

CONSPECTUS.
Biomolecules such as peptides, proteins, and nucleic acids generally cannot cross a cell membrane by passive diffusion. Nevertheless, cell-penetrating peptides (CPPs), bacterial protein toxins, certain mammalian proteins, viruses, and many synthetic drug delivery vehicles have been shown to enter the cytosol of mammalian cells with varying efficiencies. They generally enter the cell by one or more of the endocytic mechanisms and are initially localized inside the endosomes. But how they cross the endosomal membrane to reach the cytosol (i.e., endosomal escape) has been a mystery for decades, and this knowledge gap has been a major bottleneck for the development of efficient drug delivery systems. In addition, many bacterial and mammalian proteins are transported across the plasma membrane in their native states into the periplasmic/extracellular space through the twin-arginine translocation (TAT) and unconventional protein secretion (UPS) systems, respectively. Again, the mechanisms underpinning these protein export systems remain unclear.
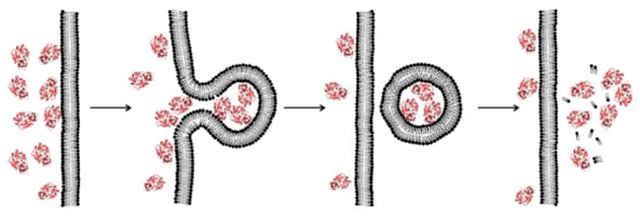
In this Account, I introduce a previously unrecognized, fundamental membrane translocation mechanism which we have termed as the “vesicle budding-and-collapse (VBC) mechanism”. Through VBC, biomolecules of diverse sizes and physicochemical properties autonomously translocate across cell membranes topologically (i.e., from one side to the other side of membrane), but not physically (i.e., without going through the membrane). We have demonstrated that CPPs and bacterial protein toxins escape the endosome by the VBC mechanism in giant unilamellar vesicles as well as live mammalian cells. This advance resulted from studies in which we labeled the biomolecules with a pH-sensitive, red-colored dye (pHAb) and phosphatidylserine with a pH-insensitive green dye (TopFluor) and monitored the intracellular trafficking of the biomolecules in real time by confocal microscopy. In addition, by enlarging the endosomes with a kinase inhibitor, we were able to visualize the structural changes of the endosomes (i.e., endosomal escape intermediates) as they went through the VBC process. I postulate that bacterial/viral/mammalian proteins, non-enveloped viruses, as well as synthetic drug delivery vehicles (e.g., polyplexes, lipoplexes, and lipid nanoparticles) may also escape the endosome by inducing VBC. Furthermore, I propose that VBC may be the mechanism that drives the bacterial TAT and mammalian UPS systems. Our findings fill a long-standing gap in cell biology and provide guiding principles for designing more efficient drug delivery vehicles.
Graphical Abstract
INTRODUCTION
The plasma membrane of a cell is a highly effective barrier that keeps cellular contents inside and foreign substances out. While small organic molecules of molecular weight <500 and a proper balance in hydrophilicity and hydrophobicity attributes can traverse the cell membrane by passive diffusion, large biomolecules (e.g., peptides, proteins, and nucleic acids) generally cannot. Nevertheless, some biomolecules are capable of autonomously entering the cytosol of mammalian cells. Examples include cell-penetrating peptides (CPPs),4 non-peptidic cell-penetrating molecules (CPMs),5 bacterial protein toxins,6 certain mammalian proteins,7 viruses,8 and synthetic drug delivery systems9,10 (Figure 1). Most of these entities enter the mammalian cell by endocytic mechanisms and are initially localized inside the endosomes. To reach the cytosol, some of these entities subsequently translocate across the endosomal membrane, a process termed “endosomal escape”.11 Other entities have been shown to directly translocate across the plasma membrane12,13 or travel further into the endoplasmic reticulum (ER) and cross the ER membrane into the cytosol by retrograde transport.14 Folded proteins also translocate across the plasma membrane in the reverse direction, i.e., from the cytosol to the periplasmic/extracellular space. The latter process has been described as the unconventional protein secretion (UPS) system in eukaryotic cells15 and the twin-arginine translocation (TAT) system in bacteria and organelles.16 In all of these scenarios, the biomolecules/entities must travel across a lipid bilayer. How biomolecules autonomously translocate across a lipid bilayer has been a long-standing mystery in several fields and the lack of mechanistic understanding has greatly hampered the development of cell-permeable biologics as next-generation therapeutics.
Figure 1.
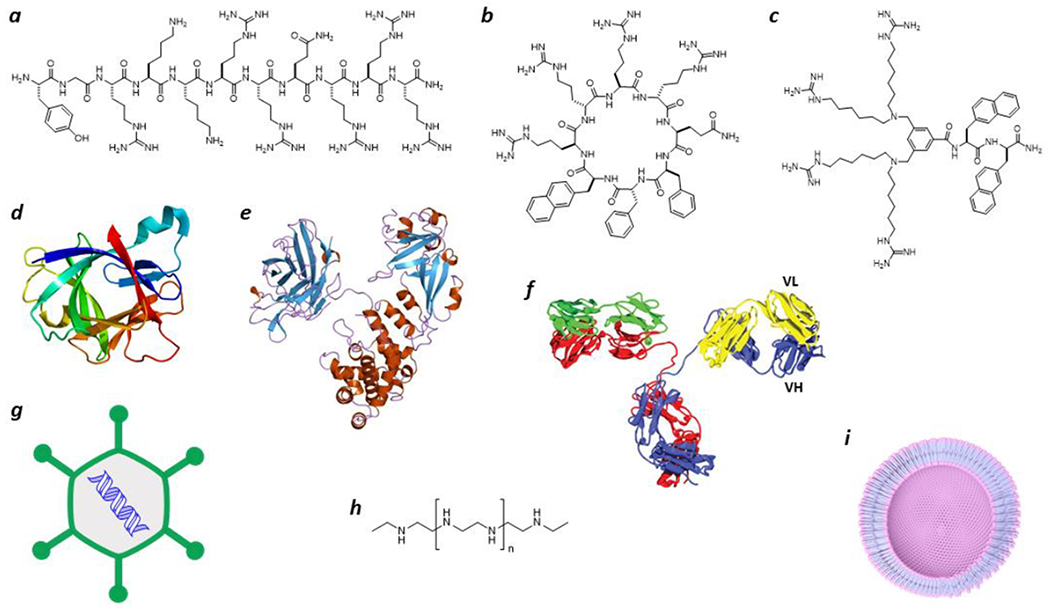
Representative biomolecules/entities that are capable of autonomously translocating across the plasma or endosomal membrane of a cell. (a) Linear CPP Tat;4 (b) cyclic CPP12;1 (c) non-peptidic CPM3;5 (d) IL-1β 55 (PDB: 31BI); (e) diphtheria toxin39 (PDB: 1xdt); (f) a cell-permeable antibody;52,53 (g) adenovirus;8 (h) polyethyleneimine;9 and (i) a lipid nanoparticle.10
Recent studies by others and us have begun to unravel this mystery. We discovered that CPPs and CPMs escape the endosome by a novel vesicle budding-and-collapse (VBC) mechanism (Figure 2).1,2 We subsequently demonstrated that two different bacterial protein toxins also exit the endosome by the VBC mechanism.3 The latter finding prompted me to conduct a literature survey on other membrane-permeable modalities. This exercise has led to the hypothesis that the VBC mechanism may be a previously unrecognized, fundamental mechanism by which many (and potentially all) of the above biomolecules translocate autonomously across the lipid bilayer. In this Account, I provide a summary of existing evidence for the VBC mechanism and how this concept reconciles many previously enigmatic observations. Protein transport via the well-characterized leader peptide-mediated secretory (Sec) pathway will not be covered here; interested readers are referred to an excellent review.17
Figure 2.
