

Abstract
Neural stem cells (NSCs) have the ability to proliferate, differentiate, undergo apoptosis, and even enter and exit quiescence. Many of these processes are controlled by the complex interplay between NSC intrinsic genetic programs with NSC extrinsic factors, local and systemic. In the genetic model organism, Drosophila melanogaster, NSCs, known as neuroblasts (NBs), switch from quiescence to proliferation during the embryonic to larval transition. During this time, larvae emerge from their eggshells and begin crawling, seeking out dietary nutrients. In response to animal feeding, the fat body, an endocrine organ with lipid storage capacity, produces a signal, which is released systemically into the circulating hemolymph. In response to the fat body-derived signal (FBDS), Drosophila insulin-like peptides (Dilps) are produced and released from brain neurosecretory neurons and glia, leading to downstream activation of PI3-kinase growth signaling in NBs and their glial and tracheal niche. Although this is the current model for how NBs switch from quiescence to proliferation, the nature of the FBDS extrinsic cue remains elusive. To better understand how NB extrinsic systemic cues regulate exit from quiescence, a method was developed to culture early larval brains in vitro before animal feeding. With this method, exogenous factors can be supplied to the culture media and NB exit from quiescence assayed. We found that exogenous insulin is sufficient to reactivate NBs from quiescence in whole-brain explants. Because this method is well-suited for large-scale screens, we aim to identify additional extrinsic cues that regulate NB quiescence versus proliferation decisions. Because the genes and pathways that regulate NSC proliferation decisions are evolutionarily conserved, results from this assay could provide insight into improving regenerative therapies in the clinic.
Introduction
Stem cells are of great interest because of their potential for use in regenerative medicine1,2. Many animals, especially those that are long-lived, maintain stem cells within their adult tissues. These resident stem cells function to maintain tissue homeostasis and are utilized for repair following physical injury or disease3,4. Most stem cells in adult animals are quiescent, a relatively dormant state characterized by cell cycle arrest and inactivation of growth signaling5. In response to extrinsic cues, stem cells exit from quiescence, enter the cell cycle and begin generating daughter progeny specific to their tissue type. For example, in order to mount an effective immune response, antigen-presenting cells induce quiescent naive T cells to enter cell cycle and clonally expand6. In response to skeletal muscle damage, muscle satellite stem cells enter the cell cycle and generate daughter myoblasts to replace damaged myofibrils5,7. While it is clear that quiescent stem cells respond to extrinsic signals, in many cases, the nature of the extrinsic cue remains unclear as well as the mechanism of cue-induced stem cell activation. Gaining a better understanding of how quiescent stem cells respond to extrinsic cues and enter the cell cycle will aid in the development of better stem cell therapies in the clinic and increase scientific knowledge.
For decades now, model organisms have been used to uncover the genes and cell signaling pathways that regulate stem cell proliferation during development and in adulthood. In Drosophila, neural stem cells (NSCs), known as neuroblasts (NBs), divide throughout development to generate all neurons and glia that ultimately integrate, forming the neural circuity required for brain function8,9. Like other stem cells, NBs divide asymmetrically to self-renew and, in some cases, symmetrically to expand the stem cell pool. NBs are specified during embryogenesis and most enter quiescence towards the end, coincident with declining maternal nutrient stores (Figure 1). After embryogenesis is complete, larvae hatch and begin feeding. In response to animal feeding, NBs reactivate from quiescence and resume cell divisions10,11,12,13,14,15,16. Because the Drosophila CNS is relatively simple and because NBs enter and exit quiescence at defined times, using Drosophila to investigate the regulation of quiescence, entry and exit, proves ideal.
Figure 1: Relative proliferation of CB NBs (central brain neuroblasts, red) and MB NBs (mushroom body neuroblasts, blue) over developmental time.
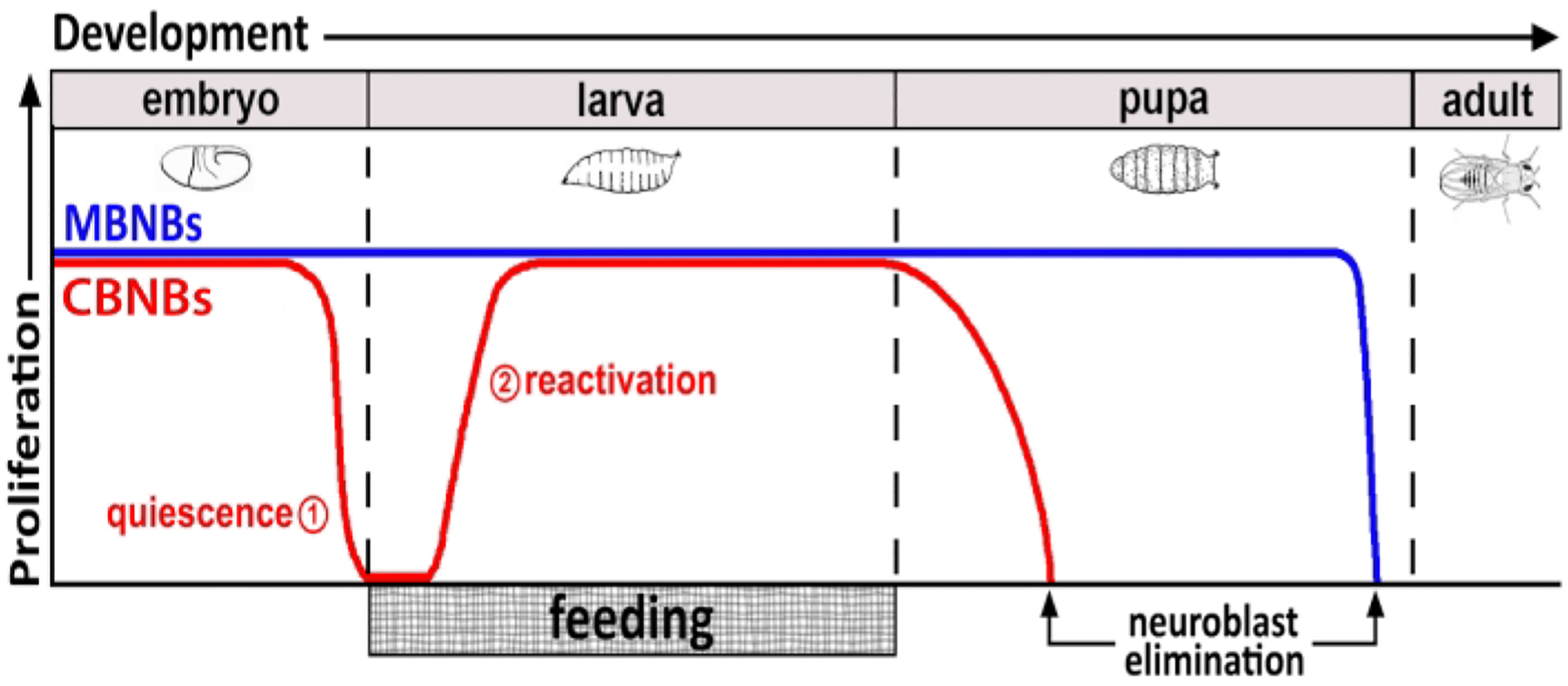
At the end of embryogenesis, most NBs (red line) cease proliferation and enter quiescence. Quiescence continues until freshly hatched larvae consume their first complete meal. The time points of focus for this methodology are denoted in red circles (1, quiescence and 2, reactivation). MB NBs (blue) are a subset of central brain NBs that divide continually throughout development (4 per brain hemisphere).
