

Abstract
Prenatal ambient particulate matter (PM2.5) exposure impacts infant development and alters placental mitochondrial DNA abundance. We investigated whether the timing of PM2.5 exposure predicts placental mitochondrial mutational load using NextGen sequencing in 283 multi-ethnic mother-infant dyads. We observed increased PM2.5 exposure, particularly during mid- to late- pregnancy and among genes coding for NADH dehydrogenase and subunits of ATP synthase, was associated with a greater amount of nonsynonymous mutations. The strongest associations were observed for participants of African ancestry. Further work is needed to tease out the role of mitochondrial genetics and its impact on offspring development and emerging disease disparities.
Keywords: mitochondria, air pollution, pregnancy, bioenergetics
Introduction
Gestational exposure to ambient fine particulate matter with a diameter of ≤ 2.5 microns (PM2.5) has been linked to correlates of chronic disease risk (e.g., low birth weight, preterm delivery)1,2 and adverse child health outcomes (e.g., poorer cognition, asthma)3–5. While mechanisms involved in the toxicity of PM are complex, evolving epidemiological and biological evidence suggests ambient air particles are capable of reaching the fetal side of the placenta6 and that PM exposure can trigger a cellular stress response and consequent oxidative damage7–11.
Mitochondria facilitate cellular energy delivery through the production of adenosine-5’-triposphate (ATP) via oxidative phosphorylation. Mitochondrial function is critical to maintaining appropriate energy supply (i.e., ATP), cell functions/signaling, and fetal vitality. Cells contain numerous mitochondria, each containing multiple copies of mitochondrial DNA (mtDNA) and mutations can affect all (homoplasmy) or a portion (heteroplasmy) of the molecules. Mutational load, specifically heteroplasmy, has been identified as a marker of oxidative damage12 and inefficient cellular respiration13 and has been linked to numerous human diseases14.
Biomarkers of mitochondrial dysfunction at the maternal-fetal interface that correlate with in utero environmental exposures can provide insight into the underlying involvement of mitochondrial bioenergetics in disease programming. Prenatal PM exposure has already been linked to changes in mtDNA copy number (mtDNAcn), a commonly used marker of mitochondrial dysfunction, in both umbilical cord blood (a marker more accurately reflecting the state of the fetus) and placenta (a key regulator of the external environmental and maternal fetal signaling15)16–18. Further, emerging data suggest that timing of PM exposure during pregnancy may be a key factor in eliciting a mitochondrial response. Recent studies demonstrate that increased exposure to PM2.5 during the third trimester (35–40 weeks gestation) of pregnancy was associated with decreased mtDNAcn in cord blood17,18. Further, mitochondrial haplogroups (a marker of genetic ancestry) have been shown to modify the effect of environmental pollutants with similar oxidative stress (OS) mechanisms of action (i.e., black carbon) on health outcomes in a non-pregnant sample19.
To our knowledge, this is the first study to examine the effect of in utero PM2.5 exposure on placental mtDNA mutational load. We leveraged daily prenatal PM2.5 exposure estimates over pregnancy in the PRogramming of Intergenerational Stress Mechanisms (PRISM) study and implemented Bayesian distributed lag interaction models (BDLIMs)20 to statistically examine and visualize the PM2.5 time-dependent pattern of associations with placental mtDNA mutational load. These models also allowed for the assessment of interactive effects with ancestry haplogroup.
Methods
Sample:
Mother-child dyads participating in the Programming of Intergenerational Stress Mechanisms (PRISM) study were included in analyses. PRISM is an urban prospective pregnancy cohort located in the northeastern United States (US) designed to investigate associations among maternal psychosocial stress, other environmental factors (i.e., air pollution, smoking, diet), and child development. Eligibility criteria included being a pregnant English- or Spanish-speaking woman, aged 18 years and older, and carrying a singleton fetus. Women meeting these criteria were recruited from prenatal clinics at the Beth Israel Deaconess Medical Center and the East Boston Neighborhood Health Center in Boston, Massachusetts between the years of 2011 and 2013 and from the prenatal clinic at Mount Sinai Hospital in New York City from 2013 to 2018. Exclusion criteria included being born prior to 37 weeks (mean 39 weeks, SD 1.3); maternal intake of ≥7 alcoholic drinks per week before and/or any alcohol after pregnancy recognition as usage above these thresholds has been associated with increased risk for adverse child development21–23; HIV positive status, which would influence/confound biomarkers of interest; pregnancy loss; or the presence of a major congenital or genetic disorder identified during pregnancy or at birth that would influence participation in future activities. Medical chart review and postnatal questionnaires were used to ascertain birth details. Study procedures were approved by the relevant institutional review boards. Written informed consent was obtained in the participant’s preferred language.
Placenta collection and DNA extraction:
Placenta samples (~1–2 cm3) were taken on the fetal side ~1 – 1.5 cm below the fetal membrane to avoid membrane contamination and approximately 4 cm from the cord insertion site, taking care to avoid large vessels. To confirm fetal placental tissue origin, 64 genotyping probes were used and near-perfect agreement of placenta and cord blood samples was observed24. Placenta DNA extraction was conducted using the Promega Wizard Genomic DNA Purification Kit (Promega – Madison, WI, USA). DNA concentration was assessed by Qubit.
Mitochondrial Sequencing:
Whole mtDNA sequencing was performed by the Genomics, Epigenomics and Sequencing Core at the University of Cincinnati. To amplify whole mtDNA genome (complete genome sequence ID AY495156.2, 16569 bp), Illumina (San Diego, CA) protocol (Document # 15037958 v01) was followed with modifications. For each sample two pairs of primers that cover the entire mtDNA sequence were used in two individual amplifications: MTL-F1 /MTL-R1 and MTL-F2/MTL-R2. TaKaRa LA Taq (Takara Bio USA, Mountain View, CA) was used in long range PCR with 2 ng human gDNA as input in each 50 μl PCR reaction. The volumes of the two PCRs were then adjusted based on their yields from agarose gel electrophoresis and pooled to result in roughly equal amounts of the two PCR products in the pool. To reduce potential differences in mtDNA copy number among samples, after DNA clean up using Wizard SV Gel and PCR Clean-Up kit (Promega), about 100 ng DNA was used as input for library preparation using NEBNext Ultra II FS DNA Library Prep kit (NEB, Ipswich, MA). After Bioanalyzer QC analysis (Agilent, Santa Clara, CA) of the library and qPCR quantification using NEBNext Library Quant Kit (NEB), individually indexed and compatible libraries were proportionally pooled and sequenced using HiSeq 1000 sequencer (Illumina). Under the sequencing setting of paired-end 2×101 bp, about 8 million pass filter reads per sample were generated. The demultiplexed fastq files were used for downstream analysis. The average depth of coverage (i.e., number of unique reads) was 1494 and this was consistent across samples (e.g., participants). Information on primer sequences, amplicon sizes, and detailed methods on checking amplification specificity/yield using this approach have been previously reported25.
Sequencing Data Processing:
A mitochondria consensus genome, based on analyses of the complete mtDNA sequence of 53 humans of diverse origins, was used for alignment (URL: https://www.ncbi.nlm.nih.gov/nuccore/AF346978.1)26. Alignment to the reference genome and removal of adapters and duplicates was done using Picard (version 2.17.8).27 Variant calls were made using the Genome Analysis Toolkit (GATK).28 Summary Phred scores, which are log-likelihood scores, indicating our confidence in assignment of each base call, were computed as the sum of depth of coverage multiplied by the Phred score at the variant site. To reduce the effects of sequencing errors, we removed variant calls with a summary Phred score < 1000029. Repeated sequencing was carried out on 20% of the samples showing high reproducibility of the base calls. Over 80% of the mutations identified using our filtering criteria can be confirmed by at least one read in repeated sequencing of the same samples.