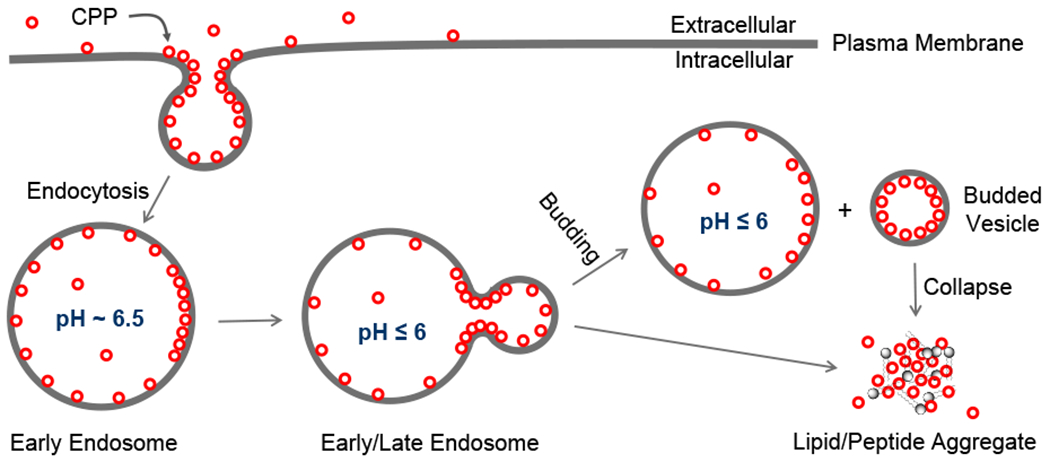
Endocytic uptake and endosomal escape of cyclic CPPs by the VBC mechanism.
ENDOSOMAL ESCAPE BY VESICLE BUDDING AND COLLAPSE
Several endosomal escape mechanisms have been proposed, including osmotic lysis caused by the “proton sponge effect”,18 membrane fusion,19,20 pore formation,21,22 local membrane disruption,23 and VBC.1–3 To my knowledge, the VBC mechanism is the only one that has been experimentally validated and is applicable to all of the modalities described above, whereas the other hypotheses are usually invoked for a specific modality and have been challenged by various experimental observations.11 The mechanism of cellular entry and endosomal escape for cyclic CPPs is illustrated in Figure 2. Cyclic CPPs bind directly to the plasma membrane phospholipids (and potentially other membrane components) and are brought into the early endosome by endocytosis. As the early endosome matures into late endosome with concomitant endosomal acidification, the CPPs bind to the endosomal membrane with increased affinity.1 Because the arginine side chains of the CPPs are not expected to undergo further protonation during endosomal acidification (pH 6.5–4.5), I posit that the protonation events occur on the phospholipids (e.g., the phosphate head group). The guanidinium group of Arg is capable of simultaneously forming bidentate hydrogen bonds with two adjacent phosphates (Figure 3a). This unique capability, together with the presence of multiple Arg residues in a typical CPP (e.g., four in CPP12), enables CPPs to “crosslink” phospholipids into CPP-enriched lipid domains. Partial protonation of the phosphates would facilitate phospholipid clustering by reducing electrostatic repulsion. Formation of a lipid domain generates a line tension between the domain and its surrounding membrane, which drives the lipid domain to bud out as a vesicle.24 During the vesicle budding process, the budding neck (the “transition state”) features negative Gaussian curvature (i.e., simultaneous positive and negative curvatures in orthogonal directions) and has high potential energy relative to the “ground states” present either before or after the budding event (Figure 3b).25 To promote the budding event, CPPs should bind selectively to the budding neck and reduce the energy barrier. CPPs and CPMs of high endosomal escape efficiencies are usually amphipathic and conformationally constrained.25 Conformational rigidity increases the binding affinity of CPPs/CPMs for the endosomal membrane, while amphipathicity is conducive to generating negative Gaussian curvature at the budding neck. Insertion of hydrophobic groups in between phospholipid molecules generates positive membrane curvature (Figure 3c), whereas Arg residues induce negative curvature by hydrogen-bonding to and bringing together the phosphate head groups of phospholipids (Figure 3a). After or during budding off the endosomal membrane, the small vesicle spontaneously and rapidly disintegrates into a peptide/lipid aggregate, which gradually dissolves into the cytosolic milieu. The driving force for vesicle collapse is not yet clear, although the small vesicle (~100 nm in diameter) is likely intrinsically unstable due to high membrane curvature and/or the presence of high concentrations of CPPs. A unique feature of the VBC mechanism is that the biomolecule crosses a membrane topologically, but without physically going through the membrane, and the endosome remains intact before, during, and after each endosomal escape event. In contrast, all other membrane transport mechanisms involve an entity physically traversing a cell membrane and therefore require partial or total disruption of the membrane.11 For different biomolecules, the details of cellular entry may differ to some extent, e.g., the nature of receptor binding at the cell surface.
Figure 3.
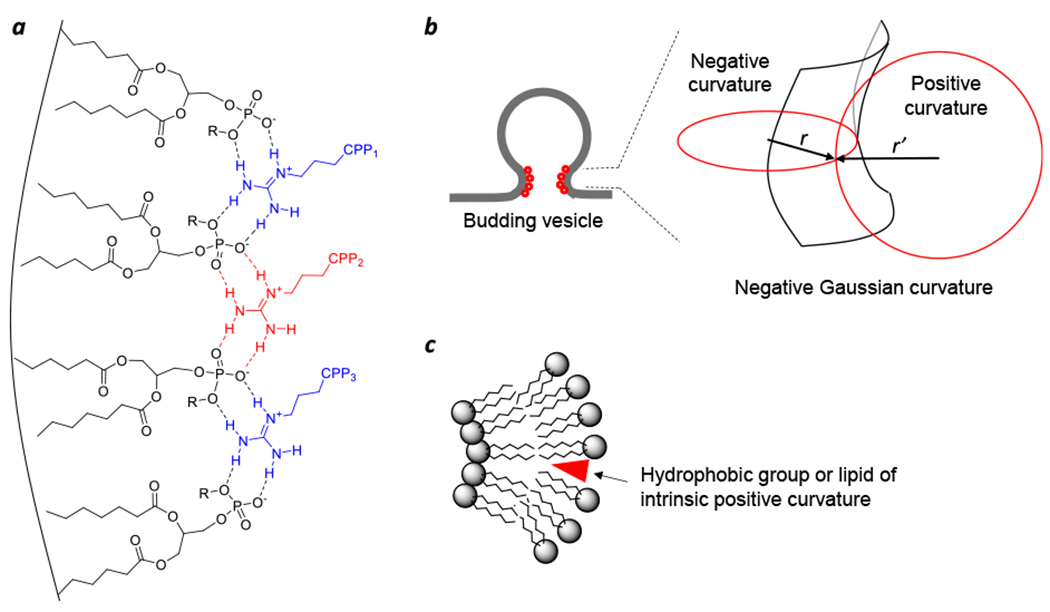
Effect of biomolecules on the membrane structure. (a) Scheme showing a cluster of four phospholipid molecules and three arginine side chains (from three different CPPs) held together through bidentate hydrogen bonds and the generation of negative membrane curvature by simultaneous coordination of phosphate head groups. (b) Schematic representation of the negative Gaussian curvature at a budding neck. (c) Generation of positive membrane curvature by the insertion of a hydrophobic moiety (e.g., the naphthalene side chain of CPP12, a myristoyl group, or LPC) into the lipid bilayer. Adapted with permission from ref. 25. Copyright 2019 American Chemical Society.