In response to animal feeding, PI3-kinase and TOR growth signaling pathways become active in NBs and in their glial and tracheal niche10,11,15,16. When dietary nutrients are withdrawn or when levels of PI3-kinase are reduced, NBs fail to reactivate and growth of glia and trachea are also reduced10,11,15,16. The current model posits that NB reactivation is coupled to larval growth by the fat body, which releases a systemic signal in response to animal feeding12,17,18. This signal, which remains elusive, likely promotes the expression and release of Drosophila insulin-like peptide (Dilps) in the brain, which leads to the downstream activation of PI3-kinase in NBs and their glial and tracheal niche. To better understand the nature of the systemic cue(s), we developed a method to reactivate quiescent NBs in cultured brain explants. With this method, reactivation of NBs can be assayed in the absence of whole animal systemic cues. Exogenous factors can be resupplied to the culture media and NB reactivation assayed based on the incorporation of the thymidine analog, EdU. Using this method, we determined that exogenous insulin is sufficient to reactivate quiescent NBs in brain explants. Future work will be aimed at identifying additional factors that, when added back, either positively or negatively regulate NB quiescence in brain explants.
Protocol
1. Drosophila larvae collection
NOTE: Prepare the yeast plate, grape paste, and the Fly condo before starting:
-
Yeast paste: In a small container, mix 5 g of active dry yeast with 10 mL of water to form a paste that has the consistency of peanut butter. Cover the yeast paste with plastic wrap and use a rubber band to firmly attach it to the container.
NOTE: Fresh yeast paste will expand in its container and will pop off the lid unless firmly attached. Yeast paste will last for several days at room temperature (RT).
- Grape plates: Follow the recipe for making grape plates (Table 1). If using plates stored at 4°C, make sure to pre-warm plates before use by placing them at RT for 1 h.
- Mix water (750 mL) and agar (18.75 g) in a 4 L flask, swirl, and autoclave for 20 min (liquid cycle).
- Mix the grape juice (250 mL) and sucrose (25 g) in a 1 L flask with a large stir bar on a heated plate (low heat). When the sucrose is dissolved, turn off the heat, wait until the flask can be touched before adding Tegosept (10%, 4 mL) and Propionic acid (5 mL). Keep the stir bar on.
- When the autoclaving is complete, let it cool until the flask can be touched (~60 °C), then mix in the grape juice mix.
- Combine all solutions in one flask and let stir on the plate.
- Pipette the solution into lids of small-sized Petri dishes (35 mm). Pipette roughly 9 mL per lid or until a convex dome is obtained.
- OPTIONAL: Flame the lids to get rid of any bubbles.
- When the plates solidify, stack the grape plates into a box with an airtight lid and place the box at 4 °C. Plates can be stored for up to 1 month.
Fly condo: Punch ~20 holes into a 6-ounce square bottom polypropylene Drosophila bottle using an 18 G needle.
Transfer adult flies (~100 OregonR or any genotype) to a fly condo and cap the condo with a grape agar plate topped with a dab of yeast paste. Place the dab towards the center of the plate and affix the plate to the condo with lab tape.
Invert the container so that the grape agar plate is on the bottom and place it in a 25 °C incubator for 24 h (Figure 2).
After 24 h, change the grape agar plate and replace it with a new plate topped with yeast paste. Quickly switch the two plates while continuously tapping the condo lightly on the bench so that the adult flies do not escape.
Examine the plate by eye and assess the number of embryos on the plate. Drosophila embryos are oblong and white with two string-like appendages.
If there are very few embryos on the plate (less than 100), discard the plate (scrape out the agar into the fly trash and save the plastic lid for reuse). In many cases, adult females will not lay many embryos on the first night in a new condo. If this is the case, give the adult flies another 24 h to acclimate.
If there is a large number of embryos on the plate (at least 100), keep it, and carefully remove the yeast paste using a flat bottom spatula.
Once the yeast paste is removed, use a metal pick to manually remove all larvae from the grape plate under a dissecting microscope. When looking at the plate under a dissecting microscope, crawling larvae should be observed as well as embryos.
-
Remove all larvae by brushing the metal pick towards the side of one larva. Larvae are sticky and will stick to the pick. Once one larva is on the pick, additional larvae can be easily picked up by using the larva on the tool to attach more.
NOTE: Larvae like to stick to each other. At this point, it does not matter if larvae get damaged. These larvae will be discarded.
After picking and removing all larvae, place the plate back into the 25 °C incubator. Ensure to place the plate in a larger container that can be sealed. Place wet paper towels in the bottom of the larger container to keep in moisture.
After 30–60 min, take the plate back to the dissecting microscope and now, carefully pick ~20–25 larvae from the same grape agar plate to ensure that the picked larvae are freshly hatched within a 30–60 min window of time.
Submerge the tip of the tool with the 20–25 freshly hatched larvae into a Petri dish (60 mm) filled with 1–2 mL of 1x Phosphate Buffered Saline (PBS) for 2 min.
After 2 min, tip the dish at an angle to pool the liquid at the bottom. Using a small paintbrush, brush the larvae out of the liquid up the bottom of the Petri dish.
Collect all larvae on the paintbrush and transfer the larvae to a new Petri dish (60 mm) containing 1–2 mL of 70% ethanol. Repeat steps 1.15 to collect larvae with a paintbrush and transfer them to a new Petri dish with 1–2 mL of 1x PBS.
Table 1: Recipe for making grape plates.
Follow the steps described in section 1.2.
| Ingredient/Amount | 1 L (~125 grape plates) |
|---|---|
| Grape juice | 250 mL |
| Sucrose | 25 g |
| Water | 750 mL |
| Bacto Agar | 18.75 g |
| Tegosept (10%) | 4 mL |
| Propionic Acid | 5 mL |
Figure 2: Visual representation of inverted fly bottle (condo) with male and female Drosophila adults.
