

Abstract
Aim: Atherosclerosis is responsible for high morbidity and mortality rates around the world. Local arterial oxidative stress is involved in all phases of atherosclerosis development. Mitochondria is a relevant source of the oxidants, particularly under certain risky conditions, such as hypercholesterolemia. The aim of this study was to test whether lowering the production of mitochondrial oxidants by induction of a mild uncoupling can reduce atherosclerosis in hypercholesterolemic LDL receptor knockout mice.
Methods: The mice were chronically treated with very low doses of DNP (2,4-dinitrophenol) and metabolic, inflammatory and redox state markers and atherosclerotic lesion sizes were determined.
Results: The DNP treatment did not change the classical atherosclerotic risk markers, such as plasma lipids, glucose homeostasis, and fat mass, as well as systemic inflammatory markers. However, the DNP treatment diminished the production of mitochondrial oxidants, systemic and tissue oxidative damage markers, peritoneal macrophages and aortic rings oxidants generation. Most importantly, development of spontaneous and diet-induced atherosclerosis (lipid and macrophage content) were significantly decreased in the DNP-treated mice. In vitro, DNP treated peritoneal macrophages showed decreased H2O2 production, increased anti-inflammatory cytokines gene expression and secretion, increased phagocytic activity, and decreased LDL-cholesterol uptake.
Conclusions: These findings are a proof of concept that activation of mild mitochondrial uncoupling is sufficient to delay the development of atherosclerosis under the conditions of hypercholesterolemia and oxidative stress. These results promote future approaches targeting mitochondria for the prevention or treatment of atherosclerosis.
Keywords: Atherosclerosis; Mitochondrial oxidants; LDL receptor knockout mice; 2,4-dinitrophenol
See editorial vol. 29: 811-813
Introduction
Atherosclerosis causes mortality mainly due to ischemic heart disease and stroke. Atherogenesis involves local oxidative stress, accumulation of oxidized LDL, and formation of macrophage-derived cholesterol-loaded foam cells and evolves as an unresolved inflammatory disease 1 , 2) . The majority of the clinical trials of antioxidants did not decrease cardiovascular diseases-related mortality. This failure is attributed to a limited ability of the antioxidant treatment protocols (types, doses, combinations, and duration) to influence key cells, cell compartments and pathways leading to atherosclerosis 3 , 4) . Furthermore, the treatment with antioxidants can disturb certain basic aspects of cell functionality since physiological levels of the oxidants are important for signaling proliferation, differentiation, migration, angiogenesis, and other processes 5) .
Mitochondrial respiration is an important source of intracellular oxidants 6) . Mitochondria present an efficient antioxidant enzyme system and may decrease the generation of the oxidants through an increase in the respiration rate due to mild uncoupling 7 , 8) . Mild uncoupling may be caused by endogenous (uncoupling protein activity) or exogenous (drugs) mechanisms that induce partial dissipation of the proton gradient across the inner mitochondrial membrane. As a consequence, mitochondrial respiratory rates are increased to reestablish the membrane electrochemical potential. Enhanced respiration rates decrease the production of the oxidants by several pathways: 1- decreasing O2 tension in the mitochondrial microenvironment; 2- favoring a more oxidized state of the respiratory chain intermediates; 3- maintaining low NADH levels; and 4- inhibiting the reverse flow of the electrons from Complex II to Complex I 8) . We have previously reported that elevated generation of mitochondrial oxidants is an independent risk factor for atherosclerosis in LDL receptor knockout mice 9 , 10) . In addition, Wang et al. 11 , 12) showed that overexpression of a mitochondria-targeted catalase in macrophages reduces atherosclerosis in LDL receptor knockout mice. Robust experimental evidence linking mitochondrial redox dysfunctions and atherosclerosis has been recently reported 13 , 14) .
Mitochondria-targeted antioxidants have been used as a promising anti-atherosclerosis strategy. A wide range of antioxidants are targeted to the mitochondria by conjugation with triphenylphosphonium cation (TPP), such as MitoE, MitoSOD, MitoQ and MitoTempo 15 , 16) . In mouse models, MitoQ reduces macrophage content and cell proliferation within atherosclerotic plaques and inhibits multiple features of metabolic syndrome 17) . Aged apoE knockout mice treated with MitoTempo have decreased vascular oxidant levels and atherosclerosis 18) ; however, mice with hypercholesterolemia due to overexpression of PCSK9 showed an increase in atherosclerosis when treated with MitoTempo 19) . Although of high therapeutic potential, targeting mitochondria for cardiovascular disorders still encounters obstacles 20) .
2,4-Dinitrophenol (DNP), a protonophore with high affinity for the mitochondrial membrane, is a potent mitochondrial uncoupler that was used for the first time as a drug to induce weight loss in the early 1930s 21) ; however, the use of DNP was abandoned due to several lethal cases 22) . More recently, very low and safe doses of DNP have been used in experimental investigations. Wild-type mice treated with a low dose of DNP showed a decrease in oxidant production in the brain, liver and heart, a reduction in the plasma levels of glucose, insulin and triglycerides and an increase in longevity 23) . Mice acclimated to thermoneutrality treated with DNP exhibited reduced fat mass and hepatic steatosis and improved glucose tolerance 24) . In addition, DNP treatment protected against motor dysfunction and neuron loss in a mouse model of Parkinson’s disease 25) .
Aim
The objectives of this study were to investigate whether chronic mild mitochondrial uncoupling induced by low doses of DNP may attenuate the development of atherosclerosis in a mouse model of familial hypercholesterolemia.
Methods
Animals
LDL receptor knockout (LDLr−/−) mouse breeders were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained at the State University of Campinas Multidisciplinary Center for Biological Research in Laboratory Animals (CEMIB/Unicamp, Brazil). Animal experiments were approved by the University Committee for Ethics in Animal Experimentation, protocol #1969-1 (CEUA/UNICAMP). Male mice had free access to a standard laboratory rodent chow diet (Nuvital CR1, Colombo, Paraná, Brazil) and were housed at 22±1℃ on a 12 h light/dark cycle. 2,4-dinitrophenol (DNP) treatment was performed daily in the drinking water containing 1 mg/L DNP 23) over 12 weeks starting from the age of 8 weeks. For diet-induced atherosclerosis experiments, five–month-old male and female LDL receptor knockout mice were treated with DNP and fed with an atherogenic diet (western diet: 22 g% fat plus 0.21 g% cholesterol, Pragsoluções, Jaú, SP, Brazil) for 4 weeks.
Indirect Calorimetry
The Oxylet System (Pamlab e Harvard Apparatus) was used for the indirect calorimetry. The mice were acclimated to the chamber for 24 hours and the metabolism was measured during the next 24 hours in mice with free access to food and water. The software metabolism v2.2.01 was used to calculate total energy expenditure (EE).