The VBC mechanism suggests that endosomal escape would require a minimum number of biomolecules inside an endosome and the efficiency of endosomal escape would be concentration dependent. Indeed, it was recently found that Tat, a prototypical linear CPP, has endosomal escape efficiencies of 0.08%, 0.38%, and 0.66% at 0.2, 10, and 20 μM concentrations, respectively.26 This finding explains why the cytosolic entry efficiency of CPPs is highly sensitive to and increases nonlinearly with the CPP concentration, as endocytic uptake is also CPP concentration dependent. I estimate the minimum number of biomolecules required to induce a VBC event to be on the order of 80-360 molecules per endosome, corresponding to an intraluminal concentration of 2-9 μM (by assuming that an average endosome has a diameter of 0.5 μm), although this number may vary somewhat depending on the nature of the biomolecule. This estimate is based on an earlier observation that each endosomal escape event releases a bolus of ~80 diphtheria toxin (DT) molecules into the cytosol27 and the fact that each adenovirus has 360 copies of capsid protein VI, which is responsible for the endosomal release of the virus (see below).28
CELL-PENETRATING PEPTIDES
It is generally accepted that at low concentrations, CPPs enter the cell primarily through one or more energy-dependent endocytosis mechanisms followed by endosomal escape, whereas at high concentrations some of the CPPs can also directly translocate across the plasma membrane in an energy-independent fashion.29 Early clues for VBC came from our in vitro studies of cyclic CPP12 and giant unilamellar vesicles (GUVs) that mimic the endosomal membrane composition.1 Dye-labeled CPP12 bound to the outer leaflet of GUVs and was initially distributed uniformly over the GUV membrane. Over time, the CPP12 molecules clustered into intensely fluorescent areas, presumably lipid domains, which subsequently budded out (or less frequently into the GUV lumen) as small vesicles (Figure 4a). Prior to budding, CPP12 was concentrated at the budding neck (Figure 4b). The budded vesicles then collapsed into irregularly shaped and intensely fluorescent aggregates, either concurrent with the budding event or shortly after budding was complete. To observe the VBC events in live cells, we labeled CPP12 with a pH-sensitive dye (pHAb) which fluoresces inside the acidic environment of endosomes/lysosomes but not in the neutral cytosol or extracellular space.2 HeLa (human cervical cancer) cells were treated with CPP12pHAb (red color) and a membrane marker, phosphatidylserine labeled with TopFluor (PSTopFluor), and imaged by live-cell confocal microscopy in real time. Endosomal escape began with the budding of a small vesicle from the endosomal membrane; at this stage, the budded vesicle as well as the remaining endosome emitted both red (CPP12pHAb) and green fluorescence (PSTopFluor) (Figure 4c). Subsequent collapse of the small vesicle (and exposure of the remnant to the cytosolic pH) was evident from the sudden disappearance of the red (CPP12pHAb) but not the green fluorescence (PSTopFluor), while the intact endosome retained both red and green fluorescence. Each endosomal escape event occurred rapidly, typically spanning <60 s. We next pretreated HeLa cells with a kinase inhibitor (YM201636) to enlarge the endosomes (from ~0.5 to ~2 pm in average diameter), prior to the addition of tetramethylrhodamine (TMR)-labeled CPP12 (CPP12TMR) and an endosomal marker, AlexaFluor488-labeled dextran (DextranAlexa).2 This allowed us to visualize the structural changes of endosomes as they underwent the VBC process by time-lapse, live-cell confocal microscopy (Figure 4d). Remarkably, the VBC intermediates captured in live cells are very similar to those observed in the earlier GUV studies1 (Figure 4a,b). Furthermore, a synchrotron small-angle X-ray scattering study showed that cyclic CPPs are highly effective in generating negative Gaussian curvatures on artificial membranes.30 To assess the generality of the VBC mechanism, we also monitored the endosomal escape of Tat (which is a prototypical linear CPP with a low endosomal escape efficiency of ≤0.66%26) and CPM3 (which escapes the endosome nearly quantitatively5) in HeLa cells.2 CPM3 induced robust VBC events from endosomes and generated similar VBC intermediates to CPP12, whereas Tat-mediated VBC events were much less frequent (and difficult to observe). These observations suggest that the endosomal escape efficiency is primarily determined by how effectively (frequently) a biomolecule induces VBC at the endosomal membrane.
Figure 4.
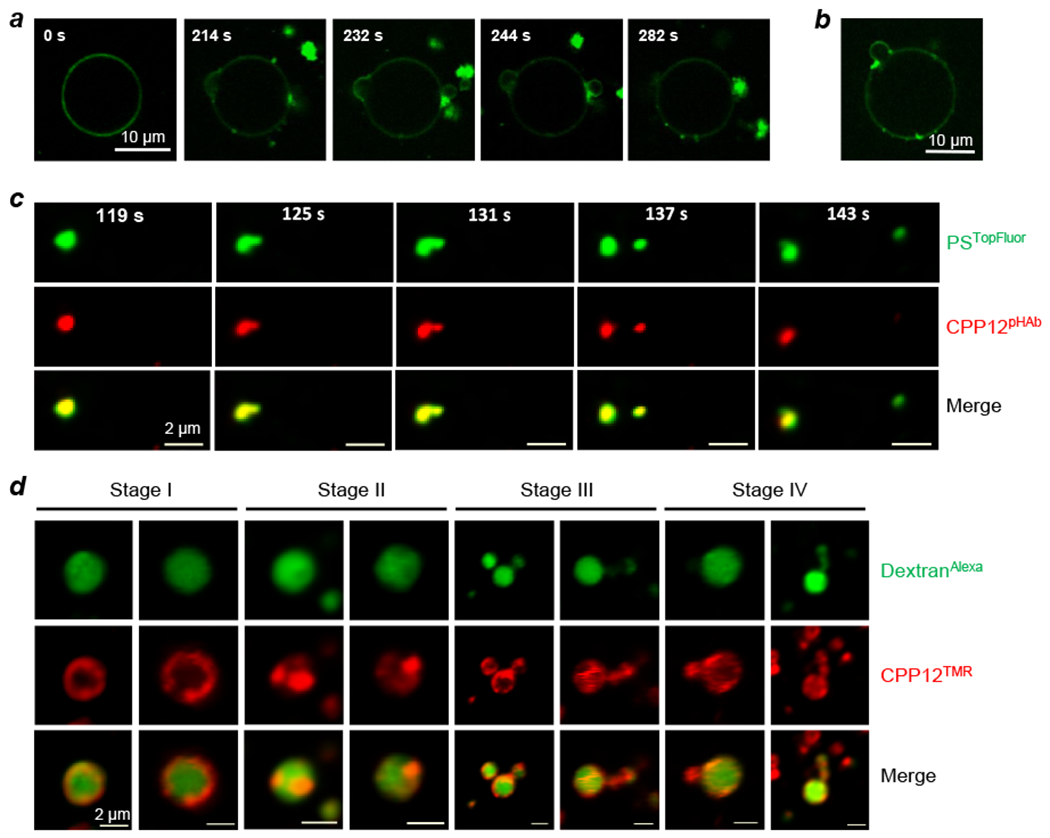
CPP12 induces vesicle budding and collapse in vitro and in live cells. (a) Time-lapse confocal images of budding-and-collapse events upon suspension of GUVs in a buffer (pH 5) containing 20 μM FITC-labeled CPP12. (b) A snapshot of a GUV from (a) showing the concentration of CPP12FITC at the budding neck. (c) Time-lapse confocal microscopy images of a single endosome inside a HeLa cell at 119–143 s after initiation of imaging. Cells were incubated with PSTopFluor (1.5 μM, green channel) for 15 min on ice, washed with DPBS, treated with CPP12pHAb (2.5 μM, red channel) for 10 min, and imaged every 6 s. The endosome split into two vesicles at 137 s and at 143 s, the smaller vesicle collapsed while the larger one remained intact. Scale bars = 2 μm. (d) CPP12-induced vesicle budding and collapse from enlarged endosomes. HeLa cells were pretreated with kinase inhibitor YM201636 (800 nM) for 2 h, and then CPP12TMR (2 μM, red channel) and DextranAlexa (50 μg/mL, green channel) were added. After incubation for 30–40 min, the cells were washed and imaged by live-cell confocal microscopy. Images of different endosomes at different stages (I-IV) of vesicle budding and collapse are shown. Scale bars = 2 μm. Reproduced with permission from ref. 1 and 2. Copyright 2016, 2020 American Chemical Society.