Insertions, deletions, and variants observed only once were ignored. Single nucleotide substitutions, which typically account for 90% of somatic mutations in the mitochondrial genome, accounted for 96.3% of mutations in our data and provided the basis for determining mutational load. Positions were considered heteroplasmic if less than 50% of the reads supported alternative alleles; all other mutations were considered homoplasmic. We chose this criteria to reduce the effects of potential homoplasmic mutations in the germline30, better representing risk alleles of complex disease31, and to obtain good concordances among cell types at the heteroplasmy level32. Total mutational load is the total number of heteroplasmic and homoplasmic mutations present in a sample. Nonsynonymous mutations were determined based on the nucleotide’s change affecting protein sequence. For secondary gene-wise analyses, protein-coding genes and ribosomal RNAs were examined independently and grouped based on involvement in the functioning of the same electron transport chain (ETC) complex (e.g., combining MT-ATP8 and MT-ATP6 coding for subunits of ATP synthase). The number of mutations observed in tRNAs was minimal (n=395) with only 5 out of 22 tRNAs accumulating more than 30 mutations. Thus, mutational loads for tRNAs were summed to create one tRNA total mutational load variable. These methods have been previously reported25.
Ambient fine particulate matter exposure:
Geographic Information Systems were used to geocode residential addresses using Arc Geographic Information Systems (ArcGIS). Records of relocation were documented and geocoding was conducted for all address histories and subsequent daily PM2.5 estimates were based on location and time at each address. To validate our geocoding we randomly sampled 15% of the data and evaluated accuracy by visually examining the locations using established map services like the Environmental Systems Research Institute (ESRI) ArcGIS street datasets. Initial completeness was approximately 90% and the erroneous few addresses were then geocoded manually. Daily PM2.5 levels were estimated using temporally- and spatially-resolved exposure models as detailed previously33. Moderate Resolution Imaging Spectroradiometer (MODIS) satellite-derived Aerosol Optical Depth (AOD) measurements based on the Multi-Angle Implementation of Atmospheric Correction (MAIAC) algorithm were used at a 1km x 1km grid spatial resolution to predict daily PM2.5 levels across New England. For each study participant, the hybrid model combined remote sensing data with spatio-temporal predictors that represents within-grid variation to yield residence-specific estimates of PM2.5 exposures. The model was run using day-specific calibrations of AOD data using ground PM2.5 measurements from 161 monitoring stations and land use regression (LUR) (e.g., traffic density, point sources, etc.) and meteorological (e.g., temperature, wind speed, visibility, elevation, distance to major roads, percent open space, point emissions and area emissions) variables using mixed models with day-specific random intercepts, and fixed and random AOD and temperature slopes. Generalized additive mixed models were used to estimate exposures on days when AOD measures were not available (due to cloud coverage, snow, etc.). The residuals from the final model for each monitor then were regressed against the local spatial and temporal variables at each monitoring site to derive 200 meter localized predictions. The mean cross validation R2 for daily values was 0.88. Given the interquartile range of PM2.5 averaged across pregnancy was 0.9 μg/m3, results are presented for a 1 μg/m3 increase in PM2.5.
Potential Covariates:
Covariates included self-reported maternal race (White, Black, Hispanic, Other/multi-race) and education status (≤ high school degree, some college or/college degree) which were ascertained at enrollment; child sex and maternal age at delivery were obtained postnatally. These covariates were included due to documented racial and SES disparities in air pollution exposure34 and our prior observed sex differences in PM2.5 effects on mitochondrial DNA copy number35. Mode of delivery (vaginal vs. C-section), pregnancy complications (i.e., gestational diabetes, hypertension, and/or preeclampsia), and maternal smoking have been shown to impact placental functioning/response leading to inflammation, OS, and mitochondrial dysfunction36–40, thus these were also considered in the early stage of the analysis. Women were classified as prenatal smokers if they reported smoking at enrollment or during the third trimester. Birth weight was extracted from delivery records and gestational age was determined by reported last menstrual period and compared to the first trimester ultrasound; if there was a discrepancy of more than two weeks, obstetrical values were used. Sex-specific Fenton birthweight for gestational age z-scores were then derived41. Prenatal daily temperature was derived using a model that calibrated MODIS satellite surface temperature measurements to air temperature monitors using LUR42. Genetic ancestry was determined by mitochondrial haplogroup and categorized as African (L), Native American/Asian (A, B, C, D, F, G, N, P, Y, Z, M, E, R), or European (H, I, J, K, T, U, V, W, X)43. Ultimately, mode of delivery, pregnancy complications, and maternal smoking were not included in the final model due to the lack of relationship with PM2.5 exposure; we also considered these variables, along with sex, as effect modifiers, but found no significant interactions (data not shown).
Data Analysis:
We estimated the association between placental mutational load and daily average prenatal PM2.5 exposure using Bayesian distributed lag interaction models (BDLIMs) to characterize overall effects of PM2.5 and haplogroup-specific effects44. BDLIM is a data-driven approach to estimating critical windows during pregnancy and to allow testing for effect modification by haplogroup. The logistic BLDIM for dyad i (i=1,…,n) who is carrying genetic ancestry haplogroup j (j=1 for African, j=2 for Native American/Asian, and j=3 for European) is
where aj is a fixed sex-specific intercept, βj is the regression coefficient characterizing the haplogroup-specific association between weighted PM2.5 exposure and mutational load of interest, is the weighted exposure, and is the covariate regression term. The wjt identify critical windows of susceptibility while βj represents the cumulative effect. When weights are constant, the model is equivalent to using exposure averaged over pregnancy. However, when the weight varies by time the model assigns greater relative weight to some periods. Time periods with weights having a 95% confidence interval omitting zero are identified as critical windows. This approach allows uncertainty in the effect to vary over time resulting in the ability for 1) sensitive windows to be identified without a cumulative effect being observed, or 2) a cumulative effect to be identified without sensitive windows. In some cases, the cumulative effect is not significant because we identify a significant effect during some time points but have uncertainty in the effect at other time points. That uncertainty results in decreased statistical significance for the cumulative effect (e.g., a signficant association at a portion of time points can be washed out when averaged over time points). On the other hand, when uncertainties behave similarly across all the time points, the summarizing cumulative effect can sometimes suggest relatively lower uncertainty and results in a narrower confidence interval.
In this analysis, genetic ancestry haplogroups is considered an effect modifier. The approach computes four different potential patterns of effect modification by allowing βj (effect magnitude) and/or the weights wjt (critical window) to be haplogroup-specific or the same for all groups: 1) Both weight and beta are constant across different haplogroups, 2) weights are constant across different haplogroups but the effect estimates are different, 3) effect estimates are constant across different haplogroups but the weight are different, 4) both weights and effect estimates are different across the haplogroups. The approach quantifies the likelihood of each pattern of heterogeneity and estimates the association between exposure and outcome under the effect modification pattern best supported by the data. Results are not normalized based on haplogroup sample size. Secondary analyses consisted of gene-wise analyses to test for associations between PM2.5 and total mutational load in gene-specific regions. All analyses were conducted using the “regimes” package in R (v4.0.2, Vienna, Austria)44. Models were adjusted for maternal race (White, Black, Hispanic, Other/multi-race], and education status, child sex, maternal age at delivery, and averaged daily temperature over pregnancy. Sensitivity analyses were conducted including a location indicator (Boston/New York) to rule out the influence of location on the association between PM2.5 and mutational load. The results were unchanged (data not shown).
Multiple imputation generating 10 iterations was implemented in R (v4.0.2, Vienna, Austria) using the package “mice”45to account for missingness on maternal race/ethnicity (1%), and education (1.8%).