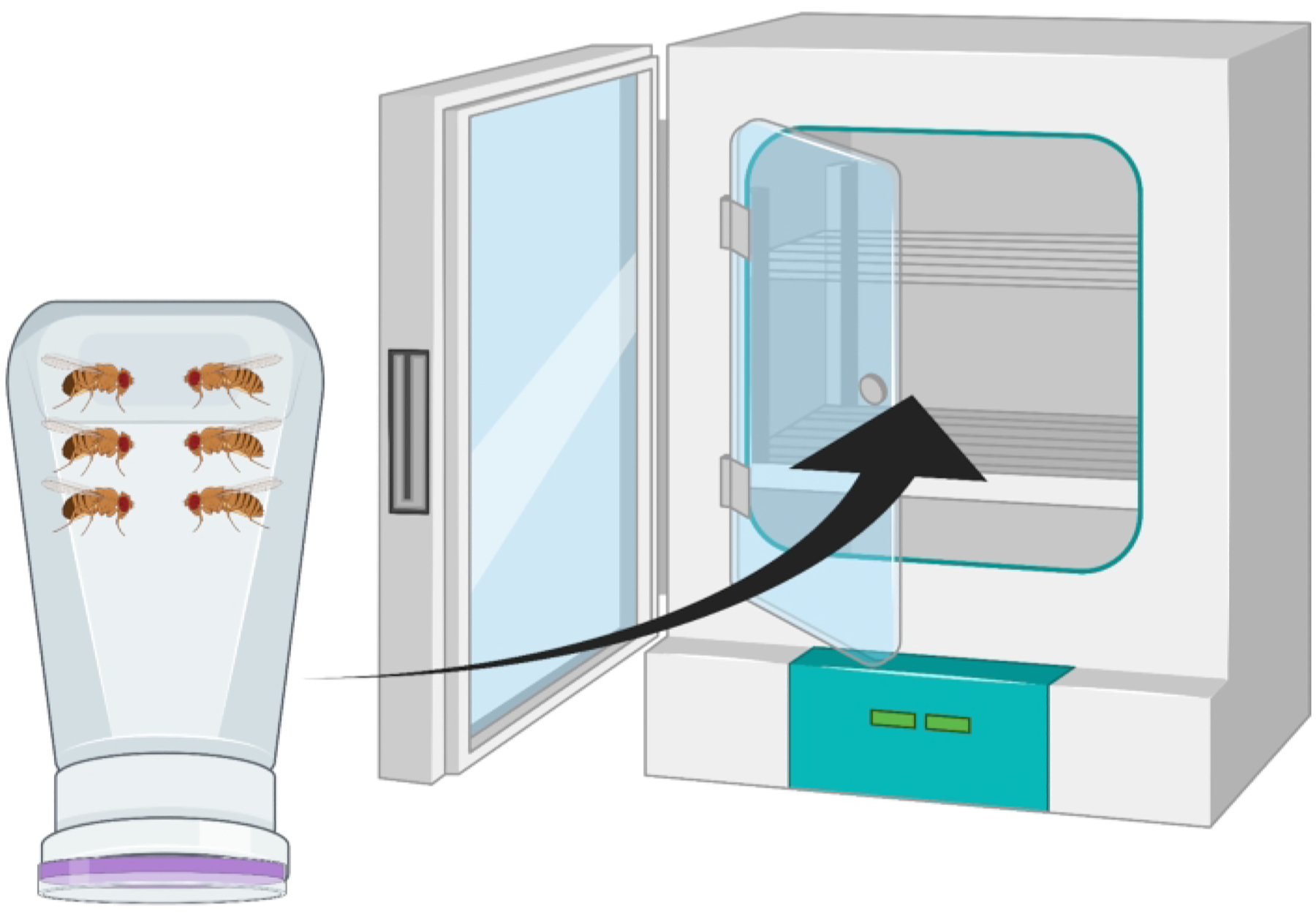
The plastic bottle has small punctures, generated with an 18 G needle, for oxygen exchange. The mouth of the bottle is sealed with an agar grape juice cap and is inverted and stored in a 25 °C incubator.
2. Culture media and tool preparation
Spray the bench and work area with 70% ethanol and let dry.
Spray the dissection tools, forceps, and two glass watch dishes, with 70% ethanol and let them dry on the bench.
Make the supplemented Schneider’s media (SSM, Table 2) and place it on ice.
Pipette 1 mL of SSM into each of the glass watch dishes.
Using a micropipette with a sterile tip, transfer the freshly hatched larvae from the plate of PBS to the SSM in the first glass watch dish. Using a micropipette with a sterile tip, transfer the freshly hatched larvae to the SSM in the second glass watch dish.
Table 2: Recipe for making supplemented Schneider’s media (SSM).
The table lists the volume of all ingredients required for preparing 5 mL of SSM.
| Ingredient (starting concentration) | To make 5 mL | Final concentration |
|---|---|---|
| Schneider’s Drosophila media | 3.89 mL | |
| Fetal bovine Serum (100%) | 500 μL | 10% |
| Glutamine (200 mM) | 500 μL | 20 mM |
| Penicillin (5000 units/mL), Streptomycin (50,000 μg/mL) |
100 μL | 1000 U/mL Pen, 1 mg/mL Strep |
| Insulin (10 mg/mL) | 10 μL | 0.02 mg/mL |
| glutathione (50 mg/mL) | 5 μL | 0.05 mg/mL |
| In a sterile hood, using sterile technique, add all ingredients together in a 14 mL conical tube. Place it on ice until use. | ||
3. Dissections and brain cultures
Once the larvae are in the second glass watch dish with SSM, dissect the brains out of the larvae using forceps and a dissecting microscope. Adjust the magnification as needed.
-
Use one forceps to grab the mouth hooks and with the other, gently grab the body halfway down and pull in the opposite direction (Figure 3) to split the larva into two pieces.
NOTE: The brain will be located right behind the mouth hooks. Note that there may be other tissues surrounding the brain. Be very careful when removing these tissues as it can result in damaging the brain
Once 15–20 brains have been dissected, add 1 mL of SSM into one well of a sterile 12-well culture tray. Transfer the freshly dissected brains into the SSM using a micropipette and a sterile tip (Figure 4A).
Place the brains in the SSM media in the 12-well culture tray into an incubator at 25 °C for 24 h (Figure 4A).
Figure 3: Drosophila larvae in a glass watch dish with SSM.