A survey of the CPP literature suggests that other CPPs may also induce VBC at the endosomal and/or plasma membrane. For example, in YM201636-treated Saos-2 cells, a cell-permeable miniature protein (ZF5.3) caused the endosome (and lysosome) to form membrane structures that are highly similar to the VBC intermediates induced by CPP 12 and CPM3.31 Linear arginine-rich CPPs such as penetratin, nonaarginine (R9), and RW9 have demonstrated the capacity to enter artificial vesicles such as GUVs and plasma membrane spheres (PMSs) autonomously by inducing membrane invagination and the formation of amorphous peptide/lipid aggregates inside the GUVs and PMSs.32–34 Synchrotron small-angle X-ray scattering studies confirmed that linear CPPs are also capable of generating negative Gaussian curvatures on artificial membranes.35 Previously perplexing observations can now be rationalized by and in turn provide additional support for the VBC mechanism, including: (1) CPPs are capable of delivering cargos of different sizes (from small molecules to large proteins) and physicochemical properties which are either covalently attached to or noncovalently associated with the CPPs;4 (2) CPPs promote the endosomal release of unassociated macromolecular cargos (e.g., cyclodextrin and antibodies);36,37 (3) fusogenic lipids and peptides improve the endosomal release of CPPs;38 and (4) the enhanced cytosolic entry efficiency of cyclic and other conformationally constrained CPPs and CPMs.25 However, it remains to be determined whether all CPPs/CPMs exit the endosome or translocate across the plasma membrane by the VBC mechanism.
BACTERIAL TOXINS
Bacterial protein toxins are diverse in their structures and cell entry mechanisms.6 Some toxins translocate directly across the plasma membrane to reach the cytosol.13 Others bind to cell-surface receptors, enter the cell by endocytosis and endosomal escape.39 Still other toxins are trafficked to the ER where they undergo retrograde translocation into the cytosol.14 Regardless of the specific cell entry pathway, however, the toxin must topologically cross a lipid bilayer at some point before reaching the cytosol. The AB class of bacterial toxins, as exemplified by DT, have been well characterized. They typically consist of two functional units: an enzymatic moiety (A, which is the actual toxin) and a nonenzymatic moiety (B, which is the delivery vehicle) that mediates receptor binding (R-domain) and membrane translocation (T-domain).39 Cellular entry starts with the R-domain recognizing a cell surface receptor on the host cell, leading to the endocytosis of the receptor-toxin complex. Acidification of the endosomal compartment dissociates the toxin from the receptor and induces a conformational change of the T-domain, which then inserts into the endosomal membrane to form an ion-conducting pore/channel. It is widely accepted that the A moiety unfolds and threads through the narrow channel formed by the T-domain to reach the cytosol.40 However, the pore/channel hypothesis has been challenged by many experimental observations over the years. For example, DT has been used to deliver hyperstable cargo proteins41 as well as noncovalently associated nucleic acids42,43 into the cytosol, suggesting that DT is capable of translocating cargos in their folded states. Mutations have been found to eliminate the ion channel activity of DT but have little effect on the translocation of the A moiety,44 while other mutations prevent translocation but not the channel activity.45 Lastly, the endosomal escape of DT exhibits “quantal” kinetics, i.e., each endosomal escape event releases ~80 DT molecules into the cytosol simultaneously, irrespective of the extracellular DT concentration,27 whereas DT release through a pore would be sequential (i.e., one DT molecule at a time).
We recently showed that DT escapes the endosome by the VBC mechanism.3 We labeled DT with pHAb or tetramethylrhodamine (TMR) and monitored the intracellular trafficking of DT by live-cell confocal microscopy. DT induced robust VBC in HeLa cells (Figure 5a,b) and generated endosomal membrane structures that were indistinguishable from those of CPPs/CPMs (Figure 5c). For example, DT™ was localized on the endosomal membrane, evenly distributed over the membrane initially but later clustered into DT-enriched lipid domains, which subsequently budded out as small vesicles and collapsed (Figure 5c). Interestingly, NleC, a metalloprotease produced by enterohemorrhagic and enteropathogenic Escherichia coli, also escapes the endosome by VBC. Unlike bacterial toxins of the AB class, NleC consists of a single catalytic domain of 330 residues and enters the host cell in its native structure.46 NleC contains two polybasic sequences in its structure,47,48 which are likely responsible for its cellular entry. However, each polybasic sequence in isolation failed to function as a CPP, consistent with the requirement of an intact 3D structure for cellular entry. This and other results (see below) demonstrate that neither acid-induced conformational change nor membrane insertion is required for protein translocation across the endosomal membrane. It thus raises the question of what function(s) membrane insertion of the T-domain and the ion-conducting channel might play during endosomal escape. I suggest that membrane insertion of the T-domain increases the binding affinity of the toxin for the endosomal membrane, ensuring that most of the 80-360 toxin molecules inside an endosome are bound to the endosomal membrane to promote VBC. This feature is likely critical for toxins which exert their pathophysiological effects at very low concentrations (e.g., pM). The ion-conducting channel is likely an offshoot of the T-domain insertion and may not have any biological function.
Figure 5.
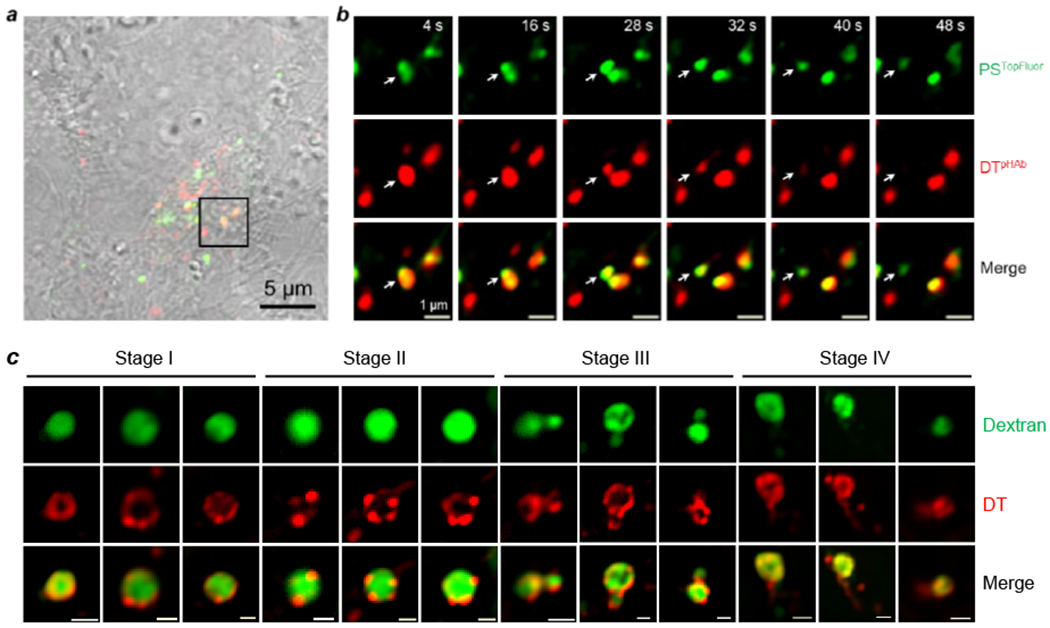
DT-induced vesicle budding and collapse from the endosomal membrane in live HeLa cells. (a) Merged image of several HeLa cells (DIC, green and red channels) at 23 min after the addition of unlabeled DT. (b) Time-lapse confocal microscopic images of the boxed area in (a) during the period of 4 - 48 s after the initiation of imaging. Cells were incubated with 1.5 μM PSTopFluor (green channel) for 10 min, washed with DPBS, and treated with 5 μM DTpHAb (red channel) for 30 min. Cells were washed with DMEM to remove DTpHAb, treated with 5 μM unlabeled DT, and imaged every 4 s. A budded vesicle emerged from an endosome (marked by a white arrow) at 28 s, completely separated from the endosome at 32 s, and collapsed at 40 s. Scale bars, 1 and 5 μm. (c) Confocal microscopic images of enlarged endosomes in HeLa cells at different stages of vesicle budding and collapse. Cells were pretreated with kinase inhibitor YM201636 (800 nM) for 2 h and DTTMR (5 μM) and DextranAlexa (50 μg/mL) were added. After incubation for 30 min, the cells were washed and imaged by live-cell confocal microscopy. Scale bars, 1 μm. Reproduced with permission from ref. 3. Copyright 2021 American Chemical Society.
