

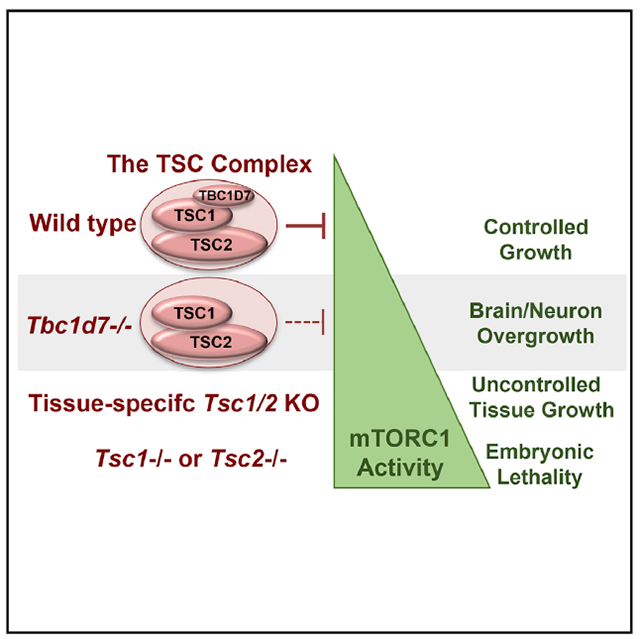
SUMMARY
The tuberous sclerosis complex (TSC) 1 and 2 proteins associate with TBC1D7 to form the TSC complex, which is an essential suppressor of mTOR complex 1 (mTORC1), a ubiquitous driver of cell and tissue growth. Loss-of-function mutations in TSC1 or TSC2, but not TBC1D7, give rise to TSC, a pleiotropic disorder with aberrant activation of mTORC1 in various tissues. Here, we characterize mice with genetic deletion of Tbc1d7, which are viable with normal growth and development. Consistent with partial loss of function of the TSC complex, Tbc1d7 knockout (KO) mice display variable increases in tissue mTORC1 signaling with increased muscle fiber size but with strength and motor defects. Their most pronounced phenotype is brain overgrowth due to thickening of the cerebral cortex, with enhanced neuron-intrinsic mTORC1 signaling and growth. Thus, TBC1D7 is required for full TSC complex function in tissues, and the brain is particularly sensitive to its growth-suppressing activities.
Graphical abstract
In brief
Schrötter et al. describe a mouse knockout model of TBC1D7, a core component of the tuberous sclerosis complex (TSC) protein complex, which serves as a central brake on mTORC1 signaling and cell and tissue growth. Tbc1d7 KO mice exhibit brain overgrowth associated with an mTORC1-dependent increase in neuronal cell size.
INTRODUCTION
The mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) integrates growth cues from nutrients and secreted growth factors to properly regulate the metabolic processes underlying cell, tissue, and organismal growth (Kim and Guan, 2019; Saxton and Sabatini, 2017; Valvezan and Manning, 2019). The importance of this growth-regulatory circuit is underscored by the sheer number of seemingly distinct pathological settings, in which alterations in the control of mTORC1 have been implicated, including tumor syndromes and cancer, autoimmune and metabolic diseases, and neurological disorders. Human genetic studies have indicated that the brain is particularly sensitive to aberrant mTORC1 signaling, with even modest changes in the control of mTORC1 leading to a variety of neurological disorders, including epilepsy, autism spectrum disorder, and neurocognitive deficits, pathologies sometimes accompanied by focal cortical dysplasia (FCD) or brain overgrowth (i.e., megalencephaly) (Andrews et al., 2020; Crino, 2016; Griffith and Wong, 2018; Kassai et al., 2014; Lipton and Sahin, 2014; Nguyen et al., 2019; Switon et al., 2017; Tarkowski et al., 2019; Winden et al., 2018).
mTORC1 is subjected to exquisite control by a regulatory network of signals impinging on two small GTPase switches directly upstream (Valvezan and Manning, 2019). The Rag GTPases engage and recruit mTORC1 to the lysosomal surface when sufficient amino acids are present (Kim and Guan, 2019; Saxton and Sabatini, 2017). A key negative regulator is the Rag GTPase-activating protein (GAP) complex GATOR1 (Bar-Peled et al., 2013), the components of which are frequently disrupted in FCD associated with epilepsy (Iffland et al., 2019; Marsan and Baulac, 2018). Once at the lysosome, mTORC1 is directly activated by the small GTPase Rheb, but only when Rheb is in its GTP-bound form. The GTP-binding state of Rheb is controlled by growth factor signaling pathways that regulate a protein complex comprised of the tuberous sclerosis complex (TSC) 1 and 2 proteins, referred to as the TSC complex, which serves as a GAP for Rheb. Growth factors activate mTORC1 by relieving the TSC complex-mediated inhibition of lysosomal Rheb, thereby allowing Rheb-GTP to accumulate and activate mTORC1 (Menon et al., 2014).
Germline or mosaic loss-of-function mutations in either TSC1 or TSC2 give rise to the pleiotropic genetic disorder TSC, which affects up to one in 6,000 births and displays autosomal dominant inheritance (Crino et al., 2006). While clinical manifestations vary widely, the presence of cortical growths in the brain, referred to as tubers, a variety of other neoplastic lesions affecting multiple organ systems, and neurological disorders, including epilepsy, autism spectrum disorder, and neurocognitive deficits, are common (Crino et al., 2006). Complete loss of TSC1 or TSC2, arising from loss of heterozygosity (LOH) in focal TSC lesions, gives rise to maximally elevated and constitutive mTORC1 activation, which is widely believed to underlie many, if not all, TSC manifestations. As such, the mTOR inhibitor rapamycin and its analogs are effective treatments for various features of TSC (Henske et al., 2016). However, the degree of ubiquitous mTORC1 activation upon complete loss of TSC1 or TSC2 appears to be incompatible with mammalian development and survival, as humans with homozygous germline loss are not known to exist and complete mouse knockout of either Tsc1 or Tsc2 yield early embryonic lethality (Kobayashi et al., 1999, 2001; Kwiatkowski et al., 2002; Onda et al., 1999).
We identified TBC1D7 as a third and final stoichiometric component of the TSC complex in all tissues (Dibble et al., 2012). Within the complex, TSC1 scaffolds TSC2 and TBC1D7 together to form a very stable, high-molecular-mass protein complex. A recent partial cryoelectron microscopy (cryo-EM) structure indicates that the TSC complex is comprised of a 2:2:1 complex of TSC2 to TSC1 to TBC1D7, with both TSC2 and TBC1D7 interacting with an extended coiled-coil dimer in TSC1 (Yang et al., 2021). While TBC1D7 is a ubiquitous core component of the TSC complex, loss-of-function mutations in the TBC1D7 gene have not been identified in TSC patients, even in those with no mutations identified in TSC1 or TSC2 (Dibble et al., 2012). However, TBC1D7 depletion from cultured cells results in modest growth-factor-independent activation of mTORC1, albeit substantially less than that observed with loss of TSC1 or TSC2, and this is attributed to a partial destabilization of the TSC complex resulting in reduced GAP activity toward Rheb (Dibble et al., 2012). In addition to its unknown function in the TSC complex, TBC1D7 also exists in a more labile pool free of the TSC complex (Dibble et al., 2012; Menon et al., 2014), the function of which is also unknown.
Subsequent to our findings, human genetic studies identified germline homozygous truncating mutations in TBC1D7 as the cause of a rare autosomal-recessive megalencephaly and neurocognition syndrome (Alfaiz et al., 2014; Capo-Chichi et al., 2013). These individuals are born without TBC1D7, further distinguishing it from the essential TSC1 and TSC2. While the limited clinical descriptions reference intellectual disabilities and a suite of neuropsychiatric abnormalities also observed in TSC, other common manifestations of TSC were not evident in the children reported. Distinct from the focal cortical tubers of TSC, individuals lacking TBC1D7 exhibit whole-brain overgrowth, reminiscent of that observed in mouse models with brain-specific deletion of TSC1 or TSC2 (Carson et al., 2012; Way et al., 2009; Zeng et al., 2008). In addition, genome-wide association studies (GWASs) have found that SNPs within the TBC1D7 locus at 6p24.1 are highly associated with Tourette’s syndrome, attention deficit/hyperactivity disorder, and migraines (Tsetsos et al., 2016; Anttila et al., 2013).
Here, we generate and characterize a mouse knockout model of Tbc1d7. Animals lacking TBC1D7 undergo normal growth and development, despite showing elevated mTORC1 signaling in multiple tissues, with a wide gait being the only overt phenotype. While Tbc1d7−/− mice have normal body weight, they exhibit a specific increase in brain size and weight by 6 weeks of age, thus recapitulating the megalencephaly phenotype of the human “TBC1D7 syndrome.” This increase in brain size is driven by cortical thickening and is accompanied by an mTORC1-dependent increase in neuronal cell size and aberrant axonal polarization. This Tbc1d7−/− mouse model provides further genetic evidence that brain tissue, and especially neurons, are particularly sensitive to even modest increases in basal mTORC1 signaling.
