNTD by integrating solid-state magic angle spinning (MAS) NMR and X-ray diffraction. Our combined approach provides atomic details of protein packing interfaces as well as information about flexible regions as the N- and C-termini and the functionally important, RNA binding, β-hairpin loop. In addition, ultrafast (100 kHz) MAS 1H-detected experiments permitted assignment of side chain proton chemical shifts, not available by other means. The present structure offers guidance for designing therapeutic interventions against the SARS-CoV-2 infection.
Keywords: COVID-19, SARS-CoV-2, nucleocapsid protein, N-terminal domain, magic angle spinning NMR, X-ray crystallography, atomic-resolution structure
Graphical Abstract
INTRODUCTION
SARS-CoV-2, a positive-sense single-stranded RNA virus from the beta-coronavirus family1 is the causative agent of the COVID-19 pandemic that killed millions of people and brought the world economy to a grinding halt2–3. The SARS-CoV-2 genome encodes four structural proteins: spike (S) glycoprotein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein4–5. All play crucial roles in the viral life cycle and pathogenicity, including host immunity evasion6. Due to its important role in genome packaging and ribonucleoprotein (RNP) formation the N protein represents a potential target for therapeutic interventions7–9.
The N protein comprises two folded domains, the N-terminal (NNTD, residues 40 to 174) and C-terminal (NCTD, residues 246 to 365) domains, connected by a ~70 amino acid linker region that contains a 13-residue serine/arginine motif, as well as extensive intrinsically disordered regions (IDRs) at the N- and C-termini10–15 (Fig. 1a). All domains including the NNTD play an important role on RNA genome interaction16–18.
Figure 1 |. Domain delineation, amino acid sequence, and MAS NMR spectra used for resonance assignment of SARS-CoV-2 NNTD.
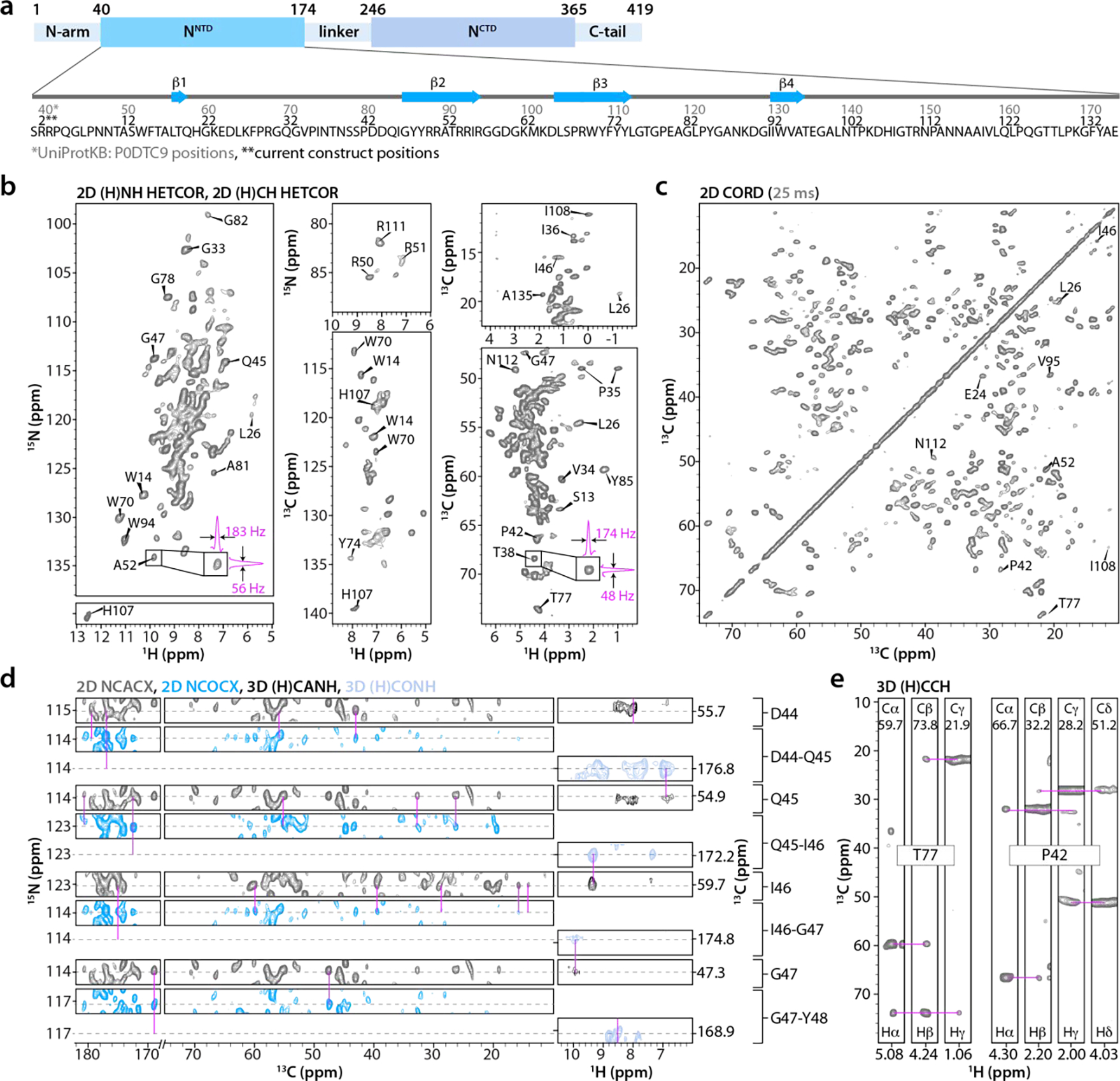
a) Top: domain organization of SARS-CoV-2 nucleocapsid (N) protein; N-terminal domain (NNTD), C-terminal domain (NCTD). Bottom: NNTD primary sequence and β-strands (blue arrows); Residues in the current NNTD construct (this work) 2–136 (black) correspond to residues 40–174 (gray) in the full-length N protein. b) Selected regions of 1H-detected 2D (H)NH HETCOR and (H)CH HETCOR spectra of U-13C,15N-NNTD. The expansions around the A52 and T38 cross peaks in (H)NH and (H)CH spectra depict 1D slices to illustrate the line widths in the two frequency dimensions. c) Aliphatic region of the 2D CORD spectrum (25 ms mixing time). d) Sequential assignments for the D44-G47 stretch of residues are illustrated with representative strips of 2D NCACX (gray), NCOCX (blue), 1H-detected 3D (H)CANH (black), and (H)CONH (paleblue) spectra. e) Selected strips from the 1H-detected 3D (H)CCH spectrum for residues T77 and P42 illustrating 13C and 1H the side chain resonance assignments. CORD, NCACX, and NCOCX spectra were recorded at MAS frequency of 14 kHz; (H)CANH and (H)CONH spectra were acquired at MAS frequency of 60 kHz; HETCOR and (H)CCH spectra were acquired at MAS frequency of 100 kHz. The number of scans, the number points in the direct and indirect dimensions are as follows: 2D (H)NH HETCOR- 32 scans, 1024 t2 points, 1034 t1 points; 2D (H)CH HETCOR- 64 scans, 1024 t2 points, 2310 t1 points; 2D CORD- 192 scans, 2048 t2 points, 840 t1 points; 2D NCACX- 2048 scans, 2048 t2 points, 96 t1 points; 2D NCOCX- 1536 scans, 3072 t2 points, 96 t1 points; 3D (H)CANH- 48 scans, 2048 t3 points, 112(15N) t2 points, 32(13C) t1 points; 3D (H)CONH- 32 scans, 2048 t3 points, 112(15N) t2 points, 32(13C) t1 points; and 3D (H)CCH- 8 scans, 1024 t3 points, 264 (13C) t2 points, 264 (13C) t1 points.
Several structures of β-coronavirus NNTD domains have been reported, all of which possess the same architecture, resembling a right hand19–24. The core structure is made of a four-stranded antiparallel β-sheet, the palm, from which the β2, β3 hairpin prominently protrudes. It contains several basic residues, and this basic finger and the palm have been implicated in RNA binding25. The loop connecting β2 and β3 is flexible, in agreement with missing density in this region of most X-ray structures (see below)19, 26. The N-terminal disordered tail projects outward, and may contribute to RNA binding19.
Here, we report atomic-resolution structure of crystalline NNTD, determined by combining X-ray crystallography and solid-state magic angle spinning (MAS) NMR spectroscopy. The protein crystallized in the P212121 space group with 4 chains in the asymmetric unit, and the X-ray structure was solved at 1.7 Å resolution. The MAS NMR structure of an individual NNTD chain, at 0.7 Å r.m.s.d. resolution, was determined using a single crystalline U-13C,15N-NNTD sample, based on 2968 non-redundant 13C-13C, 15N-13C, and 15N-1H distance restraints. Several inter-chain contacts were identified in 13C-13C correlation experiments, both for chains in the asymmetric unit as well as across asymmetric units. Side chain proton chemical shifts were assigned from high-frequency (100 kHz) MAS NMR correlation experiments and provided important structural information, such as the tautomeric state of H107 residue. Our results illustrate the power of integrating orthogonal structural techniques, here MAS NMR and X-ray diffraction, for assessing details of protein conformations. The atomic-resolution structure of crystalline NNTD reported here will guide the development of small-molecule inhibitors and biologics for treatment as well as biosensors for detection of SARS-CoV-2 infection27.
RESULTS
Resonance assignments
Chemical shift assignments and distance restraints for NNTD structure calculation were obtained using a single sample of fully protonated crystalline U-13C,15N-NNTD comprising residues 40–174 (current construct residues 2–136, Fig. 1a, see materials and methods section for experimental details). A total of eleven 2D and three 3D 1H- and 13C-detected high-frequency (100 and 60 kHz) MAS NMR experiments were recorded (Fig. 1b–e and Supporting Information Table S1). The spectra are of remarkably high resolution, with line widths as narrow as 35 Hz for 15N, 48 Hz for 13C, and 174 Hz for 1HN (Fig. 1b).
2D CORD28, NCACX, NCOCX at 25 ms mixing time, as well as 1H-detected 2D (H)NH HETCOR, 3D (H)CANH, and (H)CONH spectra (Fig. 1c, d) were used for sequential backbone assignments, and 13C and 15N chemical shifts are complete for 128 of 136 residues. For 5 residues, F28, P84, P113, P124, and E136, partial backbone chemical shift assignments were obtained, and, for 119 residues, backbone amide proton (HN) chemical shifts were assigned. The resonances of the first two residues, R2, and R3, are missing in the spectra, likely due to disorder. Overall, good agreement is observed between 1H and 15N chemical shifts determined in this work and those reported previously from solution NMR25, 29. MAS NMR assignments for a representative stretch of residues D44-G47 are illustrated in Fig. 1d.
Side chain 13C chemical shifts and inter-residue correlations were obtained from 2D NCACX, NCOCX, and CORD spectra, the latter acquired with 25, 100, and 500 ms mixing times (Fig. 1c and 2a). High spectral resolution permitted unambiguous assignment of numerous cross peaks, including those corresponding to aliphatic-to-aromatic (left panel) and aliphatic-to-aliphatic (right panel) side chain correlations (Fig. 2a). To determine side chain and backbone 1H chemical shifts, a 3D (H)CCH correlation experiment was recorded at the MAS frequency of 100 kHz (Fig. 1e). In conjunction with spectra acquired at the MAS frequency of 60 kHz, 84 side chain proton resonances for 71 residues and Hα resonances for 65 residues were assigned. For 11 Ala, 3 Val, 4 Ser, and 1 His residues complete 13C, 15N and 1H backbone and side chain chemical shifts were obtained. Overall, assignments for 132 residues were attained (Fig. S1) on the basis of 3728 cross-peaks in various spectra (Table 1). All chemical shifts are summarized in Table S2 of the Supporting Information.