Plasma Biochemical Analysis
Blood samples were collected from either the retroorbital plexus or the tail tip of anesthetized mice. Samples were obtained after 12 h of fasting. Total cholesterol, triglycerides and non-esterified fatty acids were measured in fresh plasma using standard commercial kits (Roche-Hitachi®, Germany and Wako®, Germany). Glucose levels were measured using a hand-held glucometer (Accu-Chek Advantage, Roche Diagnostics®, Switzerland). The plasma levels of insulin, leptin and adiponectin (Millipore, USA), tumor necrosis factor-α (TNF-a) (eBioscience, USA) and C-reactive protein (IBL America, USA) were measured with commercial ELISA kits. Plasma lipoproteins were determined by fast protein liquid chromatography (FPLC) (Amersham-Pharmacia Biotech, Uppsala, Sweden) as described previously 26) .
Plasma Thiobarbituric Acid Reactive Substances (TBARS)
Plasma (100 µL) was incubated with 200 µL of 0.7% thiobarbituric acid in 0.05 M NaOH and 60 µL of 50% trichloroacetic acid. Samples were incubated in boiling water for 30 min followed by centrifugation at 664 ×g for 15 min. A standard curve was prepared using several dilutions of 0.05 mM 1,1,3,3-tetramethoxypropane. The optical density of the samples and standard curve was measured in a microplate reader at 532 nm (SpectraMax M3, Molecular Devices, USA).
Body Composition and Liver Lipid Content
The mice and food intake were weighed once per week. The epididymal adipose tissue and liver mass were determined gravimetrically. Mouse carcass composition was determined as described previously in detail by Salerno et al. 27) . Liver lipids were extracted using the Folch method 28) . The liver contents of cholesterol and triglycerides were determined using colorimetric enzymatic assays (Roche-Hitachi®, Germany) after dissolving the lipid extracts in a Triton-containing buffer.
Liver Protein Carbonyl Content
The liver protein carbonyl content was estimated according to Reznick and Packer 29) , as modified by Schild et al. 30) . Briefly, samples of the liver homogenate were incubated with 10 mM dinitrophenylhydrazine in 2.5 M HCl for 1 h at room temperature. The reaction was stopped by the addition of 20% trichloroacetic acid. The pellets were washed twice with ethanol/ethyl acetate (1/1) and once with 10% trichloroacetic acid. The protein pellets were dissolved in 6 M guanidine hydrochloride, and the absorption at 370 nm was determined. Carbonyl content was calculated using the molar absorption coefficient of aliphatic hydrazones (0.022 µM−1cm−1).
Isolation of Mouse Liver Mitochondria
Mitochondria were isolated by conventional differential centrifugation at 4℃ as described previously 31) . The livers were homogenized in 250 mM sucrose, 1 mM EGTA, and 10 mM HEPES buffer (pH 7.2). The mitochondrial suspension was washed twice in the same medium containing 0.1 mM EGTA, and the final pellet was resuspended in 250 mM sucrose to a final protein concentration of 80–100 mg/ml. The protein concentration was determined by a modified biuret assay. The experiments were done in a standard medium containing 125 mM sucrose, 65 mM KCl, 2 mM inorganic phosphate, 1 mM magnesium chloride, and 10 mM HEPES buffer, pH 7.2, as described previously 32) .
Isolation of Peritoneal Macrophages
Anesthetized mice were euthanized, and the epidermal abdominal layer was carefully cut, preserving the peritoneum. Afterward, 6 mL of ice-cold PBS (phosphate-buffered saline) was injected into the peritoneum, and approximately 5 mL of cell-containing buffer was aspirated from the peritoneum. The cell-containing buffer was then centrifuged for 5 min at 400 ×g at 4℃. The cell pellet was gently resuspended in RPMI 1640 medium (Vitrocell, Brazil) supplemented with 10% fetal bovine serum. Cell counting was performed using an automatic cell analyzer (Muse® Millipore Corporation, Hayward, CA, USA.). The peritoneal cells were diluted to 1 x 107 cells/mL, seeded in 96-well (100 µL) plates and washed after 2 h to remove non-adherent cells. Macrophages were ready for experimentation after overnight incubation at 37℃ and 5% (v/v) CO2.
Global Oxidant Production
Global oxidant production was monitored using a membrane-permeable fluorescent dye, 2´,7´-dichlorodihydrofluorescein diacetate (H2DCF-DA). The global oxidant production by isolated mitochondria was monitored as described previously 9) . In the case of the aorta, two 3 mm rings from the descending aorta were placed in a well of a 96-well plate in 10 µM H2DCF-DA in PBS buffer containing 11.1 mM glucose, pH 7.4, for 1 h. Then, the fluorescence signal was measured at the excitation/emission wavelengths of 492/517 nm in a SpectraMax M3 microplate reader (Molecular Devices, USA).
H2O2 Release
H2O2 release was quantified using Amplex® red (Molecular Probes, USA) in the presence of horseradish peroxidase (HRP). Isolated-mitochondria: The H2O2 release in isolated mitochondria was determined as described previously 33) . Aorta: Two 3 mm rings of the descending aorta were placed in a well of a 96-well plate with Amplex® red (10 µM) and HRP (0.2 U/mL) in PBS buffer containing 11.1 mM glucose, pH 7.4, for 1 h. Then, the aortic rings were removed, and the fluorescence signal was measured in the buffer at the excitation/emission wavelengths of 530/590 nm (SpectraMax M3, Molecular Devices, USA) 34) . Peritoneal macrophages: Experiments were done in a 96-well plate in 100 µl of PBS buffer containing 11.1 mM glucose, pH 7.4, in the presence of Amplex® red (25 µM) and HRP (0.2 U/mL) in the dark at 37℃ for 40 min; fluorescence (excitation: 530 nm, emission: 590 nm) was measured every 10 min (SpectraMax M3- Molecular Devices, USA). Catalase (500 U/mL) was added at least in one well per group as a background control to account for nonspecific signal. Phorbol 12-myristate 13-acetate (PMA) (100 nM) was used to activate the macrophages during the assay.
Superoxide Generation
Total intracellular and mitochondrial superoxide anion generation were determined using the dihydroethidium (DHE) and Mitosox® reagents, respectively (Thermo Fisher Scientific, USA), which are fluorogenic dyes that react with the superoxide anion to produce red fluorescence. The macrophages were incubated with DHE (10 µM) or Mitosox® (2 µM) in PBS buffer containing 11.1 mM glucose, pH 7.4, in the dark at 37℃ for 30 min. Then, macrophages were washed twice with PBS, and total fluorescence was quantified in a fluorimeter (SpectraMax M3-Molecular Devices) with excitation/emission wavelengths as follows: DHE: 518/605 nm and Mitosox: 510/580 nm. Fluorescence was corrected against the background fluorescence of the cells without the probes. In the Mitosox plate, the cells were incubated with Hoechst 33342 (5 µg/mL) (Thermo Fisher Scientific, H3570) in PBS for 15 min and then fixed with paraformaldehyde 3.7% (v/v) in PBS for 10 min at room temperature. Images of the macrophages were captured by an ImageXpress Micro confocal high content imaging system (Molecular Devices) using Texas Red and DAPI fluorescence filters to obtain a representative image of mitochondrial superoxide generation.