How do 80-360 toxin molecules accumulate into a single endosome to reach a concentration of 2-9 μM, when the extracellular toxin concentration is in the pM range? Receptor-mediated endocytosis results in significant concentration of the toxin from the extracellular environment into the endosomes. Further concentration may be accomplished by endosomal fusion.49 A typical mammalian cell has hundreds of endosomes, which are interconnected through constant vesicle fusion and fission. Although individual endosomes may not initially contain the minimum number of molecules for VBC, lateral fusion of the endosomes coupled with clustering of toxins into lipid domains would gradually concentrate the toxin molecules into fewer, larger endosomes, which culminate to reach the “quantum” for VBC.27 This scenario offers a potential explanation for the previously observed concentration-dependent temporal lag between endosomal acidification and DT release into the cytosol (i.e., longer lag at lower DT concentration).18,50 It may also explain why the HOPS complex, which functions in vesicle tethering and fusion, is critical for the endosomal escape of CPPs in vivo,31 despite that CPPs alone induce robust VBC from GUVs in the absence of any cellular protein (albeit at relatively high concentrations of 5-20 μM).1
OTHER MEMBRANE-PERMEABLE PROTEINS
In addition to bacterial toxins, many other bacterial and eukaryotic proteins have been found to translocate the plasma or endosomal membrane, in either direction. For example, cell-to-cell transmission of α-synuclein is implicated in the progression of Parkinson’s disease,7 while intercellular transfer of the PTEN-long phosphatase regulates PI3K signaling in recipient cells.51 Cell-permeable autoantibodies against nuclear DNA were initially discovered in patients with lupus disease,52 a finding that led to the development of engineered antibody therapeutics against intracellular targets.53 We recently showed that grafting a short CPP sequence (e.g., RRRRWWW) into a surface loop of mammalian proteins (e.g., protein-tyrosine phosphatase 1B and purine nucleoside phosphorylase) is sufficient to render the latter cell-permeable.54 In the reverse direction, numerous bacterial and mammalian proteins are transported from the cytosol into the periplasmic or extracellular space in their native states by non-canonical secretory mechanisms. The TAT system, found in prokaryotes, chloroplasts, and some mitochondria, allows folded proteins to be moved across membranes.16 Proteins transported by the TAT system often contain complex cofactors (e.g., ion-sulfur clusters) or are components of multiprotein complexes, presumably as a way of avoiding the need to reassemble the complex structures outside the cytosol. Remarkably, this transport is achieved without significant ion leakage. The eukaryotic UPS system exports a significant number of leaderless proteins, most notably the interleukin 1β (IL-1β) family of cytokines,55 across the plasma membrane.15 Interestingly, bacteria appear to have an analogous UPS system.56 An advantage of the TAT and UPS systems (over the canonical Sec pathway) is that proteins can be rapidly transported across membranes in a regulated manner (see below), which is crucial for cytokines involved in inflammatory responses. How these proteins travel across the membranes has been an intriguing mystery.
I propose that the above proteins translocate the membranes by VBC, based on two reasons. First, transporting folded proteins of different sizes and physicochemical properties across a cell membrane without causing substantial ion leakage is inconceivable for any mechanism other than VBC. Second, most of the above proteins contain both polybasic and hydrophobic motifs that are capable of inducing VBC. Cell-permeable antibodies (e.g., TMab4) contain a hydrophobic motif, WYW (or similar sequences), in the CDR3 loop of their VL domain, which has been identified as the “endosomal escape motif’.53 Inspection of their sequences revealed that the antibodies also contain polybasic sequences in the CDR1 and CDR2 loops of the VL domain. The maturation and secretion of IL-1β requires processing by caspase-1, which is in turn regulated by the inflammasome.55 Pro-IL-1β has a pi value of 4.6; removal of the highly acidic N-terminal 117 residues by caspase-1 increases the pI of mature IL-1β to 8.8.57 Moreover, proteolytic processing exposes a C-terminal polybasic sequence which proves essential for IL-1β secretion.
The bacterial TAT machinery consists of three proteins, TatA, TatB, and TatC.16 The most critical component, TatA, is a small membrane protein consisting of a hydrophobic α-helix followed by an amphipathic (polybasic) α-helix. The hydrophobic helix inserts across the plasma membrane, while the amphipathic helix lies parallel to the inner leaflet of the plasma membrane and interacts with the membrane. Substrate proteins are recognized by TatBC through a highly conserved twin-arginine motif (SRRxFLK) in the substrates and translocation requires oligomerization of TatA. It is conceivable that TatA mediates the translocation of substrate proteins by promoting VBC, likely with help from TatBC and the twin-arginine motif of the substrates. The existing literature is consistent with this hypothesis. For example, the insertion of TatA into lipid bilayers causes “quantized”, partial, and temporary leakage of calcein (a fluorescent dye) from large unilamellar vesicles - after each protein addition, the fluorescence reached a new plateau and did not increase further until more protein was added.58 TatABC is evenly distributed on the plasma membrane in the absence of substrates; upon substrate binding, TatABC clusters into individual foci,59 as observed during CPP12- (Figure 4) and DT-induced VBC (Figure 5c).
VIRUSES
The subversion of cellular processes by viruses has long been instructive in revealing the fundamental mechanisms in cells (e.g., RNA splicing, autophagy, and cell cycle). Investigation of the cellular entry of viruses has likewise led to novel insights into the mechanism of intracellular trafficking of biomolecules. Enveloped viruses exit the endosome by fusing their membranes with the endosomal membrane, but the mechanism by which non-enveloped viruses escape the endosome is poorly understood. A general expectation is that non-enveloped viruses must disrupt the endosomal membrane to access the cytoplasm. Three major classes of “membranolytic” viral factors have been discovered: amphipathic α-helical domains (e.g., adenovirus protein VI), myristoylated proteins (e.g., N-myristoylated capsid protein μ1 of reovirus), and membrane-remodeling enzymatic domains [e.g., the phospholipase A type 2 (PLA2) domain of parvovirus VP1]7 It is currently unclear how these structurally different viral factors accomplish the same task – the release of a mostly intact virion, which may have a diameter of ~100 nm, from the endosome into the cytosol. I propose that all three viral factors promote viral endosomal escape by inducing VBC.