Figure 2 |. Correlation spectra, inter-residue distance restraints and MAS NMR structure of a single NNTD chain.
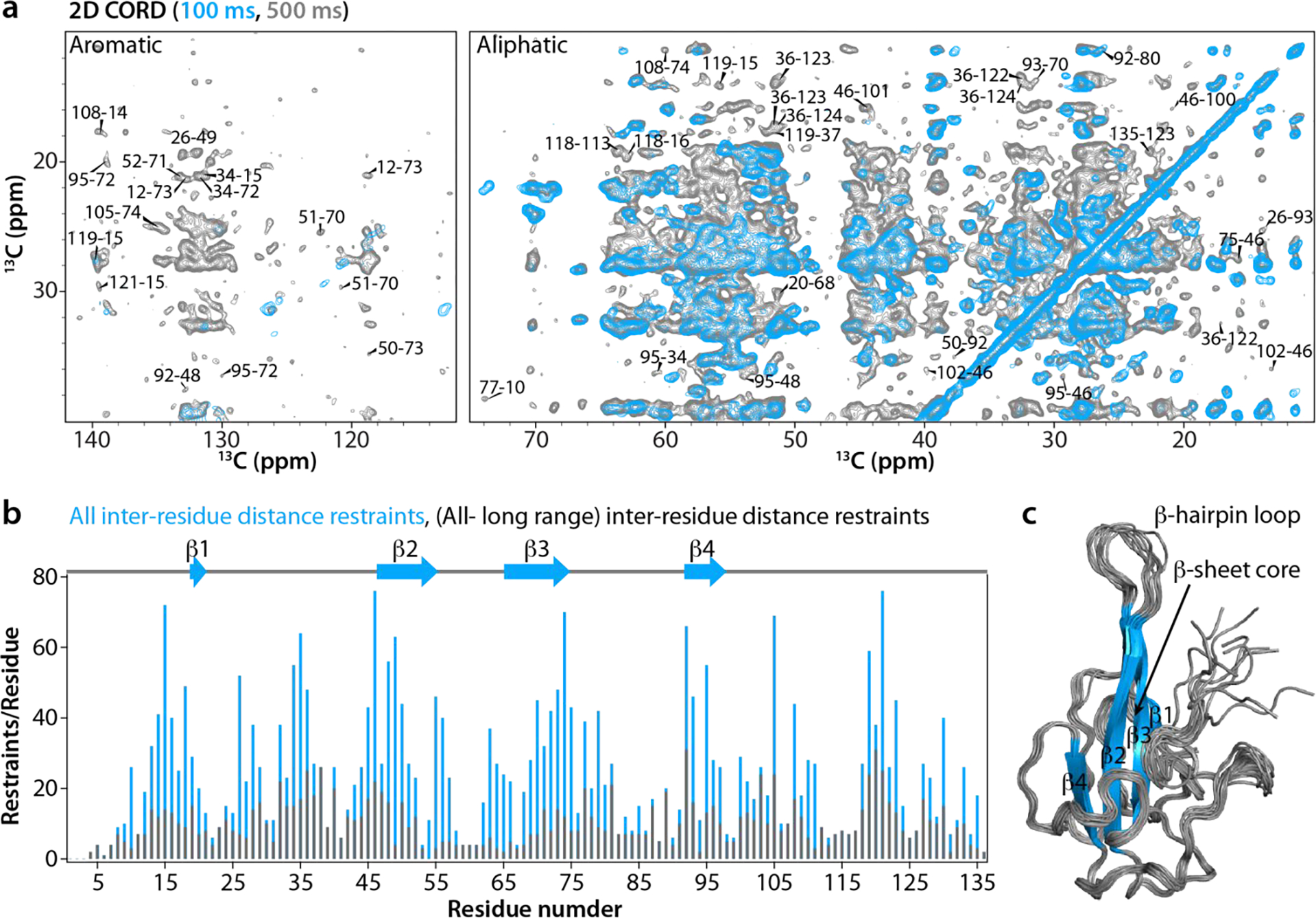
a) Superposition of representative regions of 2D CORD spectra of U-13C,15N-NNTD acquired with the mixing times of 100 ms (blue) and 500 ms (gray). Aromatic and aliphatic regions are shown in the left and right panel, respectively. Representative cross peaks between amino acids are labeled by residue numbers. b) The number of all inter-residue distance restraints and long-range inter-residue distance restraints are plotted for each residue along the polypeptide chain. c) Superposition of the ten lowest-energy MAS NMR structures of a single chain of SARS-CoV-2 NNTD. β-strands are colored in blue and labeled. The number of scans, the number points in the direct and indirect dimensions are as follows: 2D CORD (100 ms mixing time)- 96 scans, 3072 t2 points, 840 t1 points; 2D CORD (500 ms mixing time)- 192 scans, 3072 t2 points, 667 t1 points.
Table 1:
Summary of samples and the number of assigned peaks
| U-13C,15N-NNTD (MAS NMR) | No. of assigned peaks* |
|---|---|
| Intra-residue | 1943 |
| Sequential (|i-j|=1) | 495 |
| Medium range (1<|i-j|<5) | 306 |
| Long range (|i-j|≥5) | 972 |
| Long range (|i-j|∖5) (inter-chain) | 12 |
| Total assigned peaks (MAS NMR) | 3728 |
| U-15N- NNTD (solution NMR) | |
| Intra-residue | 159 |
| Total assigned peaks | 3887 |
Cross-peaks present in different experiments are counted only once.
Gratifyingly, many side chain protons of aromatic residues could be unambiguously assigned from the 1H-detected 100 kHz MAS NMR spectra (Fig. 1b). For example, for W70 and W94, located in the β-sheet core and assumed to be involved in RNA binding, side chain protons were assigned fully (W70) or partially (W94). Moreover, tautomeric state of H107 was determined (see below).
Structure of a single NNTD chain determined by MAS NMR
The structure of an NNTD single chain was calculated using 2968 non-redundant distance and 101 ϕ/ψ torsion angle restraints. Of these, 2197 are unambiguous 13C-13C, 763 15N-13C, and 4 1H-15N distance restraints, including 968 long-range (|i-j|≥5) restraints (Table 2 and Fig. S2 of the Supporting Information). The number of restraints per residue is plotted in Fig. 2b. The ten lowest-energy MAS NMR structures in the structural ensemble and an average structure of a single chain of NNTD are shown in Fig. 2c and Fig. S3 of the Supporting Information, respectively. All MAS NMR distance restraints are summarized in Table 2. With nearly 22 restraints/residue on average, the NNTD structure determined in this study represents a notable technical advance being one of only 2 MAS NMR structures of proteins larger than 100 residues per chain determined using more than 20 restraints/residue and reaching the maximum accuracy and precision attained for MAS NMR protein structures30, see below.
Table 2:
Summary of MAS NMR restraints and structure statistics
| MAS NMR distance restraints | 13C-13C | 15N-13C | 1H-15N |
|---|---|---|---|
| Unambiguous | 2197 | 763 | 4 |
| Intra-residue | 807 | 505 | 0 |
| Sequential (|i-j|=1) | 119 | 258 | 4 |
| Medium range (1<|i-j|<5) | 303 | 0 | 0 |
| Long range (|i-j|∵5) | 968 | 0 | 0 |
| Ambiguous | 4 | ||
| Total number of restraints assigned | 2968 (21.8 restraints per residue) | ||
| MAS NMR dihedral angle restraints | |||
| Φ | 101 | ||
| Ψ | 101 | ||
|
Structure statistics from 10 lowest energy subunits Violations (mean ± s.d.) | |||
| Distance restraints ≥ 7.2 Å (Å) | 0.144 ± 0.001 | ||
| Dihedral angle restraints ≥ 5° (°) | 1.528 ± 0.137 | ||
| Max. distance restraint violation (Å) | 1.254 | ||
| Max. dihedral angle restraint violation (°) | 17.267 | ||
| Deviations from idealized geometry | |||
| Bond lengths (Å) | 0.008 ± 0.000 | ||
| Bond angles (°) | 0.774 ± 0.012 | ||
| Impropers (°) | 0.516 ± 0.016 | ||
| Average pairwise r.m.s.d. (Å)* | |||
| Backbone (N, Ca, C’) | 0.7 ± 0.2 | ||
| Heavy | 1.2 ± 0.1 | ||
Disordered N-terminus (residues 1–9) excluded.
Like all coronavirus NNTD structures16, 19, 24–26, 31–32, the MAS NMR-derived structure exhibits the overall shape of a right hand, made up of a four-stranded β-sheet, comprising β1 (L18-T19), β2 (I46-R55), β3 (D65-Y74), and β4 (I92-A96). At its center, a long β-hairpin protrudes out from the palm (Fig. 2c). The irregular regions at the N- and C-termini exhibit well-defined backbone and side chain orientations in the MAS NMR structure, except for the first eight amino acids (R2-N9) and the last residue (E136) (see Fig. S2 and S3 of the Supporting Information). The lack of long-range inter-residue distance restraints for the N-terminal tail residues (P4-N9) and β-hairpin loop (I56-K64) suggests that they are dynamic (Fig. 2b). The precision of the single chain MAS NMR structure is 0.7 ± 0.2 Å, as measured by the pairwise atomic backbone r.m.s.d. for the 10 lowest-energy structures (excluding the disordered N-terminal tail, residues R2-N9) (Table 2 and Fig. S3 of the Supporting Information).
X-ray crystal structure of the NNTD
The protein crystallized in the P212121 orthorhombic space group with four monomers (chains A-D) in the asymmetric unit (Fig. 3a & Fig. S4a of the Supporting Information). Two views of the four chains are provided in Fig. 3a, and chain A is depicted in Fig. 3b. Details of the β2, β3 hairpin and loop region as well as the difference electron density map are shown in Fig. 3c. Complete statistics for X-ray data collection, phasing, and refinement are provided in Table 3. The average pairwise r.m.s.d. value between the four chains in the asymmetric unit is 0.5 ± 0.1 Å for the backbone atoms (excluding common missing residues in all four chains, R2-N9, Q20-D25, R57-P68, and P124-E136) (Fig. S5b & Table S3 of the Supporting Information). A positively charged region, comprising arginine residues in the β-sheet (R50, R51, R54, R55, R69) and at the tip of the β-hairpin finger (R57, K62), may possibly contribute to RNA binding. (Fig. S4c of the Supporting Information)19, 25.
Figure 3 |. X-ray crystal structure of SARS-CoV-2 NNTD.