Histological Analysis of Atherosclerosis Lesions
In situ-perfused hearts were excised, embedded in Tissue-Tek® OCT compound (Sakura, USA), frozen at −80℃, cut in 10 µm-sections along 480 µm aorta length from the aortic valve leaflets and stained with Oil Red O as described previously 9) . A sub-group of slides were immune-stained for CD68, a general marker of macrophages. These sections were blocked with 10% bovine serum albumin (BSA) and then incubated for 3h at room temperature with the primary antibody rat anti-CD68 (1:250; AbD Serotec). The sections were washed and incubated with fluorescent Alexa Fluor-conjugated secondary antibody (Invitrogen). Nuclei were counterstained for 10 min with DAPI. The sections were mounted with VECTASHIELD medium, and pictures were taken with a Leica DMI600B microscope. The lipid and immune-stained lesions areas were quantified using ImageJ (1.45 h) software.
Macrophage In Vitro DNP Dose-Effect and Cell Viability
1 x 105 cells were incubated in RPMI 1640 medium supplemented with 10% fetal bovine serum during 24h at 37℃, in the absence and presence of 50, 100, 200, 400 µM DNP. H2O2 medium concentrations were determined with Amplex red as described above. Cell viability was determined with crystal violet staining, which is a cation-based dye that stain DNA in attached alive cells (dying cells detached from the plate), at the end of incubation. After media removal, cells were fixed with 100 µL of paraformaldehyde 4% during 5 min and incubated with 100 µL crystal violet 0.05% during 10 min at room temperature. The stained cells were then washed thoroughly with distilled water, dried and eluted with 100 µL of acetic acid (10%). The absorbance was measure at 590 nm in a plate reader (Spectramax M3-Molecular Devices, USA).
Gene Expression by Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Peritoneal macrophages were seeded at 3 x 105 cells/well in a 24-wells plate (Nest Biotechnology, 702001, Wuxi, China) in RPMI 1640 medium supplemented with 10% fetal bovine serum during 24h at 37℃, in the presence and absence of 100 µM DNP. Total RNA was extracted using TRIzolTM reagent (ThermoFisher Scientific, 15596026, Rockford, IL) according to the manufacturer’s protocol and then quantified in NanoDropTM 2000/2000c spectrophotometer (ThermoFisher Scientific). The amount of 1 µg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystem, 4368814, Foster City, CA). The amplification step was carried out using Fast SYBRTM Green Master mix (ThermoFisher Scientific, 4385612), 100 ng of each cDNA, and 200 nM of each forward and reverse oligonucleotide as described below. The oligonucleotides were designed and tested against the Mus musculus genome deposited in the Gene Bank of the National Center for Biotechnology Information (NCBI). RT-qPCR assays were carried out in the StepOne real-time PCR system (Applied Biosystems). The mRNA relative abundance was quantified using the threshold cycle method 2(-ΔΔCT) 35) and the data were normalized against the endogenous controls (ATCB or 36B4). Oligonucleotides used to monitor gene expression by RT-qPCR are described below.
| Genes | Forward (5’→3’) | Reverse (5’→3’) |
Amplicon size (bp) |
|---|---|---|---|
| IL-1β | CCTTCCAGGATGAGGACATGA | TGAGTCACAGAGGATGGGCTC | 71 |
| IL-6 | CACGGCCTTCCCTACTTCAC | GGTCTGTTGGGAGTGGTATC | 66 |
| TNFα | CCCTCCTGGCCAACGGCATG | TCGGGGCAGCCTTGTCCCTT | 109 |
| IL-4 | CCAAACGTCCTCACAGCAAC | AAGCCCGAAAGAGTCTCTGC | 157 |
| IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG | 105 |
| ARG1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC | 185 |
| 36B4 | GAGGAATCAGATGAGGATATGGGA | AAGCAGGCTGACTTGGTTGC | 72 |
| ACTB | AGAAGCTGTGCTATGTTGCTCTA | TCAGGCAGCTCATAGCTCTTC | 91 |
IL-10 Secretion by ELISA
Peritoneal macrophages were seeded at 1 x 106 cells/well in a 24-well plate (Nest Biotechnology, 702001, Wuxi, China) in RPMI 1640 medium supplemented with 10% fetal bovine serum during 24h at 37℃, in the presence and absence of 100 µM DNP. Cytokine content was quantified in the medium using Mouse IL-10 Quantikine ELISA kit (R&D Systems, M1000B, Minneapolis, MN) according to the manufacturer’s protocol. For normalization, adhered cells were harvested and incubated with 100 µL RIPA buffer (Sigma-Aldrich, R0278, St. Louis, MO) containing cOmpleteTM Protease Inhibitor Cocktail (Roche, 11697498001, Mannhein, Germany) during 15 min at 4℃. The lysate was centrifuged at 13,000 rpm, 4℃ for 15 min. The protein content was determined in the supernatant using PierceTM 660 nm Protein Assay Reagent (ThermoFisher Scientific, 22660, Rockford, IL) and PierceTM Bovine Serum Albumin Standard pre-diluted (ThermoFisher Scientific, 23208, Rockford, IL). Absorbance was measured in SpectraMax M3 spectrophotometer (Molecular Devices) at the wavelength of 660 nm.
Macrophage Phagocytosis Assay
The control and DNP treated peritoneal macrophages (1 x 105) were incubated for 30 min with 100 µL of neutral-red stained zymosan (1 x 107 particles/mL) and fixed with Baker’s solution (4% formaldehyde, 2% sodium chloride, 1% calcium acetate) for additional 30 min at 37℃. Cells were then washed twice with PBS buffer and the neutral-red stain was solubilized with 0.1 mL of acidified alcohol (10% acetic acid, 40% ethanol in distilled water) for 30 min. The absorbance was then read at 550 nm as previously described 36) .
Macrophage Cholesterol Uptake Assay
The peritoneal macrophages (5 x 105) were incubated with 50 µg/mL of oxidized LDL (CuSO4-induced oxidation, 40 µM, 37℃, 24h) in RPMI 1640 medium supplemented with 1% fetal bovine serum during 24h at 37℃, in the presence and absence of 100 µM DNP. The cells were washed twice with PBS buffer and lysed with RIPA buffer. Then, in a black 96 well plate, 10 µL of each sample and 40 µL of catalase (20 U/mL) were mixed and incubated for 15 min at 37℃ in order to eliminate any peroxides present in reagents or samples 37) . Next, 100 µL of cholesterol oxidase/amplex-red reagent (0.1 M potassium phosphate buffer, pH 7.4, 50 mM NaCl, 5 mM cholic acid, 0.1% Triton X-100, 2 U/ml cholesterol oxidase, 0.2 U/mL cholesterol esterase, 2 U/ml HRP, and 100 µM amplex-red) was added, mixed and incubated at 37℃ for an additional 30 min. Fluorescence was then read at excitation wavelength of 530 nm and emission wavelength of 590 nm. The cholesterol mass was normalized by total lysate protein using the bicinchoninic acid (BCA) assay kit (ThermoFisher Scientific, USA).
