

Abstract
Two distinct groups of parasitic nematodes use programmed DNA elimination to silence germline-expressed genes in the somatic cells (ascarids) or for sex determination (Strongyloides spp.). In the ascarids, DNA is lost only in pre-somatic cells during early embryogenesis, leading to a reduced somatic genome compared to the intact germ cell genome. Comparative genome analysis has provided information on the retained vs. eliminated sequences, DNA breaks, a full chromosome view on DNA elimination, and the evolutionary conservation of DNA elimination among ascarids. These studies have revealed novel insights into the functions and mechanisms of DNA elimination and provided a reference for in-depth molecular analysis of DNA elimination. Here, I describe the genomics methods we used to study programmed DNA elimination, focusing on the parasitic nematode Ascaris.
Keywords: Programmed DNA elimination, DNA break, telomere healing, gene silencing, chromosome end remodeling, DNA isolation, genome assembly, long reads, genome coverage, parasitic nematode, Ascaris
1. Introduction
Programmed DNA elimination is a significant exception to the genome constancy rule. It is a normal developmental process where large-scale genome changes occur in an organism, generating distinct germline and somatic genomes (1). DNA elimination was discovered by Theodor Boveri in the horse parasitic nematode Parascaris over 130 years ago (2). It was subsequently found in single-cell ciliates (3,4) as well as in a variety of multicellular organisms (>100 known species), including both invertebrates (insects, nematodes, copepods, arachnids) and vertebrates (lampreys, hagfish, birds, and marsupials) (1). The broad phylogenetic distribution of DNA elimination and distinct differences observed suggest that DNA elimination likely evolved independently in different taxa, may serve various functions, and could be mechanistically diverse.
Although programmed DNA elimination has been the subject of much interest and speculation, little was known about metazoan DNA elimination until recently. A critical step to understanding DNA elimination is determining what sequences are removed through the elimination process. Early studies in parasitic nematodes demonstrated that a significant portion of the eliminated sequences were tandemly repeated satellite DNA - 121 bp in Ascaris (5) and 5 bp and 10 bp in Parascaris (6). Later, transposon elements (7) and three single-copy genes were also found eliminated in Ascaris (8–11). Muller et al. further demonstrated that new telomeres were added to the sites of DNA breaks in both Ascaris and Parascaris (12,13). However, given the technologies available, these studies were focused on a few genomic loci, and the overall genome picture of DNA elimination remained largely unknown.
New developments in high-throughput genome sequencing, particularly next-generation and third-generation sequencing technologies, have enabled sequencing and assembling large genomes, once the domain of sequencing centers, in individual laboratories. We have used these genomics approaches to identify retained and eliminated sequences and DNA breaks associated with DNA elimination in parasitic nematodes and provided a complete chromosome view on DNA elimination in Ascaris. Our studies revealed that DNA elimination is a gene silencing mechanism (14–16) and demonstrated that all germline chromosome ends are remodeled and redefined by DNA elimination (16). The identification of retained and eliminated sequences, as well as the sites for DNA breaks, has enabled and will allow additional studies on the mechanism of DNA elimination. For example, using light microscope and ChIP-seq analyses of Ascaris centromere and kinetochore proteins CENP-A and CENP-C, we found that changes in centromere (and kinetochore) deposition before and during a DNA elimination mitosis determines sequences that will be retained or eliminated (17).
This chapter will describe the genomics methods we used to study programmed DNA elimination in the human and pig parasite Ascaris (18). The methods described should be applicable to other related ascarids and Strongyloides species that undergo DNA elimination (19,20), as well as nematode chromosome-level genome projects. I will first describe how to prepare high-quality genomic DNA, a crucial step in building chromosome-level genome assembly using long-read technologies, such as PacBio and Nanopore sequencing. High-quality genomic DNA is also needed in other genomic approaches such as fosmid, bacterial artificial chromosome, Illumina mate-pair and synthetic long-reads, as well as optical mapping. I will then describe the data analysis that is specifically related to DNA elimination, including how to identify the retained vs. eliminated DNA, DNA breaks, and how to examine the consequences of DNA elimination in somatic cells. The related general genomics methods on library preparation, genome assembly, annotation, and transcriptomes analysis are beyond this chapter’s scope.