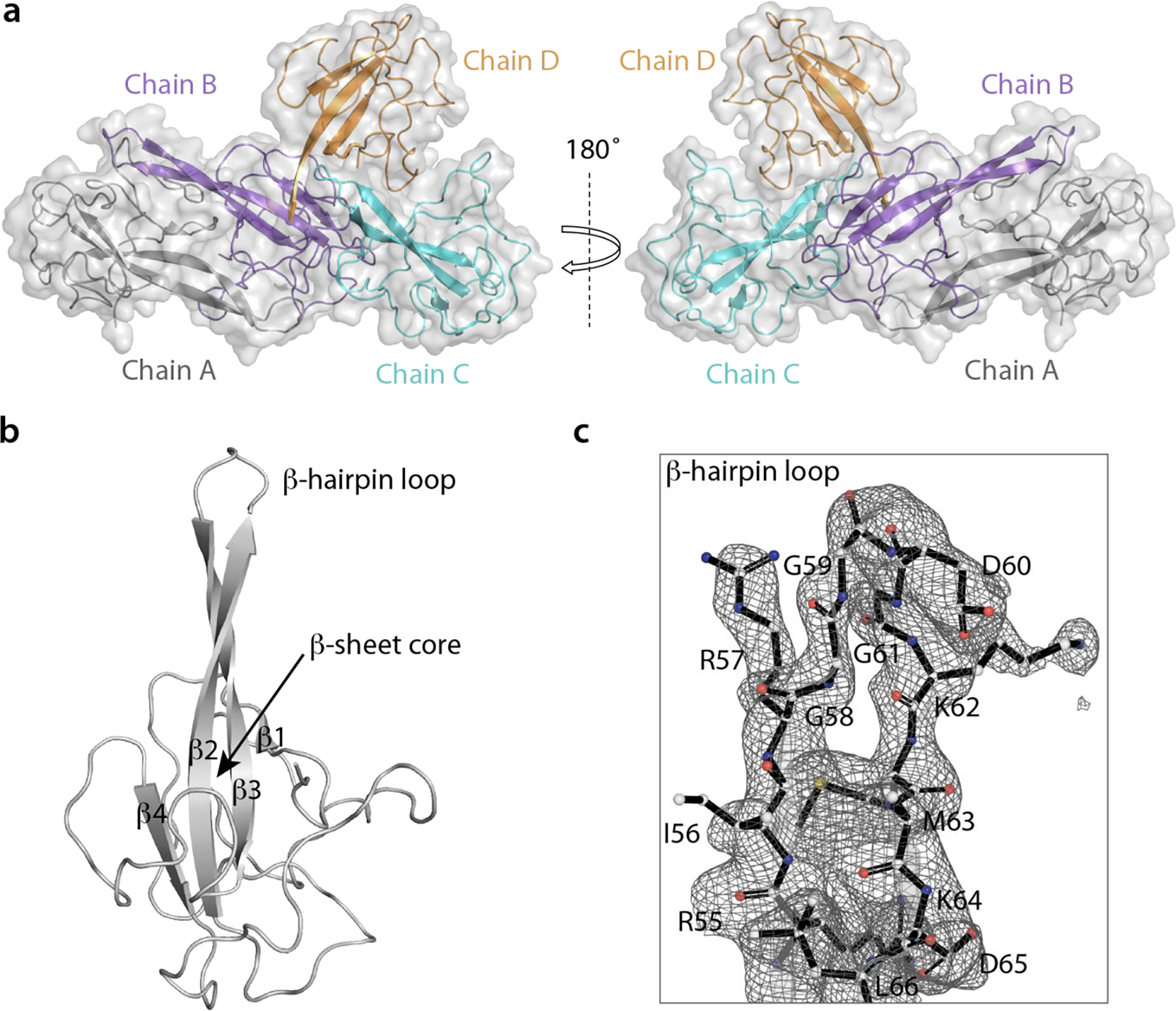
a) Ribbon and surface representation of the four NNTD chains in the asymmetric unit shown in two different orientations (PDB: 7UW3); chain A (gray), chain B (purple), chain C (cyan), chain D (orange). b) Structure of chain A (ribbon representation) with the strands in the β-sheet core labeled β1 to β4. c) Electron density map for the β-hairpin loop of chain A superimposed on the atomic model in stick representation.
Table 3:
X-ray data collection and refinement statistics (molecular replacement)
| SARS-CoV-2 NNTD | |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.9794 |
| Space group | P 212121 |
| Cell dimensions | |
| a, b, c (Å) | 58.76, 92.76, 95.59 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 37–1.70 |
| Rsym or Rmerge | 0.024(0.65)* |
| I /σ/-CC1/2 | 27.5(1.1)-99(42) |
| Completeness (%) | 98.9(98.2) |
| Redundancy | 11(7) |
| Refinement | |
| Refinement program | COOT |
| Resolution range (Å) | 37–1.70 |
| No. reflections | 59316 |
| Rwork/Rfree | 24.4/28.5 |
| No. of nonhydrogen atoms | |
| Protein | 3642 |
| Solvent (water) | 650 |
| B-factors | |
| Protein | 48.5 |
| Solvent (water) | 42 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.68 |
| PDB ID | 7UW3 |
Values in parentheses are for highest-resolution shell.
Interesting details about intra-tetramer interfaces in the crystal can be noted in the structure, with five unique types of contacts formed by the residues within the tetramer (Fig. 4a). Specifically, (i) the A-B interface comprises several residues (T18, H21, I36, R54, R56-G59, K64, L66, S67, V120, Q122, Y134, and A135) from both chain A and chain B; (ii) the A-C interface is very small and involves residues G59 and D60 of chain A contacting K131 of chain C; (iii) the B-C interface comprises several residues, such as R30-Q32, P42, D43, E98-L101, and P124-T127 of the chain B palm region, which are in contact with the C-terminal tail (residues L121-L129, K131, G132, Y134, A135), as well as H21, G22, and K23 of chain C; (iv) the B-D interface packs the palm regions of chain B and D against each other; and (v) the D-C interface comprises residues I56, and P113-A117 of chain D and T16, H21, A117, A118, V120, and Q122 of chain C.
Figure 4 |. Inter-chain interfaces and crystal packing in the NNTD structure.
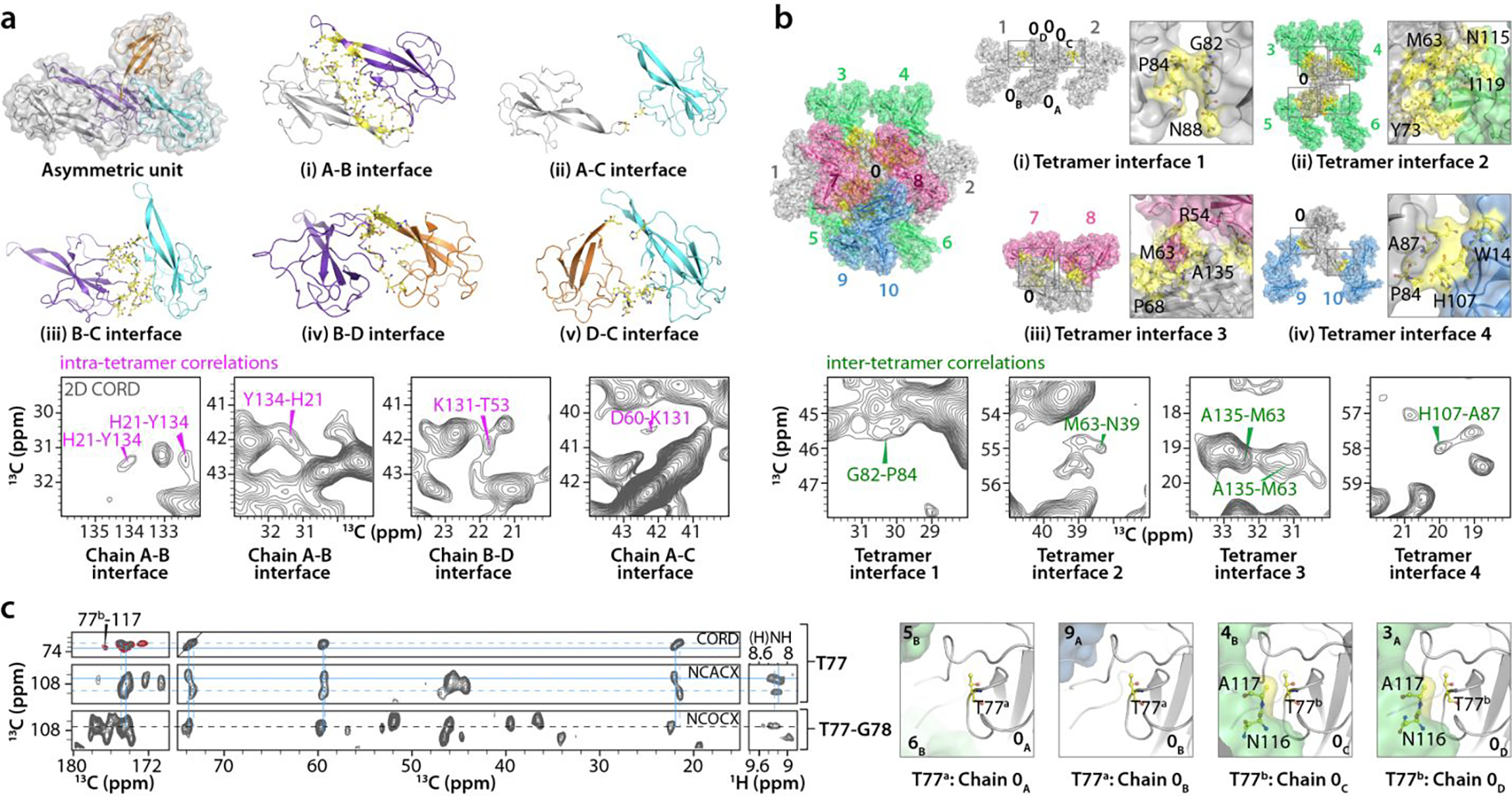
a) Intra-tetramer interfaces. Top & middle: five unique inter-chain interfaces in the asymmetric unit of NNTD crystal; chain A (gray), chain B (purple), chain C (cyan), chain D (orange). Interface residues are in yellow stick representation. b) Inter-tetramer interfaces. Top-left: each single tetramer (numbered 0) forms inter-tetramer interfaces with ten neighboring tetramers (1 to 10). Inter-tetramer interface residues are colored yellow. Top-right & middle-right: four unique inter-tetramer interfaces are formed based on symmetry operation. The nomenclature for a specific chain (A) in a tetramer (0) is 0A. Symmetry-related interfaces are boxed and expanded, with individual residues labeled and depicted in stick representation. Selected regions of a 2D CORD spectrum (100 ms mixing time) showing intra-tetramer correlations (magenta) and inter-tetramer correlations (green)(a and b bottom panels). c) Left: representative strips of the 2D CORD (top strips, 25 ms mixing time, gray; 100 ms mixing time, red), 2D NCACX (middle strip), 2D NCOCX (bottom strip), and 2D (H)NH HETCOR (right strips) spectra illustrating the sequential assignment for T77-G78. Resonances for two conformers, a and b, of T77 are indicated by dotted and solid lines, respectively. Right: inter-chain contacts of T77 for each chain are colored yellow. The number of scans, the number points in the direct and indirect dimensions are as follows: 2D CORD (25 ms mixing time)- 192 scans, 2048 t2 points, 840 t1 points; 2D CORD (100 ms mixing time)- 96 scans, 3072 t2 points, 840 t1 points; 2D NCACX- 2048 scans, 2048 t2 points, 96 t1 points; 2D NCOCX- 1536 scans, 3072 t2 points, 96 t1 points; 2D (H)NH HETCOR- 80 scans, 3072 t2 points; 400 t1 points.