Data Analysis
The results are presented as the mean±SE. Comparisons between the groups were performed by unpaired Student’s t test. The level of significance was set at p ≤ 0.05.
Results
LDL receptor knockout (LDLr−/−) mice were treated for 3 months with low doses of the mitochondrial uncoupler 2,4-dinitrophenol (DNP) in the drinking water, and atherosclerotic risk factors and the size of atherosclerotic lesions at the aortic root were analyzed. The DNP doses were low and slightly modified the whole body metabolic rates, increasing 11% the O2 consumption rate (p<0.05) in the DNP-treated mice ( Fig.1 ) . The DNP treatment did not change body weight, visceral adipose tissue mass, lean carcass mass, and liver lipid content ( Table 1 ) . DNP treatment did not change the plasma levels of total and lipoprotein-associated cholesterol, triglycerides, adipokines (leptin and adiponectin), glucose, insulin, or systemic inflammatory markers (TNFα and C-reactive protein) ( Table 2 ) . However, the plasma levels of non-esterified fatty acids were significantly reduced (22%) in the DNP-treated mice ( Table 2 ) . This reduction may represent an energy substrate preference since DNP activates AMPK in the muscles 38) , thus stimulating β-oxidation 39) . Glucose and insulin tolerance were also not modified by DNP ( Supplementary Fig.1 ) .
Fig.1. Indirect calorimetry during 24 hours (light and dark times) in LDL receptor knockout mice (LDLr−/−) and treated with low doses of 2,4-dinitrophenol (LDLr−/−DNP) .

Body O2 consumption (A), CO2 production (B) and energy expenditure (C) measurement each 2 hours during all day and an average during the light and dark time (n=7, *p=0.023, #p=0.056).
Table 1. Body weight and composition of LDL receptor knockout mice (LDLr-/-) treated with or without 2,4-dinitrophenol (DNP) and fed a chow diet .
| LDLr-/- | n | LDLr-/-DNP | n | P* | |
|---|---|---|---|---|---|
| Body weight (g) | 22.3±0.4 | 17 | 21.8±0.5 | 14 | 0.39 |
| Perigonadal adipose tissue (% body weight) | 1.00±0.07 | 17 | 0.95±0.08 | 14 | 0.68 |
| Lean mass (% carcass weight) | 86.5±1.8 | 11 | 87.9±1.0 | 10 | 0.51 |
| Liver cholesterol (mg/g) | 1.99±0.11 | 13 | 2.02±0.10 | 11 | 0.86 |
| Liver triglycerides (mg/g) | 22.5±2.2 | 13 | 19.2±2.3 | 10 | 0.32 |
Data are the mean±SE (n). *P values according to the Student’s t test.
Table 2. Plasma levels of lipids, glucose, insulin, adipokines and inflammatory proteins in LDL receptor knockout mice (LDLr-/-) treated with or without 2,4-dinitrophenol (DNP) and fed a chow diet .
| LDLr-/- | n | LDLr-/-DNP | n | P* | |
|---|---|---|---|---|---|
| Cholesterol (mg/dL) | 219±7.8 | 15 | 223±10.6 | 13 | 0.79 |
| VLDL-chol (mg/dL) | 13.1±1.5 | 8 | 14.2±2.1 | 7 | 0.62 |
| LDL-chol (mg/dL) | 104.2±3.0 | 8 | 111.0±2.9 | 7 | 0.13 |
| HDL-chol (mg/dL) | 101.3±2.1 | 8 | 97.3±2.5 | 7 | 0.23 |
| Triglycerides (mg/dL) | 120.0±9.1 | 16 | 116±8.5 | 13 | 0.75 |
| NEFAa (mmol/L) | 0.61±0.04 | 15 | 0.49±0.03 | 14 | 0.027 |
| Glucose (mg/dL) | 82.3±3.3 | 15 | 81.7±2.7 | 15 | 0.88 |
| Insulin (ng/mL) | 1.12±0.14 | 7 | 1.31±0.21 | 6 | 0.47 |
| Adiponectin (µg/mL) | 7.7±0.3 | 7 | 8.2±0.4 | 11 | 0.45 |
| Leptin (ng/mL) | 0.43±0.07 | 11 | 0.35±0.04 | 10 | 0.30 |
| TNFα (pg/mL) | 7.2±0.3 | 11 | 8.2±0.7 | 12 | 0.25 |
| C-reactive protein (ng/mL) | 7.5±0.3 | 8 | 7.3±0.6 | 7 | 0.78 |
Data are the mean±SE (n). a NEFA: Non-esterified fatty acids; *P values according to the Student t test.
Supplementary Fig.1. Glucose and insulin tolerance tests in LDL receptor knockout mice (LDLr−/−) treated with or without 2,4-dinitrophenol (DNP) .
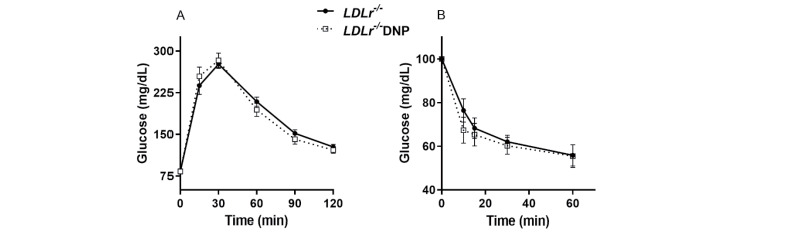
(A) Glycemia curve after an oral dose of glucose (1.5 g/kg body weight). (B) Glycemia curve after an ip insulin injection (0.5 U/Kg body weight) (n=9).
Oxidant production rates were analyzed in isolated liver mitochondria. As expected, the global mitochondrial oxidant production (probed with H2DCF-DA) and the H2O2 release rate (probed with Amplex red and HRP) were significantly diminished by 32% and 16%, respectively, in organelles obtained from DNP-treated mice ( Fig.2 A, B ) . In addition, liver protein carbonyl content and plasma levels of thiobarbituric acid-reactive substances (TBARS) were decreased by 36% and 23%, respectively, in DNP-treated mice, indicating a decrease in tissue protein oxidation and plasma lipoperoxidation in DNP-treated mice ( Fig.2 C, D ) . Reduced and oxidized glutathione contents in the liver and mitochondrial density measured as citrate synthase activity were similar between the groups ( Supplementary Fig.2 ) .
