

Abstract
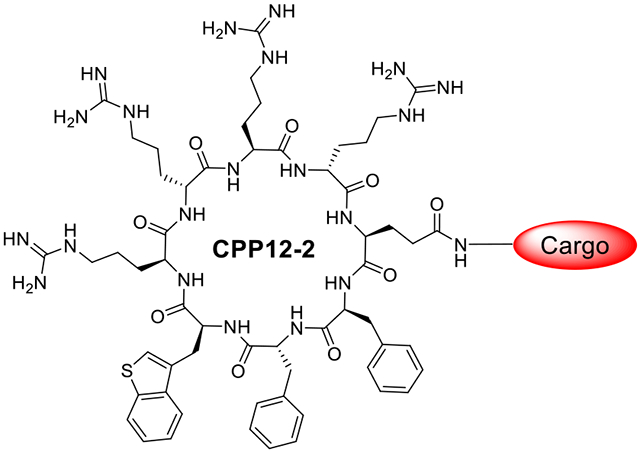
Cyclic cell-penetrating peptide 12 (CPP12) is highly efficient for the cytosolic delivery of a variety of cargo molecules into mammalian cells in vitro and in vivo. However, its cytosolic entry efficiency is substantially reduced at lower concentrations or in the presence of serum proteins. In this study, CPP12 analogs were prepared by replacing its hydrophobic residues with amino acids of varying hydrophobicity and evaluated for cellular entry. Substitution of L-3-benzothienylalanine (Bta) for L-2-naphthylalanine (Nal) resulted in CPP12-2, which exhibits up to 3.8-fold higher cytosolic entry efficiency than CPP12, especially at low CPP concentrations, thanks to improved endosomal escape efficiency. CPP12-2 is well suited for the cytosolic delivery of highly potent cargos to achieve biological activity at low concentrations.
Keywords: Cell-penetrating peptide, drug delivery, endosomal escape, intracellular biologics, peptide therapeutics
Graphical Abstract
INTRODUCTION
Cell-penetrating peptides (CPPs) are short peptides that are capable of translocating across the cell membrane without causing significant damage to the membrane structure. The first-generation CPPs were linear peptides of typically 5-30 amino acids.1-3 Although linear CPPs have been used to deliver a variety of membrane-impermeable cargos (e.g., small molecules, peptides, proteins, nucleic acids, and nanoparticles) into the mammalian cell,4-15 their clinical translation has been hampered by low cytosolic delivery efficiency (which is defined as the ratio of cytosolic versus extracellular concentration), poor metabolic stability, and frequently inadequate bioavailability.16,17 These limitations prompted researchers to develop structurally constrained peptides as second-generation CPPs.18 It was discovered that cyclization of CPPs greatly enhances their cellular uptake efficiency.19-23 For example, cyclo(Phe-D-Phe-Nal-Arg-D-Arg-Arg-D-Arg-Gln) (CPP12, where Nal is L-2-naphthylalanine) exhibits a cytosolic entry efficiency of 120% and is one of the most active CPPs discovered to date.24 In addition, cyclic CPPs generally have greater proteolytic stability than their linear counterparts and better bioavailability.24-26 Cyclic CPPs have proven highly effective for the cytosolic delivery of peptides,25-28 proteins,25,29-33 and nucleic acids34,35 into mammalian cells in vitro and in vivo. For example, a CPP12-nanobody conjugate specifically blocked tick-borne infection by an intracellular bacterium, Ehrlichia chaffeensis, in cell culture and in a mouse model.32 A CPP12-thymidine phosphorylase conjugate has demonstrated excellent in vivo efficacy as well as pharmacokinetics and is poised to enter Phase I/II human clinical trials for treatment of mitochondrial neurogastrointestinal encephalopathy (MNGIE), a rare, fatal genetic disease.33
CPPs can enter the cell by two distinct mechanisms. Direct translocation across the plasma membrane, which typically occurs at high peptide concentrations (>10 μM), involves the direct entry of the peptide into the cell cytosol. At lower concentrations (<10 μM), the predominant route of cell entry is endocytosis followed by endosomal escape. The initial endocytic uptake step is very efficient but does not translate into high cytosolic delivery, and the majority of CPPs remain entrapped inside the endolysosomal compartments. In either mechanism, the CPP must traverse a lipid bilayer in order to reach the cytosol of the cell.1-3 Following endocytic uptake, CPPs escape from the endosome into the cytosol by inducing budding and collapse of small vesicles from the endosomal membrane.24,36 The overall cytosolic entry efficiency of CPPs is thus determined