In nematodes, programmed DNA elimination may not be restricted to parasitic species. A recent study in the free-living nematode Oscheius tipulae using chromosome-level assembly and genome coverage analysis has shown that its germline chromosome ends have a consistent reduction in read coverage (21), suggesting potential DNA elimination events at all chromosome ends. The genomics methods described here should be applicable to identify genome changes and DNA loss in any organism. DNA elimination was historically identified using cytological methods, which are often serendipitous and sensitive to only very large-scale DNA loss. Comparative genome analysis provides an alternative and more sensitive way to identify DNA elimination. With the genomes of many more species being sequenced at depth, it is possible that DNA elimination will be identified in many more organisms in the near future.
2. Materials
2.1. Parasitic nematode materials
The key to identifying DNA elimination at the genome level is to determine if there are sequence differences between the germline and somatic genomes (see below). Where possible, it is best to compare the genomes from individual or a limited number of worms or stages that consist predominantly of germline or somatic cells to reduce sequence variation and heterogeneity. This was exemplified in our studies using the large size of mature Ascaris (on average 15 cm in length for males and 30 cm for females) and the ability to easily dissect, isolate, and collect germline vs. somatic tissues as described previously (14–16,18,22,23). We collect adult Ascaris at local slaughterhouses from freshly isolated pig intestines. A single male and female Ascaris are dissected, and the spermatids, testis, intestine, and remaining tissue (carcass, which includes muscle, hypodermis, pharynx, and neurons) are collected for the male. The ovary/oviduct, uterus, intestine, and carcass are collected for the female. These samples are frozen in liquid nitrogen and stored at −80 °C. To collect Ascaris eggs at discrete developmental stages, Ascaris 1-cell (0 hr) embryos are incubated at 30 °C for the desired period of time (22). For Ascaris, the ideal choice of germline tissues for DNA elimination analysis would be in the following order: spermatids, testis, ovary/oviduct, and 1-4-cell embryos (before DNA elimination); the preference of somatic tissues would be intestine, later embryos (32-cell embryos to larvae L1), and carcass.
2.2. Buffers and solutions
Nuclei buffer: 10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2
Decoating buffer: 0.5 M KOH and 1.5% of sodium hypochlorite
90% isopropanol (in 1x PBS)
Cell suspension buffer: 10 mM Tris, pH 7.4, 20 mM NaCl, 50 mM EDTA
2% CleanCut Agarose (Bio-Rad catalog #1703594)
Lysis buffer: 10 mM Tris pH 8, 25 mM EDTA, 100 mM NaCl, 0.5% SDS
20 mg/ml Proteinase K solution (200x stock)
Washing buffer: 10 mM Tris pH 8.0, 50 mM EDTA
TE buffer: 10 mM Tris pH 8.0, 1 mM EDTA
β-Agarase and buffer: from NEB (catalog # M0392)
Dialysis membrane: 0.1 μm, 47 mm from Millipore (catalog # VCWP04700)
15 mg/ml glycoblue (Ambion catalog # AM9516)
3 M Sodium acetate, pH 5.2
Ethanol, 100% and 70%
3. Methods
3.1. High-quality genomic DNA preparation
Here, I will describe a method that uses agarose plugs to limit DNA shearing to prepare megabase size genomic DNA for DNA elimination studies. Isolated nuclei or embryos can be used as starting material for agarose embedding. For Ascaris, prepare 1-2 million cells for one agarose plug (~100 μL). See Note 1 for alternative approaches to extract high molecular weight genomic DNA.
Nuclei are isolated using a general (24,25) or the specific protocol described here that removes the tough chitinous shell and inner membrane for Ascaris (26). They can be stored at −80 °C for long-term use. Frozen nuclei can be resuspended in 1x nuclei buffer upon thawing.
To prepare decoated embryos, Ascaris eggs are incubated with decoating buffer for 1.5 h at 30 °C with constant shaking. The decoated embryos are washed twice with PBS, followed by 90% isopropanol (in PBS) treatment for 1 min to remove the ascaroside layer. The residual isopropanol is removed by washing twice with PBS.
Wash the nuclei or decoated embryos twice with cold PBS.