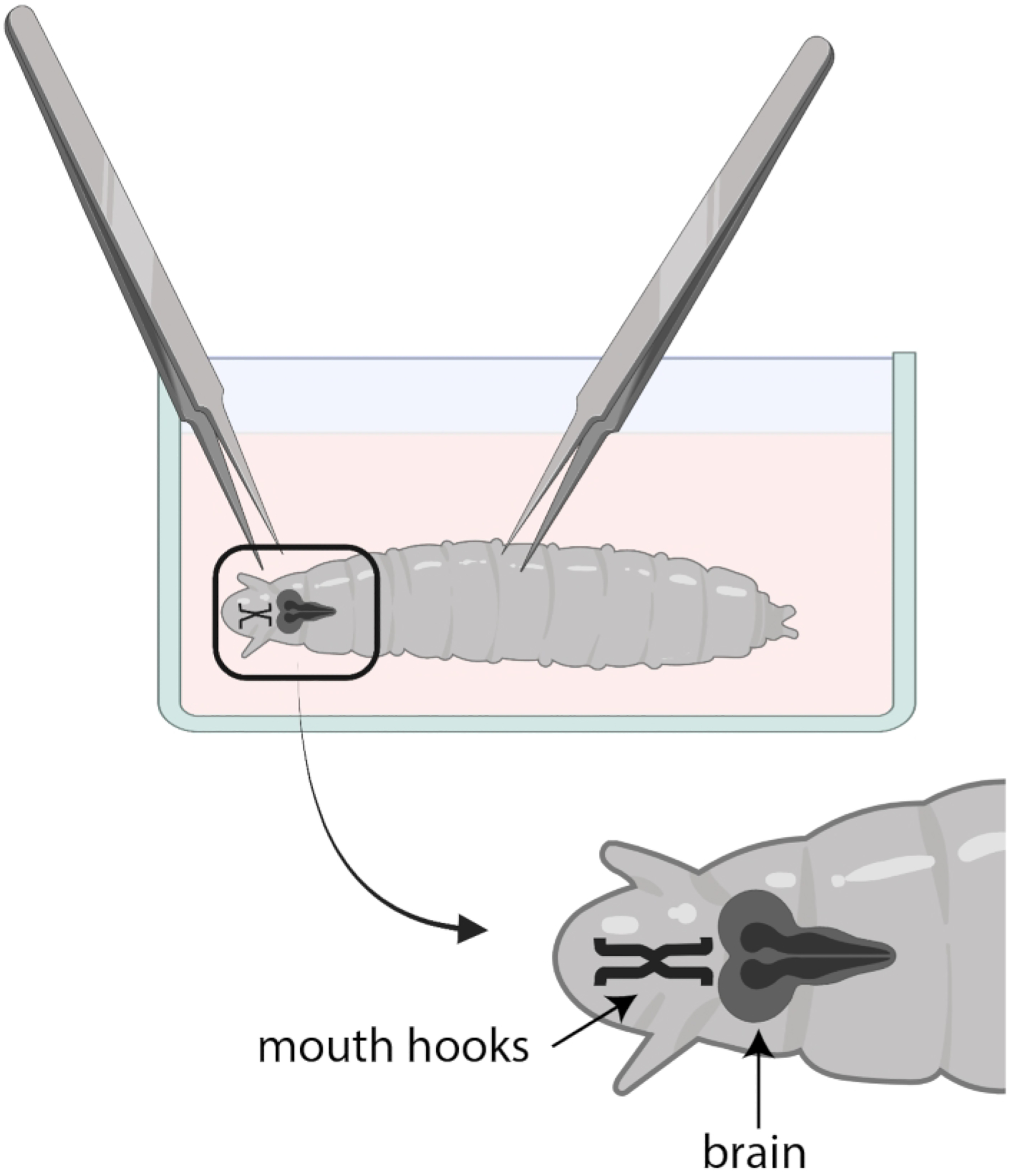
Forceps are properly positioned for dissection. The location of the larval brain (dark grey) is posterior to the mouth hooks (black), and both are shown inside the larva.
Figure 4: Brain culture and immunostaining.
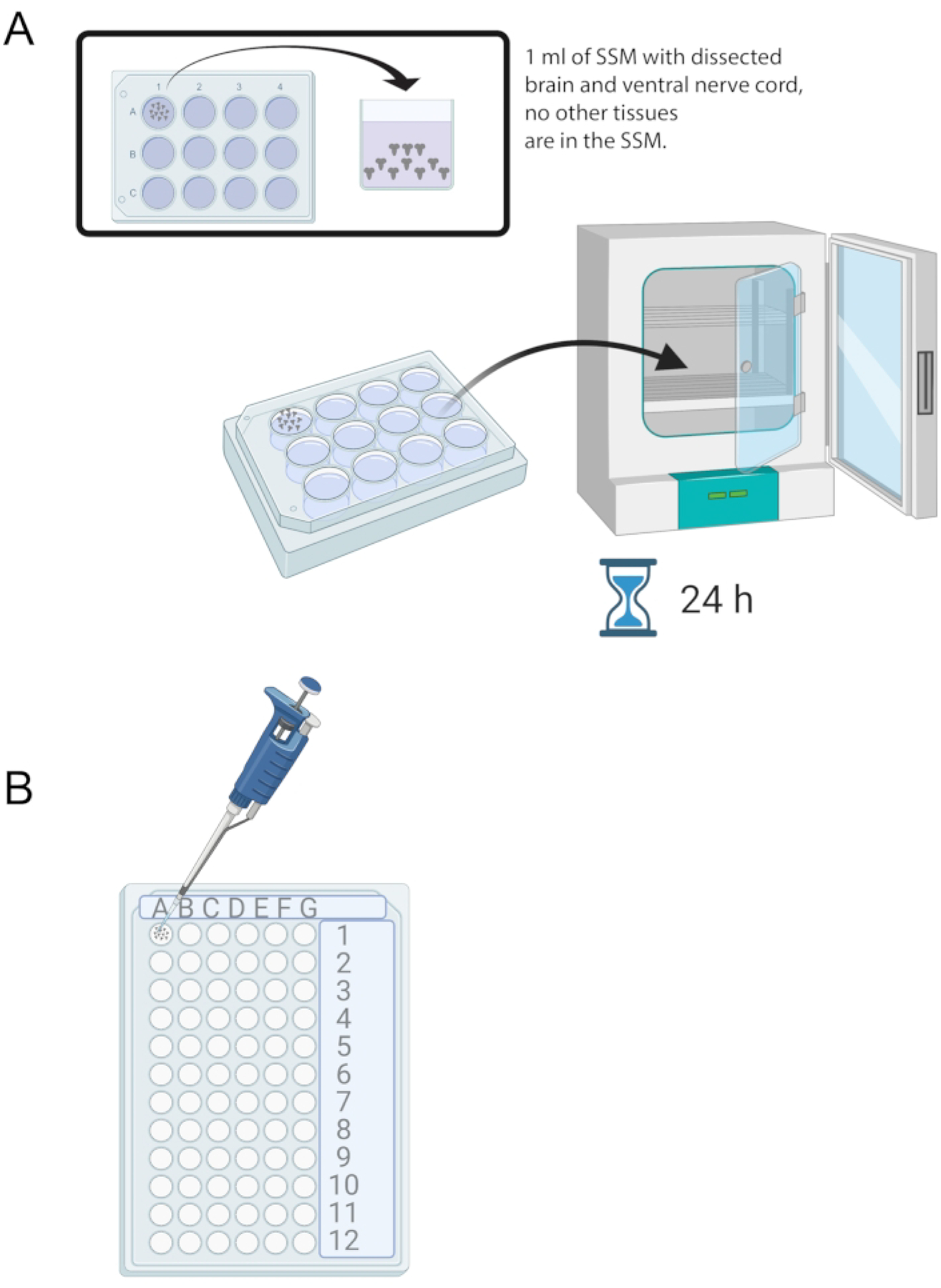
(A) Whole brains in a 12-well culture dish containing 1 mL of SSM. The culture dish is then placed in a 25 °C incubator for 24 h. (B) 72-well mini tray that holds brain explants during immunostaining. Brains are washed and solutions transferred using a P20 micropipette set to 10 μL.