Fig.2. Systemic and liver oxidative damage markers and oxidant production by isolated liver mitochondria in male LDL receptor knockout mice (LDLr−/−) treated with or without 2,4-dinitrophenol (DNP) .

(A) Global oxidant production in liver mitochondria (n=6, *p=0.044). (B) H2O2 release by liver mitochondria (n=5, *p=0.02). (C) Liver protein carbonyl content (n=8, *p=0.047). (D) Plasma thiobarbituric acid-reactive substances (TBARSs) (n=5-6, *p=0.02).
Supplementary Fig.2. Hepatic levels of reduced and oxidized glutathione and citrate synthase activity of LDL receptor knockout mice (LDLr−/−) treated with or without 2,4-dinitrophenol (DNP) .

(A) Hepatic reduced glutathione (GSH). (B) Hepatic oxidized glutathione (GSSG). (C) GSH/GSSG ratio (n=16). (D) Liver citrate synthase activity (n=9-10). Reduced (GSH) and oxidized (GSSG) glutathione levels were determined in liver homogenates as described by Teare et al (1993). Citrate synthase activity was determined by the conversion of oxaloacetate and acetyl-CoA to citrate and SH-CoA and monitored by the appearance of the thionitrobenzoic acid product at 412 nm.
Teare, JP, Punchard, NA, Powell, JJ, et al., Automated spectrophotometric method for determining oxidized and reduced glutathione in liver, Clinical chemistry, 1993; 39: 686-689.
We also analyzed the effects of DNP on oxidant production by the cells (macrophages) and tissue (aorta) directly related to atherosclerosis. The data on oxidant production in peritoneal macrophages and aortic rings from DNP-treated mice are shown in Fig.3 . Macrophages from DNP-treated mice released 30% less H2O2 than those from the untreated mice, while markedly elevated levels of H2O2 were observed after stimulating the cells with the phorbol ester PMA (which activates NADPH oxidase) ( Fig.3A ) . The aorta from the DNP-treated mice had global oxidant (DCF) and H2O2 decreased release by 25% and 18%, respectively ( Fig.3B, C ) . We then measured early spontaneous atherosclerosis in the aortic root in these mice fed with chow diet and observed a significant 36% decrease in the lipid-stained lesion areas of DNP-treated mice ( Fig.3D ) .
Fig.3. Oxidant production in peritoneal macrophages and aorta and spontaneous atherosclerosis in male LDL receptor knockout mice (LDLr−/−) treated with or without 2,4-dinitrophenol (DNP) .

(A) H2O2 release in peritoneal macrophages incubated with or without 100 nM phorbol 12-myristate 13-acetate (PMA) (n=5-9, *p=0.025). (B) Global (DCF) oxidant production in aortic rings (n=7-8, *p=0.025). (C) H2O2 release in aortic rings probed with Amplex red (n=8-11, *p=0.036). (D) Lipid-stained areas of atherosclerotic lesions in the aortic root and representative images (n=14-17, *p=0.033).
Since spontaneous atherosclerosis in chow fed mice showed only small lesions in the aorta root, we fed mice a western-type diet (22 g% fat) containing cholesterol (0.21%) to evaluate the effect of DNP treatment under diet induced atherosclerosis conditions. The LDL receptor knockout mice were treated simultaneously with DNP and the atherogenic diet for 4 weeks ( Fig.4 ) . Peritoneal macrophages from the DNP-treated mice fed with the atherogenic diet showed a 25% reduction in total superoxide production (DHE) ( Fig.4A ) and a 35% reduction in mitochondria-derived superoxide (Mitosox) ( Fig.4B ) . More importantly, the aortic atherosclerotic lesion area was 26% smaller in the DNP-treated mice fed the atherogenic diet ( Fig.4C ) . The macrophage content of lesions were determined using CD68 antibody ( Fig.4D ) . Indeed, DNP treated mice presented lower content of macrophages (-33%) in their aorta root, suggesting a less inflamed type of lesion.
Fig.4. Peritoneal macrophage superoxide production and diet-induced atherosclerosis in LDL receptor knockout male and female mice (LDLr−/−) treated with 2,4-dinitrophenol (DNP) and fed with an atherogenic diet .

(A) Superoxide production (DHE) in peritoneal macrophages incubated with or without 100 nM phorbol 12-myristate 13-acetate (PMA) (n=4-6, *p=0.01). (B) Mitochondrial superoxide production (Mitosox) and representative images of peritoneal macrophages (n=5-6, *p=0.03). (C) Lipid-stained areas of atherosclerotic lesions in the aortic root and representative images (n=9-15, *p=0.026). (D) Anti-CD68 (macrophage marker) stained areas of lesions in the aortic root and representative images (n=7-8, *p=0.024).
Similar to the results observed under low-fat chow diet, the DNP-treated mice fed the atherogenic diet had no significant changes in the adiposity and plasma lipids levels ( Supplementary Table 1 ) .
Supplementary Table 1. Adipose tissue mass and plasma lipid levels in LDL receptor knockout mice (LDLr-/-) treated with or without 2,4-dinitrophenol (DNP) and fed an atherogenic diet for 4 weeks .
| LDLr-/- | LDLr-/-DNP | |
|---|---|---|
| Perigonadal adipose tissue (% body weight) | 1.51±0.13 | 1.26±0.08 |
| Cholesterol (mg/dL) | 520±24 | 506±34 |
| Triglycerides (mg/dL) | 156±9.8 | 149±14.8 |
| NEFA (mmol/L) | 0.46±0.03 | 0.43±0.04 |
NEFA: Non-esterified fatty acids; data are the mean±SE (n = 9-15).
In order to further explore the DNP effects on macrophages, we performed in vitro experiments ( Fig.5 ) . A DNP dose-effect experiment was done to stablish the experimental conditions. The DNP dose of 100 µM during 24 hour incubation significantly decreased H2O2 release without perturbations in cell viability ( Fig.5A and B ) and, thus, this dose was chosen for further experiments. DNP in vitro treatment of peritoneal macrophages from hypercholesterolemic mice showed 40% reduced H2O2 production ( Fig.5C ) and modified the inflammatory profile ( Fig.5D, E ) , phagocytosis activity ( Fig.5F ) and lipid uptake and accumulation ( Fig.5G ) . Concerning inflammation related gene expression ( Fig.5D ) , we observe that DNP induced marked increases (3-5 fold) in the expression of anti-inflammatory genes such as IL-4, IL-10 and arginase-1. On the other hand, DNP significantly decreased TNFα and increased IL-1β and IL-6. While TNFα and IL-1β are pro-inflammatory genes, IL-6 is a cytokine found to be elevated in both pro- or anti-inflammatory settings. This finding is related with the time frame and specific context in which the cytokine is measured, because one classical role of IL-6 is to signal and activate the expression of the anti-inflammatory response 40) . These results thus suggest a transition state towards a less inflammatory phenotype. DNP induced IL-10 expression was confirmed by a 3-fold elevation in IL-10 secretion into the cell media ( Fig.5E ) . Functionally, DNP induced phagocytosis activity in treated macrophages ( Fig.5F ) . This is compatible with M2 (anti-inflammatory) macrophage phenotype that usually exhibit high phagocytic activity 41) . Finally, LDL-cholesterol uptake and accumulation in macrophages, as a measure of foam cell formation, was 43% decreased in DNP treated cells ( Fig.5G ) .