Resuspend the sample in cold cell suspension buffer for a total volume of 62.5 μL for each plug, equilibrated for 5 min at room temperature.
For each plug, mix with 37.5 μL of 2% melted CleanCut agarose prewarmed at 50 °C, and transfer immediately into plug molds (Bio-Rad) on a metal block on ice or move quickly to a cold room. The final agarose concentration will be 0.75% in the plugs. Plugs are allowed to solidify at 4 °C for >20 min.
Up to 4 plugs are incubated in 2.5 mL of lysis buffer with 150 μL proteinase K stock solution in a 17 mm plastic polystyrene tube in a 50 °C water bath for 2 h and then overnight (using fresh proteinase K solution), with intermittent mixing.
Transfer the proteinase K digested plugs and the solution to a 50 ml conical tube. Rinse the plugs 3 times with >10 mL washing buffer.
This solution is kept at room temperature overnight. Alternatively, wash 5 times in washing buffer for 15 min on a horizontal platform mixer at 180 rpm at room temperature.
Rinse 2 times in TE buffer, followed by 2 washes in TE buffer for 15 min on a horizontal platform mixer at 180 rpm at room temperature. It is essential to follow the extensive proteinase K digestion and washing procedures to ensure high-quality plugs (see Note 2). The plugs are then ready to use. RNase and restriction digestion of the plugs can be performed when necessary (see Note 3). Plugs can be stored at 4 °C for at least 2 weeks.
To extract genomic DNA, the plugs are transferred to 2.0 mL microcentrifuge tubes.
Plugs are equilibrated with 250 μL 1x beta-agarase gel buffer for 10 min at room temperature. Discard the buffer.
Add 10 μL/plug of 10x-agarase gel buffer, incubate 2 min at 70 °C to melt the gel. Then equilibrate for 5 min in a water bath at 43 °C.
Add 2U/plug (2 μL) of β-agarase, incubate for 45 – 60 min at 43 °C to hydrolyze the agarose and liberate the genomic DNA.
The liberated DNA is cleaned by drop-dialysis using Millipore dialysis membranes (shiny side up during the dialysis) in a 35 mm petri dish with 15 ml TE buffer for 45 – 60 min. Cover the Petri dish to avoid evaporation of the DNA solution.
To the collected DNA solution following dialysis, SDS is added to a final concentration of 0.1% and the DNA treated with 80 μg of proteinase K for 15 min at 50 °C.
The DNA is brought to a volume of 130 μL with TE buffer. DNA is precipitated with 1 μL of glycoblue, 0.1 volume of 3 M sodium acetate, and 2.5 volumes of 100% ethanol with incubation on dry ice for 15 min, and centrifugation at full speed in a standard microcentrifuge at 4 °C for 15 min.
The pellet is washed twice with 70% ethanol, lightly air-dried, and solubilized in 50 - 100 μL of low TE buffer (10mM Tris-HCl pH 8.0, 0.1mM EDTA) or water.
Run a 0.5-0.7% agarose gel or pulsed-field gel electrophoresis (PFGE) to evaluate the molecular weight (intactness) of the DNA. The DNA should be in 200 kb to 10 Mb size range (varies depend on the size of the chromosomes in the species). The DNA can be quantified using a Qubit fluorometer or a NanoDrop (ThermoFisher). See Note 4 on the quantification of viscous DNA.
3.2. Genomic data analysis on DNA elimination
Eliminated sequences are present in the germline and absent in the somatic genome. For non-repetitive eliminated sequences, their copy number (normalized genome coverage based on sequencing depth) is one in the germline and zero in the somatic cell genome (see Figure 1). For repetitive sequences, copy number differences between the germline and somatic cells reveal the number of repeat copies lost during DNA elimination. See Note 5 on bioinformatics tools for data analysis.
Figure 1. Identification of retained and eliminated sequence, DNA break, and telomere addition in Ascaris DNA elimination.