Results
Descriptive Statistics:
Maternal and child characteristics for the analytic sample with complete PM, covariate, and sequencing data (n = 283) are presented in Table 1. The average maternal age at delivery was 29.6 years; the majority of women were racial/ethnic minorities (Black, 44%; Hispanic, 24%; multi-racial, 6%) with 32% reporting less than or at most a high school degree, 12.5% reporting smoking during pregnancy, and 25% delivering their child via C-section; 53% of the children were male. All three haplogroups are represented with the majority (43%) belonging to the African haplogroup followed by 32% European and 25% Native American/Asian. Descriptive data on variants mapping to MitoMap and subsequent disease associations can be found in the online supplement of Brunst et al. (2020)25. The mean number of mutations per subject is 32, standard deviation is 16 (Table S1).
Table 1.
Maternal and child characteristics of the study population
| Characteristic | n, mean | %, SD |
|---|---|---|
| Maternal Racea | ||
| White | 74 | 26 |
| Hispanic | 67 | 24 |
| Black | 123 | 44 |
| Other | 16 | 6 |
| Mitochondrial Haplogroup | ||
| African (Haplogroup 1) | 121 | 43 |
| European (Haplogroup 3) | 90 | 32 |
| Native American/Asian (Haplogroup 2) | 72 | 25 |
| Maternal Educationa | ||
| High school degree or less | 89 | 32 |
| Mode of deliverya | ||
| C-Section | 71 | 25 |
| Vaginal Delivery | 211 | 75 |
| Smoking during pregnancy | 35 | 12 |
| Male child sex | 151 | 53 |
| Fenton Birthweight Z-score (mean, SD) | −0.19 | 0.81 |
| Maternal age (years) at birth (mean, SD) | 29.7 | 5.75 |
| Mean PM2.5 exposure by trimester (μg/m3) | ||
| PM2.5 (T1) | 9.08 | 1.92 |
| PM2.5 (T2) | 8.74 | 1.62 |
| PM2.5 (T3) | 8.61 | 1.90 |
Three participants missing maternal race, one missing mode of delivery, and five missing maternal education.
Associations between prenatal PM2.5 and total and nonsynonymous mutational loads:
The cumulative and time-varying associations per μg/m3 increase in prenatal PM2.5 and total and nonsynonymous placental mutational loads were examined in separate BDLIMs, adjusting for maternal race (White, Black, Hispanic, Other/multi-race), and education status, child sex, maternal age at delivery, and averaged daily temperature over pregnancy (Figure 1A and B). The estimated cumulative effect of PM2.5 over the entire pregnancy was significantly associated with a greater amount of nonsynonymous mutations detected in the placenta (cumulative effect estimate = 0.58; 95% CI 0.12, 1.07); a statistically significant cumulative effect of PM2.5 on total mutational load was not observed. Further, two statistically significant windows in mid- and late- pregnancy were observed where increased PM2.5 exposure was associated with a higher mtDNA nonsynonymous mutational load (Figure 1B); the first sensitive window was identified in the second trimester (162–183 days) and the second sensitive window was observed in the third trimester (224–243 days). No statistically significant cumulative effect or sensitive window was found for the effect of PM2.5 on total mutational load (Figure 1A).
Figure 1. Cumulative and time-varying associations between daily PM2.5 levels over gestation and placental mtDNA total (A) and nonsynonymous (B) mutational loads.
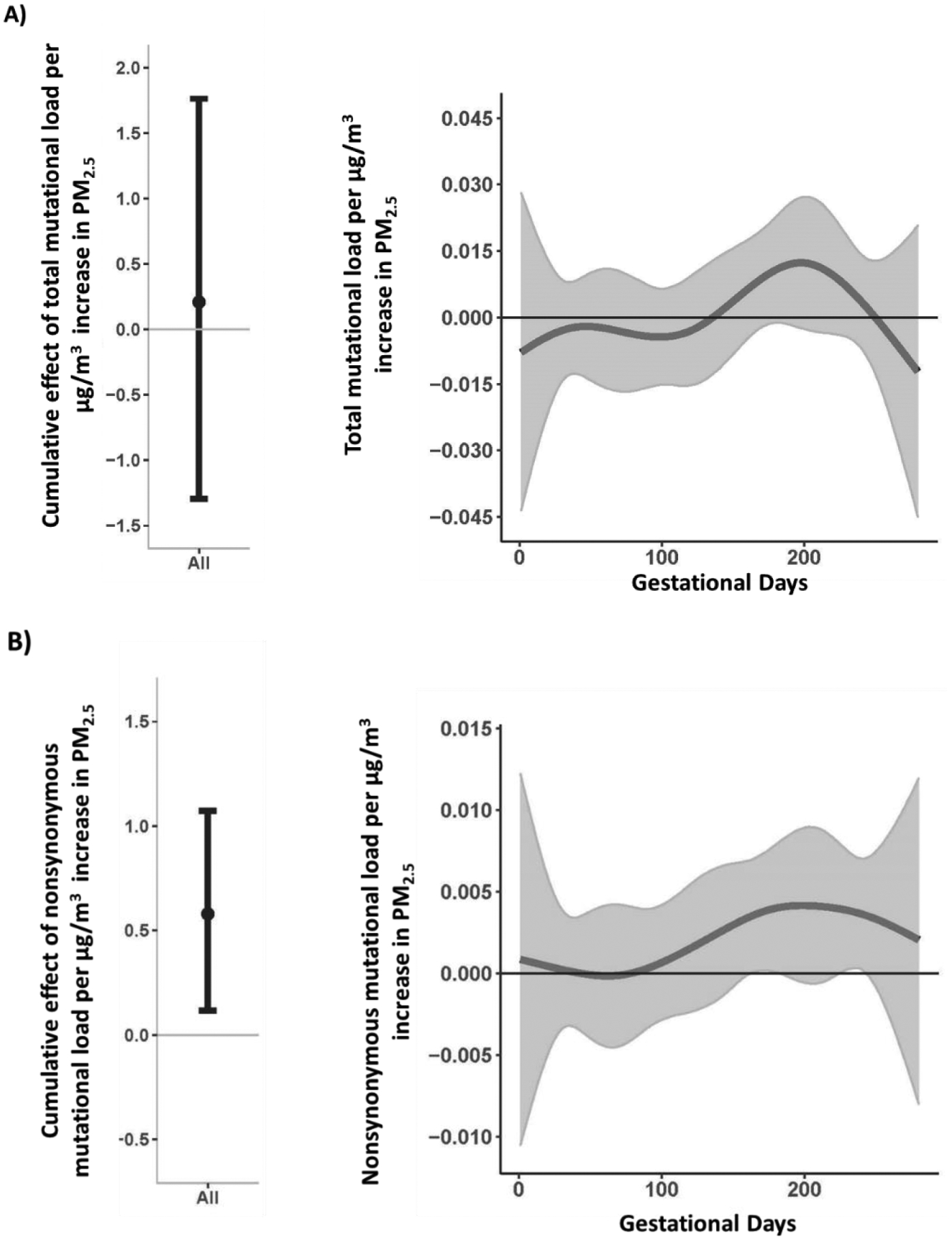
This figure demonstrates the cumulative and time-varying associations between PM2.5 exposure over pregnancy and change in mtDNA mutational load: (A) total, and (B) nonsynonymous mutational load using BDLIM assuming day-specific effects. The y-axis represents the change in mtDNA mutational load corresponding to a μg/m3 increase in PM2.5; the x-axis is gestational age in days. Solid lines for time-varying associations show the predicted change in mtDNA mutational load. Gray areas indicate 95% confidence intervals (CIs). A sensitive window is identified for the days where the estimated pointwise 95% CI (shaded area) does not include zero. The models were adjusted for maternal age, education, race/ethnicity, child sex, and ambient temperature.