RESULTS
Generation of a Tbc1d7 knockout mouse
A mouse knockout (KO) was generated via CRISPR-Cas9 gene editing of Tbc1d7 exon 3 (see STAR Methods). A mouse with a 35-bp out-of-frame deletion in exon 3 was chosen for this study due to its genomic alteration facilitating subsequent PCR genotyping of offspring (Figures 1A and 1B). To eliminate potential CRISPR off-target effects, the final Tbc1d7+/− line was obtained after 6 backcrosses with wild-type C57BL/6J mice. Tbc1d7−/− offspring from subsequent crosses had no detectable TBC1D7 protein across all tissues examined, with no consistent changes in expression of the other two TSC complex components (Figures 1C and S1A). Consistent with cell-culture studies and partial loss of TSC complex function (Dibble et al., 2012), many, but not all, tissues from Tbc1d7−/− mice displayed elevated basal mTORC1 signaling after a 6 h daytime fast, as scored by increased phosphorylation of the downstream targets ribosomal S6 and 4EBP1 (Figures 1C, 1D, and S1A).
Figure 1. Generation and initial characterization of Tbc1d7 knockout mice.
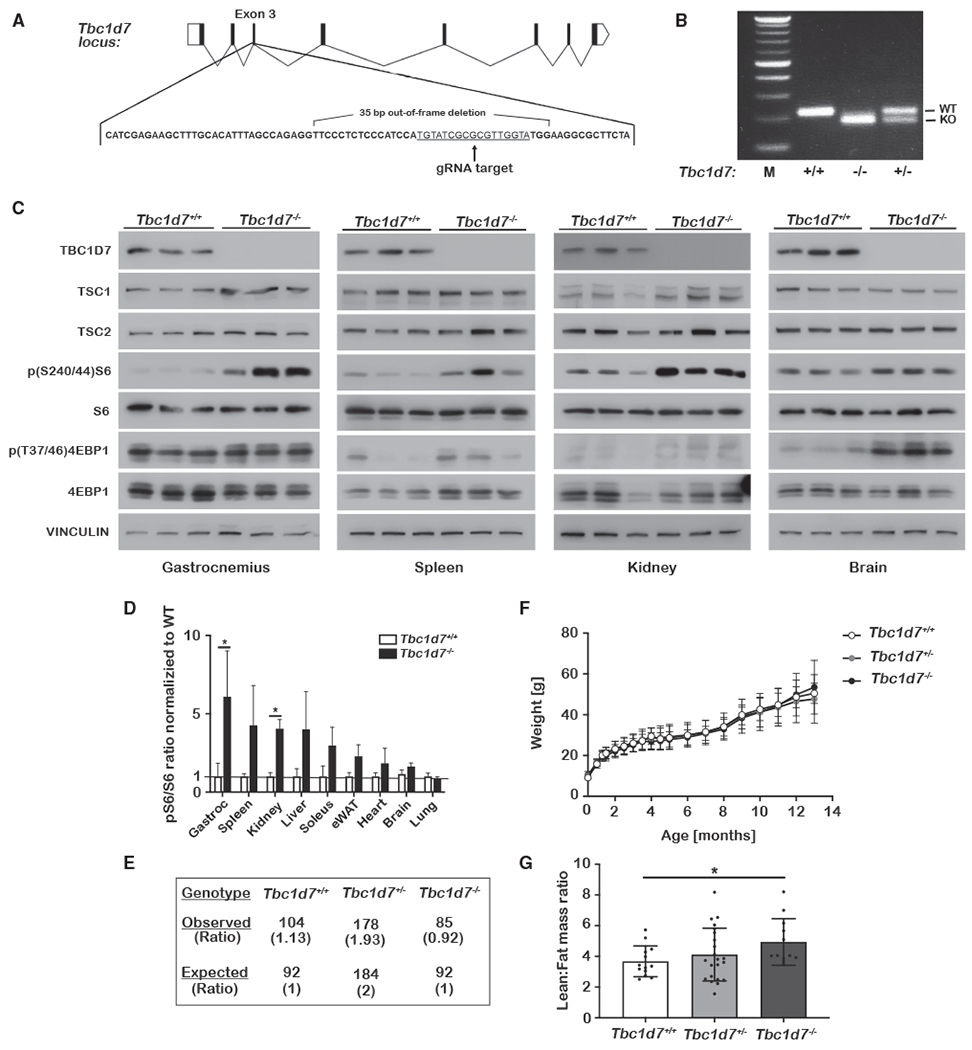
(A and B) Scheme for CRISPR-Cas9 targeting of Tbc1d7, with the identified 35-bp out-of-frame deletion (A) denoted and (B) detected by PCR genotyping.
(C and D) Immunoblots of tissue lysates from littermate Tbc1d7+/+ and Tbc1d7−/− mice at 6 weeks of age following a 6 h daytime fast. Phospho-S6 to total S6 ratios are quantified in (D) as mean ± SD. n = 3.
(E) Genotypes of 368 offspring of crosses between Tbc1d7+/− mice.
(F) Body weights from a cohort of littermate offspring from Tbc1d7+/− crosses over 13 months graphed as mean ± SD at each age. n = 13 Tbc1d7+/+, 19 Tbc1d7+/−, and 12 Tbc1d7−/−. Age 0 is the weight at weaning (21 days).
(G) DEXA scan analysis of the mice in (F) at 6 months of age, with the lean-to-fat-mass ratio graphed as mean ± SD.
Statistical analysis by one-way ANOVA, *p < 0.05.
Intercrosses between Tbc1d7+/− mice yielded viable Tbc1d7−/− offspring at near-Mendelian ratios (Figure 1E, Chi2 value of 0.318). Despite increased mTORC1 signaling in multiple organs, animals lacking TBC1D7 exhibited development and growth that was grossly indistinguishable from their wild-type or heterozygous littermates over 13 months (Figures 1F and S1B–S1D). However, a dual-energy X-ray absorptiometry (DEXA) scan at 6 months revealed that Tbc1d7−/− mice have a higher lean-to-fat-mass ratio (Figure 1G.). A previous study of Drosophila TBC1D7 suggested that its loss enhances systemic insulin signaling (Ren et al., 2017). However, no change was observed in fasting insulin and glucose levels (Figures S1E and S1F) or glucose and insulin tolerance (Figures S1G and S1H) with mouse Tbc1d7 KO.
Tbc1d7 KO mice have increased skeletal muscle fiber size, reduced hind limb strength, and abnormal motor activity and gait
Consistent with the elevation in skeletal muscle mTORC1 signaling in Tbc1d7 KO mice (Figures 1C, 1D, and S1A), a significant increase in muscle fiber size (i.e., cross-sectional area) was measured for both gastrocnemius and soleus muscles in Tbc1d7 KO mice (Figures 2A and 2B). However, there was no apparent difference in fiber-type composition between genotypes (Figure 2C). While front-limb grip strength was normal, hind-limb grip strength was significantly decreased in Tbc1d7−/− mice in distinct cohorts at 6 weeks, 3 months, and 6 months (Figures 2D, 2E, S2A, and S2B). Thus, despite elevated mTORC1 signaling and a measurable increase in muscle fiber size in hind-limb muscles, loss of TBC1D7 results in decreased strength. Furthermore, 1 month of rapamycin treatment failed to significantly restore hind-limb grip strength to the 3-month-old cohort (Figures S2C and S2D).
Figure 2. Muscle and gait analysis of Tbc1d7 knockout mice.
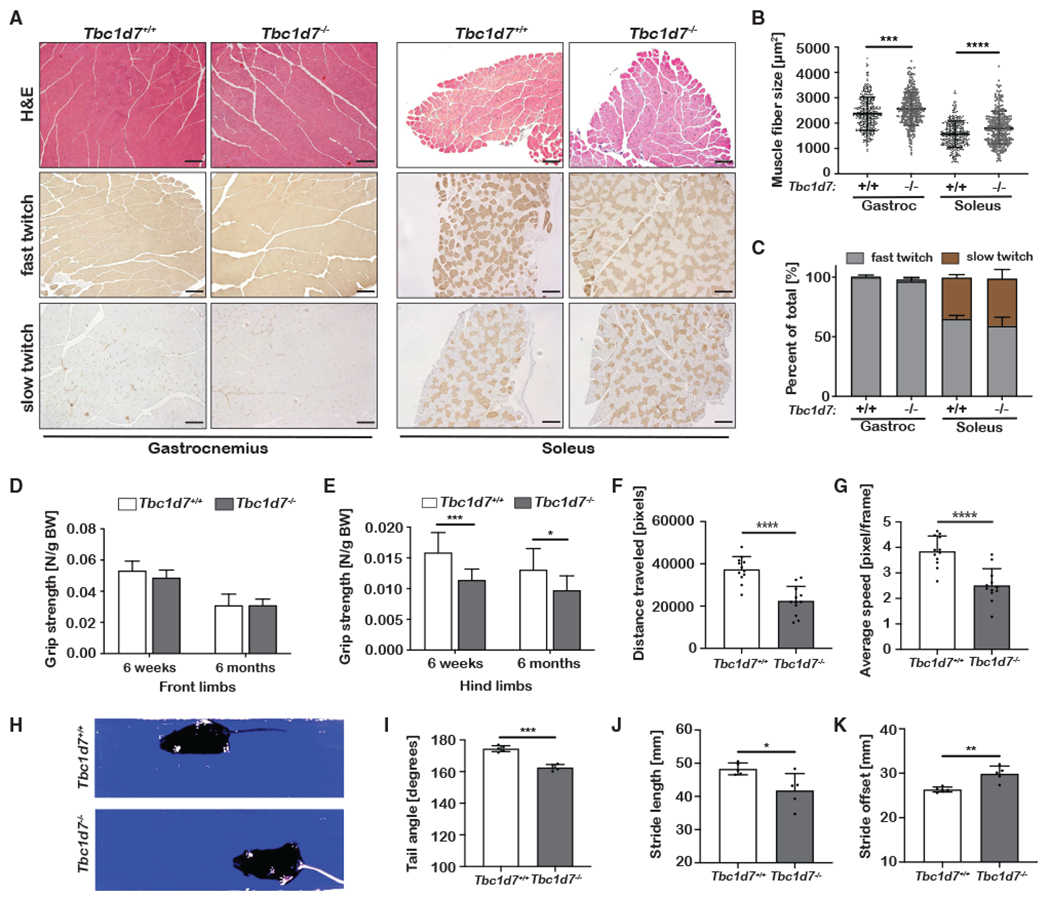
(A–C) Histological cross-sections of muscle from 6-week-old mice stained as indicated. Scale bars, 100 μm. (B) Cross-sectional muscle-fiber area graphed as mean ± SD.