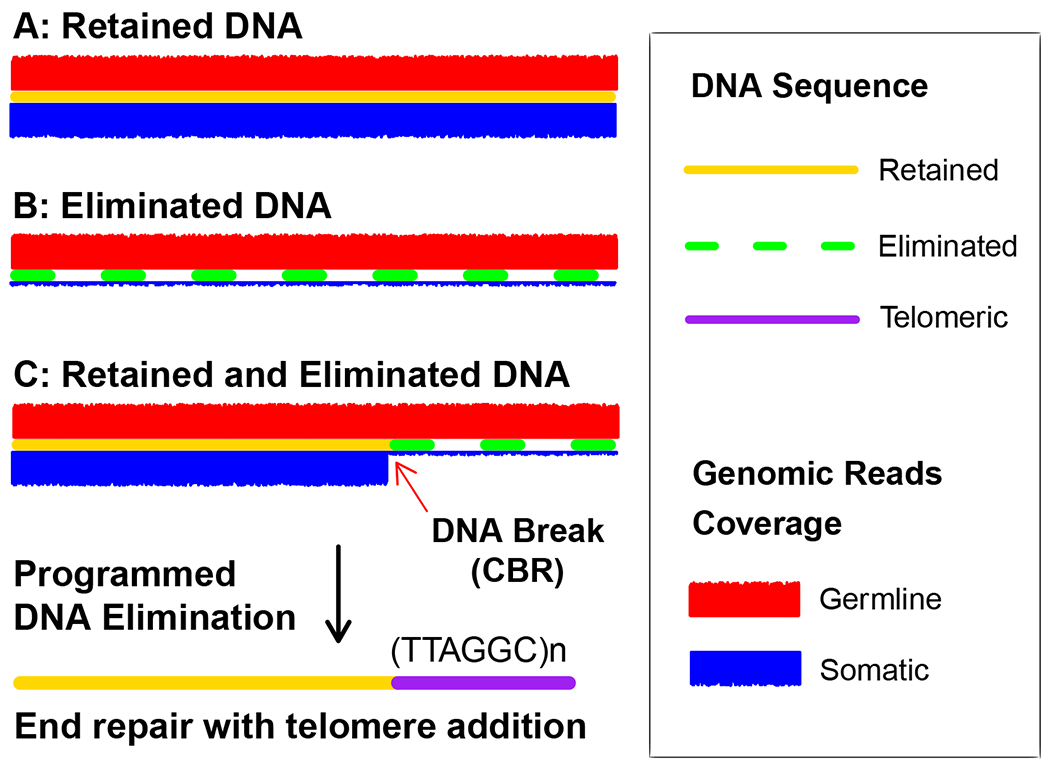
Illustrated are three assembled DNA pieces (A-C, contigs or scaffolds) without repetitive sequence. To determine if a piece of DNA is retained or eliminated, we can map germline (red) and somatic (blue) reads to the DNA. Retained DNA will have the same read coverage from both the germline and somatic cells (A), while eliminated DNA will have little to no somatic reads coverage (B), although some background coverage from germline contamination is commonly seen. At the junction between retained and eliminated DNA is where a presumable DNA break occurs (C). The DNA break end is healed with the addition of new telomere sequences (tandem repeats of TTAGGC, with no preference for the starting nucleotide) during Ascaris DNA elimination (15).
3.2.1. Identify retained and eliminated sequences
Assemble (de novo) germline reads into contigs (or scaffolds).
Map genomic reads from germline and somatic tissues to the assembled germline contigs.
For each contig, determine the normalized copy number for both the germline and somatic tissues.
Compare the copy number difference between the germline and somatic tissues for each contig to determine if the contig is retained, eliminated, or partially eliminated (see Figure 1). See Note 6 on possible issues and data interpretations.
For repetitive sequences, make sure the assembled genomic region is validated, and the mapping approach is appropriate before concluding. The results often need to be manually checked (see Note 7).
3.2.2. Identify DNA break regions
For contigs that are partially eliminated (Figure 1C), examine the junctions between the retained and eliminated regions. A sharp drop in somatic reads coverage is usually an indication of a potential DNA break.
Look for paired-end or long reads that span the potential break regions. In the germline, there should be many reads covering both sides of the breaks, while in the somatic cell, these reads should be confined only to the retained DNA regions.
If necessary, perform a PCR assay to validate the result. For a break to be confirmed, one PCR primer should be in the retained DNA region, while the other in the eliminated DNA region. The PCR should yield a product with the expected size only for the germline DNA, not the somatic DNA. See Note 8 on the nature of break regions in ascarids.