Effect modification by haplogroup:
The BDLIM also suggested an interaction between PM2.5 and haplogroup on nonsynonymous placental mutational load (Figure 2B). The estimated cumulative effect of PM2.5 over the entire pregnancy was significantly associated with a greater amount of nonsynonymous mutations for the African Haplogroup (cumulative effect estimate = 0.59; 95% CI 0.03, 1.25) but not Native American/Asian (cumulative effect estimate =0.31; 95% CI −0.28, 0.98) or European (cumulative effect estimate = −0.003; 95% CI −0.51, 0.51) haplogroups; windows of susceptibility were not detected. The BDLIM did not detect a significant interaction between the effect of PM2.5 exposure and haplogroup in analyses of total mutational load (Figure 2A).
Figure 2. Cumulative and time-varying associations between daily PM2.5 levels over gestation and placental mtDNA total (A) and nonsynonymous (B) mutational loads by haplogroup.
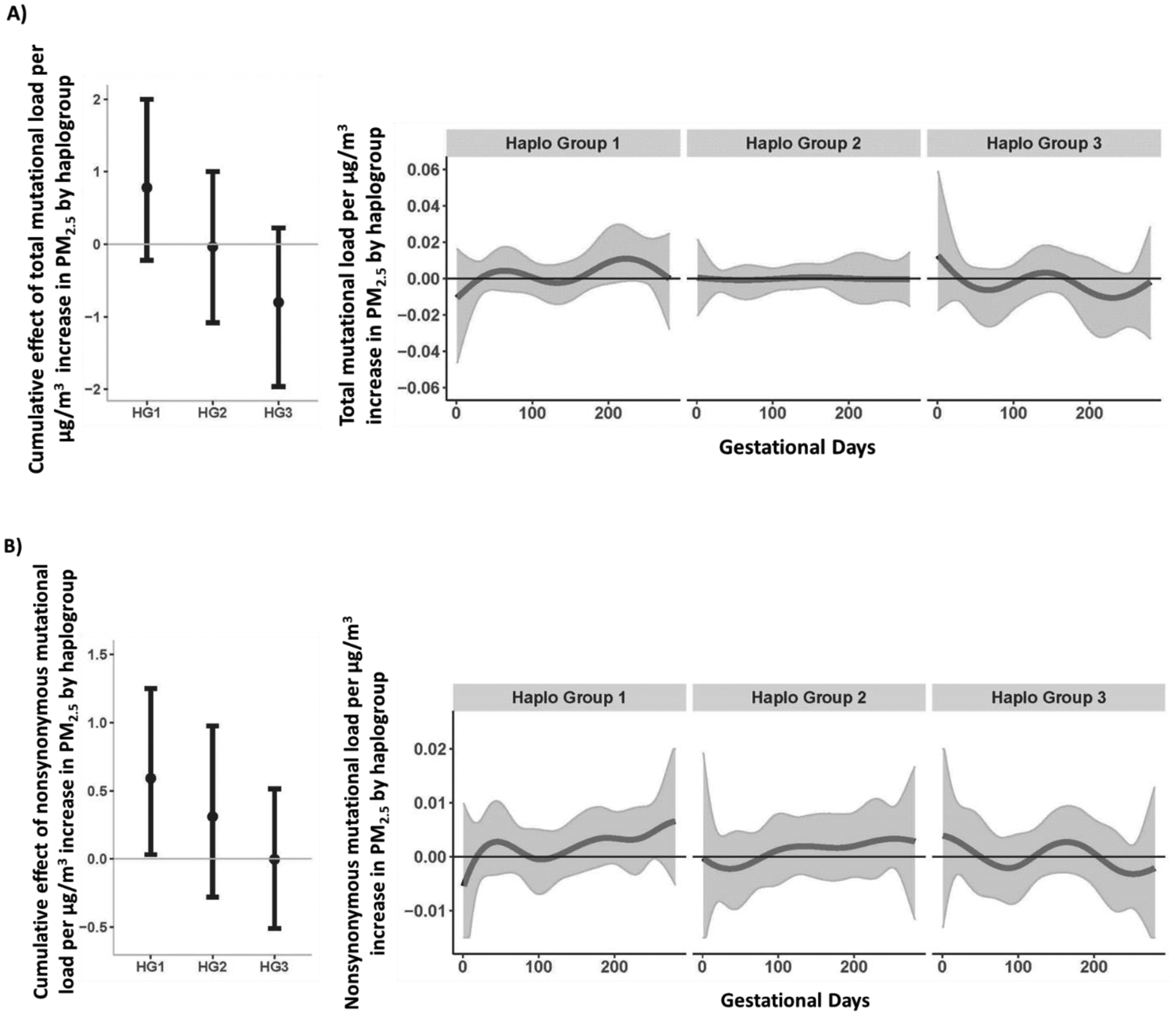
This figure demonstrates the effect modification by haplogroup on the association between PM2.5 exposure over pregnancy and mtDNA mutational load in placenta, using BDLIM assuming daily-specific effects. The models were adjusted for maternal age, education, race/ethnicity, child sex, and ambient temperature. The y-axis represents the combined cumulative effect of time-varying prenatal PM2.5 exposure on increased mtDNA mutational loads for each haplogroup for which the BDLIM detected an interaction. Solid lines for time-varying associations show the predicted change in mtDNA mutational load. Gray areas indicate 95% confidence intervals (CIs). A sensitive window is identified for the days where the estimated pointwise 95% CI (shaded area) does not include zero. We did not observe any sensitive windows by haplogroup. Abbreviations [Haplogroup1 (African); Haplogroup 2 (Native American/Asian); Haplogroup 3 (European)].
Gene-wise effects of PM2.5 on mutational load:
In order to determine if increased PM2.5 was more strongly related to mutations in a particular region, we conducted gene-wise BDLIM analyses across genes (e.g., MT-ND5) and gene families (e.g., MT-ATP, sum of all mutations from MT-ATP genes) focusing on total mutations given the low frequency of nonsynonymous mutations in specific-gene regions. While cumulative effects were not observed, significant windows of susceptibility were identified in gene-wise associations between PM2.5 exposure during late second/early third trimester and increased total mutational load across all MT-ATP genes (164–223 days, Figure 3A) and MT-ND5 (158–205 days, Figure 3B). PM2.5 was not associated with mutational load in other gene regions (Figure S2 A-F of the online supplement).
Figure 3. Time-varying associations between daily PM2.5 levels over gestation and placental mtDNA total mutational load in (A) MT-ATP (B) MT-ND5 gene regions.