The X-ray and the MAS NMR structures of the individual chains are in good agreement, with a backbone r.m.s.d. of 1.1 Å between the X-ray structure (averaged over the four chains in the asymmetric unit) and the MAS NMR structure (averaged over the ensemble of 10 lowest-energy structures) (Fig. S5 & Table S3 of the Supporting Information). Upon exclusion of chain D, which possesses the highest degree of disorder in the X-ray structure, the corresponding value becomes 0.7 Å. The average pairwise backbone r.m.s.d. between the four chains in the X-ray structure and within the ensemble of 10 lowest-energy MAS NMR structures are, both, 0.5 ± 0.1 Å (Table S3 of the Supporting Information). Side chain conformations for most residues in all four chains of the X-ray structure vary little, except for residues N10, R30, R55 and I56, which exhibit major differences (Fig. S6 of the Supporting Information). Unlike in the MAS NMR structure, which was determined using distance restraints or/and chemical shifts for most residues, except R2, R3, E136, density is either missing or very weak for residues R2-N9 and E136 in all chains and, for residues Q20-D25, R57-P68, and P124-E136 in chain D.
In addition to the five intra-tetramer interfaces, each tetramer (arbitrarily designated as “tetramer 0”) contacts 10 neighboring tetramers (tetramers 1 to 10) resulting in four distinct inter-tetramer interfaces, classified by symmetry operators. The nomenclature for a specific chain (A) in a tetramer (0) is denoted as 0A. The packing of tetramers in the crystallographic lattice is depicted in Fig. 4b. Inter-tetramer interface 1 is formed by two tetramers, 1 and 2, adjacent to tetramer 0. This interface comprises residues G82-P84 and A87-K89 of chain C (0C) and D (0D) of tetramer 0, and equivalent residues G82-P84 and A87-K89 in 2D and 1C, respectively (colored light gray in Fig. 4b(i)). Inter-tetramer interface 2 is formed by four tetramers, 3, 4, 5 and 6 (colored green in Fig. 4b(ii)) around tetramer 0. Several residues in 0A (N10-W14, T16, T38-S41, D44, R50-R57, K62, M63, L66, R69, Y71, Y73, P79, D106-V120), 0B (N10-S13, T16, T38-S40, R50-R57, G58-D60, M63, L66, R69, Y71, Y73, P79, G82-K89, R111-V120, Q122), 0C (N10, T11, W14, R30, N37-I46, Y48, L75-A87, V95-I119), and 0D (N10, T11, W14, N39, S41-D44, I46, Y48, L75-A87, A96-N116) are involved in crystallographically inequivalent interfaces (see Table S4). Inter-tetramer interface 3 is formed by two tetramers, 7 and 8 (colored pink in Fig. 4b(iii)), adjacent to tetramer 0. Residues H21-D25, K27, P29-Q32, A96-G99, Q122-A135 in 0A, Q20-K23, R57, K62, K64, L66-P68, Y134 in 0B, and residues R50, R51, T53-R57, G61-M63, D65-L66 in 0C, I36 and V120-L123 of 0D have contacts with residues comprising tetramers 7 and 8. Inter-tetramer interface 4 comprises two tetramers, 9 and 10 (colored blue in Fig. 4b(iv)), adjacent to tetramer 0. The residues involved in this interface are E80, G82-P84, A87-K89 of 0A and 9A and W14, D106-R112 in 10B and 0B, respectively. All intra- and inter-tetramer contacts for each NNTD residue are summarized in Table S4.
These unique intra- and inter-tetramer interfaces are reflected in distinct correlations in the MAS NMR spectra. In the (H)NH spectrum recorded at the MAS frequency of 100 kHz multiple resolved peaks, or broad (unresolved) peaks are observed for residues that are found in several different local environments, with 15N peak widths of ~85–110 Hz (Fig. 5b), whereas those for amino acids in single conformations are ~40–60 Hz (Fig. 5a,c). Examples of correlations corresponding to intra-tetramer A-B, A-C, and B-D interfaces, as well as to the inter-tetramer interfaces are shown in Fig. 4a,b, bottom panels.
Figure 5 |. Selected amino acids that exhibit multiple backbone amide resonances in the 2D CORD spectra of crystalline SARS-CoV-2 NNTD.
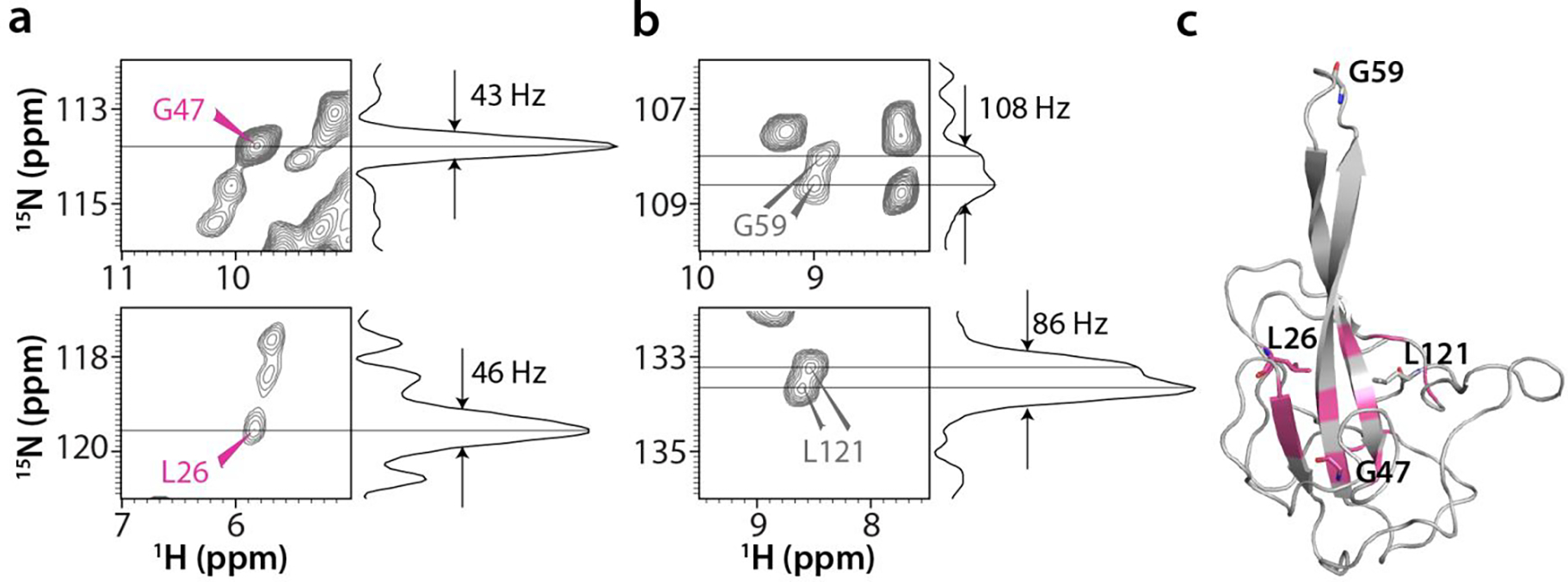
a, b) Individual single cross peaks reporting on a unique environment (a, pink labels) and doubled cross peaks reporting on different environments for the different chains (b, gray labels) with their corresponding 1D 15N slices. c) The location of amino acids (pink) possessing single amide backbone cross peaks, mapped onto the structure of chain A in the X-ray structure.
One notable example of unique intra-tetramer contacts, evidenced by multiple cross peaks in the MAS NMR spectra, is seen for the H21-Y134 pair of residues (Fig. 4a, bottom panel). It is evident from the X-ray structure that the inter-chain distances are much shorter than the intra-chain ones (3.5–4.6 Å and 7.0–8.0 Å, respectively); hence only inter-chain correlations are expected in the spectra (Fig. 6). Another interesting example involves residues D60 and M63 in the β2, β3-hairpin loop, for which intra- and inter-tetramer correlations were identified based on the following considerations: i) D60 from chain A with no inter-tetramer contacts has a unique intra-tetramer correlation with K131 at the A-C interface. In contrast, there are no intra-tetramer correlations involving D60 from other chains with K131, and only inter-tetramer correlations are present; ii) M63 has no intra-tetramer interactions; iii) M63 from chains A and B is in close proximity to N39 in the symmetry-related tetramer interface 2; and iv) M63 from chain C forms contacts with A135 across the tetramer interface 3 (Fig. 4b, bottom panel and Fig. 7).
Figure 6 |. H21-Y134 intra- and inter-chain contacts at the A-B interface in the X-ray structure.
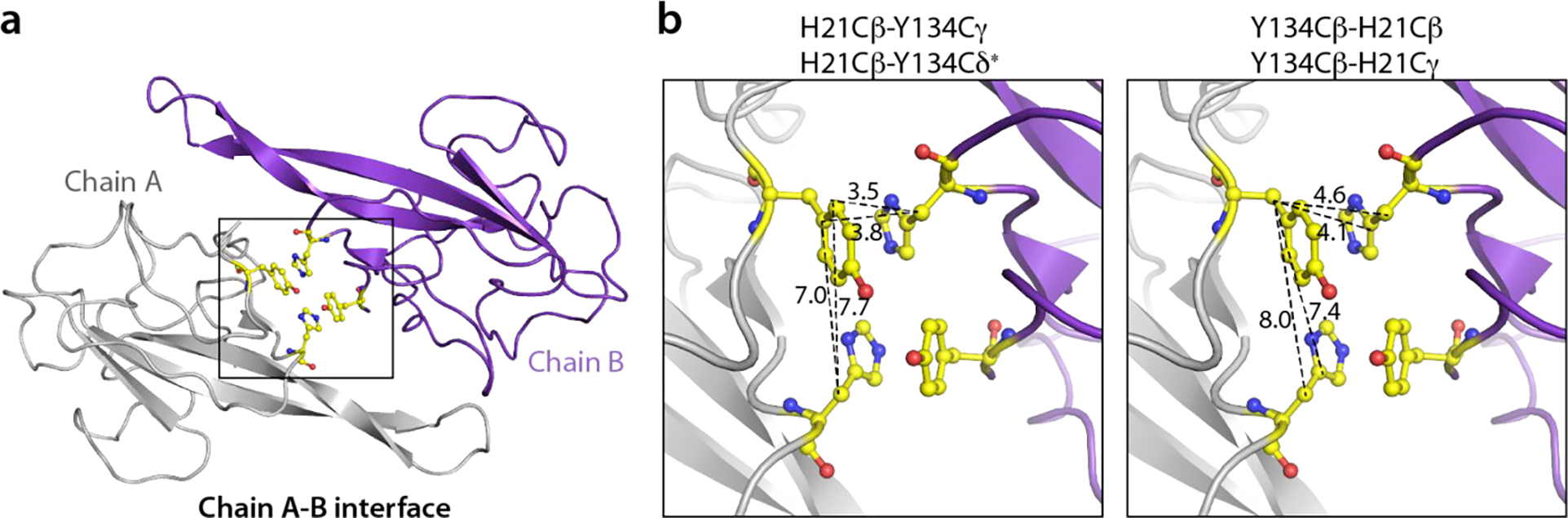
a) The A-B dimer in the asymmetric unit. b) Close-up view of the inter-chain contacts formed between H21 and Y134 from chains A and B. Inter-chain contacts (dotted lines) are shorter than the intra-chain contacts.