(C) Quantification of muscle-fiber types graphed as mean percentage of total ± SD. n = 4.
(D and E) Front-limb (D) and hind-limb (E) grip strength measured at 6 weeks and 6 months of age graphed as mean ± SD. 6 week: n = 9 Tbc1d7+/+ and 13 Tbc1d7−/− 6 month: n = 13 Tbc1d7+/+ and 12 Tbc1d7−/−.
(F and G) Open-field test of 6-month-old mice, with (F) distance traveled and (G) speed graphed as mean ± SD. n = 12.
(H–K) Video-recorded gait analysis of 6-month-old mice with (I) tail angle, (J) stride length, and (K) stride offset graphed as mean ± SD. n = 5.
Statistical analysis (B, E–G, and I–K) by Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
In handling the Tbc1d7−/− mice in a blinded fashion, we observed abnormalities in hind-limb clasping, stance, and gait of movement relative to their littermates. In an open-field test, 6-month-old Tbc1d7−/− mice traveled significantly less distance over time due to reduced walking speed, without significant changes in time spent moving or in the inner region of the testing area (Figures 2F, 2G, S2E, and S2F). Using a transparent platform to video capture and quantify parameters of their movement (Figure 2H), Tbc1d7−/− mice were found to display a loss of streamline body movement, with angled tail (Figure 2I) and laterally extended hind limbs. The abnormal gait of Tbc1d7−/− mice was reflected in decreased stride length and increased stride offset and stance length, with no change in sway length (Figures 2J, 2K, S2G, and S2H). However, no defect in performance on an accelerating rotarod was observed at 6 months, suggestive of normal gross motor skills and coordination (Figure S2E). Social and behavioral assays also revealed no significant difference between Tbc1d7+/+ and Tbc1d7−/− mice (Figures S2J–S2L).
Loss of TBC1D7 leads to brain overgrowth and a cell-intrinsic increase in neuronal size
While no gross anatomical or histological changes were identified in the brains of Tbc1d7−/− mice, both male and female KO brains were visibly larger, with a significant increase in weight at 13 months (Figures 3A and 3B). An increase in brain weight was also detected in 6-week-old Tbc1d7 KO mice but not in embryonic day 16.5 (E16.5) embryos, suggesting that this phenotype reflects an increase in postnatal brain growth (Figures 3C and 3D). Brain overgrowth, or megalencephaly, is generally caused by an increase in cortical thickness. Indeed, measurements of the primary motor cortex in sagittal brain sections revealed an increase in cortical thickness in Tbc1d7 KO brains (Figures 3E and 3F). The thickness of cortical layer IV/V (CTIP2), layer VI (TBR1), and the entire cortex (NeuN) were all measured as significantly and proportionally increased in the Tbc1d7 KO brains (Figures 3G–3M). Thus, the cortical overgrowth of Tbc1d7−/− brains reflects an overall enlargement of the cerebral cortex proportionally across its laminar structure, with no significant change in neuronal density (Figures 3N and 3O). While a similar distribution of IBA1+ microglia was found, an expansion of cortical GFAP+ astrocytes (e.g., astrogliosis) was evident in the KO brains, accompanying their overgrowth (Figure 3P).
Figure 3. Loss of TBC1D7 leads to brain overgrowth.
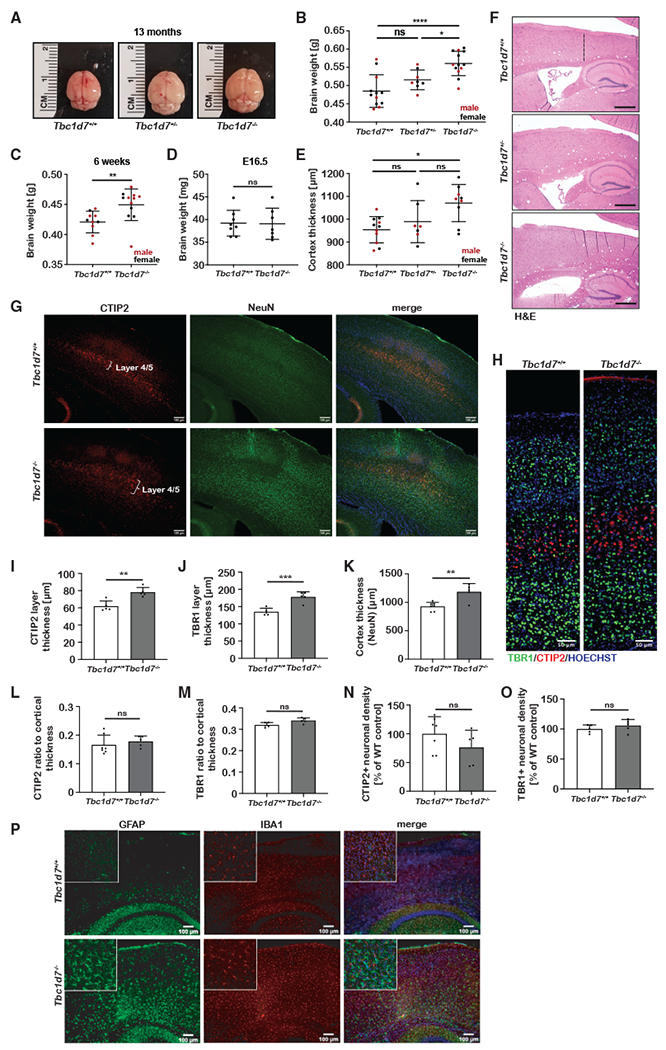
(A and B) Representative brains from each genotype at 13 months (A) with brain weights graphed as mean ± SD. (B). n = 12 Tbc1d7+/+, 8 Tbc1d7++/−, and 13 Tbc1d7−/−.
(C) Brain weights at 6 weeks graphed as mean ± SD. n = 11 Tbc1d7+/+ and 13 Tbc1d7−/−.
(D) Brain weights at E16.5 graphed as mean ± SD. n = 8 Tbc1d7+/+ and 7 Tbc1d7−/−.
(E and F) Cortical thickness measured at the primary motor cortex in sagittal brain sections from 13-month-old mice is graphed as mean ± SD (E). n = 11 Tbc1d7+/+, 7 Tbc1d7+/− and 9 Tbc1d7−/−. Representative H&E-stained sections are shown, with the cortical region measured denoted with a dashed line (F, top). Scale bars, 500 μm.
(G and H) Immunofluorescence imaging of brain sections from 8-month-old mice stained as indicated (G and H). Scale bars, 100 (G) and 50 μm (H).
(I–O) Measurement of thickness for layer IV/V (I), layer VI (J), and total cortex (K) and ratio of layer IV/V (L) or layer VI (M) to total cortex, and quantification of neuronal density in these layers (N and O) were graphed as mean ± SEM. n = 7 Tbc1d7+/+ and 5 Tbc1d7−/−.
(P) Immunofluorescence imaging of brain sections from 8-month-old mice stained as indicated. Inset shows a 5× magnification. Scale bar, 100 μm.
Statistical analysis by one-way ANOVA (B and E) and Student’s t test (C, D, and I–K), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
As observed previously in cell culture (Dibble et al., 2012; Menon et al., 2014), size-exclusion chromatography (SEC) on whole mouse brain extracts demonstrated that TBC1D7 exists as two pools, one that co-fractionates with TSC1 and TSC2 (peaking in fraction 8) and another that fractionates at a smaller molecular mass (peaking at fraction 21), referred to as “free” TBC1D7 (Figures 4A and S3A). While much of TSC2 still co-fractionates with TSC1 in brain extracts from Tbc1d7 KO mice, there is an increase in the TSC1-free pool of TSC2 upon loss of TBC1D7 (peaking in fraction 15), suggesting that TSC complex integrity is partially compromised. This is consistent with previous studies (Dibble et al., 2012) and likely underlies the modest increase in mTORC1 signaling in Tbc1d7 KO brain extracts (Figure 1C.). This aberrant signaling appears to be predominantly neuronal in origin, as Tbc1d7 KO brains displayed elevated phospho-S6 staining in neuronal cell bodies (Figure 4B). Furthermore, measurements of soma area revealed an increase in soma size of both cortical neurons and cerebellar Purkinje neurons in Tbc1d7 KO brains (Figures 4C, S4B, and S3B).