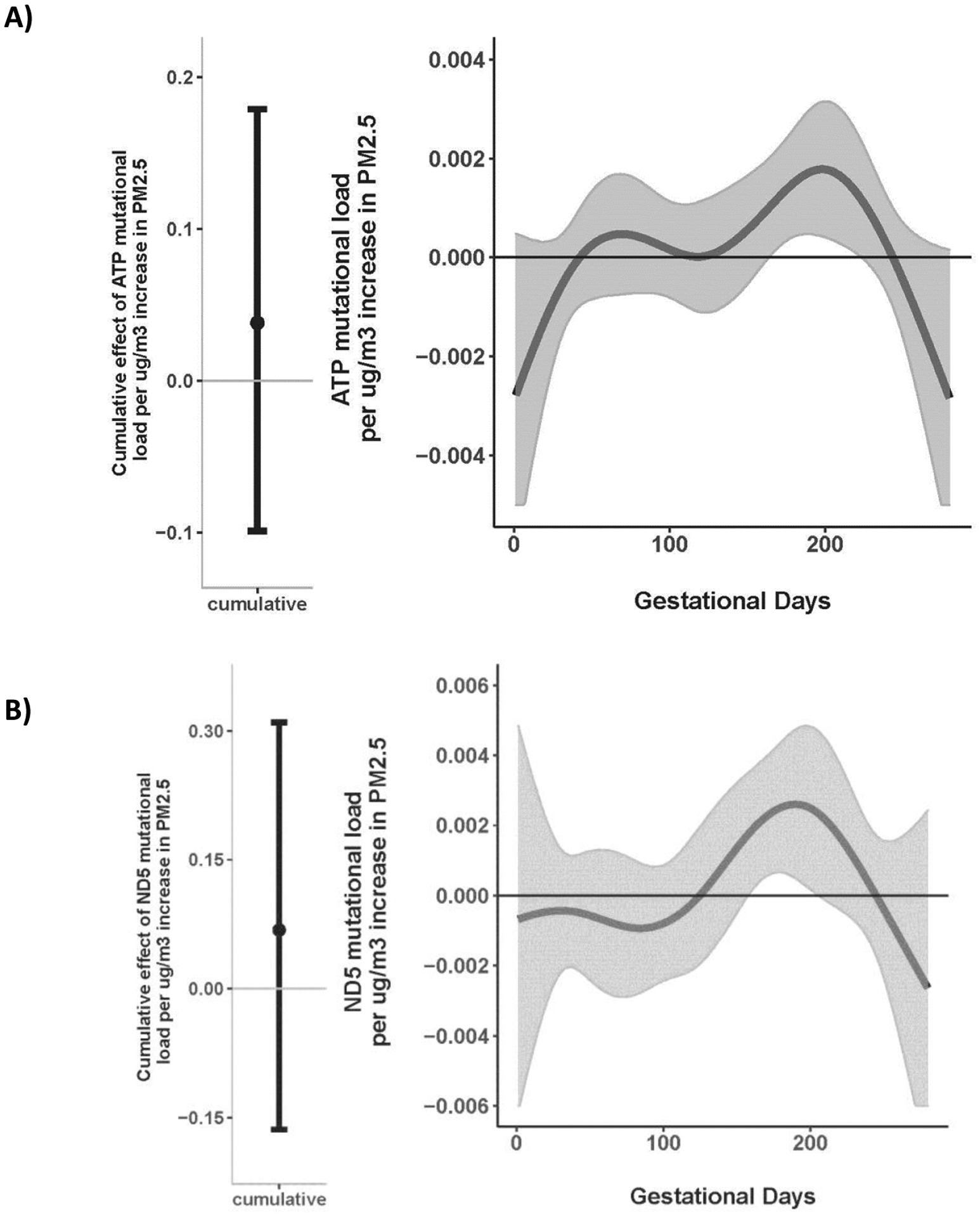
This figure demonstrates the association between PM2.5 exposure over pregnancy and change in mtDNA total mutational load: (A) MT-ATP, and (B) MT-ND5 gene regions using BDLIM assuming day-specific effects. The y-axis represents the change in mtDNA total mutational load corresponding to a μg/m3 increase in PM2.5; the x-axis is gestational age in days. Solid lines show the predicted change in mtDNA total mutational load. Gray areas indicate 95% confidence intervals (CIs). A sensitive window is identified in the late second/early trimester for the days where the estimated pointwise 95% CI (shaded area) does not include zero. The models were adjusted for maternal age, education, race/ethnicity, child sex, and ambient temperature.
BDLIM analyses suggest that PM2.5 has a stronger magnitude of effect on MT-ATP mutational load in haplogroup 1 (African) compared to haplogroup 2 and 3 but the exposure weighting is similar over time (Figure 4). Similar findings can be observed across other gene regions as outlined in the online supplement. Briefly, the magnitude of effect of PM2.5 varied by haplogroup for models interrogating MT-CYB and MT-tRNA regions (Figure S3 B and D), exposure weighting varied for MT-CO and MT-RNR regions (Figure S3 A and C), and both magnitude of effect and weighting varied for the D Loop region (Figure S3 E) although no signficant windows of exposure or cumulative effects were observed.
Figure 4. Cumulative and time-varying associations between daily PM2.5 levels over gestation and placental MT-ATP mutational load by haplogroup.
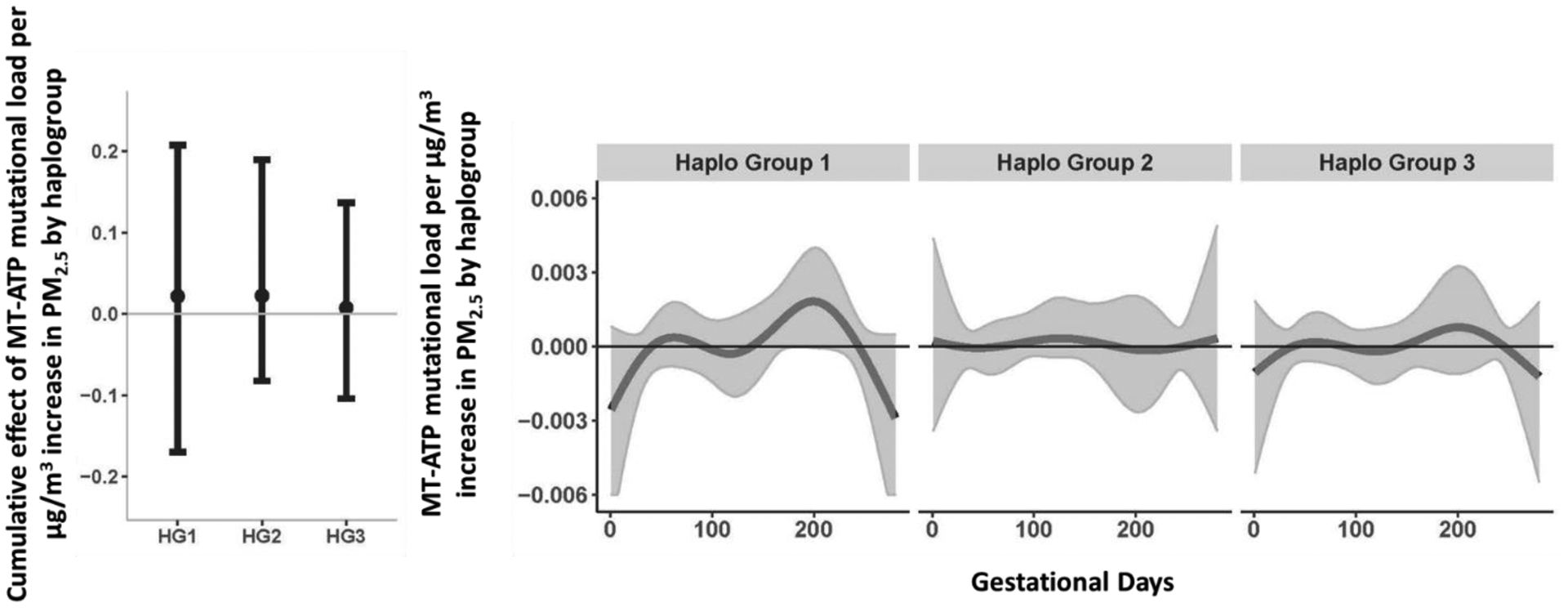
This figure demonstrates the effect modification by haplogroup on the association between PM2.5 exposure over pregnancy and mtDNA mutational load in the MT-ATP gene region, using BDLIM assuming daily-specific effects. The model was adjusted for maternal age, education, race/ethnicity, child sex, and ambient temperature. The y-axis represents the combined cumulative effect of time-varying prenatal PM2.5 exposure on increased mtDNA mutational loads for each haplogroup for which the BDLIM detected an interaction. Solid lines for time-varying associations show the predicted change in mtDNA mutational load. Gray areas indicate 95% confidence intervals (CIs). A sensitive window is identified for the days where the estimated pointwise 95% CI (shaded area) does not include zero. We did not observe any sensitive windows, but did observe varying magnitudes of effect by haplogroup. Abbreviations [Haplogroup1 (African); Haplogroup 2 (Native American/Asian); Haplogroup 3 (European)].