4. Proliferation assay, brain fixation, and antibody staining
The following day, make 1 mL of EdU SSM solution. Pipette 10 μL of a 10 mM stock of 5-Ethynyl-2′-deoxyuridine (EdU) with 990 μL of SSM (final EdU concentration equals 0.1 mM) into a sterile microcentrifuge tube and mix. After the 24 h incubation is complete, pipette the 1 mL of EdU SSM into one well of the sterile 12-well culture tray.
Transfer the brains using a micropipette with a sterile tip from the well containing SSM only to the new well containing the EdU SSM solution. Incubate for 1 h at 25 °C.
-
Next, transfer the EdU-labeled brains to another well in the same culture tray containing 1 mL of fixative (4% paraformaldehyde, see Table 3 for recipe) for 20 min.
CAUTION: Paraformaldehyde is a biological hazard and must be disposed of properly.
-
After fixation, quickly transfer the brains to the wells of a 72-well mini tray using a micropipette. Each well can hold 10 brains and 10–15 μL of liquid (Figure 4B). Once the brains are transferred to the mini tray (no more than 10 brains per well), remove the fix and rinse the brains 3 times in 10 μL of 1x PBT (phosphate buffer, pH 7.4 containing 0.1% Triton-X 100).
NOTE: Rinsing means pipetting 10 μL of 1x PBT on the brains, removing, and repeating 3 times.
Next, wash the brains 3 times for 10 min each, again in 10 μL 1x PBT. Ensure that the brains are covered with some liquid at all times.
After the washes are complete, pipette 10 μL of blocking solution (1x PBT with 10% normal goat serum) onto the brains. Cover the tray and seal it using a strip of parafilm around the edge.
Once sealed, place the mini tray into a sealed box with wet towels to provide a moist environment to prevent evaporation. Place the box containing the tray at 4 °C overnight.
-
The following day, make a primary antibody solution.
NOTE: In this protocol, rabbit anti-scribble was used to label cell membranes and rat anti-deadpan to label neuroblasts, although any number of other primary antibodies could be used.-
To make the primary antibody solution, first, make dilutions of primary antibodies in the blocking solution. For example, rabbit anti-scribble is used at a final concentration of 1:1000. Therefore, first, dilute the rabbit anti-scribble antibody at 1:100 (1 μL of antibody plus 99 μL of blocking solution). Rat-deadpan is used at a final concentration of 1:100. Therefore, first, dilute the rat-deadpan antibody at 1:10 (1 μL of antibody plus 9 μL of blocking solution).NOTE: These dilutions can be stored long term at 4 °C if sodium azide (0.05%) is also added to inhibit bacterial growth.
-
Next, count the number of wells that contain brains. The number of wells determines the volume of primary antibody solution to make. For example, if there are 2 wells of brains, prepare 20 μL of primary antibody solution (for 10 wells, 100 μL, etc.). To make a 20 μL primary antibody solution, add 2 μL of each primary antibody dilution and 16 μL of the blocking solution.NOTE: The final concentration of each of the primary antibodies is 1:1000 and 1:100, respectively. In short, make the first dilution of primary antibodies at a concentration so that the second dilution is always 1:10 to arrive at the respective final concentrations. In this case, 1:1000 for rabbit anti-scribble and 1:100 for rat anti-deadpan.
-
Remove the blocking solution with the micropipette set to 10 μL and pipette 10 μL of primary antibody solution into each well.
-
Cover and seal the tray using parafilm and place it back into the sealed box with wet towels. Incubate overnight at 4 °C.
NOTE: Shaking is not required and is strongly discouraged. Antibodies will penetrate the brains without shaking or mixing.
The following day, remove the primary antibody solution using a micropipette and rinse the brains 3 times with 10 μL of 1x PBT. Next, wash the brains 4 times with 10 μL of 1x PBT for 10 min each. During the 10 min washes, prepare the secondary antibody solution.
- To make the secondary antibody solution, choose secondary antibodies that recognize the primary antibodies. In this protocol, goat anti-rabbit Alexa Fluor 488 and goat anti-rat Alexa 555 were used.
- Pipette 1 μL of each of the secondary antibodies into a microcentrifuge tube with 298 μL of the blocking solution to make the final concentration 1:300 for each secondary antibody.
-
After the last 10 min wash, remove the 1x PBT and pipette 10 μL of the secondary antibody solution into each well. Seal the tray using parafilm and place it back into the box with moist towels. Incubate overnight at 4 °C.
NOTE: Do not worry about removing every last μL in the wells between rinses, washes, or when adding primary and secondary antibody solutions. The brains will always remain submerged in a few μL of liquid, which is just fine.
The following day, remove the secondary antibody solution using a micropipette and rinse the brains 3 times with 10 μL each of 1x PBT. Next, wash the brains 4 times with 10 μL each of 1x PBT for 10 min each.
During the 10 min washes, prepare the EdU reaction mix to detect EdU incorporation. Prepare the EdU reaction mix according to the manufacturers’ guidelines.