For adenoviruses, capsid protein VI is primarily responsible for the endosomal escape of the viruses.28 Wiethoff and co-workers further determined that a 20-aa amphipathic α-helix at the N-terminus of protein VI, which is highly conserved among adenoviral species, is responsible for endosomal escape.60 This peptide binds to GUVs of the endosomal membrane composition with high affinity (apparent KD ~3 μM), induces curvature on the vesicles, and causes the GUVs to “fragment” into smaller vesicles, tubular structures, and peptide/lipid aggregates. These properties are reminiscent of those of CPP12.1 It is conceivable that upon endosomal entry, an adenovirus releases up to ~360 copies of protein VI, which bind to the endosomal membrane and induce budding and collapse of a virus-loaded vesicle.
N-myristoylated capsid protein μ1 is responsible for the endosomal escape of reoviruses. An N-terminal 41-aa fragment of the protein, μ1N, is sufficient to cause the release of 40 kDa dextran from red blood cell ghosts.61 N-myristoylation may facilitate VBC by two different mechanisms. First, by inserting into the endosomal membrane, the myristoyl group targets the viral protein to the endosomal membrane and increases its binding affinity for the membrane. Second, insertion of a short acyl group (the myristoyl group has a 14-carbon chain) into the luminal leaflet generates positive membrane curvature (Figure 3c), which is required at the budding neck. Interactions between the rest of μ1N and the endosomal membrane likely induce negative membrane curvature.
How might the viral PLA2 activity induce VBC? The intraluminal membrane of the early endosome, which is similar in composition to the outer leaflet of the plasma membrane, is rich in phosphatidylcholine (PC). PC has an intrinsic lipid curvature of ~0 and favors the formation of lamellar membranes. Hydrolysis of PC by PLA2 produces lysophosphatidylcholine (LPC) and fatty acids. LPC has a large polar head group and a single hydrocarbon chain and, when inserted into a lipid bilayer, promotes positive membrane curvature (Figure 3c). Fatty acids, on the other hand, generate negative curvature. Thus, the PLA2 action produces lipid molecules that support the formation of simultaneous positive and negative curvatures on the endosomal membrane, as required for the budding neck. Viral PLA2s are promiscuous in substrate specificity, also hydrolyzing other phospholipids found on the endosomal membrane.62 The use of membrane-remodeling enzymes appears to be a common strategy for viral entry and life cycle, as adenoviruses leverage the host acid sphingomyelinase to achieve rapid cytosolic entry.63 Some bacterial protein toxins also employ phospholipase A activity to remodel the plasma membrane of host cells to gain cellular entry.13
SYNTHETIC DRUG DELIVERY SYSTEMS
Numerous artificial systems have been developed to deliver biomolecules into mammalian cells as research tools and therapeutics. This Account will limit the discussion to polyplexes, lipoplexes, and lipid nanoparticles (LNPs) which are used for nucleic acid delivery.9,10 While some of these delivery vehicles have achieved remarkable success in the clinic, as illustrated by the delivery of mRNA vaccines against SARS-CoV-2 with LNPs, the mechanistic understanding of how they work has lagged significantly behind. The latter has prevented the design of delivery vehicles with improved endosomal escape efficiencies, which will likely be necessary for therapeutic applications.
Several research groups recently investigated the endosomal escape of polyplexes, lipoplexes, and LNPs by using advanced live-cell confocal microcopy techniques. ur Rehman et al.64 discovered that during lipoplex- and polyplex-mediated siRNA delivery, both nucleic acids and the carrier were suddenly released from the endosomes, followed by rapid diffusion of the nucleic acids throughout the cytoplasm and nucleus (Figure 6). In the case of polyplexes, an instantaneous and complete discharge of nucleic acids and the carrier from the “endosomes” was observed. These observations were interpreted as direct evidence for the “proton sponge effect”. A later study by Wittrup et al.65 confirmed many of the findings of ur Rehman et al. However, Wittrup et al. found that free siRNAs, rather than intact lipoplexes or LNPs, were released into the cytosol and that the endosomal escape was incomplete, suggesting that the endosomes did not completely rupture. They further observed that the release of siRNA coincided, in many cases, with small cytosolic Ca2+ transients of variable magnitude.
Figure 6.
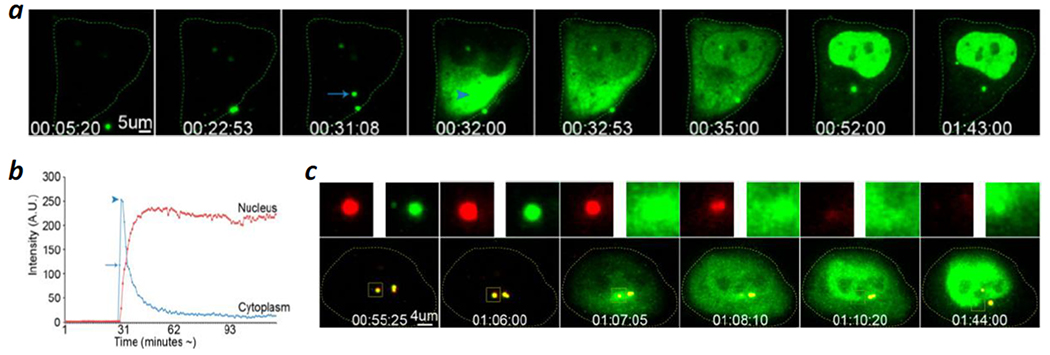
Polyethyleneimine (PEI)-mediated cytosolic delivery of oligonucleotides (ODNs). (a) Time-lapse live-cell confocal microscopic images of a HeLa cell which was incubated with PEI polyplexes containing FITC-labeled ODN. A single endosome split into two vesicles at ~31 min and one of them (indicated by an arrow) collapsed at 32 min. The ODN content is rapidly released into the cytoplasm (arrowhead in fourth panel), followed by a ready accumulation into the nucleus (panels 5–8). (b) Line graph of (a) showing the fluorescence at a region of interest (ROI) within the cytoplasm and within the nucleus at the time of the endosomal escape of ODNs, followed by their accumulation in the nucleus. The arrow indicates the fluorescence intensity of the ODNs within the endosome (cf. arrow in (a), panel 3), while the arrowhead indicates the cytoplasmic fluorescence intensity upon endosomal escape (cf. arrowhead in (a), panel 4). (c) Polyplexes composed of FluoR-labeled PEI (red) and FITC-ODNs (green) were incubated with HeLa cells and monitored by live-cell imaging. Representative frames are shown, together with the signals (upper panels) from the individual fluorescence channels for the boxed area. The boxed area in (c) is yellow due to the colocalization of the PEI and ODN fluorescence (panels 1–4) and disappears in time due to loss of both signals after bursting (panels 5 and 6). Note that a pair of vesicles are already present in panel 1 and are likely derived from a prior budding event. Scale bars, 5 μm in (a) and 4 μm in (c). Time in (a) and (c) is indicated in hh/mm/ss. Reproduced with permission from ref. 64. Copyright 2013 American Chemical Society.