Figure 7 |. Intra- and inter-tetramer contacts involving the β-hairpin loop.
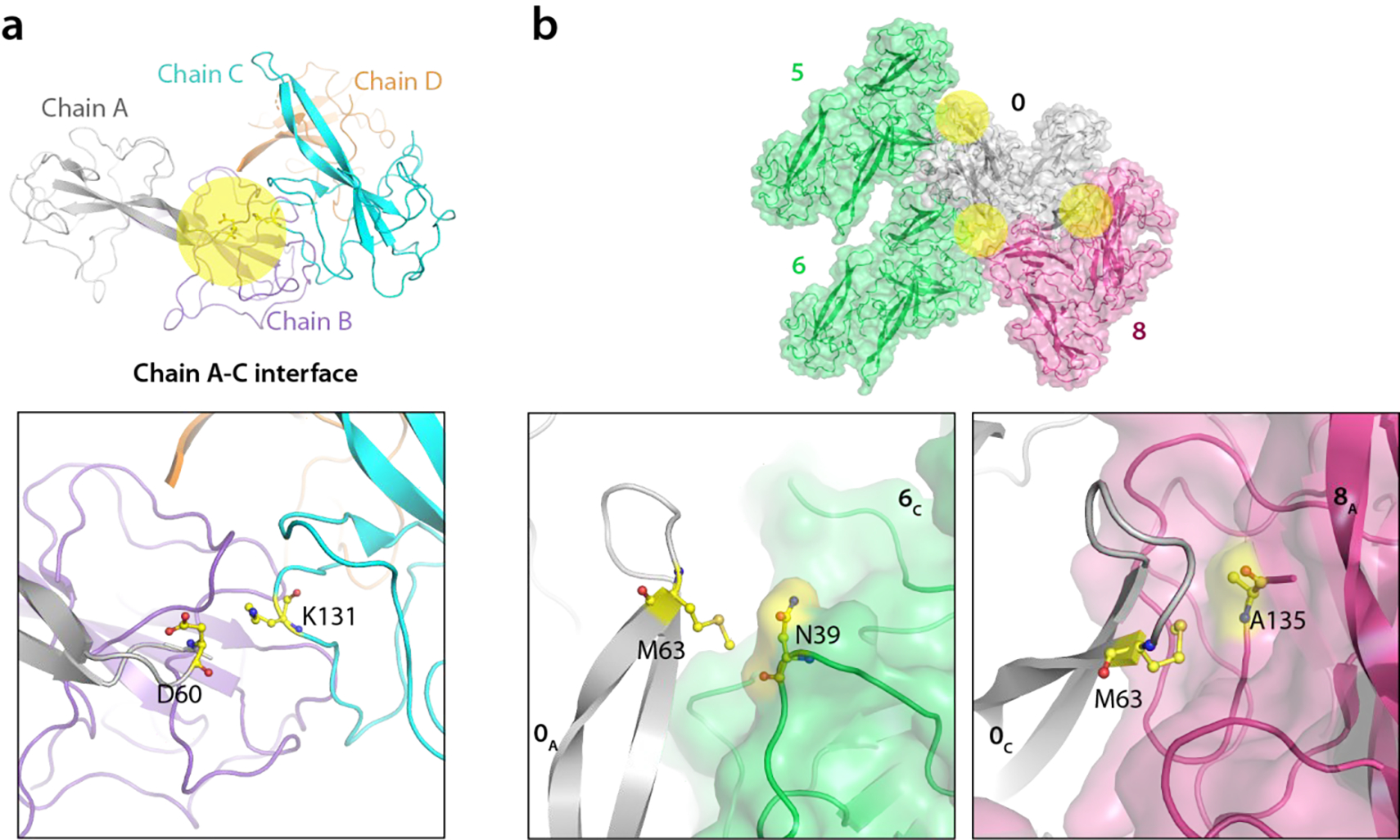
a) The A-C interface in the asymmetric unit of X-ray structure is colored yellow (top) and the D60-K131 contact for which correlations are observed in MAS NMR spectra are shown at the bottom. b) The three inter-tetramer interfaces around M63 in the crystal lattice are colored yellow (top) with a detailed view provided in the bottom two panels (bottom). The numbering of the tetramers and colors are as in Fig. 4b.
In addition to the intra- and inter-tetramer contacts discussed above, we also observed multiple conformers for many residues, as is evident from their distinct backbone and side chain chemical shifts. For example, T77 exhibits several resolved resonances with unique chemical shifts each (Fig. 4c, left panel). A unique T77Cβ - A117C’ cross peak is found for one of the two conformers (designated as conformer b, T77b) in the 100 ms mixing time CORD spectrum. This correlation is missing for the second conformer, T77a. Both conformers exhibit significant chemical shift differences (ΔN=1.4 ppm, ΔCβ=0.3 ppm, ΔCγ=0.3 ppm, ΔC’=0.6 ppm), consistent with distinct local environments for T77. Indeed, in the X-ray structure, T77 in 0A and 0B have no intra-or inter-tetramer contacts within 7 Å, consistent with this conformer being T77a. In contrast, T77 in chains 0C and 0D exhibits inter-tetramer contacts with A117 from chain 4B and 3A, respectively, therefore suggesting that these resonances correspond to the T77b conformer (Fig. 4c, right panels). Likewise, at least two distinct conformations are seen for A52, whose backbone chemical shifts are different (ΔN=2.0 ppm, ΔCα=0.4 ppm), suggesting that they exist in unique local environments (Fig. S7, top panel, Supporting Information). This finding is fully consistent with the X-ray structure (Fig. S7, bottom panel, Supporting Information), where A52 in 0A and 0B form inter-tetramer contacts with D43 and L101 from chains 6C and 5D, respectively, while A52 in chains 0C and 0D form contacts with E24 and K27 from chain 8A and chain 0B, respectively.
Tautomeric state of Histidine-107 in the crystal
In the (H)NH spectrum acquired with a CP contact time of 0.3 ms, H107 gives rise to two distinct 15Nε2-Hε2 cross peaks, at δ(15N)=170.2 ppm / δ(1H)=12.4 ppm and δ(15N)=170.4 ppm / δ(1H)=12.6 ppm, (Fig. 1b and Fig. 8a). In 2D (H)CH spectra, multiple Cε1-Hε1 and Cδ2-Hδ2 correlations are also observed (Fig. 8a). Taken together, these results suggest the presence of local heterogeneity around H107, consistent with two distinct local environments seen in the X-ray crystal structure (Table S4 of the Supporting Information).
Figure 8 |. Side chain imidazole state of H107 in NNTD crystal.
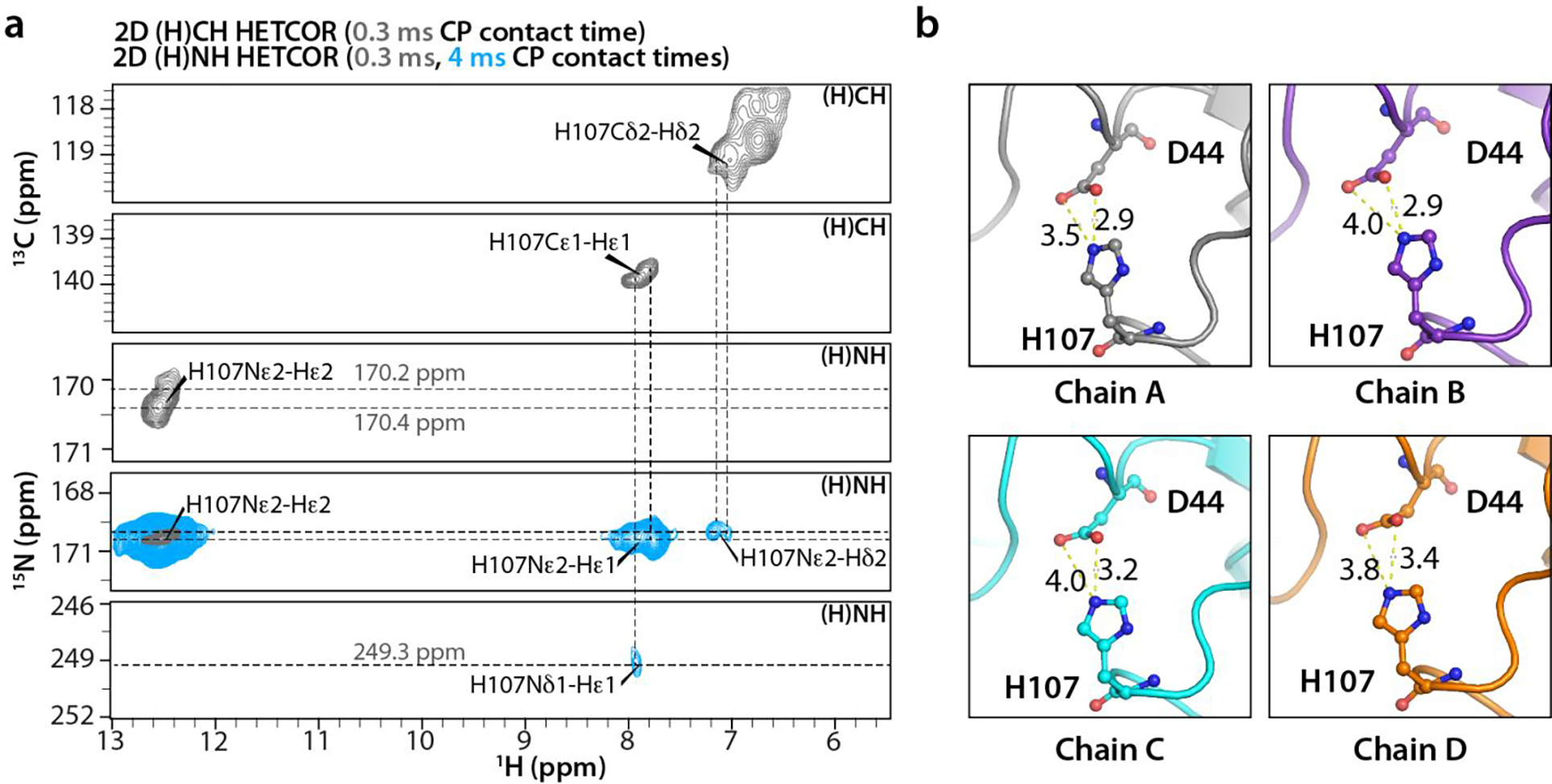
a) Strips extracted from 2D (H)CH and (H)NH HETCOR spectra acquired with CP contact times of at 0.3 ms (gray) and 4 ms (blue) (2D (H)NH HETCOR spectra only). H107 side chain 13C, 15N, and 1H resonances are labeled. b) Conformations of H107 and the neighboring D44 residues in Chains A-D of the crystal. The inter-atomic H107Nε2-D44Oδ1,δ2 distances vary between 2.9 Å to 3.4 Å. The number of scans, the number points in the direct and indirect dimensions are as follows: 2D (H)CH HETCOR- 64 scans, 1024 t2 points, 2310 t1 points; 2D (H)NH HETCOR- 16 scans, 1024 t2 points, 512 t1 points.