After the final wash, remove the 1x PBT and pipette 10 μL of the EdU reaction mix into each well with brains. Seal the microwell plate with parafilm and cover with aluminum foil. Leave plate on the bench for 30 min.
After 30 min, rinse the brains 3 times with 10 μL each of 1x PBT and wash the brains 3 times with 10 μL each of 1x PBT for 5 min each.
After the last wash, remove the 1x PBT and pipette 10 μL of a glycerol-based mounting media solution. Seal the plate and place at 4 °C overnight.
Table 3: Recipe for making fixative and PEM buffer.
The table lists the chemicals and their respective concentrations for preparing fixative and PEM buffer.
| Recipe for making fixative | ||
|---|---|---|
| Ingredient (starting concentration) | To make 40 mL | Final concentration |
| EM grade formaldehyde 16% | 10 mL | 4% |
| PEM buffer pH 7.0 | 29.96 mL | |
| Triton X-100 | 40 mL | 0.10% |
| Mix all ingredients together. Aliquot 1 mL into microcentrifuge tubes and store at −20 °C. Do not refreeze the aliquots after thawing. | ||
| Recipe for PEM buffer | ||
| Ingredient (starting concentration) | To make 100 mL | Final concentration |
| PIPES pH 6.8 (500 mM) | 20 mL | 100 mM |
| EGTA (500 mM) | 2 mL | 10 mM |
| MgSO4 (1 M) | 100 mL | 1 mM |
| Water | 77.9 mL | |
5. Mounting and imaging the brains
The following day, prepare microscope slides (25 mm × 75 mm × 1 mm): Glue (e.g., superglue) one 22 mm × 22 mm × 1 mm square cover glass to each end of the microscope slide to create a ‘bridge’ over which the larger 22 mm × 50 mm × 1 mm coverslip will be placed to make a space between the slide and the larger coverslip (Figure 5A). This space will allow the brain just enough movement to be correctly oriented while preventing it from being crushed.
-
After gluing the 22 mm × 22 mm × 1 mm cover glasses to the microscope slide, pipette 9.3 μL of the glycerol-based mounting media solution containing one brain from a well of the microwell plate and place it onto the center of the slide (Figure 5A).
NOTE: Larval brains may adhere to the pipette tip, so be careful. To avoid adhesion, begin by aspirating the antifade and then aspirating the single brain towards the end of the 9.3 μL volume.
Once the brain is on the slide, gently place the 22 mm × 50 mm × 1 mm coverslip on top. Position the brain as seen in Figure 5B. Gently move the coverslip around to orient the brain. The sample is then ready for imaging.
- Use a confocal microscope equipped with high magnification and a high numerical aperture objective (Figure 5C) to acquire the best images. For example: 60x or 63x, 1.4 NA oil-immersion lens.
-
Image the brains with the dorsal surface closest to the coverslip (and objective). Acquire the Z stacks through the entire brain hemisphere starting at the ventral surface (furthest away from the objective) at 1 μm intervals or Z step size.NOTE: Lasers used depend on the secondary antibodies. In this protocol, the laser lines used were 488 nm to detect Scribble staining, 555 nm to detect Deadpan, and 633 nm to detect EdU.
-
Figure 5: Schematic showing microscope slide, orientation, and cell types in the larval brain.
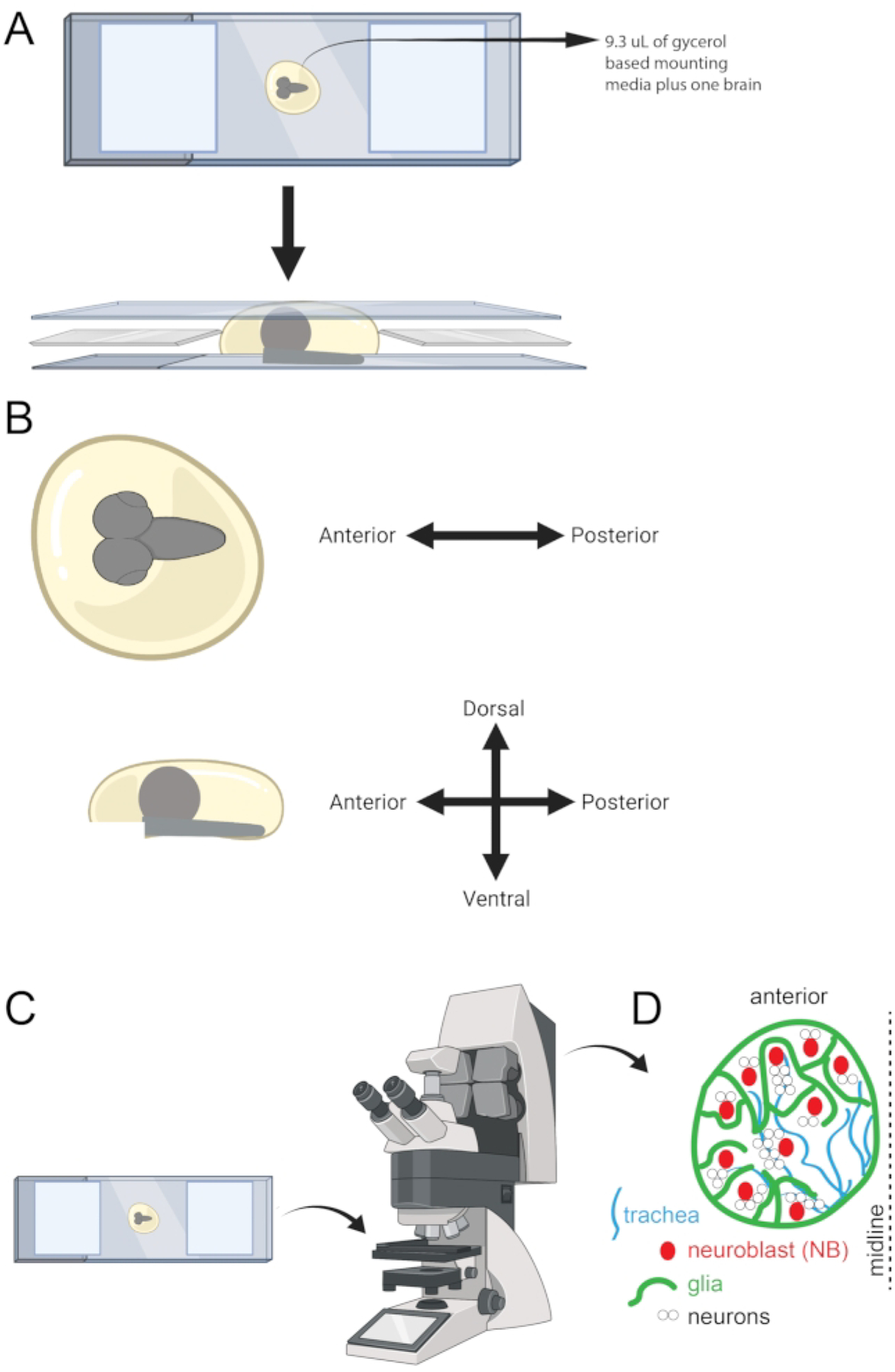
(A) Visual representation of microscope slide upon which a larval brain is mounted and is ready to be imaged. (B) A guideline is also shown to use for tissue orientation. (C) Microscope slide ready for imaging on a confocal microscope. (D) Cartoon showing some of the cell types in the larval brain.