Figure 4. Analysis of neuronal cell size and polarization of Tbc1d7 knockout mice.
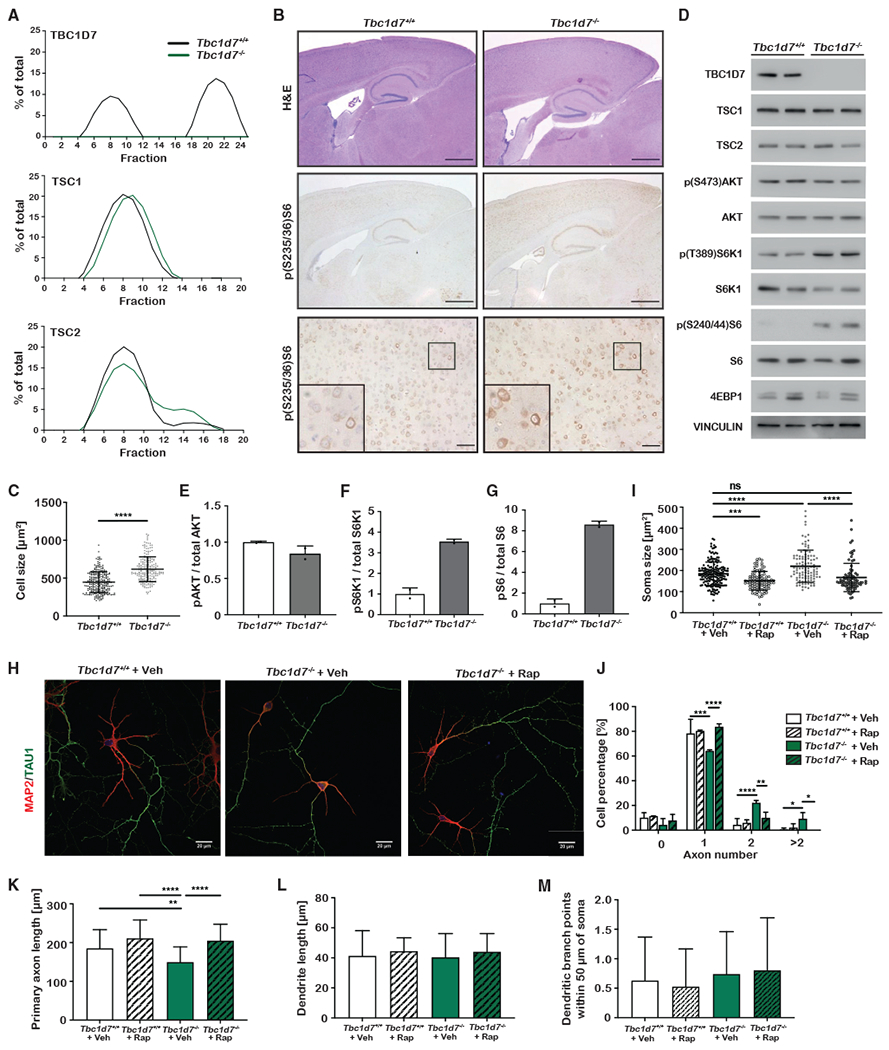
(A) Percentage total for the indicated proteins in size-exclusion chromatography fractions of whole-brain lysates from mice aged 1 year.
(B) Histological analysis of sagittal sections from brains of 6-week-old mice stained as indicated. Scale bars, 1 mm (top/middle) and 100 μm (bottom), with inset showing a 2.5× zoom of the marked area.
(C) Quantification of soma area in phospho-S6-stained cortical neurons from (B) in 5 mice from each genotype, graphed as mean ± SD. n = 262 Tbc1d7+/+ and 213 Tbc1d7−/− cells.
(D–G) mTORC1 signaling in primary cortical neurons (DIV7) following 4 h of growth factor and amino-acid withdrawal. Immunoblots (D) and quantification (E-G) graphed as mean ± SD. n = 2.
(H–L) Immunofluorescence imaging of primary hippocampal neurons (DIV7) treated from DIV4 with vehicle or rapamycin (100 nM). Data are from DIV7 neurons, with soma area (I), axon number (J), axon length (K), dendrite length (L), and dendritic branch points (M) measured and graphed as mean ± SD for 2 technical replicates in each of 3 independent experiments. n = 179 Tbc1d7+/+ with vehicle, 93 Tbc1d7+/+ with rapamycin, 109 Tbc1d7−/− with vehicle, and 87 Tbc1d7−/− with rapamycin.
Statistical analysis by Student’s t test (C) and one-way ANOVA (H–K), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further explore the neuron-intrinsic effects of TBC1D7 loss, primary cortical neurons were cultured from litters of wild-type and Tbc1d7−/− E16.5 embryos. Under growth-factor-withdrawal conditions, elevated mTORC1 signaling was detected in Tbc1d7−/− neurons, as seen by increased phosphorylation of both S6K1 and its substrate S6 (Figures 4D–4G). The apparent levels of another mTORC1 substrate, 4E-BP1, varied between samples, but analysis of electrophoretic mobility likewise revealed increased phosphorylation in Tbc1d7−/− neurons (Figure S3D). While the activating phosphorylation of Akt was similar in wild-type and KO neurons (Figures 4D and 4E), phospho-Akt was decreased in whole-brain extracts from Tbc1d7−/− mice (Figures S3E and S3F), in accordance with previous studies (Dibble et al., 2012). Primary Tbc1d7−/− hippocampal neurons were significantly larger, and rapamycin restored soma size to that of wild-type neurons (Figures 4H and 4I). Taken together, these results indicate that an increase in mTORC1 signaling upon loss of TBC1D7 enhances neuronal cell growth, providing a likely mechanism for the brain-overgrowth phenotype of mice and humans lacking TBC1D7.
Neuronal loss of TBC1D7 alters axon specification
Developing neurons polarize to form a single axon and multiple dendrites, a specification process recapitulated in cultured hippocampal neurons (Polleux and Snider, 2010). To investigate if loss of TBC1D7 effects neuronal polarity, primary hippocampal neurons were analyzed at 4 and 7 days in vitro (DIV4 and DIV7, respectively) (Figures 4H and S3G). While Tbc1d7 KO neurons had significantly increased soma size at DIV4, there was no difference in Tau1-positive axon number between genotypes at this stage, when only approximately 50% of neurons have polarized to form a single axon (Figures S3H and S3I). In wild-type neurons, single-axon specification increased to 80% by DIV7, with the majority of the remainder lacking a determined axon. However, Tbc1d7-deficient neurons exhibited a significant reduction in single-axon specification, with approximately 30% having multiple axons at this stage, a phenotype detected in less than 10% of wild-type neurons (Figure 4J). Importantly, axon specification was returned to wild-type levels upon treatment with rapamycin from DIV4 to DIV7. Tbc1d7 KO neurons also displayed a decrease in axon length that was reversed with rapamycin (Figure 4K). However, neither dendritic length nor soma-proximal branching were significantly changed in Tbc1d7 KO neurons (Figures 4L and 4M). Thus, TBC1D7 loss, similar to neurons lacking TSC1 or TSC2 (Choi et al., 2008; Tavazoie et al., 2005), results in defects in neuronal growth and polarity that are driven by dysregulated mTORC1 signaling.
DISCUSSION
This study provides key genetic evidence that TBC1D7, while not essential, is required for full TSC complex function as it relates to suppression of mTORC1 signaling in mammalian tissues. While full deletion of most other mTOR pathway components is embryonically lethal, phenotypic overlap between the Tbc1d7 KO mice and a subset of tissue-specific genetic models is evident. For instance, skeletal-muscle-specific KO of Depdc5 (a component of the GATOR1 complex) also leads to increased fiber size, while transgenic overexpression of human TSC1 in skeletal muscle leads to a decrease in fiber size (Graber et al., 2017; Wan et al., 2006). The wide-gait phenotype of Tbc1d7 KO mice is similar to that reported for mice with Purkinje-cell-specific Tsc1 KO or transgenic expression of an activated allele of mTOR (Sakai et al., 2019; Tsai et al., 2012). However, while Purkinje cells are larger in the Tbc1d7 KO cerebellum, no change in cell number was detected, contrasting the decrease described in Purkinje cell KOs of Tsc1 or Tsc2 (Reith et al., 2013; Tsai et al., 2012). Furthermore, loss of Tsc1 or Tsc2 in Purkinje cells yields specific autistic-like behaviors, which were not evident in Tbc1d7 KO mice using similar assays. Gait phenotypes are also common in mouse models of Parkinson’s disease (Amende et al., 2005; Noda et al., 2020), so future studies should explore potential shared molecular defects, such as impaired induction of autophagy (Dibble et al., 2012; Komatsu et al., 2006). Perhaps the most remarkable feature of the Tbc1d7 KO mice is the lack of overt growth phenotypes affecting the whole body or multiple tissues, despite a detectable elevation in tissue mTORC1 signaling. This finding highlights the differential sensitivity of specific cell types or tissues to distinct thresholds of mTORC1 signaling for enhanced growth.