3.2.3. Determine the consequence of DNA break ends in the somatic cells.
Get the sequences flanking the DNA break regions (e. g. 5 kb on each side) from the germline contigs.
The germline break regions are used to compare to either a de novo assembled somatic genome or somatic reads.
In Ascaris, chromosome breaks are healed by telomere addition (Figure 1). Thus, for each break, examine whether it is healed with telomere addition (telomere healing), fused to another break end (chromosome fusion), or connected to a different portion of the genome (genome rearrangement). Chromosome fusion or rearrangement should be validated with PCR assay and/or long reads. No apparent chromosome fusion or rearrangement was detected in Ascaris (14–16).
Acknowledgements
I thank the members of the Davis and Wang labs for critical reading and helpful discussions. Work in the Wang lab is supported by NIH grant AI155588 and the University of Tennessee Knoxville Startup Funds.
Footnotes
High molecular weight genomic DNA can also be isolated using other methods, such as the QIAGEN Genomic-tip (20 – 150 kb) or phenol-chloroform extraction (50 – 200 kb). To use the Genomic-tip, follow QIAGEN instructions. For phenol-chloroform extraction, the samples are digested overnight at 55 °C with 0.5% SDS and 150 μg/mL proteinase K in buffer (50 mM Tris-HCl pH 7.4, 100 mM EDTA, and 100 mM NaCl). The DNA is extracted twice with phenol/chloroform (1:1) and once with chloroform/isoamyl alcohol (24:1), followed by ethanol precipitation. Use extra care to prepare and handle high molecular weight genomic DNA, including using wide-bore pipet tips and gentle inversion/mixing to prevent DNA shearing.
Incomplete digestion and insufficient washing often yield low-quality plugs and thus little genomic DNA. A high-quality proteinase K, such as from Ambion or QIAGEN, is recommended to ensure good digestion. When rinsing and washing the plugs, use large amounts of solution (>10 mL). If necessary, count the plugs when changing buffers to make sure no plugs are sticking to tube’s side, unsubmerged.
In-plug enzymatic treatments are necessary for certain applications. To remove RNA, the plugs are incubated in 2.5 mL of TE buffer with 50 μL of RNase A (final concentration of 20 μg/mL) for 1 h at 37 °C with intermittent mixing, followed by 4 washes with washing buffer. For restriction digestion, the plugs are first equilibrated in 1x appropriate restriction buffer with two 15 min washes in a 500 μL volume. The plugs are incubated with buffer and restriction enzymes at 4 °C overnight, then at 37 °C for > 4 h. Wash the digested plugs in TE buffer 3 times.
High molecular weight genomic DNA is often very viscous, making it hard to pipette accurately for quantification. If possible, pipette more than 2 μL; a smaller volume is more likely to be error-prone. Be patient as viscous DNA may take 10-30 seconds to fill the tip to the 2 μL mark. An aliquot of the DNA may be diluted, sonicated, or restriction digested to make it less viscous before quantification. If necessary, measure the concentrations of the top, middle, and bottom of the DNA solution. The variation from the three samplings should be less than 25%. The DNA yield from high-quality plugs is 60-80% of the total amount of DNA in the cells or nuclei. For example, 1 million Ascaris germline cells (0.6 pg DNA/cell) will yield 300 – 500 ng of genomic DNA.
Bioinformatics tools for genomic data analysis are improving every day. For each task described, there may be a handful of robust tools available. Therefore, only the goals of the tasks are presented with no specific tools recommended. Most standard tools will do. Check out the methods and tools used in our previous work (14–16) to see if similar approaches could be used. When in doubt, search or ask colleagues to see what tools are available before reinventing the wheel.
Possible issues with copy number analysis for DNA elimination may include 1). Patchy genomic coverage due to a low-quality genomic library, often caused by low amounts of genomic DNA and over-amplification (> 15 PCR cycles); 2). Repetitive regions that cause genome assembly and mapping issues (see Note 7); 3). False-positive identification of eliminated DNA caused by foreign contamination in the germline but not in the somatic tissues; and 4). High background levels of eliminated DNA in somatic cells due to contamination with germ cells (Figure 1). Preparing a large amount of high-quality DNA from samples with little contamination will overcome most of these issues.