by the efficiencies of both endocytic uptake and endosomal escape. Since both endocytic uptake37 and endosomal escape38 are CPP concentration dependent, the cytosolic entry efficiency of CPPs increases nonlinearly with the CPP concentration, with poor efficiencies at low CPP concentrations. Indeed, a drawback of CPP12 and other cyclic CPPs is that their cellular entry kinetics and efficiency are substantially reduced at low CPP concentrations (⩽1 μM) or in the presence of high concentrations of serum proteins, likely because binding of the CPPs to serum proteins reduces the concentration of free CPPs. For example, the cytosolic entry efficiency of CPP12 into HeLa (human cervical cancer) cells was decreased by 25-fold in the presence of 10% fetal bovine serum (FBS).40
To potentially alleviate protein binding and improve the cytosolic entry efficiency, we have explored the structure-activity relationship (SAR) of cyclic CPP12 by modifying the number as well as the sidechain structures of its hydrophobic residues. We previously reported that replacement of the three hydrophobic aromatic residues of CPP12 with a single amino acid containing a long alkyl side chain (e.g., a decyl group) resulted in a cyclic CPP of 2.8-fold higher cytosolic entry efficiency than CPP12 under high serum protein concentrations.40 However, the modified CPP enters the cell predominantly by direct translocation across the plasma membrane (which, in our opinion, is less desirable as a drug delivery vehicle) and appears to be less effective for delivering macromolecular cargos (e.g., proteins). In this study, we replaced the hydrophobic residues of CPP12 with amino acids of varying hydrophobicity and discovered an analog of substantially improved cytosolic entry efficiency, especially at low CPP concentrations. To our surprise, the improvement was primarily the result of enhanced endosomal escape efficiency, instead of decreased protein binding.
MATERIALS AND METHODS
Materials.
Reagents for peptide synthesis were purchased from Chem-Impex (Wood Dale, IL), NovaBiochem (La Jolla, CA), or Anaspec (San Jose, CA). Rink amide resin (100-200 mesh, 0.43 mmol/g) and fluorescein isothiocyanate (FITC), isomer I, were from Chem-Impex (Wood Dale, IL). 5(6)-Carboxynaphthofluorescein succinimidyl ester (NF-NHS), was from Setareh Biotech (Eugene, OR). 5(6)-Carboxytetramethylrhodamine succinimidyl ester (TMR-NHS) and Geneticin™ were from ThermoFisher Scientific (Waltham, MA). Tetrakis(triphenylphosphine) palladium(0) [Pd(PPh3)4] was from Sigma-Aldrich (St. Louis, MO) and phenylsilane was from TCI America (Portland, OR). O-Benzotriazole-N,N,N,N-tetramethyluronium hexafluorophosphate (HATU) and (benzotriazol-1-yloxy)-tripyrrolidinophosphonium hexafluorophosphate (PyBOP) were from Matrix Scientific (Columbia, SC). All solvents and other chemical reagents were from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and were used without further purification. Cell culture media, fetal bovine serum (FBS), penicillin-streptomycin, 0.25% trypsin-EDTA, DPBS, 100x non-essential amino acids, and sodium pyruvate solution were from Sigma-Aldrich. The cell proliferation kit [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)] was from Roche (Indianapolis, IN). Nano-Glo® Live Cell assay system and LgBit expression vector were purchased from Promega (Madison, WI). HeLa cells were from ATCC (Manassas, VA). Human embryonic kidney 293T (HEK293T) cell line was a generous gift from Dr. K. Nakanishi’s group (The Ohio State University). NRF2/ARE responsive luciferase reporter HepG2 cell line and One-Step™ luciferase assay system were from BPS Bioscience (San Diego, CA). All cell lines were maintained in a humidified chamber with 5% CO2 at 37 °C.