The brain-overgrowth phenotype of Tbc1d7 KO mice further emphasizes that this organ is particularly sensitive to changes in mTORC1 signaling. Postnatal megalencephaly with an increase in cortical thickness and enlarged dysplastic neurons has been described in various mouse models with loss of TSC1 or TSC2 in neurons (Carson et al., 2012; Crowell et al., 2015; Magri et al., 2011; Meikle et al., 2007; Mietzsch et al., 2013; Way et al., 2009). This phenotype also recapitulates the predominant phenotype described for the human TBC1D7 syndrome (Alfaiz et al., 2014; Capo-Chichi et al., 2013). mTORC1-dependent increases in soma size and defects in axonal polarization in primary hippocampal neurons are also phenotypes shared between Tbc1d7 KO neurons and those with loss of TSC1 or TSC2 (Choi et al., 2008; Tavazoie et al., 2005). These findings highlight the critical nature of full TSC complex function for restraining neuronal and whole-brain growth.
Limitations of the study
The bases of the wide gait and hind-limb-specific decrease in grip strength of the Tbc1d7 KO mice are currently unknown. While rapamycin treatment from 3 to 4 months of age failed to restore grip strength, it is possible that this and other phenotypes require younger starting ages or longer durations of treatment to reveal potential dependence on aberrant mTORC1 signaling. Furthermore, the developmental nature and molecular basis of the brain-overgrowth phenotype, including the thickening of the CTIP2 layer, warrants further investigation, as we only focused on structural changes in postnatal brains, which could miss potential effects on the proliferation and abundance of specific cell types in embryonic brains. An increase in neuronal number in embryonic and neonatal brains of Pten+/− mice has been found to underlie early increases in brain size, whereas an increase in glial cells appears to contribute to adult brain overgrowth in this model (Chen et al., 2015). This is interesting given the increase in cortical astrocytes observed in adult Tbc1d7 KO brains, which accompanies enhanced mTORC1 signaling and growth of cortical neurons. Finally, other neuron-intrinsic changes induced by Tbc1d7 loss and the contributions of the TSC complex, mTORC1 signaling, and the free pool of TBC1D7 await further study using this model, including a full Sholl analysis of dendritic arborization, morphological analyses of dendritic spines, and the formation and function of primary cilia.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Brendan Manning (bmanning@hsph.harvard.edu).
Materials availability
Unique materials generated in this study are available from the Lead contact without restriction.
Data and code availability
All original data reported in this paper will be shared by the Lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal studies were reviewed and approved by the Harvard Medical Area Standing Committee on Animals IACUC, an AAALAC International and USDA-accredited facility. All animal care and protocols were in accordance with the principles of animal care and experimentation in the Guide for the Care and Use of Laboratory Animals. Mice were group housed (3-4 per cage) in temperature controlled, pathogen-free facilities with a 12:12 h light:dark cycle in standard static microisolator top cages. Autoclaved food (Lab Diet #5058, St Louis MO) and water were provided ad libitum. For all experiments shown, mixed cohorts of male and female mice were used as we observed no phenotypic or behavioral differences between genders. Animals were used between 6 weeks and 6 months of age, as indicated in the corresponding figure legends.
Cultured primary neurons
Mouse cortices and hippocampi were dissected from E16.5 wild-type and Tbc1d7−/− mouse embryos and dissociated in 0.25% trypsin (Thermo #25200114) for 15-min at 37°C. Four washing steps were performed, two in Hank’s balanced salt solution buffer (Life Tech #14170-120), one in growth medium (Neurobasal A medium (Life Tech #10888-022) supplemented with 1% Glutamine, 2% B27 (Invitrogen #17504-044), 1% penicillin/streptomycin and 100 μM β-mercaptoethanol), and one in Neurobasal A supplemented with 10% horse serum (Sigma #H1138). Tissue pieces were then triturated in growth medium using sterile fire-polished Pasteur pipettes (VWR #14672-380). Single-cell suspensions were plated on glass coverslips previously washed with 100% methanol, 70% ethanol and 100% ethanol, dried, and coated with 30 ng/μL poly-ornithine (Sigma #P3655). For biochemical experiments, cortical neurons were plated in 12-well plates at a density of 300,000 cells per well. For microscopy experiments, hippocampal neurons were plated in 24-well plates at 40,000 cells per well. Neurons were maintained in a humidified incubator at 37°C and 5% CO2 for the indicated amount of time. Where indicated, neurons were treated with 100 nM rapamycin or vehicle (DMSO) for 72 h, from DIV4 to DIV7.
METHOD DETAILS
Animals
The Tbc1d7−/− mouse line was generated at the Harvard Medical School Transgenic Core by CRISPR/Cas9 injection into fertilized eggs, which were subsequently transplanted into pseudo-pregnant C57BL/6 mice. PCR analysis was used to identify KO offspring from genomic DNA isolated from ear punches. The primer set flanking the targeted region in exon 3 of Tbc1d7 was AAGTCGGCTGCTGATCATG (forward) and TCTCATACCTAGAAGCGCCTTC (reverse). PCR products were sequenced by Genewiz (Cambridge, MA, USA). A mouse harboring a 35bp out-of-frame deletion in exon 3 was backcrossed to wild-type C57BL/6J mice for 6 generations. Colonies of mice were established, bred and maintained as Tbc1d7+/− heterozygotes.
Body weight and size (snout to tail length) of Tbc1d7+/+, Tbc1d7+/− and Tbc1d7−/− littermates were recorded at weaning, as well as every week until P42, then bi-weekly until P140 and then monthly until 13 months of age, where the cohort had to be sacrificed due to a COVID-19-related laboratory shutdown at Harvard. Prior to harvesting tissues for histological or biochemical analyses, mice were subjected to a 6 h daytime fast to limit variation from feeding during the normal fasting cycle for mice. In the afternoon following the fast, mice were sedated with isoflurane and euthanized, with tissues were dissected promptly resected and snap frozen in liquid nitrogen or fixed in 10% Formalin.
Protein extracts, immunoblotting, and fast protein liquid chromatography (FPLC)
Tissues, following homogenization, and primary neurons were lysed in ice-cold NP-40 lysis buffer (40 mM HEPES [pH 7.4], 120 mM NaCl, 1 mM EDTA, 1% NP-40 [Igepal CA-630], 5% glycerol, 10 mM sodium pyrophosphate, 10 mM glycerol 2-phosphate, 50 mM NaF, 0.5 mM sodium orthovanadate, and protease inhibitors (Sigma #P8340)). Samples were centrifuged at 20,000 × g for 20-min at 4°C, and protein concentration in the supernatant was determined by Bradford assay (Bio-Rad, #5000202) and normalized across samples. For FPLC analysis of the TSC complex, whole mouse brains were homogenized in ice-cold NP-40 lysis buffer without glycerol. 500 μg protein from each supernatant were fractioned by size exclusion chromatography in lysis buffer on a Superose 6 Increase column attached to an Äkta Pure FPLC sytem (Cytiva, Sweden). 300 μL fractions were collected and analyzed via immunoblot for TSC complex components. Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with indicated antibodies. Primary antibodies used at 1:1000: p(T389)S6K1 (CST #9234), S6K1 (CST #2708), TSC2 (CST #4308), TSC1 (CST #6935), TBC1D7 (CST #14949), p(240/44)S6 (CST #2215), S6 (CST #2217), AKT (CST #4691), p(S473)AKT (CST #4060), 4EBP1 (CST #9644), p(T37/46)4EBP1 (CST #2855), VINCULIN (CST #4650); secondary antibody used at 1:5000: HRP-conjugated anti-rabbit secondary antibody (CST #7074). Immunoblots were imaged using KwikQuant Imager (Kindle Biosciences, USA) and quantified using Fiji.
Dual-energy X-ray absorptiometry (DEXA) scan
Body composition analysis was performed using a Lunar PIXImus DEXA machine (GE Healthcare, North Richland Hills, TX). The machine was calibrated using the phantom according to the manufacturer’s instructions. Mice were anesthetized with 4% isoflurane-oxygen via an induction box and then maintained at 2% for the duration of the scan via a nose cone. Mice were placed in the bed of the scanner and the body (excluding the head) was scanned to determine fat and lean mass.
Fasting blood glucose and insulin measurement
Mice were fasted for 6 h during the day and blood from the tail vein was collected. Glucose was measured using an OneTouch® Ultra glucometer (LifeScan, Milpitas, CA). For insulin measurements, following the manufactures protocol, the blood insulin concentrations were determined using Ultra-Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Elk Grove Village, IL).