Fig.5. In vitro effects of DNP on LDLr-/- peritoneal macrophage H2O2 production (A, C), cell viability (B), inflammation related gene expression (D), IL-10 secretion (E), phagocytic activity (F) and cholesterol uptake upon incubation with oxidized LDL (50 µg/mL) (G) .

In all experiments cells were incubated 24h at 37℃. Panels C to G show cells treated with 100 µM DNP. See Material and Methods Sections for details. IL-1β, IL-4 and IL-10 gene expressions were normalized with ACTB (Actin Beta) while TNF-a, IL-6 and ARG1 genes were normalized with 36B4 (acidic ribosomal phosphoprotein). Experiments show mean±SE and individual experimental points (n=5-7 LDLr-/- macrophage donor mice), *p<0.05.
Discussion
The oxidative modification hypothesis of atherosclerosis 42) has been continuously supported since its proposal. Therefore, the efforts to counteract oxidant stress have been tested in several trials using antioxidant supplementation; however, none of the trials have successfully reduced atherosclerosis 3) . In this study, we hypothesized that counteracting endogenous production of oxidants, specifically oxidants derived from mitochondria, can beneficially impact atherosclerosis development. Indeed, we showed that inducing a mild mitochondrial uncoupling and thus reducing oxidant generation results in a decrease in spontaneous and diet-induced atherosclerosis development in the hypercholesterolemic LDLr−/− model. The treatment with low doses of DNP did not alter classical risk factors for cardiovascular diseases, such as plasma lipids, glucose homeostasis, adiposity and systemic inflammation markers in the LDLr−/− mice, reinforcing that mitochondrial reactive oxygen species in the arterial wall have a key pro-atherogenic role.
Low dose DNP treatment also promotes other beneficial metabolic effects in other experimental models. In normolipidemic Swiss mice, a long treatment with DNP (>5 months) improved plasma glucose and triglyceride levels and reduced body weight 23) . Goldgof et al. showed that DNP treatment attenuated obesity in mice acclimated at thermoneutrality, but no changes were observed in body weight, adiposity and glucose homeostasis in mice maintained at the standard 22℃ 24) . There is evidence that mitochondrial uncoupling may indeed protect against metabolic diseases. Niclosamide ethanolamine, an anthelmintic drug, uncouples mitochondria and protects against high-fat diet-induced hepatic steatosis and insulin resistance. Moreover, the drug improved glycemic control of db/db mice 43) . Yet, a controlled-release mitochondrial protonophore that produces mild liver-targeted mitochondrial uncoupling reversed diabetes and steatohepatitis in rats 44) and decreased hypertriglyceridemia and reverse nonalcoholic steatohepatitis (NASH) and diabetes in a mouse model of severe lipodystrophy and diabetes 45) . Another mitochondrial uncoupler, BAM15, induced anti-obesity effects and increased insulin sensitivity in mice fed a western type diet 46) . In addition, Sorafenib, a drug used to treat hepatocellular carcinoma that elicits serious side effects, when employed in a 1/10 of the clinical dose, induces mild mitochondrial uncoupling and prevents the progression of NASH in mice and monkeys without adverse effects 47) . However, no previous studies have evaluated the association of mitochondrial uncoupling and atherosclerosis.
We have shown previously that the hypercholesterolemic LDL receptor knockout mice exhibited increased mitochondrial oxidant production in several tissues 32 , 33) . In this study, we demonstrated that treatment with the mitochondrial uncoupler DNP decreases the mitochondrial oxidant production and oxidative damage in various tissues (liver, plasma, macrophages and aorta) of LDLr−/− mice. Similar results in other tissues were found in Swiss mice treated in vivo with low doses of DNP 23) . The authors demonstrated that DNP treatment enhanced tissue respiration rates due to mild mitochondrial uncoupling. Moreover, muscle cells treated with DNP exhibit a reduction in mitochondrial oxidant production 48) .
Attenuation of spontaneous and diet-induced development of atherosclerosis by DNP treatment, independently of classical risk factors, indicates that mitochondrial superoxide and H2O2 production are causally related to atherosclerosis. These findings confirm and expand our previous reports on a positive correlation between mitochondrial oxidants and severity of atherosclerosis 9 , 10) . In line with our results, but using a genetic approach, Wang et al. improved the mitochondrial antioxidant capacity in macrophages by ectopically overexpressing catalase in mitochondria and observed reduced atherosclerotic lesion area in LDLr−/− mice 11 , 12) . Since DNP is a lipophilic highly specific proton translocator, it is likely that, in vivo, DNP acts in several tissue mitochondria. As we showed, DNP reduced oxidants production in liver, macrophages and aorta. These effects probably also include smooth muscle cells and endothelial cells, which are very relevant for atherosclerosis development. Thus, in vivo treatment with DNP contributes to local (arterial) and systemic antioxidant environment, attenuating atherosclerosis development.
Mild mitochondrial uncoupling has long ago been proposed as part of a cellular defense system to prevent the formation of superoxide 49) . More recently, mitochondrial uncoupling has been identified as a cytoprotective strategy under conditions of oxidative stress, including diabetes, drug-resistance in tumor cells, ischemia-reperfusion (IR) injury or aging 50) and, as proposed here, atherosclerosis. In terms of mechanisms, attenuation of mitochondrial oxidant production in the arterial wall may directly prevent the early atherogenic events, such as LDL oxidation, endothelial cell activation and recruitment of immune cells to arterial wall. In endothelial cells, oxidative stress induces the expression of several adhesion and chemotactic molecules that recruit immune cell and initiate local inflammation and atherogenesis 51) . In vascular smooth muscle cells, genes involved in migration (fibronectin), proliferation (p105 coactivator) and apoptosis (ECA39) were shown to be upregulated by H2O2 52) . After overexpressing mitochondrial catalase in macrophages of LDLr-/- mice, Wang et al. (2014) observed attenuation of NF-kB pathway and suppression of MCP-1 and other inflammatory markers. Thus, NF-kB is a pathway likely deactivated in macrophages of DNP treated hypercholesterolemic mice, as well as JNK and p38 and downstream inflammatory cytokines expression. We showed here that DNP treated macrophages exhibit a response compatible with a transition towards a less inflammatory phenotype shown by marked increases in IL-4, IL-10 and Arg-1 gene expression, 3 fold increase in IL-10 secretion and higher phagocytic activity. More importantly, DNP treated macrophages take up less LDL-cholesterol thus decreasing foam cell formation rates.