Peptide Synthesis.
Peptides were synthesized manually on Rink amide resin (0.43 mmol/g) using standard Fmoc chemistry. The typical coupling reaction contained 5 eq. of Fmoc-amino acid, 5 eq. of HATU, and 10 eq. of diisopropylethylamine (DIPEA) and was allowed to proceed with mixing for 30 min at room temperature (RT). For cyclic CPPs, after the addition of the last (N-terminal) residue, the allyl group on the C-terminal Glu residue was removed by treatment with 0.3 eq. of Pd(PPh3)4 and 10 eq. of phenylsilane in anhydrous DCM in the dark (3 x 15 min). The resin was washed twice with sodium dimethyldithiocarbamate (SDDCM, 0.5 M in DMF) and the N-terminal Fmoc group was removed by treatment with 20% piperidine in DMF. The resin was extensively washed with DMF and DCM and incubated in 1 M 1-hydroxybenzotriazole (HOBt) in DMF for 20 min. The peptide was cyclized by using 10 eq. of PyBOP, 10 eq. of HOBT, 20 eq. of DIPEA in DMF for 1 h at RT. For CPP-P1 conjugates, Fmoc-Lys(Mtt)-OH was first added at the C-terminus, to serve as a linker for conjugation of cyclic CPP and cyclic peptide P1. The linear CPP sequence was synthesized using the standard Fmoc chemistry. The CPP sequence was cyclized as described above. The 4-methyltrityl (Mtt) group on the C-terminal Lys was removed by treatment with 2% trifluoroacetic acid (TFA and 1% triisopropylsilane in DCM (6 x 5 min). Fmoc-8-amino-3,6-dioxaoctanoic acid (miniPEG) was coupled to the Lys side chain, followed by the synthesis of the linear sequence of P1. The allyl protecting group on the L-Glu and the Fmoc group of the N-terminal Gly residue were removed as described above. The linear sequence of P1 was cyclized on resin as described above. Cleavage and deprotection of the peptide were performed on resin using 92.5/2.5/2.5/2.5 (v/v) TFA/triisopropylsilane/1,3-dimethoxybenzene/water for 3 h at RT. Peptide HiBiT (VSGWRLFKKIS) was obtained from Promega. HiBit peptide with an N-terminal cysteine (CVSGWRLFKKIS) was synthesized as described above and activated with 2, 2’-dipyridyl disulfide (PyS-HiBit). Cyclic CPPs containing a C-terminal cysteine were synthesized as described above. Cleavage and deprotection of the peptide were performed using 90/2.5/2.5/2.5/2.5 (v/v) TFA/triisopropylsilane/1,3-dimethoxybenzene/water/1,2-ethanedithiol for 3 h at RT. The peptides were triturated with cold diethyl ether (3 times) and purified by reversed-phase HPLC on a semi-preparative Waters XBridge C18 column. For CPP-HiBit conjugates, CPP containing a C-terminal Cys was incubated with 1 eq. of PyS-HiBit in MeOH containing 2 % acetic acid (v/v) for 3 h at RT and purified by reversed-phase HPLC on a semi-preparative Waters XBridge C18 column. The purity of the peptides ≥95%) was assessed by reversed-phase HPLC equipped with an analytical Waters XBridge C18 column (Figure S1). Peptide authenticity was confirmed by MALDI FT-ICR mass spectrometry at Campus Chemical Instrumentation Center of The Ohio State University (Table S1).