My analysis of the results of ur Rehman et al.64 and Wittrup et al.65 (as well as other literature data) led to an alternative interpretation and conclusion that the synthetic delivery systems exit the endosome by inducing VBC. The study of ur Rehman et al. showed that for every vesicle that burst, there was usually another vesicle nearby that stayed intact during the entire experiment. In fact, the two adjacent vesicles in Figure 6a (marked by an arrow in panel 3) were originally derived from a single endosome (see Movie 1a in Ref. 64). In the alternative interpretation, the vesicle that remained intact during experimentation is the actual endosome after a budding event, whereas the burst “endosome” is the budded vesicle that subsequently collapsed. The multi-step, partial siRNA release observed by Wittrup et al.65 may represent multiple VBC events from the same endosome and/or different endosomes. The cytosolic Ca2+ transients are expected, as each VBC event results in the release of a small endosomal volume, including Ca2+ ions and any unassociated cargo, into the cytosol.2
CONCLUDING REMARKS
We have experimentally demonstrated that five structurally diverse biomolecules including a linear CPP (Tat),2 a cyclic CPP (CPP12),2 a non-peptidic CPM (CPM3),2 a bacterial protein toxin of the AB class (DT),3 and a single-domain protein toxin (NleC)3 escape from the endosome into the cytosol by inducing VBC from the endosomal membrane. There is strong literature evidence that polyplexes,64 lipoplexes,64,65 and LNPs65 exit the endosome by the same mechanism. In addition, literature data suggest that other bacterial and mammalian proteins and non-enveloped viruses may also cross cell membranes in either direction by the VBC mechanism. By invoking the VBC mechanism, we can now rationalize and reconcile many of the previously enigmatic and conflicting observations. In sum, the VBC mechanism is a previously unrecognized and potentially general membrane transport mechanism, which functions in parallel to the well-characterized passive diffusion and energy-dependent secretory pathway. However, further studies are necessary to determine whether the VBC mechanism is broadly applicable to diverse biomolecules/systems. I hope that this Account will encourage other researchers to test our hypothesis in their respective systems and apply the mechanistic insights to design more effective drug delivery systems.
ACKNOWLEDGMENT
I thank the Pei group members for their contributions to the work covered in this article, A. Sahni for the Table of Contents art, and V. Gopalan and R. Dalbey for their comments on the article.
Funding Sources
This work was supported by the National Institutes of Health (GM122459 and CA234124).
ABBREVIATIONS
- CPM
cell-penetrating molecules
- CPP
cell-penetrating peptide
- DT
diphtheria toxin
- GUV
giant unilamellar vesicle
- LNP
lipid nanoparticle
- TAT
twin-arginine translocation
- UPS
unconventional protein secretion
- VBC
vesicle budding and collapse
BIOGRAPHICAL INFORMATION
Dehua Pei is Kimberly Professor of Chemistry and Biochemistry at The Ohio State University. He received his BS degree in chemistry from Wuhan University (China) and PhD degree in organic chemistry from University of California, Berkeley, and was a Damon Runyon-Winchell Walter Cancer Fund postdoctoral fellow at Harvard Medical School before joining the faculty at The Ohio State University (1995). His research group is currently investigating how biomolecules cross cell membranes and developing cell-penetrating molecules as research tools and potential therapeutics against previously undruggable targets, such as intracellular protein-protein interactions. He is a co-Founder of Entrada Therapeutics, Inc.
Footnotes
The author declares no competing financial interests.
REFERENCES
- 1.Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Cross C; Phelps MA; Rossman JS; Pei D Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study first proposed the VBC mechanism and demonstrated it in vitro with CPPs and giant unilamellar vesicles.
- 2.Sahni A; Qian Z; Pei D Cell-penetrating peptides escape the endosome by inducing vesicle budding and collapse. ACS Chem. Biol 2020, 15, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work first demonstrated that CPPs escape the endosome by the VBC mechanism in live cells.
- 3.Sahni A; Pei D Bacterial toxins escape the endosome by inducing vesicle budding and collapse. ACS Chem. Biol 2021, 16, ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrated that diphtheria toxin and enteropathogenic NleC escape the endosome by the VBC mechanism.
- 4.Kurrikoff K; Vunk B; Langel Ü Status update in the use of cell-penetrating peptides for the delivery of macromolecular therapeutics. Expert Opin. Biol. Ther 2021, 21, 361–370. [DOI] [PubMed] [Google Scholar]
- 5.Appiah Kubi G; Qian Z; Amiar S; Sahni A; Stahelin RV; Pei D Non-peptidic cell-penetrating motifs for mitochondrion-specific cargo delivery. Angew. Chem., Int. Ed 2018, 57, 17183–17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams JM; Tsai B Intracellular trafficking of bacterial toxins. Curr. Opin. Cell Biol 2016, 41, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallegos S; Pacheco C; Peters C; Opazo CM; Aguayo LG Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci 2015, 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staring J; Raaben M; Brummelkamp TR Viral escape from endosomes and host detection at a glance. J. Cell Sci 2018, 131, jcs216259. [DOI] [PubMed] [Google Scholar]
- 9.Roberts TC; Langer R; Wood M Advances in oligonucleotide drug delivery. Nat. Rev. Drug Disc 2020, 19, 673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajj K; Whitehead K Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater 2017, 2, 17056. [Google Scholar]
- 11.Pei D; Buyanova M Overcoming endosomal entrapment in drug delivery. Bioconjugate Chem. 2019, 30, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi T; Futaki S Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull 2016, 64, 1431–1437. [DOI] [PubMed] [Google Scholar]
- 13.González-Bullón D; Uribe KB; Martín C; Ostolaza H Phospholipase A activity of adenylate cyclase toxin mediates translocation of its adenylate cyclase domain. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E6784–E6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandvig K; Skotland T; van Deurs B; Klokk TI Retrograde transport of protein toxins through the Golgi apparatus. Histochem. Cell Biol 2013, 140, 317–326. [DOI] [PubMed] [Google Scholar]
- 15.Kim J; Gee HY; Lee MG Unconventional protein secretion - new insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci 2018, 131, jcs213686. [DOI] [PubMed] [Google Scholar]
- 16.Berks BC The twin-arginine protein translocation pathway. Annu. Rev. Biochem 2015, 84, 843–864. [DOI] [PubMed] [Google Scholar]
- 17.Beckwith J The Sec-dependent pathway. Res. Microbiol 2013, 164, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behr J-P The proton sponge: a trick to enter cells the viruses did not exploit. CHIMIA Int. J. Chem 1997, 51, 34–36. [Google Scholar]
- 19.White JM; Whittaker GR Fusion of enveloped viruses in endosomes. Traffic 2016, 17, 593–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zelphati O; Szoka FC Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11493–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herce HD; Garcia AE; Litt J; Kane RS; Martin P; Enrique N; Rebolledo A; Milesi V Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J 2009, 97, 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tilley SJ; Saibil HR The mechanism of pore formation by bacterial toxins. Curr. Opin. Struct. Biol 2006, 16, 230–236. [DOI] [PubMed] [Google Scholar]
- 23.Bus T; Traeger A; Schubert US The great escape: how cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [DOI] [PubMed] [Google Scholar]
- 24.Jülicher F; Lipowsky R Domain-induced budding of vesicles. Phys. Rev. Lett 1993, 70, 2964–2967. [DOI] [PubMed] [Google Scholar]
- 25.Dougherty PG; Sahni A; Pei D Understanding cell penetration of cyclic peptides. Chem. Rev 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucchino M; Billet A; Bai SK; Dransart E; Hadjerci J; Schmidt F; Wunder C; Johannes L Absolute quantification of drug vector delivery to the cytosol. Angew. Chem. Int. Ed 2021, 60, 14824–14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudson TH; Neville DM Jr. Quantal entry of diphtheria toxin to the cytosol. J. Biol. Chem 1985, 260, 2675–2680. [PubMed] [Google Scholar]