6. Data analysis
Use Fiji open-source software to analyze brain hemispheres and use the Fiji cell counter plugin to count the cells.
Representative Results
Freshly hatched OregonR wild-type brains were dissected and cultured for 24 h in supplemented Schneider’s media (SSM) with insulin. Tissues were fixed and stained according to the protocol. Primary antibodies generated against Deadpan (Dpn) to detect NBs and Scribble to label cell membranes were used. The thymidine analog 5-Ethynyl-2′-deoxyuridine (Edu) was added to detect S-phase entry and NB reactivation. We found large sized Edu positive and Dpn positive NBs after 24 h in culture (Figure 6A–C). Next, freshly hatched OregonR wild-type brains were cultured for 24 h in supplemented Schneiders media without insulin. After 24 h in culture, we found no large-sized Edu positive and Dpn positive NBs other than the four mushroom body NBs and one ventrolateral NB (Figure 6D–F). The mushroom body and the ventrolateral neuroblasts are a subset of the central brain neuroblasts that divide continuously during the embryonic to larval transition. We conclude that exogenous insulin is sufficient to reactivate neuroblasts from quiescence in brain explants cultured in Schneider’s media.
Figure 6: Confocal imaging.
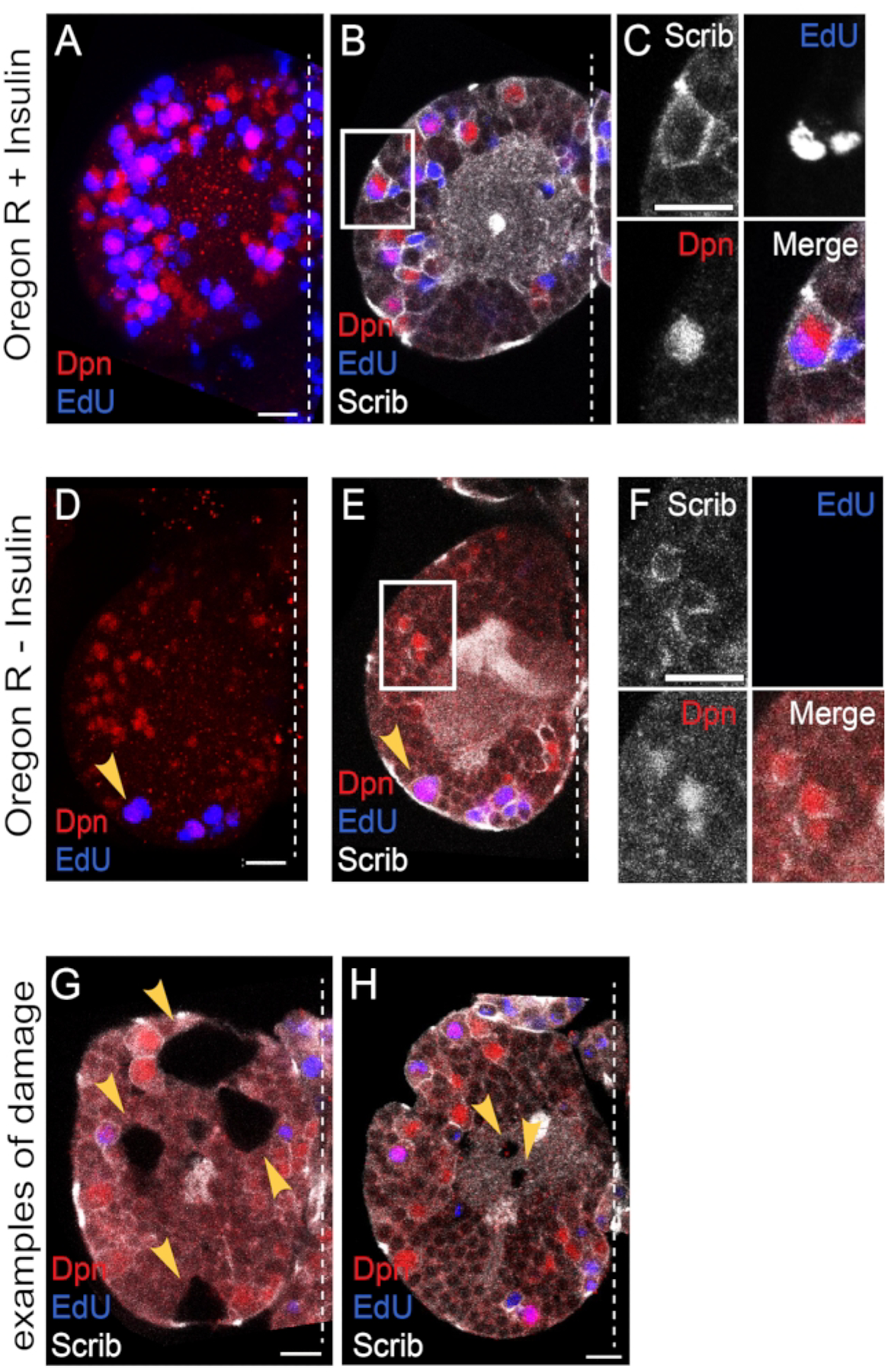
(A–C) Exogenous insulin is sufficient to reactivate quiescent NBs. (A) A maximum intensity projection of one brain hemisphere showing Deadpan (Dpn) and Edu positive NBs after 24 h of culture in the presence of insulin. (B) A single Z slice image of the same brain hemisphere with Scribble (Scrib) immunostaining marking cell membranes. (C) High magnification of NB in the inset in panel B. Single-channel grayscale images with color merge bottom right. (D–F) NBs remain quiescent in the absence of exogenous insulin. (D) A maximum intensity projection of one brain hemisphere showing Dpn and Edu positive NBs after 24 h of culture in the absence of insulin. (E) A single Z stack of the same brain hemisphere with Scrib immunostaining. (F) High magnification of NB in the inset in panel E. Single-channel grayscale images with color merge bottom right. Arrowhead denotes one of the 4 MB NBs, which divide continuously and do not enter and exit quiescence (refer to Figure 1). (G,H) Examples of damaged brain explants, not used in the analysis. (G) A single Z slice of one brain hemisphere with large holes in the tissue. (H) A single Z slice of a brain hemisphere shows small holes in the tissue. Scale bar: 10 μm. The white dashed line indicates the midline. Anterior is up, and posterior is down.