The concentration of peptides was determined by measuring the absorbance of the peptide at 280 nm using the following molar extinction coefficients: ε (CPP12) = 3550 cm−1 M−1, ε (CPP12-2) = 3450 cm−1 M−1
Fluorescent Labeling of Peptides.
Peptide labeling with NF-NHS, TMR-NHS, or FITC was carried out in the solution phase. Lyophilized CPP (1 mg) was dissolved in 25 μL of DMF and the pH was adjusted to 8.0 (for NF-NHS or TMR-NHS labeling) or 8.5 (for FITC labeling) by the addition of a 0.1 M sodium bicarbonate solution. Three eq. of NF-NHS, TMR-NHS, or FITC was dissolved in 25 μL of DMF and added to the peptide solution. The mixture was incubated at RT for 2 h on a shaker. The labeled peptides were purified by HPLC and analyzed by MALDI FT-ICR mass spectrometry as previously described.
The concentration of fluorescently-labeled peptides was determined by measuring the absorbance at λmax of the dye using the following λmax and molar extinction coefficients: λmax = 595 nm and ε = 47,500 cm−1 M−1 for NF; λmax = 555 nm and ε = 80,000 cm−1 M−1 for TMR; and λmax = 495 nm and ε = 75,000 cm−1 M−1 for FITC.
Cell Culture.
HeLa cells and HEK293T cells were cultured in Dulbecco's modified eagle's medium (DMEM, Sigma-Aldrich, D1145) supplemented with 10% FBS and 1% penicillin-streptomycin sulfate. ARE reporter (Luc)-HepG2 cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin sulfate, 1% non-essential amino acids, 1 mM sodium pyruvate, and 600 μg/mL Geneticin™. Cells were cultured in a humidified incubator at 37 °C in the presence of 5% CO2.
Flow Cytometry.
HeLa cells were seeded in a 12-well plate at a density 1.5 x 105 cells per well and cultured overnight. Next day, fluorescently labeled peptide at varying concentrations (0.5, 1, 2, or 5 μM) was added in DMEM supplemented with 1% or 10% FBS and 1% penicillin-streptomycin sulfate and incubated at 37 °C for 2 h. After incubation, the cells were washed with cold DPBS twice, detached from the plate with 0.25% trypsin, diluted into cold DPBS and pelleted at 300 g for 5 min at 4 °C. The supernatant was discarded and the cells were washed twice with cold DPBS and resuspended in 200 μL of cold DPBS. The samples were analyzed on a BD FACS LSR II flow cytometer. Data were analyzed using Flow Jo software. Flow cytometry at lower pH (to quench extracellular fluorescence) was carried out by adding a glycine-HCl buffer (pH 3.0) to the samples to attain a final pH of 6.0 immediately before the cytometry analysis. For NF-labeled peptides, a 633-nm laser was used for excitation and the fluorescence emission was analyzed in the APC channel. For FITC-labeled peptides, a 488-nm laser was used for excitation and the fluorescence was analyzed in the FITC channel. For TMR-labeled peptides, a 561-nm laser was used for excitation and the fluorescence was analyzed in the PE channel.
Endosomal Escape Efficiency.
The apparent endosomal escape efficiency of CPP12-2, relative to that of CPP12, was calculated by using equation
where MFI12NF and MFI12-2NF represent the mean fluorescence intensity (MFI) values of HeLa cells treated with NF-labeled CPP12 and CPP12-2, respectively, while MFI12TMR and MFI12TMR are the corresponding values for cells treated with TMR-labeled CPPs.
Confocal Microscopy.