Further redox mechanisms triggered by DNP may be raised. Vilne et al. have shown that, under hypercholesterolemia, there is a marked down-regulation of mitochondrial genes in the aortic arches of the Ldlr−/−Apob100/100 mouse model 53) . The authors identified the transcription factor estrogen related receptor (ERR)-α and its co-factors PGC1-α and –β as key regulatory genes that were inversely correlated with atherosclerosis expansion. These genes are known to promote mitochondrial biogenesis and antioxidant responses. Thus, by decreasing aorta mitochondrial derived oxidants in hypercholesterolemia, mild uncoupling may counter regulate these and other key genes related with redox homeostasis and atherosclerosis. A large number of genes responsive to oxidative stress could be modulated by mild mitochondrial uncoupling. For instance, mitochondrial oxidants activate Nrf2 that induce the expression of antioxidant genes. However, in response to severe oxidative stress, Nrf2 induces Klf9 expression, which represses antioxidant enzymes and enhances cell death 54) . Thus, eliminating excessive oxidants by mild mitochondrial uncoupling could rescue Nrf2 expression and decrease the development of the disease. The identification of more relevant genes that could be regulated by mild mitochondrial uncoupling would require an “omics” approach in each specific vascular cell type.
Altogether, these findings support the research for new drug design targeting mild mitochondrial uncoupling as an effective strategy of stimulating endogenous antioxidant mechanism. In addition, available drugs with known mild uncoupling activity, such as the anthelmintic niclosamide ethanolamine, might be tested as a potential repurposed drug for attenuating arterial oxidative stress and atherosclerosis.
Conclusion
The present findings are a proof of concept that activation of mild mitochondrial uncoupling is sufficient to delay atherosclerosis development in hypercholesterolemia. These results promote future approaches targeting mitochondria for prevention or treatment of atherosclerosis.
Acknowledgements AND Notice of Grant Support
We are grateful for the excellent technical assistance of Andressa Fagundes.
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo to AEV and HCFO (FAPESP #2013/07607-8, #2017/17728-8) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq #300937/2018-0). GGD was supported by CNPq (# 151345/2013-9) and FAPESP (#2017/02903-9) fellowships; JCR (#2014/02819-0), BAP (# 2009/53762-0), LHPA (# 2017/03402-3) were supported by FAPESP fellowships.
Conflicts of Interest
Authors have no conflicts of interest to declare.
References
- 1).Moore KJ, Sheedy FJ and Fisher EA: Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol, 2013; 13: 709-721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Geovanini GR and Libby P: Atherosclerosis and inflammation: overview and updates. Clin Sci, 2018; 132: 1243-1252 [DOI] [PubMed] [Google Scholar]
- 3).Thomson MJ, Puntmann V and Kaski JC: Atherosclerosis and oxidant stress: the end of the road for antioxidant vitamin treatment? Cardiovasc Drugs Ther, 2007; 21: 195-210 [DOI] [PubMed] [Google Scholar]
- 4).Steinhubl SR: Why have antioxidants failed in clinical trials? Am J Cardiol, 2008; 101: 14D-19D [DOI] [PubMed] [Google Scholar]
- 5).Sies H and Jones DP: Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol, 2020; 7: 363-383 [DOI] [PubMed] [Google Scholar]
- 6).Boveris A and Chance B: The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J, 1973; 134: 707-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, Sluse FE, Souza-Pinto NC, Vercesi AE: Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal, 2013; 18: 2029-2074 [DOI] [PubMed] [Google Scholar]
- 8).Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE: Mitochondria and reactive oxygen species. Free Radic Biol Med, 2009; 47: 333-343 [DOI] [PubMed] [Google Scholar]
- 9).Dorighello GG, Paim BA, Kiihl SF, Ferreira MS, Catharino RR, Vercesi AE, Oliveira HC: Correlation between Mitochondrial Reactive Oxygen and Severity of Atherosclerosis. Oxid Med Cell Longev, 2016; 2016: 7843685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Dorighello GG, Paim BA, Leite ACR, Vercesi AE, Oliveira HCF: Spontaneous experimental atherosclerosis in hypercholesterolemic mice advances with ageing and correlates with mitochondrial reactive oxygen species. Exp Gerontol, 2018; 109: 47-50 [DOI] [PubMed] [Google Scholar]
- 11).Wang Y, Wang GZ, Rabinovitch PS, Tabas I: Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res, 2014; 114: 421-433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Wang Y, Wang W, Wang N, Tall AR, Tabas I: Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arterioscler Thromb Vasc Biol, 2017; 37: e99-e107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, Finigan A, Figg N, Pung YF, Logan A, Murphy MP, Bennett M: Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler Thromb Vasc Biol, 2017; 37: 2322-2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Oliveira HCF and Vercesi AE: Mitochondrial bioenergetics and redox dysfunctions in hypercholesterolemia and atherosclerosis. Mol Aspects Med, 2020; 71: 100840 [DOI] [PubMed] [Google Scholar]
- 15).Oyewole AO and Birch-Machin MA: Mitochondria-targeted antioxidants. FASEB J, 2015; 29: 4766-4771 [DOI] [PubMed] [Google Scholar]
- 16).Smith RA, Hartley RC and Murphy MP: Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Signal, 2011; 15: 3021-3038 [DOI] [PubMed] [Google Scholar]
- 17).Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR: The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic Biol Med, 2012; 52: 841-849 [DOI] [PubMed] [Google Scholar]
- 18).Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR: NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxid Redox Signal, 2015; 23: 1389-1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Vozenilek AE, Vetkoetter M, Green JM, Shen X, Traylor JG, Klein RL, Orr AW, Woolard MD, Krzywanski DM: Absence of Nicotinamide Nucleotide Transhydrogenase in C57BL/6J Mice Exacerbates Experimental Atherosclerosis. J Vasc Res, 2018; 55: 98-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Bonora M, Wieckowski MR, Sinclair DA, Kroemer G, Pinton P, Galluzzi L: Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat Rev Cardiol, 2019; 16: 33-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Tainter ML, Stockton AB and Cutting WC: Dinitrophenol in the treatment of obesity - Final report. JAMA, 1935; 105: 0332-0337 [Google Scholar]