- 28.Wiethoff CM; Wodrich H; Gerace L; Nemerow GR Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol 2005, 79, 1992–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palm-Apergi C; Lönn P; Dowdy SF Do cell-penetrating peptides actually “penetrate” cellular membranes? Mol. Ther 2012, 20, 695–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao K; Choe U-J; Kamei DT; Wong GCL Enhanced activity of cyclic transporter sequences driven by phase behavior of peptide–lipid complexes. Soft Matter 2012, 8, 6430–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinauer A; LaRochelle JR; Knox SL; Wissner RF; Berry S; Schepartz A HOPS-dependent endosomal fusion required for efficient cytosolic delivery of therapeutic peptides and small proteins. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maniti O; Blanchard E; Trugnan G; Lamaziere A; Ayala-Sanmartin J Metabolic energy-independent mechanism of internalization for the cell penetrating peptide penetratin. Int. J. Biochem. Cell Biol 2012, 44, 869–875. [DOI] [PubMed] [Google Scholar]
- 33.Maniti O; Piao HR; Ayala-Sanmartin J Basic cell penetrating peptides induce plasma membrane positive curvature, lipid domain separation and protein redistribution. Int. J. Biochem. Cell Biol 2014, 50, 73–81. [DOI] [PubMed] [Google Scholar]
- 34.Säälik P; Niinep A; Pae J; Hansen M; Lubenets D; Langel Ü; Pooga M Penetration without cells: membrane translocation of cell-penetrating peptides in the model giant plasma membrane vesicles. J. Controlled Release 2011, 153, 117–125. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt N; Mishra A; Lai GH; Wong GC Arginine-rich cell-penetrating peptides. FEES Lett. 2010, 584, 1806–1813. [DOI] [PubMed] [Google Scholar]
- 36.Erazo-Oliveras A; Najjar K; La Dayani L; Wang TY; Johnson GA; Pellois JP Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014, 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu H-H; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem 2017, 9, 751–761. [DOI] [PubMed] [Google Scholar]
- 38.El-Sayed A; Futaki S; Harashima H Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parveen S; Bishai WR; Murphy JR Corynebacterium diphtheriae: diphtheria toxin, the tox operon, and its regulation by Fe2+ activation of apo-DtxR. Microbiol. Spectrum 2019, 7, GPP3-0063-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gatsogiannis C; Lang AE; Meusch D; Pfaumann V; Hofnagel O; Benz R; Aktories K; Raunser S A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature 2013, 495, 520–523. [DOI] [PubMed] [Google Scholar]
- 41.Auger A; Park M; Nitschke F; Minassian LM; Beilhartz GL; Minassian BA; Melnyk RA Efficient delivery of structurally diverse protein cargo into mammalian cells by a bacterial toxin.Mol. Pharm 2015, 12, 2962–2971. [DOI] [PubMed] [Google Scholar]
- 42.Kakimoto S; Hamada T; Komatsu Y; Takagi M; Tanabe T; Azuma EL; Shinkai S; Nagasaki T The conjugation of diphtheria toxin T domain to poly(ethylenimine) based vectors for enhanced endosomal escape during gene transfection. Biomaterials 2009, 30, 402–408. [DOI] [PubMed] [Google Scholar]
- 43.Arnold AE; Smith LJ; Beilhartz GL; Bahlmann LC; Jameson E; Melnyk RA; Shoichet MS Attenuated diphtheria toxin mediates siRNA delivery. Sci. Adv 2020, 6, eaaz4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ladokhin AS; Vargas-Uribe M; Rodnin MV; Ghatak C; Sharma O Cellular entry of the diphtheria toxin does not require the formation of the open-channel state by its translocation domain. Toxins 2017, 9, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratts R; Trujillo C; Bharti A; vanderSpek I; Harrison R; Murphy JR A conserved motif in transmembrane helix 1 of diphtheria toxin mediates catalytic domain delivery to the cytosol. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 15635–15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stolle AS; Norkowski S; Körner B; Schmitz J; Lüken L; Frankenberg M; Rüter C; Schmidt MA T3SS-independent uptake of the short-trip toxin-related recombinant NleC effector of enteropathogenic Escherichia coli leads to NF-κB p65 cleavage. Front. Cell. Infect. Microbiol 2017, 13, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turco MM; Sousa MC The structure and specificity of the type III secretion system effector NleC suggest a DNA mimicry mechanism of substrate recognition. Biochemistry 2014, 53, 5131–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W; Liu Y; Sheng X; Yin P; Hu F; Liu Y; Chen C; Li Q; Yan C; Wang J Structure and mechanism of a type III secretion protease, NleC. Acta Crystallogr. D Biol. Crystallogr 2014, 70, 40–47. [DOI] [PubMed] [Google Scholar]
- 49.Antignani A; Youle RJ Endosome fusion induced by diphtheria toxin translocation domain. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 8020–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marnell MH; Shia SP; Stookey M; Draper RK Evidence for penetration of diphtheria toxin to the cytosol through a prelysosomal membrane. Infect. Immun 1984, 44, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hopkins BD; Fine B; Steinbach N; Dendy M; Rapp Z; Shaw J; Pappas K; Yu JS, Hodakoski C; Mense S; Klein J; Pegno S; Sulis ML; Goldstein H; Amendolara B; Lei L; Maurer M; Bruce J; Canoll P; Hibshoosh H; Parsons R A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivadeneyra-Espinoza L; Ruiz-Arguelles A Cell-penetrating anti-native DNA antibodies trigger apoptosis through both the neglect and programmed pathways. J. Autoimmun 2006, 26, 52–56. [DOI] [PubMed] [Google Scholar]
- 53.Kim JS; Choi DK; Shin JY; Shin SM; Park SW; Cho HS; Kim YS Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J. Controlled Release 2016, 235, 165–175. [DOI] [PubMed] [Google Scholar]
- 54.Chen K; Pei D Engineering cell-permeable proteins through insertion of cell-penetrating motifs into surface loops. ACS Chem. Biol 2020, 15, 2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monteleone M; Stow JL; Schroder K Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine 2015, 74, 213–218. [DOI] [PubMed] [Google Scholar]
- 56.Ebner P; Götz F Bacterial excretion of cytoplasmic proteins (ECP): occurrence, mechanism, and function. Trends Microbiol. 2019, 27, 176–187. [DOI] [PubMed] [Google Scholar]
- 57.Monteleone M; Stanley AC; Chen KW; Brown DL; Bezbradica JS; von Pein JB; Holley CL; Boucher D; Shakespear MR; Kapetanovic R; Rolfes V; Sweet MJ; Stow JL; Schroder K Interleukin-1β maturation triggers its relocation to the plasma membrane for Gasdermin-D-dependent and -independent secretion. Cell Rep. 2018, 24, 1425–1433. [DOI] [PubMed] [Google Scholar]
- 58.Pettersson P; Patrick J; Jakob M; Jacobs M; Klösgen RB; Wennmalm S; Mäler L Soluble TatA forms oligomers that interact with membranes: Structure and insertion studies of a versatile protein transporter. Biochim. Biophys. Acta. Biomembranes 2021, 1863, 183529. [DOI] [PubMed] [Google Scholar]
- 59.Alcock F; Baker MA; Greene NP; Palmer T; Wallace ML; Berks BC Live cell imaging shows reversible assembly of the TatA component of the twin-arginine protein transport system. Proc. Natl. Acad. Sci. U. S. A 2013, 110, e3650–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maier O; Galan DL; Wodrich H; Wiethoff CM An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 2010, 402, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ivanovic T; Agosto MA; Zhang L; Chandran K; Harrison SC; Nibert ML Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008, 27, 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Canaan S; Zadori Z; Ghomashchi F; Bollinger J; Sadilek M; Moreau ME; Tijssen P; Gelb MH Interfacial enzymology of parvovirus phospholipases a(2). J. Biol. Chem 2004, 279, 14502–14508. [DOI] [PubMed] [Google Scholar]
- 63.Bilkova E; Forstova J; Abrahamyan L Coat as a dagger: The use of capsid proteins to perforate membranes during non-enveloped DNA viruses trafficking. Viruses 2014, 6, 2899–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.ur Rehman Z; Hoekstra D; Zuhorn IS Mechanism of polyplex- and lipoplex-mediated delivery of nucleic acids: real-time visualization of transient membrane destabilization without endosomal lysis. ACS Nano 2013, 7, 3767–3777. [DOI] [PubMed] [Google Scholar]
- 65.Wittrup A; Ai A; Liu X; Hamar P; Trifonova R; Charisse K; Manoharan M; Kirchhausen T; Lieberman J Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotech 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]