Glucose and insulin tolerance test (GTT/ITT)
For glucose tolerance tests, mice were fasted for 16 h overnight and injected with glucose at 1 g/kg BW (Dextrose, Phoenix Pharmaceuticals, Burlingame, CA). For insulin tolerance tests, mice were fasted for 6 h during the day and injected with insulin 0.25 U/kg BW (HumulinR, Eli Lilly, Cambridge, MA) dissolved in PBS plus protease-free BSA. Blood glucose was monitored over time using the OneTouch® Ultra glucometer.
Immunohistochemistry (IHC)
Freshly resected brains and muscles were fixed in 10% formalin for 24 h and then paraffin-embedded and sectioned at the Harvard Medical School Rodent Pathology Core. IHC staining was performed on sagittal brain sections and cross sections of muscle tissue (5 μm) mounted on microscope slides. Sections were washed 3 times in Histoclear (National Diagnostics #HS-200) for 5-min each, followed by 2 washes in 100% ethanol and 2 washes in 95% ethanol for 10-min each, then 2 washes in ddH2O for 5-min each. For antigen retrieval, sections were boiled for 10-min in 10 mM sodium citrate, pH 6.0, followed by washing in ddH2O, then 3% hydrogen peroxide for 10-min, followed by ddH2O again. Tris buffered saline with Tween-20 (TBST; 25 mM Tris Hydroxymethyl Aminomethane, 137 mM NaCl, 2.7 mM KCl, 0.1% Tween-20) plus 5% normal goat serum (NGS; CST #5425) was used as a blocking solution for 1 h at room temperature. Primary antibodies, p(S235/236)S6 (CST #4858, 1:400), fast skeletal Myosin (Sigma #M4276, 1:400), and slow skeletal Myosin (Abcam #Ab11083, 1:400), were diluted in SignalStain Antibody Diluent (CST #8112) and incubated overnight at 4°C. Slides were washed three times for 5 min each in TBST and incubated in SignalStain Boost IHC Detection Reagent (CST #8114) for 30-min at room temperature, followed by three 5-min washes in TBST. Signal was detected using the SignalStain DAB Substrate Kit (CST #8059) according to manufacturer’s instructions. Slides were counterstained with hematoxylin (Sigma #GHS132) and washed twice for 5-min each in ddH2O, followed by 2 washes in 95% ethanol, 2 washes in 100% ethanol, then 2 washes in Histoclear, for 10 s each. Coverslips were mounted with Permount Mounting solution (Fisher #SP15). H&E staining was performed by the Harvard Medical School Rodent Pathology core.
Brain H&E and lower magnification phospho-S6 images were acquired with an Olympus SZX7 stereomicroscope (Olympus Corporate, Center Valley, PA) at 2.5x magnification equipped with a Lumenera Infinity 2 camera (Teledyne Lumenera, Ottawa, Canada). Higher magnification brain phospho-S6, cerebellum H&E and all muscle IHC images were acquired with an inverted Olympus CKX41 microscope equipped with a Lumenera Infinity 2 camera. H&E images were used to measure muscle fiber size area. Soma area of neurons was measured in phospho-S6-stained images of the brain cortex and in H&E-stained images of the cerebellum using Fiji. Cortical thickness in H&E-stained sagittal sections of brain was measured at the primary motor cortex as previously described (Collins et al., 2018).
For fluorescence IHC, coronal brain sections of 8 month-old mice were collected, fixed, and vibratome-sectioned at 35 μm using standard methods and stored in 10% sodium azide solution. Prior to primary antibody staining, sections were incubated in blocking buffer (5% NGS and 0.25% triton-X-100 in PBS) for 1 h at room temperature. Floating sections were immunolabeled using the following primary antibodies: NeuN (Millipore #MAB377), CTIP2 (Abcam #ab18465), TBR1 (Abcam #ab183032), IBA1 (Fujifilm Wako #019-19741) and GFAP (CST #3670) at a 1:1000 concentration in blocking buffer overnight at room temperature. Primary antibodies were detected with the following secondary antibodies: anti-mouse 488 (ThermoFisher #A-11001), anti-rat 555 (ThermoFisher #A-21428) and anti-rabbit 555 (ThermoFisher #A-21428). Following three 5-min washes with PBS, secondary antibodies were also prepared in blocking buffer at a 1:1000 concentration and incubated for 2 h at room temperature in the dark. After 10-min wash in PBS, nuclei were stained with Hoechst 33342 (ThermoFisher) at a 1:10000 concentration for 5 min and subsequently washed for 10-min with PBS. Floating sections were then mounted onto glass microscope slides using Fluoromount G and then stored in the dark at 4°C. Images were acquired using a Zeiss LSM700 Laser Scanning Confocal Microscope equipped with Plan Apochromat × 10/0.3 and ×25/0.8 objectives or a Nikon 80i microscope using x4, x10, and x20 objectives. Images were analyzed using the ImageJ software. Blinded cortical thickness measurements were taken in a minimum of 4 measurements per section. A minimum of 3 anatomically matched sections per animal were used from 7 Tbc1d7+/+ and 5 Tbc1d7−/− mice. Cortical thickness was measured using the ImageJ angle and line measurement function for section images taken at 4x magnification as previously described (Yuskaitis et al., 2018). For sections stained with CTIP2, cell density at cortical layer IV and ratio of the CTIP2+ layer to cortical thickness were measured in images at 10x and 4× magnification, respectively. Using the ImageJ draw rectangle tool, a box was drawn to include the top and bottom boundaries of the CTIP2+ layer. Measurements were taken a minimum of four times using the line tool to measure distance from the top and bottom boundaries of the CTIP2+ layer and cortical thickness.
Grip strength
A digital grip strength meter (#1027SM, Columbus Instruments, Columbus, OH) was used to measure grip strength. Mice were held by the nape of the neck for hind limb measurements, and the base of the tail for forelimb measurements. Mice were placed on the tension bar and pulled back gently until releasing their hold on the tension bar, with resistance being measured with the digital force-gauging apparatus. Each mouse was tested five times, the highest and lowest resistance measurements removed, and the average of the other three measurements reported as the final grip strength after normalizing to body weight (N/g BW). The same procedure was repeated with a new cohort at 3 months of age to get pretreatment measurements. The mice were then injected i.p. with vehicle (5% Tween-80, 5% PEG-400, 5% Ethanol in 1x PBS) or rapamycin (1 mg/kg BW, LC Laboratories, Woburn, MA) three times a week (MWF) for one month, prior to final round of grip strength measurements.
Open field test
Mice were placed in a squared chamber (50 × 50 cm) for a 10-min trial during which videos were captured from above using a Sony PlayStation Eye camera. Videos were analyzed using the open-source ezTrack software package (Pennington et al., 2019) and used to score distance traveled (in pixels), time spent in motion, and velocity of movement.
Gait analysis
Qualitative and quantitative measurements of motor performance of free moving mice were recorded using a platform setup similar to that described by Mendes et al., 2015. Briefly, a transparent acrylic platform was lined with LED lights to propagate light at the site of foot contact via internal reflection. Footprints were visualized with high spatial and temporal resolution by a high-speed camera (Flir Blackfly S BFS-U3-13Y3M-C Mono with Fujinon 25 mm 2/3 inch C mount lens) under the acrylic platform. Videos of each animal were captured for three passes across the platform. Frames from the video were overlaid using Adobe Photoshop for measurement of stride length, stance length, sway length, stride offset and tail angle.
Accelerating rotarod
This functional motor coordination test was performed by the Animal Behavior and Physiology Core Facility at Boston Children’s Hospital. The mice were tested five times on a single day, with the training sessions the day before. Mice were kept in testing room for 1 h beforehand to acclimatize and minimize effects of stress prior to testing. For subject training, animals were placed on rod rotating at 5 rpm for 60 s and then returned to home cage. The procedure was repeated three times, separated by 10-min intervals. For testing on the next day, the rotarod (Columbus Instruments, OH, USA) was set to accelerate from 4 to 40 rpm over 300 s, and animals from same cage were placed in separate lanes on the rod initially rotating at 4 rpm, with timed trial ending when the animal falls off the rod. With the timed trials being repeated five times, separated by 10-min intervals, to obtain a mean latency to fall for each mouse.
Three-chamber social approach and social recognition
This sociability and social memory test was performed by the Animal Behavior and Physiology Core Facility at the Boston Children’s Hospital using methods similar to those previously described (Dhamne et al., 2017). Social approach and recognition was tested in an automated three-chambered apparatus. Noldus Etho-Vision XT videotracking software (version 15.0, Noldus Information Technologies, Leesburg, VA) was used to track the mice and score the trials. The apparatus was a rectangular three-chambered box, 40 × 60 × 23 cm, with retractable doors as entryways between the chambers. Interaction zones were defined as extending 2 cm from the novel object or novel mouse enclosure. The subject mouse was able to explore all three empty chambers during a 10-min habituation session. The mouse was then able to explore the three chambers containing a novel object in one side chamber and a novel mouse in the other side chamber for 30 min, the first 10 min of which was scored for sociability and the final 20 min to build up familiarity with the enclosed mouse. The novel object was then replaced with a novel mouse and the test mouse was able to explore and interact with the novel mouse or familiar mouse. Lack of innate side preference was confirmed during the initial 10 min of habituation to the entire arena and novel object and novel mouse sides were alternated each trial. Novel stimulus mice were age and sex matched to the subject mice by sex. The apparatus was cleaned with Peroxigard before the beginning of the first test session and after each subject mouse. Room illumination was ~30 lux.