During confocal imaging, some brain hemispheres with damage were occasionally observed. The damage we found consisted of small to large-sized holes in the explanted brain tissue (Figure 6G,H). These tissues were excluded from the analysis. In addition to the 24 h time point, brains were also successfully cultured for 48 h (data not shown). After 48 h in culture, we found a further increase in the number of Dpn positive EdU positive CB NBs and an increase in the number of brains with tissue damage. This suggests that culturing brains long-term is likely feasible; however, great care must be taken to avoid tissue damage.
Discussion
The method described here to culture brain explants can be carried out in most lab environments. The tools required, as well as the procedure and data collection, are simple and straightforward. With this method, one can test a variety of hypotheses, including those related to the cell signaling cascades and extrinsic factors that regulate NB reactivation and proliferation. Here, using wild-type OregonR animals, we found that exogenous insulin was sufficient to reactivate NBs from quiescence independent of other animal-specific systemic cues. Using the GAL4/UAS system, one could also knockdown or overexpress insulin pathway components in a cell type-specific manner to better understand the role of PI3-kinase signaling in NB reactivation. In addition to using genetics, the components in the media also could be manipulated to further test extrinsic cues or signals that promote NB reactivation and proliferation. For example, one could test the hypothesis that the addition of the steroid hormone ecdysone would increase the percentage of NB reactivation and proliferation in culture.
Although this method is straightforward, we experienced several technical difficulties early on. The first technical difficulty was damage to dissected, unfixed brains during media changes. We found that transferring brains to a new well of the culture dish instead of changing the media within the well resulted in less tissue damage because holes in brains arose when media was removed from the well and the explants stuck to the bottom of the well. To solve this problem, brains were transferred in solution using a pipette and, therefore, remained continuously submerged. Another technical aspect that was changed was the sterilization of dissecting tools. A mask and gloves were worn at all times. Ethanol (70%) was used to spray the dissection tools and dissection trays. This allowed for the bacterial load to be kept to a minimum. The steps before fixation should be performed in an environment as sterile as possible, preferably a sterile hood, and with precise and extremely gentle movements. The last difficulty we encountered was that the brains were sticky. Oftentimes brains would adhere to the inner walls of the pipette tip when we attempted to place them on the microscope slide for imaging. To fix this problem, we modified the brain transferring method between the microwell mini tray to the microscope slide as described above, filling the pipette tip with mounting media before aspirating the brain into the pipette tip. This method lubricated the pipette tip with the glycerol-based mounting media and greatly reduced sticking.
Methods to culture Drosophila brain explants have been published previously12,19,20,21,22. A method published by Prithviraj et al. reports the culturing of late larval and early pupal brains. In this case, the steroid hormone ecdysone was added to the culture media and brains were maintained in culture for up to 10 h20. Bobstock et al. report culturing of brains from late L1/early L2 animals and live imaging NBs for several hours in the ventral nerve cord. They also report difficulty in handling brains from young larvae21. Siller et al. report culturing larval brains and using live-cell imaging to assay spindle dynamics during neuroblast divisions in mid to late larval stages19. All methods published previously focus on time points after the freshly hatched larval stage before animal feeding, with one exception. In Britton et al., it is reported that NBs reactivate from quiescence when brain explants are co-cultured with fat body12. Surprisingly, Britton et al. also report that exogenous insulin was not sufficient to reactivate quiescent NBs, in contrast to what is reported here. Hence, laying the foundation for the idea that a FBDS is required for NB reactivation. At the time of the Britton paper, NB specific molecular markers were not yet available and it is clear that brain morphology is compromised after 3 days in culture. Here, we provide a straightforward method to reactivate NBs in brain explants by simply adding exogenous insulin to the culture media. One important note of caution is that the amount of insulin added to the culture far exceeds physiological conditions. High levels of insulin could lead to higher levels of PI3-kinase pathway activity in brain cell types in vitro compared to in vivo conditions.
Because the culture media can be easily manipulated by adding different factors, this technique can be used to address future hypotheses regarding extrinsic signaling and NB quiescence, entry and exit. This method could also be used for large scale screening, either RNAi or drug-based. For screening large scale, it would be best to use transgenic animals that express fluorescent reporters. This could allow one to bypass antibody staining, which is time-intensive. In general, after practice, one could set up 10 cultures of 3 brains each in 30 min. In a week, it may be reasonable to screen 150 different conditions.
Acknowledgments
We acknowledge the LSAMP Bridges to Doctorate program for funding (CNK) as well as NIH/NIGMS (R01-GM120421 and R35-GM141886). We are grateful to Dr. Conor Sipe for Figure 1. We also thank all Siegrist lab members for their continued support and mentorship. We especially thank Chhavi Sood and Gary Teeters for their careful reading of the manuscript and for providing comments.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/63189.
Disclosures
The authors have no competing interests.
References
- 1.Suman S, Domingues A, Ratajczak J, Ratajczak MZ Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1–22 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Tabar V, Studer L Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daley GQ Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrigues M, Kosaric N, Bonham CA, Gurtner GC Wound healing: A cellular perspective. Physiological Reviews. 99, 665–706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Velthoven CTJ, Rando TA Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213–225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman NM, Boothby MR, Chi H Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55–70 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Wosczyna MN, Rando TA A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Homem CC, Knoblich JA Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297–4310 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Kang KH, Reichert H Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33–45 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Chell JM, Brand AH Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161–1173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sousa-Nunes R, Yee LL, Gould AP Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508–512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Britton JS, Edgar BA Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149–2158 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Lin S et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908–1913 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sipe CW, Siegrist SE Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, eLife.26343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speder P, Brand AH Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, eLife.30413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan X, Sipe CW, Suzawa M, Bland ML, Siegrist SE Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, e3000721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colombani J et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739–749 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Geminard C, Rulifson EJ, Leopold P Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199–207 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Siller KH, Serr M, Steward R, Hays TS, Doe CQ Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127–5140 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prithviraj R, Trunova S, Giniger E Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. 65, 4270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bostock MP et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Datta S Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77–83 (1999). [DOI] [PubMed] [Google Scholar]