Repetitive regions are a challenge in genome assembly, particularly when using short reads. For most organisms with DNA elimination, a large portion of eliminated sequences are repeats (1). Major issues caused by repeats in DNA elimination analysis include 1). Large numbers of very short contigs making data analysis and interpretation cumbersome; 2). Mis-assembled repetitive regions leading to inaccurate read coverage and false identification of retained and eliminated sequences or DNA breaks; 3). Challenges in mapping the reads to the repetitive regions due to the similarity in the repeats and the inaccurate number of repeats in the genome assembly. Here are a few suggestions for working with repetitive regions. First, repeats can be identified and clustered based on their categories and sequence similarity; each clustered group can then be analyzed independently. Second, the repeats can be excluded from the coverage analysis to focus on the genome’s non-repetitive regions. Last, use long reads and other scaffolding methods (such as optical mapping and Hi-C) to resolve repetitive regions and improve the quality of the assembled genome.
In ascarids, where chromosomes break and telomere healing occurs can vary between individual worm over a 3-6 kb regions known as a chromosomal break region (CBR) (15,16). Thus, if PCR is used to confirm a break in a chromosome predicted from genome sequencing and assembly, the primer pairs should span the whole 3-6 kb CBR (the primer in the eliminated region should be beyond any possible retained regions) to ensure that no PCR amplicon will result from the somatic DNA.
References
- 1.Wang J, Davis RE (2014) Programmed DNA elimination in multicellular organisms. Curr Opin Genet Dev 27:26–34. doi: 10.1016/j.gde.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boveri T (1887) Ueber Differenzierung der Zellkerne wahrend der Furchung des Eies von Ascaris megalocephala. Anat Anz 2:688–693 [Google Scholar]
- 3.Chalker DL, Yao MC (2011) DNA elimination in ciliates: transposon domestication and genome surveillance. Annu Rev Genet 45:227–246. doi: 10.1146/annurev-genet-110410-132432 [DOI] [PubMed] [Google Scholar]
- 4.Bracht JR, Fang W, Goldman AD, Dolzhenko E, Stein EM, Landweber LF (2013) Genomes on the edge: programmed genome instability in ciliates. Cell 152 (3):406–416. doi: 10.1016/j.cell.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller F, Walker P, Aeby P, Neuhaus H, Felder H, Back E, Tobler H (1982) Nucleotide sequence of satellite DNA contained in the eliminated genome of Ascaris lumbricoides. Nucleic Acids Res 10 (23):7493–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niedermaier J, Moritz KB (2000) Organization and dynamics of satellite and telomere DNAs in Ascaris: implications for formation and programmed breakdown of compound chromosomes. Chromosoma 109 (7):439–452 [DOI] [PubMed] [Google Scholar]
- 7.Aeby P, Spicher A, de Chastonay Y, Muller F, Tobler H (1986) Structure and genomic organization of proretrovirus-like elements partially eliminated from the somatic genome of Ascaris lumbricoides. EMBO J 5 (12):3353–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etter A, Aboutanos M, Tobler H, Muller F (1991) Eliminated chromatin of Ascaris contains a gene that encodes a putative ribosomal protein. P NATL ACAD SCI USA 88 (5):1593–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Etter A, Bernard V, Kenzelmann M, Tobler H, Muller F (1994) Ribosomal heterogeneity from chromatin diminution in Ascaris lumbricoides. Science 265 (5174):954–956 [DOI] [PubMed] [Google Scholar]
- 10.Spicher A, Etter A, Bernard V, Tobler H, Muller F (1994) Extremely stable transcripts may compensate for the elimination of the gene fert-1 from all Ascaris lumbricoides somatic cells. Dev Biol 164 (1):72–86. doi: 10.1006/dbio.1994.1181 [DOI] [PubMed] [Google Scholar]