Elevated plus-maze
Elevated plus-maze anxiety-related testing was performed by the Animal Behavior and Physiology Core Facility at the Boston Children’s Hospital, according to previously described procedures (Dhamne et al., 2017) using a standard mouse apparatus (Med Associates, St. Albans, VT). The maze had two open arms (35.5 × 6 cm) and two closed arms (35.5 × 6 cm) radiating from a central area (6 × 6 cm). A 0.5-cm-high lip surrounded the edges of the open arms. 20-cm-high walls surrounded the closed arms. The arms were underlit with infrared light and mice were tracked and scored using Noldus Etho-Vision XT videotracking software (version 15.0, Noldus Information Technologies, Leesburg, VA). The apparatus was cleaned with Peroxigard before the beginning of the first test session and after each subject mouse. Room illumination was ~30 lux.
Confocal immunofluorescence microscopy of primary neurons
Cultured primary hippocampal neurons were fixed on glass coverslips in warm 4% paraformaldehyde (PFA) in PBS for 15-min at room temperature and permeabilized and blocked in Intercept PBS blocking buffer (Licor #927-70001) with 0.1% Triton for 1 h at room temperature. Coverslips were incubated in blocking buffer containing primary antibodies 1hr at room temperature. Following three 5-min washes with PBS, coverslips were incubated in blocking buffer with secondary antibodies for 30-min at room temperature. Following three 10-min washes with PBS, with DAPI (Thermo #62248) added to the last wash, coverslips were mounted to slides using Fluoromount-G (Southern Biotech #0100-01). Primary antibodies used at 1:1000: TAU1 (Millipore #MAB3420), MAP2 (Synaptic Systems #188004); secondary antibodies used: mouse-Alexafluor488 (1:1000, Invitrogen #A32723) and guinea pig-Cy3 (1:500; Jackson ImmunoResearch #706-165-148). The slides were imaged with a Nikon Ti-E inverted microscope (Nikon Instruments, Melville, NY), using a 60x objective lens with a Zyla 4.2 cMOS camera and NIS elements software for acquisition parameters, shutters, filter positions and focus control. Soma size area was determined by manually measuring MAP2 staining in Fiji. Axon number and axon length was determined by scoring TAU1-positive extensions, dendrite length by scoring TAU1-negative extensions. Dendritic branch points were manually counted in Fuji measuring along dendrites for the first 50 μm away from the soma. All quantifications were repeated from 3 independent biological experiments with 2 technical replicates each, on 20 images of each condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 7 software. Data are presented as mean ± standard error of mean (SEM) or standard deviation (SD), as indicated. Unpaired two-tailed Student’s t-test was used for comparisons between two groups. For comparisons between three or more groups, one-way ANOVA with Tukey’s multiple comparison test and post hoc analysis was used for parametric data and a Kruskal–Wallis test for non-parametric data. For the behavioral tests (EPM and three-chamber test) two-way ANOVA with Sidak’s multiple comparisons test was used. Alpha was set at 0.05 for significance and ranges for p-values are reported for significant results.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-Myosin (Skeletal, Fast) | Sigma-Aldrich | Cat. #M4276, RRID:AB_477190 |
| Mouse monoclonal anti-slow skeletal Myosin | Abcam | Cat. #Ab11083, RRID:AB_297734 |
| Rabbit monoclonal anti-phospho-S6 ribosomal protein (Ser235/236) | Cell Signaling | Cat. #14733, RRID:AB_2721245 |
| Rabbit monoclonal anti-phospho-S6 ribosomal protein (Ser240/244) | Cell Signaling | Cat. #2215, RRID:AB_331682 |
| Rabbit monoclonal anti-S6 ribosomal protein | Cell Signaling | Cat. #2217, RRID:AB_331355 |
| Rabbit Monoclonal anti-phospho-p70 S6 Kinase (Thr389) | Cell Signaling | Cat. #9234, RRID:AB_2269803 |
| Rabbit monoclonal anti-p70 S6 Kinase | Cell Signaling | Cat. #2708, RRID:AB_390722 |
| Rabbit monoclonal anti-Tuberin/TSC2 | Cell Signaling | Cat. #4308, RRID:AB_10547134 |
| Rabbit monoclonal anti-Hamartin/TSC1 | Cell Signaling | Cat. #6935, RRID:AB_10860420 |
| Rabbit monoclonal anti-TBC1D7 | Cell Signaling | Cat. #14949, RRID:AB_2749838 |
| Rabbit polyclonal anti-VINCULIN | Cell Signaling | Cat. #4650, RRID:AB_10559207 |
| Rabbit monoclonal anti-AKT | Cell Signaling | Cat. #4919, RRID:AB_915783 |
| Rabbit monoclonal anti-phospho-S473-AKT | Cell Signaling | Cat. #4060, RRID:AB_2315049 |
| Rabbit polyclonal anti-phospho-T37/46-4EBP1 | Cell Signaling | Cat. #2855, RRID:AB_560835 |
| Rabbit monoclonal anti-4EBP1 | Cell Signaling | Cat. #9644, RRID:AB_2097841 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | Cat. #7074, RRID:AB_2099233 |
| Mouse polyclonal anti-TAU-1 | Millipore | Cat. #MAB3420, RRID:AB_11213630 |
| Guinea pig polyclonal anti-MAP2 | Synaptic Systems | Cat. #188 004, RRID:AB_2138181 |
| Mouse monoclonal anti-NeuN | Millipore | Cat. #MAB377, RRID: AB_2298772 |
| Rat monoclonal anti-CTIP2 | Abcam | Cat. #ab18465, RRID:AB_2064130 |
| Rabbit monoclonal anti-TBR1 | Abcam | Cat. #ab183032 |
| Rabbit polyclonal anti-IBA1 | Fujifilm Wako | Cat. #019-19741, RRID:AB_839504 |
| Mouse monoclonal anti-GFAP | Cell Signaling | Cat. #3670, RRID:AB_561049 |
| Goat anti-Mouse IgG, Alexa Fluor 488 | Thermo | Cat. #A-11001, RRID:AB_2534069 |
| Goat anti-Rabbit IgG, Alexa Fluor 555 | Thermo | Cat. #A-21428, RRID:AB_2535849 |
| Goat anti-Rat IgG, Alexa Fluor 555 | Thermo | Cat. #A-21428, RRID:AB_141784 |
| Goat anti-Mouse IgG, Alexa Fluor Plus 488 | Invitrogen | Cat. #A32723, RRID:AB_2633275 |
| Cy3 AffiniPure Donkey Anti-Guinea Pig IgG | Jackson Immuno | Cat. #706-165-148, RRID:AB_2340460 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Rapamycin, mTORC1 inhibitor | Millipore | Cat. #553210 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem | Cat. #90080 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J wild-type | The Jackson Laboratory | JAX: 000664 |
| Mouse: C57BL/6J Tbc1d7−/− | this paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer for genotyping (forward) AAGTCGGCTGCTGATCATG | this paper | N/A |
| Primer for genotyping (reverse) TCTCATACCTAGAAGCGCCTTC | this paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji | Schindlein et al., 2012 | https://imagej.net/software/fiji/; RRID: SCR_002285 |
| GraphPad Prism 7 | GraphPad Software | www.graphpad.com/; RRID: SCR_002798 |
| NIS Elements | Nikon Software | https://www.nikoninstruments.com/Products/Software; RRID:SCR_014329 |
Highlights.
A mouse knockout of Tbc1d7 encoding a core component of the TSC complex is presented
Tbc1d7 KO mice display increased tissue mTORC1 signaling but normal development
Tbc1d7 KO mice exhibit brain overgrowth with cortical thickening
Tbc1d7 KO neurons display mTORC1-dependent increases in growth and defects in polarity
ACKNOWLEDGMENTS
We thank Meghan Wilkinson, Lina Du, and colleagues at the Harvard Medical School Transgenic Mouse Core, the Animal Behavior and Physiology Core at Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (NIH P50HD105351), and the Dana-Farber/Harvard Cancer Center (P30CA06516) Rodent Histopathology Core for technical assistance. This work was supported by grants from the German Research Foundation/DFG (SCHR 1611/1-1, Project 403153940) to S.S., the TSC Alliance to G.H., and the NIH (R35-CA197459 and P01-CA120964) to B.D.M.
DECLARATION OF INTERESTS
B.D.M. is a member of the scientific advisory board and a shareholder of Navitor Pharmaceuticals. M.S. reports grant support from Novartis, Biogen, Astellas, Aeovian, Bridgebio, and Aucta and is on the scientific advisory boards for Novartis, Roche, Regenxbio, SpringWorks Therapeutics, Jaguar Therapeutics, and Alkermes. All other authors declare no competing financial interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110824.