Discussion
This study presents novel findings linking prenatal PM2.5 exposure to placental mtDNA mutations in a multiethnic urban US cohort. Women exposed to higher PM2.5 levels over pregnancy (mainly mid- to late- pregnancy) exhibited a higher number of placental mitochondrial mutations; a greater degree of mutations was observed among genes coding for NADH dehydrogenase and subunits of ATP synthase. Further, the effect of PM2.5 exposure was more robust among participants of African ancestry.
Mitochondria are vital organelles that are found in almost every cell of all organisms except bacteria and produce nearly all of the energy needed to sustain life. A key role of mitochondria is to sense, assimilate, and relay environmental information. Mitochondrial DNA is particularly sensitive to damage caused by exogenous reactive oxygen species for which exposure to various air pollutants are known to generate including, carbon monoxide (CO), nitrogen dioxide (NO2), volatile organic compounds (VOCs), polycyclic aromatic hydrocarbons (PAHs), as well as PM46. Animal and in vitro studies have confirmed that air particles can reach the placenta6 and penetrate cells and damage the mitochondria and its genome, subsequently impacting electron transport chain and respiratory function47,48. Although studies of this nature have not been conducted in humans, research suggests that damaged mitochondria could reduce the proliferation and migration of trophoblast cells, thus affecting placental formation and development49,50. Studies have shown PM exposure to be associated with altered mtDNAcn in multiple tissues including neuroblastoma cells, peripheral blood, cord blood, and placenta51–54. While mitochondrial mutations have been reported in tissues at the maternal-fetal interface in the context of aging and maternal lifetime stress25,55; this is the first study to show in utero PM exposure to be associated with placental mitochondrial mutational load and suggests two critical windows of exposure in mid to late pregnancy. Interestingly, several species (including humans) also exhibit gestational changes in placental OS and expression of the mitochondrial-related proteins in mid to late pregnancy to support fetal growth near term56–58. Consequently, environmental exposures during this period that also induce excessive OS could result in dysfunctional ETC activity leading to pregnancy complications such as early onset preeclampsia and preterm birth59,60.
It is well known that in a scenario where oxygen is restricted, such as hypoxia occurring at high altitudes, the placenta will remodel its metabolism to increase ATP production via glycolysis rather than the ETC. Under conditions of excessive OS, however, research suggests ATP levels drop due to the inhibition of both mitochondrial (i.e., ETC) and glycolytic ATP synthesis61. Although there are metabolic adaptive responses in place to allow for continued ATP production, as of now, we have a limited mechanistic understanding of how PM-related mutations impacts these processes or what the changes/adaptations mean for long-term health outcomes. Our study showed that greater PM2.5 exposure was predominantly associated with a higher placental mutational load in one gene (i.e., MT-ND5) that codes for a subunit of NADH dehydrogenase [Complex 1 of the electron transport chain (ETC)] and among two genes coding for subunits of ATP synthase (i.e., MT-ATP6, MT-ATP8) sometimes referred to as complex V of the ETC. The first step of the ETC is the oxidation of NADH by Complex 1. While ATP synthase is part of the ETC, it is not involved in transporting ions; rather it uses the proton gradient to synthesize ATP. Mutations in MT-ND5 and MT-ATP genes have been linked to metabolic markers such as body mass index, waist-hip-ratio, and fasting insulin levels62 as well as neurological outcomes including developmental delays, autism Spectrum Disorder, Bipolar Disorder, Major Depressive Disorder, and Schizophrenia63,64. While it is difficult to prove the pathophysiological significance of certain mtDNA mutations in epidemiological studies, we have previously shown that greater than 50% and 64% of the mutations in MT-ND5 and MT-ATP, respectively, are nonsynonymous25,65. Nonsynonymous mutations with functional consequences could lead to reduced energy production capacity and systemic metabolic dysregulation that could have later-life health implications. Further work is needed to confirm this hypothesis.
Higher PM2.5 -related mtDNA mutational loads were observed for participants of African ancestry compared to those of European and Native American/Asian background. In the US, it is well documented that racial-ethnic minorities are disproportionately exposed to higher levels of PM2.5 compared to their white counterparts66. Given the disproportionate exposure to PM and the increased mutational loads observed among those of African ancestry, it is possible that PM-related variations in mtDNA could be contributing to disparities in health as has been observed for various outcomes such as cardiovascular disease67, cancer68, and insulin sensitivity69 with African American’s being at increased risk. Further, African American women are at increased risk for inflammatory and vascular pathologies compared to White women experiencing uncomplicated births70. On a molecular level, the placentas of African American women also exhibit advanced cellular aging71and diversity in immune responses72. Haplogroups have previously been shown to modify the effects of air pollution on biomarkers of systemic inflammation73. Additional analyses were suggestive but not signficant; thus, it is possible we could have detected stronger and more signficant effects if our sample sizes within strata were larger (i.e., increased power). Thus, despite our intriguing observations, larger studies are needed to tease out the ancestral differences in exposure-related outcomes and their association to placental mitochondrial function.
This study has a number of strengths. We leverage an extant prospective birth cohort of ethnically diverse mother-infant dyads with greater exposure to ambient air pollution. The assessment of PM is conducted with a validated spatiotemporal model that has been used in many studies to assess daily air pollution exposures over pregnancy. Our group is one of the first to apply whole mitochondrial genome sequencing of the placenta in an epidemiological study design25. Further, our applied data-driven statistical methods to identify sensitive windows of exposure and cumulative effects of PM on mitochondrial mutational load is also a first. This methodology also allows for enhanced power to determine effect modification by genetic ancestry. We also acknowledge some limitations. First, our effect sizes are relatively small and this is a common finding in environmental omic studies74. While this does not negate the significance (statistically speaking) of our findings, in order to understand whether biologically a “real” association has been identified validation and/or replication studies must be employed. Second, it is possible that the PM-related associations with mutational load are cell-type specific. To our knowledge, placental cell reference-based estimates for mitochondrial heteroplasmy are not available and thus we were unable to adjust for the cell-type heterogeneity. Third, we cannot confirm whether the mutations are inherited or acquired. Although some studies suggest most are inherited75, given the extensive mutagenesis of placental nuclear and mitochondrial genomes76,77, it’s possible that knowing the maternal mtDNA genome landscape would not lead to conclusive results without further knowledge on the mechanism of transmission. Fourth, it is also unclear at what level of PM exposure placental mitochondrial function will substantially be affected. Given that we detected relatively low levels of heteroplasmy, it’s likely that the perturbations associated with PM exposure would not lead to gross mitochondrial dysfunction in the placenta25,78; however, the point mutations (inherited or acquired) could contribute to the accumulation of mtDNA point mutations over time as a result of an accelerating mutation rate or clonal expansion79,80. Thus, it’s possible that exposure to environmental toxins, such as PM2.5, could impact child development by 1) increasing the level of heteroplasmy to the point of altering the functional capacity of mitochondria, or 2) lowering the threshold for the effect of an environmental toxin on cell function. Lastly, we recognize that other environmental factors may also influence mutational load and thus we cannot rule out unmeasured confounding. However, we did adjust for a number of important sociodemographic factors (race, socioeconomic status, sex and maternal age) as well as climate-related factors (temperature) that may relate to differential exposure to other toxins (e.g., black carbon, ozone, PAH) as well as oxidative stress and mitochondrial function.
To our knowledge this is the first epidemiological study to sequence the mitochondrial genome and examine the impact of prenatal PM exposure on mtDNA mutations at the maternal-fetal interface. Our urban US minority population also provides the opportunity to examine this relationship among women who may be at increased risk for PM exposure. This work also underscores the need for the inclusion of diverse population in genomic research to ensure equity and scientific progress. Lastly, further work is needed to delineate transmission mechanisms, better understand the critical timing of PM exposure on mitochondrial placental function, and examine the impact PM-related mitochondrial dysfunction has on both maternal and child short- and long-term health.