- 11.Huang YJ, Stoffel R, Tobler H, Mueller F (1996) A newly formed telomere in Ascaris suum does not exert a telomere position effect on a nearby gene. Mol Cell Biol 16 (1):130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller F, Wicky C, Spicher A, Tobler H (1991) New telomere formation after developmentally regulated chromosomal breakage during the process of chromatin diminution in Ascaris lumbricoides. Cell 67 (4):815–822 [DOI] [PubMed] [Google Scholar]
- 13.Bachmann-Waldmann C, Jentsch S, Tobler H, Muller F (2004) Chromatin diminution leads to rapid evolutionary changes in the organization of the germ line genomes of the parasitic nematodes A. suum and P. univalens. Mol Biochem Parasitol 134 (1):53–64 [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Mitreva M, Berriman M, Thorne A, Magrini V, Koutsovoulos G, Kumar S, Blaxter ML, Davis RE (2012) Silencing of germline-expressed genes by DNA elimination in somatic cells. Dev Cell 23 (5):1072–1080. doi: 10.1016/j.devcel.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Gao S, Mostovoy Y, Kang Y, Zagoskin M, Sun Y, Zhang B, White LK, Easton A, Nutman TB, Kwok PY, Hu S, Nielsen MK, Davis RE (2017) Comparative genome analysis of programmed DNA elimination in nematodes. Genome Res. doi: 10.1101/gr.225730.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Veronezi GMB, Kang Y, Zagoskin M, O’Toole ET, Davis RE (2020) Comprehensive chromosome end remodeling during programmed DNA elimination. Curr Biol 30 (17):3397–3413. doi: 10.1016/j.cub.2020.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang Y, Wang J, Neff A, Kratzer S, Kimura H, Davis RE (2016) Differential chromosomal localization of centromeric histone CENP-A contributes to nematode programmed DNA elimination. Cell Rep 16 (9):2308–2316. doi: 10.1016/j.celrep.2016.07.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Davis RE (2020) Ascaris. Curr Biol 30 (10):R423–R425. doi: 10.1016/j.cub.2020.02.064 [DOI] [PubMed] [Google Scholar]
- 19.Streit A, Wang J, Kang Y, Davis RE (2016) Gene silencing and sex determination by programmed DNA elimination in parasitic nematodes. Curr Opin Microbiol 32:120–127. doi: 10.1016/j.mib.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunt VL, Tsai IJ, Coghlan A, Reid AJ, Holroyd N, Foth BJ, Tracey A, Cotton JA, Stanley EJ, Beasley H, Bennett HM, Brooks K, Harsha B, Kajitani R, Kulkarni A, Harbecke D, Nagayasu E, Nichol S, Ogura Y, Quail MA, Randle N, Xia D, Brattig NW, Soblik H, Ribeiro DM, Sanchez-Flores A, Hayashi T, Itoh T, Denver DR, Grant W, Stoltzfus JD, Lok JB, Murayama H, Wastling J, Streit A, Kikuchi T, Viney M, Berriman M (2016) The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat Genet 48 (3):299–307. doi: 10.1038/ng.3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-de-la-Rosa PM, Thomson M, Trivedi U, Tracey A, Tandonnet S, Blaxter ML (2020) A telomere to telomere assembly of Oscheius tipulae and the evolution of rhabditid nematode chromosomes. bioRxiv. doi: 10.1101/2020.09.04.283127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Garrey J, Davis RE (2014) Transcription in pronuclei and one- to four-cell embryos drives early development in a nematode. Curr Biol 24 (2):124–133. doi: 10.1016/j.cub.2013.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, Davis RE (2011) Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res 21 (9):1462–1477. doi: 10.1101/gr.121426.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han M, Wei G, McManus CE, Hillier LW, Reinke V (2019) Isolated C. elegans germ nuclei exhibit distinct genomic profiles of histone modification and gene expression. BMC Genomics 20 (1):500. doi: 10.1186/s12864-019-5893-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabbi A, Riabowol K (2015) Isolation of nuclei. Cold Spring Harb Protoc 2015 (8):731–734. doi: 10.1101/pdb.top074583 [DOI] [PubMed] [Google Scholar]
- 26.Kang Y, Wang J, Davis RE (2017) Nuclei isolation from nematode Ascaris. Bio Protoc 7 (9). doi: 10.21769/BioProtoc.2262 [DOI] [PMC free article] [PubMed] [Google Scholar]