REFERENCES
- Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, Xenarios I, Zelante L, Merla G, and Reymond A (2014). TBC1D7 mutations are associated with intellectual disability, Macrocrania, patellar dislocation, and celiac disease. Hum. Mutat 35, 447–451. 10.1002/humu.22529. [DOI] [PubMed] [Google Scholar]
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, and Hampton TG (2005). Gait dynamics in mouse models of Parkinson’s disease and Huntington’s disease. J. Neuroeng. Rehabil 2, 20. 10.1186/1743-0003-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews MG, Subramanian L, and Kriegstein AR (2020). Mtor signaling regulates the morphology and migration of outer radial glia in developing human cortex. Elife 9, 1–21. 10.7554/elife.58737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Winsvold BS, Gormley P, Kurth T, Bettella F, McMahon G, Kallela M, Malik R, de Vries B, Terwindt G, et al. (2013). Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet 45, 912–917. 10.1038/ng.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, and Sabatini DM (2013). A tumor suppressor complex with GAP activity for the rag GTPases that signal amino acid sufficiency to mTORC1. Science 340, 1100–1106. 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo-Chichi J-M, Tcherkezian J, Hamdan FF, Décarie JC, Dobrzeniecka S, Patry L, Nadon M-A, Mucha BE, Major P, Shevell M, et al. (2013). Disruption of TBC1D7, a subunit of the TSC1-TSC2 protein complex, in intellectual disability and megalencephaly. J. Med. Genet 50, 740–744. 10.1136/jmedgenet-2013-101680. [DOI] [PubMed] [Google Scholar]
- Carson RP, Van Nielen DL, Winzenburger PA, and Ess KC (2012). Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis 45, 369–380. 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Huang W-C, Sejourne J, Clipperton-Allen AE, and Page DT (2015). Pten mutations alter brain growth trajectory and allocation of cell types through elevated -Catenin signaling. J. Neurosci 35, 10252–10267. 10.1523/jneurosci.5272-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, and He X (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev. 22, 2485–2495. 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Wagner C, Gagliardi L, Kretz PF, Fischer MC, Kessler P, Kannan M, and Yalcin B (2018). A method for parasagittal sectioning for neuroanatomical quantification of brain structures in the adult mouse. Curr. Protoc. Mouse Biol 8, e48. 10.1002/cpmo.48. [DOI] [PubMed] [Google Scholar]
- Crino PB (2016). The mTOR signalling cascade: paving new roads to cure neurological disease. Nat. Rev. Neurol 12, 379–392. 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, and Henske EP (2006). The tuberous sclerosis complex. N. Engl. J. Med 355, 1345–1356. 10.1056/nejmra055323. [DOI] [PubMed] [Google Scholar]
- Crowell B, Hwa Lee G, Nikolaeva I, Dal Pozzo V, and D’Arcangelo G (2015). Complex neurological phenotype in mutant mice lacking Tsc2 in excitatory neurons of the developing forebrain. ENeuro 2, 1–15. 10.1523/eneuro.0046-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamne SC, Silverman JL, Super CE, Lammers SHT, Hameed MQ, Modi ME, Copping NA, Pride MC, Smith DG, Rotenberg A, et al. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 8, 26. 10.1186/s13229-017-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, and Manning BD (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546. 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TG, Borack MS, Reidy PT, Volpi E, and Rasmussen BB (2017). Essential amino acid ingestion alters expression of genes associated with amino acid sensing, transport, and mTORC1 regulation in human skeletal muscle. Nutr. Metab. (Lond) 14, 35. 10.1186/s12986-017-0187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JL, and Wong M (2018). The mTOR pathway in treatment of epilepsy: a clinical update. Future Neurol. 13, 49–58. 10.2217/fnl-2018-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, and Thiele EA (2016). Tuberous sclerosis complex. Nat. Rev. Dis. Prim 2, 16035. 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- Iffland PH, Carson V, Bordey A, and Crino PB (2019). GATORopathies: the role of amino acid regulatory gene mutations in epilepsy and cortical malformations. Epilepsia 60, 2163–2173. 10.1111/epi.16370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, and Aiba A (2014). Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 7,1626–1639. 10.1016/j.celrep.2014.04.048. [DOI] [PubMed] [Google Scholar]
- Kim J, and Guan KL (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71. 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, and Noda T (1999). Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 59, 1206–1211. [PubMed] [Google Scholar]
- Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, and Hino O (2001).A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. U S A 98, 8762–8767. 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K (2006). Loss of auto-phagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, and Onda H (2002). A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11, 525–534. 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Lipton JO, and Sahin M (2014). The neurology of mTOR. Neuron 84, 275–291. 10.1016/j.neuron.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, Bulfone A, Garcìa-Verdugo J, Leocani L, Minicucci F, et al. (2011). Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell 9, 447–462. 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Marsan E, and Baulac S (2018). Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol 44, 6–17. 10.1111/nan.12463. [DOI] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, and Kwiatkowski DJ (2007). A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced Myelination, seizure activity, and limited survival. J. Neurosci 27, 5546–5558. 10.1523/jneurosci.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes CS, Bartos I, Márka Z, Akay T, Márka S, and Mann RS (2015). Quantification of gait parameters in freely walking rodents. BMC Biol. 13, 50. 10.1186/s12915-015-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mtorc1 at the lysosome. Cell 156, 1771–1785. 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietzsch U, McKenna J, Reith RM, Way SW, and Gambello MJ (2013). Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J. Comp. Neurol 521, 3817–3831. 10.1002/cne.23380. [DOI] [PubMed] [Google Scholar]
- Nguyen LH, Mahadeo T, and Bordey A (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci 39, 2762–2773. 10.1523/jneurosci.2260-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda S, Sato S, Fukuda T, Tada N, Uchiyama Y, Tanaka K, and Hattori N (2020). Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis 136, 104717. 10.1016/j.nbd.2019.104717. [DOI] [PubMed] [Google Scholar]
- Onda H, Lueck A, Marks PW, Warren HB, and Kwiatkowski DJ (1999). Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J. Clin. Invest 104, 687–695. 10.1172/jci7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington ZT, Dong Z, Feng Y, Vetere LM, Page-Harley L, Shuman T, and Cai DJ (2019). ezTrack: an open-source video analysis pipeline for the investigation of animal behavior. Sci. Rep 9, 19979. 10.1038/s41598-019-56408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, and Snider W (2010). Initiating and growing an axon. Cold Spring Harb. Perspect. Biol 2, a001925. 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, and Gambello MJ (2013). Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis 51, 93–103. 10.1016/j.nbd.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Ren S, Huang Z, Jiang Y, and Wang T (2017). dTBC1D7 regulates systemic growth independently of TSC through insulin signaling. J. Cell Biol 217, 517–526. 10.1083/jcb.201706027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Kassai H, Nakayama H, Fukaya M, Maeda T, Nakao K, Hashi-moto K, Sakagami H, Kano M, and Aiba A (2019). Hyperactivation of mTORC1 disrupts cellular homeostasis in cerebellar Purkinje cells. Sci. Rep 9, 2799. 10.1038/s41598-019-38730-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism and disease. Cell 168, 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J,Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-kaminska A, Zmorzynska J, and Jaworski J (2017). Molecular neurobiology of mTOR. Neuroscience 341, 112–153. 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- Tarkowski B, Kuchcinska K, Blazejczyk M, and Jaworski J (2019). Pathological mTOR mutations impact cortical development. Hum. Mol. Genet 28, 2107–2119. 10.1093/hmg/ddz042. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, and Sabatini BL (2005). Regulation of neuronal morphology and function by the tumor sup-pressors Tsc1 and Tsc2. Nat. Neurosci 8, 1727–1734. 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, and Sahin M (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsos F, Padmanabhuni SS, Alexander J, Karagiannidis I, Tsifintaris M, Topaloudi A, Mantzaris D, Georgitsi M, Drineas P, and Paschou P (2016). Meta-analysis of tourette syndrome and attention deficit hyperactivity disorder provides support for a shared genetic basis. Front. Neurosci 10, 1–7. 10.3389/fnins.2016.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, and Manning BD (2019). Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab 1, 321–333. 10.1038/s42255-019-0038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Wu X, Guan KL, Han M, Zhuang Y, and Xu T (2006). Muscle atrophy in transgenic mice expressing a human TSC1 transgene. FEBS Lett. 580, 5621–5627. 10.1016/j.febslet.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Way SW, Mckenna J, Mietzsch U, Reith RM, Wu HCJ, and Gambello MJ (2009). Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum. Mol. Genet 18,1252–1265. 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winden KD, Ebrahimi-Fakhari D, and Sahin M (2018). Abnormal mTOR activation in autism. Annu. Rev. Neurosci 41, 1–23. 10.1146/annurev-neuro-080317-061747. [DOI] [PubMed] [Google Scholar]
- Yang H, Yu Z, Chen X, Li J, Li N, Cheng J, Gao N, Yuan HX, Ye D, Guan KL, and Xu Y (2021). Structural insights into TSC complex assembly and GAP activity on Rheb. Nat. Commun 12, 339–410. 10.1038/s41467-020-20522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Roten-berg A, Sabatini DM, Sahin M, and Poduri A (2018). A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 111, 91–101. 10.1016/j.nbd.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L-H, Xu L, Gutmann DH, and Wong M (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Annu. Neurol 63, 444–453. 10.1002/ana.21331 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data reported in this paper will be shared by the Lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.