Supplementary Material
Acknowledgments:
We would like to thank the participants of the PRISM cohort and our funding agencies. Specifically, this work was supported by the National Heart, Lung, and Blood Institute under grants R01HL095606 (RJ Wright) and R01HL114396 (RJ Wright); the National Institute of Environmental Health Sciences (NIEHS) under grants R00ES024116 (KJ Brunst), P30ES006096 (KJ Brunst), and P30ES023515 (RJ Wright); derivation of air pollution exposure estimates was supported by UG3/UH3 OD023337 (RJ Wright). Biobanking infrastructure was supported by the Mount Sinai Health System Clinical Translational Science Award from the National Center for Advancing Translational Sciences under grant UL1 TR001433 (RJ Wright).
References
- 1.Malmqvist E, Rignell-Hydborn A, Tinnerberg H, et al. Maternal exposure ot air pollution and birth outcomes. Environmental Health Perspectives. 2011;119:553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamichhane DK, Leem JH, Lee JY, Kim HC. A meta-analysis of exposure to particulate matter and adverse birth outcomes. Environ Health Toxicol. 2015;30:e2015011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunst KJ, Ryan PH, Brokamp C, et al. Timing and Duration of Traffic-related Air Pollution Exposure and the Risk for Childhood Wheeze and Asthma. Am J Respir Crit Care Med. 2015;192(4):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basagana X, Esnaola M, Rivas I, et al. Neurodevelopmental Deceleration by Urban Fine Particles from Different Emission Sources: A Longitudinal Observational Study. Environ Health Perspect. 2016;124(10):1630–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu HH, Chiu YH, Coull BA, et al. Prenatal Particulate Air Pollution and Asthma Onset in Urban Children. Identifying Sensitive Windows and Sex Differences. Am J Respir Crit Care Med. 2015;192(9):1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bove H, Bongaerts E, Slenders E, et al. Ambient black carbon particles reach the fetal side of human placenta. Nat Commun. 2019;10(1):3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Traboulsi H, Guerrina N, Iu M, Maysinger D, Ariya P, Baglole CJ. Inhaled Pollutants: The Molecular Scene behind Respiratory and Systemic Diseases Associated with Ultrafine Particulate Matter. Int J Mol Sci. 2017;18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L, Hou XY, Wei Y, Thai P, Chai F. Biomarkers of the health outcomes associated with ambient particulate matter exposure. Sci Total Environ. 2017;579:1446–1459. [DOI] [PubMed] [Google Scholar]
- 9.Irie M, Asami S, Nagata S, Miyata M, Kasai H. Classical conditioning of oxidative DNA damage in rats. Neuroscience letters. 2000;288(1):13–16. [DOI] [PubMed] [Google Scholar]
- 10.Dai Y, Huo X, Cheng Z, Faas MM, Xu X. Early-life exposure to widespread environmental toxicants and maternal-fetal health risk: A focus on metabolomic biomarkers. Sci Total Environ. 2020;739:139626. [DOI] [PubMed] [Google Scholar]
- 11.Chen H, Oliver BG, Pant A, et al. Particulate Matter, an Intrauterine Toxin Affecting Foetal Development and Beyond. Antioxidants (Basel). 2021;10(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sondheimer N, Glatz CE, Tirone JE, Deardorff MA, Krieger AM, Hakonarson H. Neutral mitochondrial heteroplasmy and the influence of aging. Hum Mol Genet. 2011;20(8):1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopinski PK, Janssen KA, Schaefer PM, et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc Natl Acad Sci U S A. 2019;116(32):16028–16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16(9):530–542. [DOI] [PubMed] [Google Scholar]
- 15.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clemente DB, Casas M, Vilahur N, et al. Prenatal Ambient Air Pollution, Placental Mitochondrial DNA Content, and Birth Weight in the INMA (Spain) and ENVIRONAGE (Belgium) Birth Cohorts. Environ Health Perspect. 2016;124(5):659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen BG, Munters E, Pieters N, et al. Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect. 2012;120(9):1346–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosa MJ, Just AC, Guerra MS, et al. Identifying sensitive windows for prenatal particulate air pollution exposure and mitochondrial DNA content in cord blood. Environ Int. 2017;98:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colicino E, Power MC, Cox DG, et al. Mitochondrial haplogroups modify the effect of black carbon on age-related cognitive impairment. Environmental health : a global access science source. 2014;13(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson A, Chiu YM, Hsu HL, Wright RO, Wright RJ, Coull BA. Bayesian distributed lag interaction models to identify perinatal windows of vulnerability in children’s health. Biostatistics. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kesmodel US, Kjaersgaard MI, Denny CH, et al. The association of pre-pregnancy alcohol drinking with child neuropsychological functioning. BJOG. 2015;122(13):1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray J, Burgess S, Zuccolo L, Hickman M, Gray R, Lewis SJ. Moderate alcohol drinking in pregnancy increases risk for children’s persistent conduct problems: causal effects in a Mendelian randomisation study. J Child Psychol Psychiatry. 2016;57(5):575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandoli G, Coles CD, Kable JA, et al. Patterns of Prenatal Alcohol Use That Predict Infant Growth and Development. Pediatrics. 2019;143(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunst KJ, Tignor N, Just A, et al. Cumulative lifetime maternal stress and epigenome-wide placental DNA methylation in the PRISM cohort. Epigenetics. 2018;13(6):665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunst KJ, Zhang L, Zhang X, Baccarelli AA, Bloomquist T, Wright RJ. Associations Between Maternal Lifetime Stress and Placental Mitochondrial DNA Mutations in an Urban Multiethnic Cohort. Biol Psychiatry. 2021;89(6):570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingman M, Kaessmann H, Paabo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408(6813):708–713. [DOI] [PubMed] [Google Scholar]
- 27.Picard Tools. GitHub Repository; 2018. http://broadinstitute.github.io/picard/.