The tautomeric state of H107 was ascertained by a 1H-detected 2D (H)NH experiment acquired with a CP contact time of 4 ms (Fig. 8a). The cross peaks at δ(15N)~170 ppm/δ(1H)=7.9 ppm, δ(15N)~170 ppm / δ(1H)=7.7 ppm, and δ(15N)~249 ppm / δ(1H)=7.9 ppm were unambiguously assigned as Nε2-Hε1, Nε2-Hδ2, and Nδ1-Hε1 correlations, respectively, based on the Hε1 and Hδ2 chemical shift assignment from 2D (H)CH and 3D (H)CCH experiments (Fig. 8a). These results suggest that H107 is the Nε2-H tautomer33–36. Further evidence comes from a solution HMBC spectrum recorded at the pH of the crystallization (pH 6.3) (Fig. S8 of the Supporting Information), where Nε2-Hε1 and Nε2-Hδ2 correlations were observed at δ(15N)=170 ppm / δ(1H)=7.7 ppm and δ(15N)=170 ppm / δ(1H)=6.9 ppm, respectively. The Nδ1-Hε1 correlation at δ(15N)=243.4 ppm / δ(1H)=7.7 ppm is consistent with the H107 being the Nε2-H tautomer35–36.
The deshielded Hε2 resonance at δ(1H)~12.5 ppm suggests that the H107 imidazole may be involved in hydrogen bonding or close to a negatively charged group34, 37. The presence of a 15Nε2-Hε2 correlation in the (H)NH spectrum acquired with 0.3 ms CP contact time also suggests a short nitrogen-hydrogen distance, possibly a directly bonded Nε2-H. Indeed, in the X-ray crystal structure H107 is in close proximity to D44, and the interatomic H107Nε2-D44Oδ1,δ2 distances are 2.9, 2.9, 3.2 and 3.4 Å in chains A, B, C, and D, respectively (Fig. 8b). Taken together, these data indicate that the (H107)Nε2-H is close to the carboxylate side chain of D44.
DISCUSSION
The NTD domain of the of SARS-CoV-2 virus N protein has been previously structurally characterized by X-ray crystallography and solution NMR16, 19, 25. Here we present a structure that was determined by integrating MAS NMR and X-ray diffraction, providing important novel findings about distinct conformers, made possible by the remarkably high resolution of the MAS NMR spectra. The structural heterogeneity of NNTD is an outcome of crystallization, as seen in other NNTD crystal structures,19, 26, 31 and underscores the ability of the protein to form multiple types of contacts involving distinct conformers with unique local environments.
From the technical standpoint, the current study represents a notable advance for determining protein structures by MAS NMR since a single sample of only 3.6 mg of U-13C,15N-labeled NNTD packed in a 1.3 mm MAS rotor was sufficient to obtain all necessary spectra. The same sample (~0.5 mg) was subsequently packed in a 0.7 mm MAS rotor for 1H-detected experiments at 100 kHz MAS and 20.0 T, and these additional experiments yielded unique information on the side chain protons. Resonances for 98% of all amino acids were assigned and a large number of correlations corresponding to 2968 distance restraints, including 968 long-range restraints were obtained from 14 2D and 3D data sets. As a result, no preparations of isotopically diluted samples were necessary. At 21.8 restraints per residue, the NNTD single chain structure reported here is one of the highest precision and accuracy MAS NMR structures determined to date.
Notable is the complementarity of information obtained by MAS NMR and X-ray diffraction. In the X-ray structure atomic level details for individual chains and information on the quaternary arrangement in the crystal are obtained, while the strength of MAS NMR lies in providing information about dynamically disordered regions, proton positions, protonation and tautomeric states, and contacts with water molecules. To understand dynamics of the different regions of NNTD in solution and in the crystalline forms and their role in RNA binding, it will be interesting in future studies to perform measurements of relaxation rates, chemical shift and dipolar anisotropy tensors.
CONCLUSIONS
We have determined the structure of SARS-CoV-2 NNTD by integrating MAS NMR and X-ray diffraction. Our combined approach provided atomic details of packing interfaces as well as information about disordered residues at the N- and C-termini and the functionally important, RNA binding, β-hairpin loop. In addition, 1H-detected experiments at MAS frequency of 100 kHz permitted assignment of side chain proton chemical shifts, not available by other means. The present structure offers guidance for designing therapeutic interventions against the SARS-CoV-2 infection.
MATERIALS AND METHODS
Expression and purification of NNTD
The recombinant plasmid for expressing SARS-CoV-2 NNTD (residues 40–174, current construct residue numbering 2–136) was prepared form GenScript based on the sequence previously reported for NNTD19 and E.coli codon optimized. The template coding for SARS-CoV-2 NNTD was sub-cloned into a pET28a(+) vector fused with an N-terminal hexahistidine tag (His6), followed by a Tobacco Etch Virus (TEV) protease cleavage site, His6-TEV-NNTD. For the expression of U-13C,15N-NNTD and U-15N-NNTD, transformed E. coli BL21 (DE3) cells were cultured in 5 mL of Luria-Bertani (LB) medium containing 100 μg/mL kanamycin. LB pre-culture was incubated at 37 °C with agitation until the OD600 reached 1.0–1.2. 50 mL of M9 medium, supplemented with 1 g/L 15NH4Cl (U-15N-NNTD), or 1 g/L 15NH4Cl and 2 g/L U-13C6-glucose (U-13C-15N-NNTD), was inoculated with 1 mL of the LB pre-culture and incubated overnight at 37 °C. Following the overnight growth, 50 mL of M9 medium was transferred to 1 L of isotopically labeled M9 medium and incubated at 37 °C. Cells were grown to an OD600 of 1.0 and induced with a final isopropylthio-β-galactoside (IPTG) concentration of 400 μM. Protein was expressed at 20 °C for 16–18 hours and cells were harvested by centrifugation at 4000 × g for 10 min at 4 °C. The cell pellet was resuspended in lysis buffer (20 mM Tris-HCl, 500 mM NaCl, 20 mM imidazole, 0.02% NaN3, pH 8.0) and flash-frozen (−80 °C) for short-term storage.
Cells were opened after treatment with 1 mM phenylmethylsulfonyl fluoride (PMSF) by sonication at 40% power for 5 min (15 s pulse on and 45 s pulse off) on ice. The cellular lysate was clarified by centrifugation at 14,000 × g for 1 hr at 4 °C. His6-tagged NNTD was purified by affinity chromatography over a 5 mL HisTrap HP column (GE Healthcare). For elution, a gradient of 20–500 mM imidazole in 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 0.02% NaN3 was employed. The His6-tag was cleaved with TEV protease (1:25 ratio of TEV protease to His6-U-13C,15N-NNTD) for 12–16 hr at 4 °C and again fractionated over a 5 mL HisTrap HP column (GE Healthcare). Fractions were eluted in 20 mM Tris-HCl, 500 mM NaCl, 20 mM Imidazole, 0.02% NaN3, pH 8.0, and pure protein was buffer exchanged into crystallization buffer (20 mM Tris-HCl, 50 mM NaCl, pH 6.0) and NMR buffer (20 mM Tris-HCl, 150 mM NaCl, 90:10 H2O/D2O, pH 8.0). Buffer-exchanged NNTD was concentrated to 12 mg/mL for solution NMR and 30 mg/mL for crystallization, respectively.
Crystallization of NNTD
Small-scale crystallization was carried out at room temperature (~20 °C) using sitting-drop vapor diffusion. 2 μL of U-13C,15N-NNTD (20 mM Tris-HCl, 50 mM NaCl, pH 6.0) were mixed with 2 μL of crystallization buffer (100 mM MES, 30% PEG 4000, pH 6.5), modified from a previously published crystallization condition (PDB:6WKP)26.
U-13C,15N-NNTD for MAS NMR experiments was crystallized using a large-scale sitting-drop method based on the volumetric proportions of 500 μL sitting-drop crystallization wells (Fig. S9 of the Supporting Information). A series of pre-sterilized Petri dishes were used to form a concentric sitting-drop vessel with a reservoir (volume capacity of 25–75 mL) and three droplet wells (optimal volume of 300–1000 μL). Similar to the small-scale crystallization, the crystallization droplet mixture comprised 250 μL of NNTD and 250 μL crystallization buffer. The large-scale sitting-drop vessel was sealed using vacuum grease and left undisturbed at 20 °C for 5 days. Once crystallization was complete, the protein crystals were harvested and packed into a Bruker 1.3 mm rotor by ultracentrifugation at 10,000 × g for 15 minutes at 4 °C. The fully packed 1.3 mm rotor contained 3.6 mg of hydrated protein crystals. The same sample (~0.5 mg) was subsequently packed in a 0.7 mm rotor for experiments at 100 kHz MAS and 20.0 T.
Diffraction data collection and structure determination
X-ray diffraction data of protein crystals were collected at beamline 12–2 at the Stanford Synchrotron Radiation Lightsource (SRRL). All diffraction data used for analysis were collected from crystals grown in 100 mM MES, 30% PEG4000, pH 6.0 at 100K. All diffraction data were indexed and integrated using the program XDS38, and scaled using the program AIMLESS from the CCP4 suite39. The structure was solved by molecular replacement (MOLREP, CCP4 suite) using one monomer of PDB:ID 6M3M. Structure refinement was carried out in Phenix40 with manual building in COOT41 (PDB: ID 7UW3).
Solution NMR spectroscopy
A 2D 1H-15N HSQC spectrum of 850 mM U-15N-NNTD in 20 mM Tris-HCl, 150 mM NaCl, 90:10 H2O/D2O buffer (pH 8.0) was recorded 25 °C on a 14.1 T Bruker Neo spectrometer equipped with a triple-resonance inverse detection (TXI) probe. The Larmor frequencies were 600.13 MHz (1H), 150.9 MHz (13C), and 60.8 MHz (15N). Backbone and sidechain 1H and 15N chemical shift assignments (Fig. S10 and Table S5 of the Supporting Information) were obtained by comparison with SARS-CoV-2 NNTD (BMRB:34511) and SARS-CoV-1 NNTD (BMRB:6372) chemical shifts in the BMRB25, 29. 1H-15N HMBC spectra were recorded at pH 6.3 to match the crystallization pH, with delays set to 5.4 ms, 25 ms, and 50 ms, corresponding to 1/2 of 1J and 2,3J coupling constants of 92 Hz, and 10 and 20 Hz, respectively.
MAS NMR spectroscopy
MAS NMR spectra of U-13C,15N-NNTD protein crystals were recorded on a 14.1 T Bruker AVIII spectrometer outfitted with a 1.3 mm HCN probe. The Larmor frequencies were 599.8 MHz (1H), 150.8 MHz (13C), and 60.7 MHz (15N). The MAS frequencies were controlled to within ±10 Hz by a Bruker MAS controller. The actual sample temperature was maintained at ~25 °C throughout the experiments.
Typical 90° pulse lengths were 1.3–1.5 μs for 1H, 2.6–2.9 μs for 13C, and 3.2–3.5 μs for 15N. 1H-13C and 1H-15N cross-polarizations were performed with an 80–100% linear amplitude ramp on 1H, with contact times of 1–1.5 ms and 2–2.5 ms, respectively. The center of the ramp was matched to the Hartmann–Hahn condition at the first spinning sideband. 2D 13C-13C CORD28, 2D NCACX, and 2D NCOCX spectra were recorded at the MAS frequency of 14 kHz. CORD mixing times were 25 ms, 100 ms, 250 ms, and 500 ms, and the 1H radio frequency (rf) field strength during CORD mixing was 14 kHz. Band-selective 15N-13C spectrally induced filtering in combination with cross polarization (SPECIFIC-CP)42 with a contact time of 6.0–7.5 ms. SPINAL-6443 decoupling (90–100 kHz) was used during the evolution and acquisition periods.