- 22).Grundlingh J, Dargan PI, El-Zanfaly M, Wood DM: 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol, 2011; 7: 205-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ: Mild mitochondrial uncoupling in mice affects energy metabolism redox balance and longevity. Aging Cell, 2008; 7: 552-560 [DOI] [PubMed] [Google Scholar]
- 24).Goldgof M, Xiao C, Chanturiya T, Jou W, Gavrilova O, Reitman ML: The chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy homeostasis in mice at thermoneutrality. J Biol Chem, 2014; 289: 19341-19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Kishimoto Y, Johnson J, Fang W, Halpern J, Marosi K, Liu D, Geisler JG, Mattson MP: A mitochondrial uncoupler prodrug protects dopaminergic neurons and improves functional outcome in a mouse model of Parkinson’s disease. Neurobiol Aging, 2020; 85: 123-130 [DOI] [PubMed] [Google Scholar]
- 26).Jiao S, Cole TG, Kitchens RT, Pfleger B, Schonfeld G: Genetic heterogeneity of lipoproteins in inbred strains of mice: analysis by gel-permeation chromatography. Metabolism, 1990; 39: 155-160 [DOI] [PubMed] [Google Scholar]
- 27).Salerno AG, Silva TR, Amaral ME, Alberici LC, Bonfleur ML, Patrício PR, Francesconi EP, Grassi-Kassisse DM, Vercesi AE, Boschero AC, Oliveira HC: Overexpression of apolipoprotein CIII increases and CETP reverses diet-induced obesity in transgenic mice. Int J Obes, 2007; 31: 1586-1595 [DOI] [PubMed] [Google Scholar]
- 28).Folch J, Lees M and Sloane Stanley GH: A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem, 1957; 226: 497-509 [PubMed] [Google Scholar]
- 29).Reznick AZ and Packer L: Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol, 1994; 233: 357-363 [DOI] [PubMed] [Google Scholar]
- 30).Schild L, Reinheckel T, Wiswedel I, Augustin W: Short-term impairment of energy production in isolated rat liver mitochondria by hypoxia/reoxygenation: involvement of oxidative protein modification. Biochem J, 1997; 328: 205-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Kaplan RS and Pedersen PL: Characterization of phosphate efflux pathways in rat liver mitochondria. Biochem J, 1983; 212: 279-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Oliveira HC, Cosso RG, Alberici LC, Maciel EN, Salerno AG, Dorighello GG, Velho JA, de Faria EC, Vercesi AE: Oxidative stress in atherosclerosis-prone mouse is due to low antioxidant capacity of mitochondria. FASEB J, 2005; 19: 278-280 [DOI] [PubMed] [Google Scholar]
- 33).Paim BA, Velho JA, Castilho RF, Oliveira HC, Vercesi AE: Oxidative stress in hypercholesterolemic LDL (low-density lipoprotein) receptor knockout mice is associated with low content of mitochondrial NADP-linked substrates and is partially reversed by citrate replacement. Free Radic Biol Med, 2008; 44: 444-451 [DOI] [PubMed] [Google Scholar]
- 34).Dikalov S, Griendling KK and Harrison DG: Measurement of reactive oxygen species in cardiovascular studies. Hypertension, 2007; 49: 717-727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001; 25(4): 402-408 [DOI] [PubMed] [Google Scholar]
- 36).de Lima C, Alves LE, Iagher F, Machado AF, Bonatto SJ, Kuczera D, de Souza CF, Pequito DC, Muritiba AL, Nunes EA, Fernandes, LC: Anaerobic exercise reduces tumor growth, cancer cachexia and increases macrophage and lymphocyte response in Walker 256 tumor-bearing rats. Eur J Applied Physiol, 2008; 104: 957-964 [DOI] [PubMed] [Google Scholar]
- 37).Robinet, P., Wang, Z., Hazen, S.L., and Smith, J.D: A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res, 2010; 51: 3364-3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ: Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes, 2000; 49: 527-531 [DOI] [PubMed] [Google Scholar]
- 39).Hardie DG: AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes, 2008; 32 Suppl 4: S7-12 [DOI] [PubMed] [Google Scholar]
- 40).Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S: The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta, 2011; 1813: 878-888 [DOI] [PubMed] [Google Scholar]
- 41).Yunna C, Mengru H, Lei W, Weidong C: Macrophage M1/M2 polarization. Eur J Pharmacol, 2020; 877: 173090 [DOI] [PubMed] [Google Scholar]
- 42).Chisolm GM, Steinberg D: The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med, 2000; 28: 1815-1826 [DOI] [PubMed] [Google Scholar]
- 43).Tao H, Zhang Y, Zeng X, Shulman GI, Jin S: Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med, 2014; 20: 1263-1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI: Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science, 2015; 347: 1253-1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Abulizi A, Perry RJ, Camporez JPG, Jurczak MJ, Petersen KF, Aspichueta P, Shulman GI: A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J, 2017; 31: 2916-2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Alexopoulos SJ, Chen SY, Brandon AE, Salamoun JM, Byrne FL, Garcia CJ, Beretta M, Olzomer EM, Shah DP, Philp AM, Hargett SR, Lawrence RT, Lee B, Sligar J, Carrive P, Tucker SP, Philp A, Lackner C, Turner N, Cooney GJ, Santos WL, Hoehn KL: Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nat Commun, 2020; 11: 2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Jian C, Fu J, Cheng X, Shen LJ, Ji YX, Wang X, Pan S, Tian H, Tian S, Liao R, Song K, Wang HP, Zhang X, Wang Y, Huang Z, She ZG, Zhang XJ, Zhu L, Li H: Low-Dose Sorafenib Acts as a Mitochondrial Uncoupler and Ameliorates Nonalcoholic Steatohepatitis. Cell Metab, 2020; 31(5): 892-908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).MacLellan JD, Gerrits MF, Gowing A, Smith PJ, Wheeler MB, Harper ME: Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes, 2005; 54: 2343-2350 [DOI] [PubMed] [Google Scholar]
- 49).Skulachev VP: Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta, 1998; 1363: 100-124 [DOI] [PubMed] [Google Scholar]
- 50).Cadenas S: Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta Bioenerg, 2018; 1859: 940-950 [DOI] [PubMed] [Google Scholar]
- 51).Stocker R, Keaney JF Jr: Role of oxidative modifications in atherosclerosis. Physiol Rev, 2004; 84: 1381-1478 [DOI] [PubMed] [Google Scholar]
- 52).Sakamoto K, Yamasaki Y, Kaneto H, Fujitani Y, Matsuoka T, Yoshioka R, Tagawa T, Matsuhisa M, Kajimoto Y, Hori M: Identification of oxidative stress-regulated genes in rat aortic smooth muscle cells by suppression subtractive hybridization. FEBS Lett, 1999; 461: 47-51 [DOI] [PubMed] [Google Scholar]
- 53).Vilne B, Skogsberg J, Foroughi Asl H, Talukdar HA, Kessler T, Björkegren JLM, Schunkert H: Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis, 2017; 267: 39-48 [DOI] [PubMed] [Google Scholar]
- 54).Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K: Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules, 2020; 10: 320 [DOI] [PMC free article] [PubMed] [Google Scholar]