- 28.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38(6):1767–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaidi AA, Wilton PR, Su MS, et al. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc Natl Acad Sci U S A. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kido T, Sikora-Wohlfeld W, Kawashima M, et al. Are minor alleles more likely to be risk alleles? BMC Med Genomics. 2018;11(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang P, Samuels DC, Wang J, Zhao S, Shyr Y, Guo Y. Mitochondria single nucleotide variation across six blood cell types. Mitochondrion. 2016;28:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Just AC, Arfer KB, Rush J, et al. Advancing methodologies for applying machine learning and evaluating spatiotemporal models of fine particulate matter (PM2.5) using satellite data over large regions. Atmos Environ (1994). 2020;239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hajat A, Hsia C, O’Neill MS. Socioeconomic Disparities and Air Pollution Exposure: a Global Review. Curr Environ Health Rep. 2015;2(4):440–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunst KJ, Sanchez-Guerra M, Chiu YM, et al. Prenatal particulate matter exposure and mitochondrial dysfunction at the maternal-fetal interface: Effect modification by maternal lifetime trauma and child sex. Environ Int. 2017;112:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Huang K, Sun Y, et al. Placenta response of inflammation and oxidative stress in low-risk term childbirth: the implication of delivery mode. BMC Pregnancy Childbirth. 2017;17(1):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huuskonen P, Amezaga MR, Bellingham M, et al. The human placental proteome is affected by maternal smoking. Reprod Toxicol. 2016;63:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segura MT, Demmelmair H, Krauss-Etschmann S, et al. Maternal BMI and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta. 2017;57:144–151. [DOI] [PubMed] [Google Scholar]
- 39.Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative Stress in Preeclampsia and Placental Diseases. Int J Mol Sci. 2018;19(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holland O, Dekker Nitert M, Gallo LA, Vejzovic M, Fisher JJ, Perkins AV. Review: Placental mitochondrial function and structure in gestational disorders. Placenta. 2017;54:2–9. [DOI] [PubMed] [Google Scholar]
- 41.Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013;13:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kloog I, Nordio F, Coull BA, Schwartz J. Predicting spatiotemporal mean air temperature using MODIS satellite surface temperature measurements across the Northeastern USA. Remote Sensing of the Environment. 2014;150:132–139. [Google Scholar]
- 43.Mitchell SL, Goodloe R, Brown-Gentry K, Pendergrass SA, Murdock DG, Crawford DC. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Hum Genet. 2014;133(7):861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson A, Chiu YM, Hsu HL, Wright RO, Wright RJ, Coull BA. Bayesian distributed lag interaction models to identify perinatal windows of vulnerability in children’s health. Biostatistics. 2017;18(3):537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate imputation by chained equations in R. Journal of Statistical Software. 2011;45(3):1–67. [Google Scholar]
- 46.Lodovici M, Bigagli E. Oxidative stress and air pollution exposure. J Toxicol. 2011;2011:487074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gualtieri M, Ovrevik J, Mollerup S, et al. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA, mitochondria, AhR binding and spindle organization. Mutat Res. 2011;713(1–2):18–31. [DOI] [PubMed] [Google Scholar]
- 48.Delgado-Buenrostro NL, Freyre-Fonseca V, Cuellar CM, et al. Decrease in respiratory function and electron transport chain induced by airborne particulate matter (PM10) exposure in lung mitochondria. Toxicol Pathol. 2013;41(4):628–638. [DOI] [PubMed] [Google Scholar]
- 49.Anton L, DeVine A, Polyak E, et al. HIF-1alpha Stabilization Increases miR-210 Eliciting First Trimester Extravillous Trophoblast Mitochondrial Dysfunction. Front Physiol. 2019;10:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisher JJ, McKeating DR, Cuffe JS, Bianco-Miotto T, Holland OJ, Perkins AV. Proteomic Analysis of Placental Mitochondria Following Trophoblast Differentiation. Front Physiol. 2019;10:1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Hart JE, Liu Q, Wu S, Nan H, Laden F. Association of particulate matter air pollution with leukocyte mitochondrial DNA copy number. Environ Int. 2020;141:105761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu C, Sheng X, Li Y, et al. Effects of prenatal exposure to particulate air pollution on newborn mitochondrial DNA copy number. Chemosphere. 2020;253:126592. [DOI] [PubMed] [Google Scholar]
- 53.Brunst KJ, Sanchez-Guerra M, Chiu YM, et al. Prenatal particulate matter exposure and mitochondrial dysfunction at the maternal-fetal interface: Effect modification by maternal lifetime trauma and child sex. Environ Int. 2018;112:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breton CV, Song AY, Xiao J, et al. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion. 2019;46:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rebolledo-Jaramillo B, Su MS, Stoler N, et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 2014;111(43):15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fowden AL, Forhead AJ, White KL, Taylor PM. Equine uteroplacental metabolism at mid- and late gestation. Exp Physiol. 2000;85(5):539–545. [PubMed] [Google Scholar]
- 57.Jones ML, Mark PJ, Lewis JL, Mori TA, Keelan JA, Waddell BJ. Antioxidant defenses in the rat placenta in late gestation: increased labyrinthine expression of superoxide dismutases, glutathione peroxidase 3, and uncoupling protein 2. Biol Reprod. 2010;83(2):254–260. [DOI] [PubMed] [Google Scholar]
- 58.Holland OJ, Hickey AJR, Alvsaker A, et al. Changes in mitochondrial respiration in the human placenta over gestation. Placenta. 2017;57:102–112. [DOI] [PubMed] [Google Scholar]
- 59.Xu Z, Jin X, Cai W, et al. Proteomics Analysis Reveals Abnormal Electron Transport and Excessive Oxidative Stress Cause Mitochondrial Dysfunction in Placental Tissues of Early-Onset Preeclampsia. Proteomics Clin Appl. 2018;12(5):e1700165. [DOI] [PubMed] [Google Scholar]
- 60.Rappazzo KM, Nichols JL, Rice RB, Luben TJ. Ozone exposure during early pregnancy and preterm birth: A systematic review and meta-analysis. Environ Res. 2021;198:111317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullarky E, Cantley LC. Diverting Glycolysis to Combat Oxidative Stress. In: Nakao K, Minato N, Uemoto S, eds. Innovative Medicine: Basic Research and Development. Tokyo: 2015:3–23. [PubMed] [Google Scholar]
- 62.Kraja AT, Liu C, Fetterman JL, et al. Associations of Mitochondrial and Nuclear Mitochondrial Variants and Genes with Seven Metabolic Traits. Am J Hum Genet. 2019;104(1):112–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cruz ACP, Ferrasa A, Muotri AR, Herai RH. Frequency and association of mitochondrial genetic variants with neurological disorders. Mitochondrion. 2019;46:345–360. [DOI] [PubMed] [Google Scholar]
- 64.Sequeira A, Rollins B, Magnan C, et al. Mitochondrial mutations in subjects with psychiatric disorders. PLoS One. 2015;10(5):e0127280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cowell W, Brunst K, Colicino E, et al. Placental mitochondrial DNA mutational load and perinatal outcomes: Findings from a multi-ethnic pregnancy cohort. Mitochondrion. 2021;59:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tessum CW, Paolella DA, Chambliss SE, Apte JS, Hill JD, Marshall JD. PM2.5 polluters disproportionately and systemically affect people of color in the United States. Sci Adv. 2021;7(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Erqou S, Clougherty JE, Olafiranye O, et al. Particulate Matter Air Pollution and Racial Differences in Cardiovascular Disease Risk. Arterioscler Thromb Vasc Biol. 2018;38(4):935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choudhury AR, Singh KK. Mitochondrial determinants of cancer health disparities. Semin Cancer Biol. 2017;47:125–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeLany JP, Dube JJ, Standley RA, et al. Racial differences in peripheral insulin sensitivity and mitochondrial capacity in the absence of obesity. J Clin Endocrinol Metab. 2014;99(11):4307–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Assibey-Mensah V, Parks WT, Gernand AD, Catov JM. Race and risk of maternal vascular malperfusion lesions in the placenta. Placenta. 2018;69:102–108. [DOI] [PubMed] [Google Scholar]
- 71.Jones CW, Gambala C, Esteves KC, et al. Differences in placental telomere length suggest a link between racial disparities in birth outcomes and cellular aging. Am J Obstet Gynecol. 2017;216(3):294 e291–294 e298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menon R, Merialdi M, Lombardi SJ, Fortunato SJ. Differences in the placental membrane cytokine response: a possible explanation for the racial disparity in preterm birth. Am J Reprod Immunol. 2006;56(2):112–118. [DOI] [PubMed] [Google Scholar]
- 73.Wittkopp S, Staimer N, Tjoa T, et al. Mitochondrial genetic background modifies the relationship between traffic-related air pollution exposure and systemic biomarkers of inflammation. PLoS One. 2013;8(5):e64444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Breton CV, Marsit CJ, Faustman E, et al. Small-Magnitude Effect Sizes in Epigenetic End Points are Important in Children’s Environmental Health Studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ Health Perspect. 2017;125(4):511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma J, Purcell H, Showalter L, Aagaard KM. Mitochondrial DNA sequence variation is largely conserved at birth with rare de novo mutations in neonates. Am J Obstet Gynecol. 2015;212(4):530 e531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coorens THH, Oliver TRW, Sanghvi R, et al. Inherent mosaicism and extensive mutation of human placentas. Nature. 2021;592(7852):80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marchington DR, Scott-Brown M, Barlow DH, Poulton J. Mosaicism for mitochondrial DNA polymorphic variants in placenta has implications for the feasibility of prenatal diagnosis in mtDNA diseases. Eur J Hum Genet. 2006;14(7):816–823. [DOI] [PubMed] [Google Scholar]
- 78.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5(11):a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Greaves LC, Nooteboom M, Elson JL, et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 2014;10(9):e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1(8639):642–645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.