2D 13C-13C RFDR, 1H-detected 2D (H)NH and (H)CH HETCOR as well as 3D (H)CANH and (H)CONH spectra were recorded at the MAS frequency of 60 kHz with a 2.4 ms RFDR mixing time. 15 kHz swept-low power TPPM (slTPPM)44 was used for 1H-heteronuclear decoupling during acquisition. 10 kHz WALTZ-1645 broadband decoupling was used for 13C and 15N decoupling during 1H acquisition. For 3D 1H-detected (H)CANH and (H)CONH spectra, CA-N and CO-N CP contact times were 6–7.5 ms with a contact-amplitude spin lock of about 25 kHz on 13C and a tangent-modulated amplitude spin lock of mean rf field amplitude of about 35 kHz on 15N46.
Additional MAS NMR spectra were recorded on a 20.0 T Bruker AVIII spectrometer outfitted with a 0.7 mm HCND and a 1.3 mm HCN probes. The Larmor frequencies were 850.4 MHz (1H), 213.9 MHz (13C), and 86.2 MHz (15N). The MAS frequency was 100 kHz, controlled to within ±50 Hz by a Bruker MAS controller. The sample temperature was maintained at ~25 °C throughout the experiments. 90° pulse lengths were 1.3 μs for 1H, 3.15 μs for 13C, and 3 μs for 15N. The (H)NH spectrum was recorded using a back CP (HN) of 800 μs contact time with a 80–100% linear amplitude ramp on 1H; the rf field strengths were 145 kHz for 1H and 48 kHz for 15N. The forward CP (NH) used 200 μs contact time, with an 80–100% linear amplitude ramp on 1H; the rf field strengths were 134 kHz for 1H and 48 kHz for 15N. For (H)CH and (H)CCH experiments, the 13C CP rf field strength was set to 30 kHz; for forward and back CP, linear amplitude ramps on 1H were 80–100% and 100–80%; the 1H rf field strengths were set at 138 kHz and 132 kHz; the contact times were 600 μs and 175 μs, respectively. CC RFDR mixing time was 0.56 ms. For all spectra, the 1H rf field strengths for water suppression and proton decoupling were set at ¼ ωr, and a WALTZ sequence at 10 kHz was used for heteronuclear decoupling of both 13C and 15N. An additional 2D (H)NH spectrum was recorded at 60 kHz MAS, using a 1.3 mm HCN probe. The CP contact time was 4 ms and the remainder of all conditions were identical to those at 14.1 T (see above).
Data processing
All MAS NMR data were processed using Bruker TopSpin and NMRPipe47. 1H resonances are referenced with respect to water at 4.7 ppm and 13C and 15N to the external standards adamantane and ammonium chloride, respectively. All 2D and 3D data sets were processed by applying 30°, 45°, 60°, and 90° shifted sine bell apodization, followed by a Lorentzian-to-Gaussian transformation in all dimensions. Forward linear prediction to twice the number of original data points was applied in the indirect dimension for some data sets, followed by zero filling. 2D and 3D 1H-detected data sets were processed with Gaussian and/or square sine window apodization and quadrature baseline correction.
Resonance assignments
Spectra were analyzed using CCPN48 and Sparky49–50 and MAS NMR backbone and side chain 1H-15N resonance assignments were initially carried out by comparison with solution NMR chemical shifts25, 29 and verified by de novo backbone assignment based on 2D 13C-13C CORD (25 ms mixing time) and RFDR spectra, combined with 2D NCACX (25 ms mixing time), 2D NCOCX (25 ms mixing time), 1H-detected 2D (H)NH HETCOR, 3D (H)CANH, and 3D (H)CONH spectra. Side chain carbon and nitrogen resonances were assigned using 2D CORD, 2D NCACX, 2D NCOCX and 2D (H)NH spectra, and side chain and backbone hydrogens were assigned using 1H-detected 2D (H)NH, (H)CH HETCOR and 3D (H)CCH experiments.
Structure calculation of SARS-Cov2 NNTD
The MAS NMR structure of a single NNTD chain was calculated in Xplor-NIH version 2.5351−53 using 13C-13C, 15N-13C, and 1H-15N distance restraints, extracted from 2D CORD (100 ms, 250 ms, and 500 ms mixing times), NCACX, NCOCX and (H)NH HETCOR spectra and backbone dihedral angles predicted by TALOS-N54 from the experimental 1H, 13C, and 15N chemical shifts. The bounds for the distance restraints were set to 1.5–6.5 Å (4.0 ± 2.5 Å) and 2.0–7.2 Å (4.6 ± 2.6 Å) for intra- and inter-residue restraints, consistent with our previous studies30, 55.
Calculations were seeded using the primary sequence as extended strands. 1000 structures were generated with molecular dynamics simulated annealing in torsion angle space with two successive annealing schedules and a final gradient minimization in Cartesian space, essentially as described previously30, 55 and detailed below.
Two successive annealing schedules were used, the first in vacuum with the REPEL module and the second with an implicit solvent refinement using the EEFx module56. The ten lowest-energy structures were selected and served as input for the second schedule, and the ten lowest energy structures of this as input for the final ensemble (PDB: 7SD4). Standard terms for bond lengths, bond angles, and impropers were applied to enforce correct covalent geometry.
The first annealing calculation was essentially identical to that reported previously 30, 55, with initial random velocities at 3,500 K constant-temperature molecular dynamics run for the shorter of 800 ps or 8,000 steps, with the time step size allowed to float to maintain constant energy. Subsequently, simulated annealing calculations at reduced temperatures in steps of 25 K to 100 K were carried out for the shorter of 0.4 ps or 200 steps. Force constants for distance restraints were ramped from 10 to 50 kcal/mol•Å2. Dihedral angle restraints were disabled for high-temperature dynamics at 3,500 K and subsequently applied with a force constant of 200 kcal/mol•rad2. The force constant for the radius of gyration was geometrically scaled from 0.002 to 1, and a hydrogen bond term, HBPot, was used to improve hydrogen bond geometries57. After simulated annealing, structures were minimized using a Powell energy minimization scheme.
For the second schedule performed in implicit solvent, all parameters were set as in the example EEFx of Xplor-NIH. Annealing was performed at 3,500 K for 15 ps or 15,000 steps, whichever was completed first. The starting time step was 1 fs and was self-adjusted in subsequent steps to ensure conservation of energy. Random initial velocities were assigned about a Maxwell distribution at the starting temperature of 3,500 K. Subsequently the temperatures were reduced to 25 K in steps of 12.5 K. At each temperature, 0.4 ps dynamics were run with an initial time step 1 fs. Force constants for distance restraints were ramped from 2 to 30 kcal/mol•Å2. The dihedral restraint force constants were set to 10 kcal/mol•rad2 for high-temperature dynamics at 3,000 K and 200 kcal/mol•rad2 during cooling. After the EEFx module, structures were minimized using a Powell energy minimization scheme.
Structure analysis and visualization
Atomic r.m.s.d. values were calculated using routines in Xplor-NIH (version 2.53)51–53. The visualization of structural ensembles was rendered in PyMOL58, using in-house shell/bash scripts. Secondary structure elements were classified according to STRIDE59 and manual inspection.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Shi Bai for assistance with acquiring solution HMBC spectra and Roman Zadorozhnyi for helpful discussions. This work was supported by the National Institutes of Health (NIH Grant P50AI1504817, Technology Development Project 2). We acknowledge the National Institutes of Health (NIGMS Grant P30 GM110758) for the support of core instrumentation infrastructure at the University of Delaware. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health (NIH Grant P30GM133894).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
SUPPORTING INFORMATION
Summary of resonance assignment of SARS-CoV-2 NNTD; X-ray crystal structure; ten lowest-energy conformers of MAS NMR structures; all and selected side chain conformations of MAS NMR and X-ray structures; multiple resonances for A52 in the MAS NMR spectra; histidine protonation state in solution NMR; mass spectrum, crystallization, and purification of NNTD; 2D 1H-15N HSQC NMR spectrum of U-15N-NNTD; summary of all NMR experiments; MAS NMR and solution NMR chemical shifts of NNTD; r.m.s.d. values between X-ray and MAS NMR structures; intra- and inter-tetramer contacts in NNTD crystals; author contributions. This information is available online at http://pubs.acs.org.
DATA AVAILABILITY
The MAS NMR atomic structure coordinates and X-ray structure coordinates of SARS-CoV-2 NNTD have been deposited in the Protein Data Bank under accession codes 7SD4 and 7UW3, respectively. MAS NMR chemical shifts of SARS-CoV-2 NNTD has been deposited in the Biological Magnetic Resonance Data Bank under accession codes 30955.
REFERENCES:
- 1.Gorbalenya AE; Baker SC; Baric RS; de Groot RJ; Drosten C; Gulyaeva AA; Haagmans BL; Lauber C; Leontovich AM; Neuman BW; Penzar D; Perlman S; Poon LLM; Samborskiy DV; Sidorov IA; Sola I; Ziebuhr J, The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5 (4), 536–544, doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibn-Mohammed T; Mustapha KB; Godsell J; Adamu Z; Babatunde KA; Akintade DD; Acquaye A; Fujii H; Ndiaye MM; Yamoah FA; Koh SCL, A critical analysis of the impacts of COVID-19 on the global economy and ecosystems and opportunities for circular economy strategies. Resour. Conserv. Recycl. 2021, 164, 105169, doi: 10.1016/j.resconrec.2020.105169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicola M; Alsafi Z; Sohrabi C; Kerwan A; Al-Jabir A; Iosifidis C; Agha M; Agha R, The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int. J. Surg. 2020, 78, 185–193, doi: 10.1016/j.ijsu.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brian DA; Baric RS, Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30, doi: 10.1007/3-540-26765-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui J; Li F; Shi ZL, Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17 (3), 181–192, doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beyer DK; Forero A, Mechanisms of antiviral immune evasion of SARS-CoV-2. J. Mol. Biol. 2021, 167265, doi: 10.1016/j.jmb.2021.167265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masters PS; Sturman LS, Background paper functions of the coronavirus nucleocapsid protein. Adv. Exp. Med. Biol. 1990, 276, 235–238, [DOI] [PubMed] [Google Scholar]
- 8.McBride R; van Zyl M; Fielding BC, The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6 (8), 2991–3018, doi: 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savastano A; Ibáñez de Opakua A; Rankovic M; Zweckstetter M, Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat. Commun. 2020, 11 (1), 6041, doi: 10.1038/s41467-020-19843-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CK; Sue SC; Yu TH; Hsieh CM; Tsai CK; Chiang YC; Lee SJ; Hsiao HH; Wu WJ; Chang WL; Lin CH; Huang TH, Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 2006, 13 (1), 59–72, doi: 10.1007/s11373-005-9035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lo YS; Lin SY; Wang SM; Wang CT; Chiu YL; Huang TH; Hou MH, Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013, 587 (2), 120–127, doi: 10.1016/j.febslet.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen IJ; Yuann JMP; Chang YM; Lin SY; Zhao JC; Perlman S; Shen YY; Huang TH; Hou MH, Crystal structure-based exploration of the important role of Arg106 in the RNA-binding domain of human coronavirus OC43 nucleocapsid protein. Biochim. Biophys. Acta. 2013, 1834 (6), 1054–1062, doi: 10.1016/j.bbapap.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang CK; Chen CMM; Chiang MH; Hsu YL; Huang TH, Transient oligomerization of the SARS-CoV N protein - implication for virus ribonucleoprotein packaging. PloS ONE 2013, 8 (5), e65045, doi: 10.1371/journal.pone.0065045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cubuk J; Alston JJ; Incicco JJ; Singh S; Stuchell-Brereton MD; Ward MD; Zimmerman MI; Vithani N; Griffith D; Wagoner JA; Bowman GR; Hall KB; Soranno A; Holehouse AS, The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12 (1), 1936, doi: 10.1038/s41467-021-21953-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CK; Hou MH; Chang CF; Hsiao CD; Huang TH, The SARS coronavirus nucleocapsid protein - Forms and functions. Antivir. Res. 2014, 103, 39–50, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Y; Du N; Lei YQ; Dorje S; Qi JX; Luo TR; Gao GF; Song H, Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020, 39 (20), e105938, doi: 10.15252/embj.2020105938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang M; He SH; Chen XX; Huang ZX; Zhou ZL; Zhou ZC; Chen QY; Chen SD; Kang SS, Structural insight Into the SARS-CoV-2 nucleocapsid protein C-terminal domain reveals a novel recognition mechanism for viral transcriptional regulatory sequences. Front. Chem. 2021, 8, 624765, doi: 10.3389/fchem.2020.624765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiavina M; Pontoriero L; Uversky VN; Felli IC; Pierattelli R, The highly flexible disordered regions of the SARS-CoV-2 nucleocapsid N protein within the 1–248 residue construct: sequence-specific resonance assignments through NMR. Biomol. NMR Assign. 2021, 15 (1), 219–227, doi: 10.1007/s12104-021-10009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang S; Yang M; Hong ZS; Zhang LP; Huang ZX; Chen XX; He SH; Zhou ZL; Zhou ZC; Chen QY; Yan Y; Zhang CS; Shan H; Chen SD, Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B. 2020, 10 (7), 1228–1238, doi: 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossoehme NE; Li LC; Keane SC; Liu PH; Dann CE; Leibowitz JL; Giedroc DP, Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 2009, 394 (3), 544–557, doi: 10.1016/j.jmb.2009.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayaram H; Fan H; Bowman BR; Ooi A; Jaaram J; Collisson EW; Lescar M; Prasady BVV, X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J. Virol. 2006, 80 (13), 6612–6620, doi: 10.1128/jvi.00157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CY; Chang CK; Chang YW; Sue SC; Bai HI; Riang L; Hsiao CD; Huang TH, Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368 (4), 1075–1086, doi: 10.1016/j.jmb.2007.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang QL; Yu LP; Petros AM; Gunasekera A; Liu ZH; Xu N; Hajduk P; Mack J; Fesik SW; Olejniczak ET, Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 2004, 43 (20), 6059–6063, doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- 24.Saikatendu KS; Joseph JS; Subramanian V; Neuman BW; Buchmeier MJ; Stevens RC; Kuhn P, Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007, 81 (8), 3913–3921, doi: 10.1128/jvi.02236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinesh DC; Chalupska D; Silhan J; Koutna E; Nencka R; Veverka V; Boura E, Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PloS Pathog. 2020, 16 (12), e1009100, doi: 10.1371/journal.ppat.1009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang C; Michalska K; Jedrzejczak R; Maltseva N; Endres M; Godzik A; Kim Y; Joachimiak A, Crystal structure of RNA-binding domain of nucleocapsid phosphoprotein from SARS CoV-2, monoclinic crystal form. Worldwide Protein Data Bank, 2020, doi: 10.2210/pdb6wkp/pdb. [DOI] [Google Scholar]
- 27.Elledge SK; Zhou XX; Byrnes JR; Martinko AJ; Lui I; Pance K; Lim SA; Glasgow JE; Glasgow AA; Turcios K; Iyer NS; Torres L; Peluso MJ; Henrich TJ; Wang TT; Tato CM; Leung KK; Greenhouse B; Wells JA, Engineering luminescent biosensors for point-of-care SARS-CoV-2 antibody detection. Nat. Biotechnol. 2021, 39 (8), 928–935, doi: 10.1038/s41587-021-00878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou G; Yan S; Trebosc J; Amoureux J-P; Polenova T, Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids. J. Magn. Reson. 2013, 232, 18–30, doi: 10.1016/j.jmr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong N; Huang Q; Jin C; Xia B, H-1, C-13, and N-15 resonance assignments of the N-terminal domain of the SARS CoV nucleocapsid protein. J. Biomol. NMR 2005, 31 (1), 79–80, doi: 10.1007/s10858-004-6890-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell RW; Fritz MP; Kraus J; Quinn CM; Polenova T; Gronenborn AM, Accuracy and precision of protein structures determined by magic angle spinning NMR spectroscopy: for some ‘with a little help from a friend’. J. Biomol. NMR 2019, 73 (6–7), 333–346, doi: 10.1007/s10858-019-00233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang S; Yang M; He SH; Wang YM; Chen XX; Chen YQ; Hong ZS; Liu J; Jiang GM; Chen QY; Zhou ZL; Zhou ZC; Huang ZX; Huang X; He HH; Zheng WH; Liao HX; Xiao F; Shan H; Chen SD, A SARS-CoV-2 antibody curbs viral nucleocapsid protein-induced complement hyperactivation. Nat. Commun. 2021, 12 (1), 2697, doi: 10.1038/s41467-021-23036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Q; Lu S; Corbett KD, Structural basis for SARS-CoV-2 nucleocapsid protein recognition by single-domain antibodies. Front. Immunol. 2021, 12, 719037, doi: 10.3389/fimmu.2021.719037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y; de Dios AC; McDermott AE, Solid-state 15N NMR chemical shift anisotropy of histidines: experimental and theoretical studies of hydrogen bonding. J. Am. Chem. Soc. 1999, 121 (44), 10389–10394, doi: 10.1021/ja9919074. [DOI] [Google Scholar]
- 34.Li SH; Hong M, Protonation, tautomerization, and rotameric structure of histidine: a comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 2011, 133 (5), 1534–1544, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelton JG; Torchia DA; Meadow ND; Roseman S, Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 1993, 2 (4), 543–558, doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung J; Byeon I-JL; Wang Y; King J; Gronenborn AM, The structure of the cataract-causing P23T mutant of human γD-crystallin exhibits distinctive local conformational and dynamic changes. Biochemistry 2009, 48 (12), 2597–2609, doi: 10.1021/bi802292q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong M; Fritzsching KJ; Williams JK, Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed from 1H chemical shifts. J. Am. Chem. Soc. 2012, 134 (36), 14753–14755, doi: 10.1021/ja307453v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabsch W, XDS. Acta. Crystallogr. D Biol. Crystallogr. 2010, 66 (Pt 2), 125–132, doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta. Crystallogr. D Biol. Crystallogr. 2011, 67 (Pt 4), 235–42, doi: 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD; Grosse-Kunstleve RW; Hung LW; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC, PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954, doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132, doi: 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Baldus M; Petkova AT; Herzfeld J; Griffin RG, Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 1998, 95 (6), 1197–1207, [Google Scholar]
- 43.Brauniger T; Wormald P; Hodgkinson P, Improved proton decoupling in NMR spectroscopy of crystalline solids using the SPINAL-64 sequence. Monatsh. Chem. 2002, 133 (12), 1549–1554, doi: 10.1007/s00706-002-0501-z. [DOI] [Google Scholar]
- 44.Lewandowski JR; Sein J; Blackledge M; Emsley L, Anisotropic collective motion contributes to nuclear spin relaxation in crystalline proteins. J. Am. Chem. Soc. 2010, 132 (4), 1246–1248, doi: 10.1021/ja907067j. [DOI] [PubMed] [Google Scholar]
- 45.Shaka AJ; Keeler J; Frenkiel T; Freeman R, An improved sequence for broad-band decoupling - WALTZ-16. J. Magn. Reson. 1983, 52 (2), 335–338, doi: 10.1016/0022-2364(83)90207-x. [DOI] [Google Scholar]
- 46.Barbet-Massin E; Pell AJ; Retel JS; Andreas LB; Jaudzems K; Franks WT; Nieuwkoop AJ; Hiller M; Higman V; Guerry P; Bertarello A; Knight MJ; Felletti M; Le Marchand T; Kotelovica S; Akopjana I; Tars K; Stoppini M; Bellotti V; Bolognesi M; Ricagno S; Chou JJ; Griffin RG; Oschkinat H; Lesage A; Emsley L; Herrmann T; Pintacuda G, Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J. Am. Chem. Soc. 2014, 136 (35), 12489–12497, doi: 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6 (3), 277–293, [DOI] [PubMed] [Google Scholar]
- 48.Stevens TJ; Fogh RH; Boucher W; Higman VA; Eisenmenger F; Bardiaux B; van Rossum BJ; Oschkinat H; Laue ED, A software framework for analysing solid-state MAS NMR data. J. Biomol. NMR 2011, 51 (4), 437–447, doi: 10.1007/s10858-011-9569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goddard TD; Kneller DG SPARKY 3, Univ. of California, San Francisco, 2004. [Google Scholar]
- 50.Lee W; Tonelli M; Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31 (8), 1325–1327, doi: 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwieters CD; Kuszewski JJ; Tjandra N; Clore GM, The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003, 160 (1), 65–73, doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 52.Schwieters CD; Kuszewski JJ; Clore GM, Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48 (1), 47–62, doi: 10.1016/j.pnmrs.2005.10.001. [DOI] [Google Scholar]
- 53.Schwieters CD; Bermejo GA; Clore GM, Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 2018, 27 (1), 26–40, doi: 10.1002/pro.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen Y; Bax A, Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. 2015, 1260, 17–32, doi: 10.1007/978-1-4939-2239-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu M; Russell RW; Bryer AJ; Quinn CM; Hou GJ; Zhang HL; Schwieters CD; Perilla JR; Gronenborn AM; Polenova T, Atomic-resolution structure of HIV-1 capsid tubes by magic-angle spinning NMR. Nat. Struct. Mol. Biol. 2020, 27 (9), 863–869, doi: 10.1038/s41594-020-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tian Y; Schwieters CD; Opella SJ; Marassi FM, A practical implicit solvent potential for NMR structure calculation. J. Magn. Reson. 2014, 243, 54–64, doi: 10.1016/j.jmr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwieters CD; Bermejo GA; Clore GM, A three-dimensional potential of mean force to improve backbone and sidechain hydrogen bond geometry in Xplor-NIH protein structure determination. Protein Sci. 2019, 29, 100–110, doi: 10.1002/pro.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.The PyMOL Molecular Graphics System, 2.0; Schrödinger, LLC: 2000. [Google Scholar]
- 59.Heinig M; Frishman D, STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502, doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MAS NMR atomic structure coordinates and X-ray structure coordinates of SARS-CoV-2 NNTD have been deposited in the Protein Data Bank under accession codes 7SD4 and 7UW3, respectively. MAS NMR chemical shifts of SARS-CoV-2 NNTD has been deposited in the Biological Magnetic Resonance Data Bank under accession codes 30955.