HeLa cells were seeded in a 35-mm glass-bottomed microwell dish with 4 wells (Greiner) at a density of 5 x 104 cells/mL (300 μL in each well) and cultured overnight. Next day, the cells were gently washed with DPBS twice, and treated for 2 h with FITC- or TMR-labeled peptides (5, 1 or 0.2 μM as indicated) in phenol-red free DMEM containing 1% or 10% FBS and 1% penicillin-streptomycin sulfate. After removal of the medium, the cells were gently washed with DPBS twice and imaged on a Nikon A1R live-cell confocal laser scanning (ECLIPSE Ti-E automated, inverted) microscope equipped with a 100x oil objective (1.45 N.A.) and a heated (37 °C) chamber supplied with 5% CO2. For the red channel (TMR), the laser line with λEx 561 nm was set at 0.6% laser power (for 5 μM 1% or 10% FBS peptide treatment), 1.6% laser power (for 1 μM 1% FBS peptide treatment), 10.8% laser power (for 0.2 μM 1% FBS peptide treatment). For the green channel (FITC), the laser line with λEx 487 nm was set at 0.6 % laser power. The data were analyzed using NIS-Elements AR.
For semi-quantification of cytosolic/nuclear CPP concentration, HeLa cells (5 x 104 cells/mL) were treated with NF-labeled peptides (5 μM) for 2 h in phenol-red free DMEM containing 10% FBS and 1% penicillin-streptomycin sulfate and imaged on a Nikon A1R live-cell confocal laser scanning (ECLIPSE Ti-E automated, inverted) microscope equipped with a 60x oil objective (1.4 N.A.) and a heated (37 °C) chamber supplied with 5% CO2. For the far-red channel (NF), the laser line with λEx 642 nm was set at 6% laser power. All images were captured with the same settings. Mean fluorescence intensities (MFIs) of cytosolic/nuclear region of individual cells were quantified using NIS-Elements AR software. First, images were denoised using Denoise.ai. A square region of interest (ROI) (3 x 3 μm or 9 μm2) in an evenly diffuse nuclear/cytosolic region in an individual cell was manually selected and assigned for every cell. For each image, the background signal, quantified as the average of MFI for three background-assigned ROIs, was subtracted from the cellular MFI values of the cytosolic/nuclear ROIs. At least 140 cells from 20-25 images, obtained in 3 trials on different days, were analyzed for each peptide. The MFI values for each peptide were plotted and values (<0.0001) were calculated using two-tailed paired Student’s t test using GraphPad.
MTT Cell Viability Assay.
HeLa cells were seeded in a transparent 96-well plate at a density of 5 x 103 cells/well (100 μL in each well) in DMEM containing 10% FBS and 1% penicillin-streptomycin sulfate and cultured overnight. Next day, cells were treated with varying concentrations of peptide (0-50 μM) and incubated at 37 °C with 5% CO2 for 72 h. Ten μL of MTT solution (0.5 mg/mL) was added to each well and incubated for 4 h, followed by addition of 100 μL of the SDS-HCl solubilizing buffer with thorough mixing and overnight incubation at 37 °C. The absorbance of the formazan product was measured at 565 nm on a Tecan M1000 plate reader.
Fluorescence Polarization (FP) Analysis of CPP Binding to FBS.
Fluorescein-labeled peptide (100 nM) was incubated with serial dilutions of FBS (0-100 μM) in DPBS for 1 h. The solutions were transferred to a 384-black microplate (Greiner), and FP values were measured on a Tecan Infinite M1000 Pro plate reader, with excitation and emission wavelengths at 470 and 535 nm, respectively, for FITC, 530 and 580 nm for TMR, and 595 and 660 nm for NF. The titration curves were fitted using GraphPad Prism to the following equation:
where FP is the measured polarization, Amin is the minimum FP value, Amax is the maximum FP value, Qb is the quantum yield of the bound fluorophore, Qf is the quantum yield of the free fluorophore, L is the ligand concentration, KD is the dissociation constant, and x is the protein concentration. Data presented are the mean ± SD of three independent experiments.
FP-based Competition Assay.
The binding affinity of P1 and CPP-P1 conjugates to Keap1 was determined by FP-based competition assay. Fluorescein-labeled peptide 234 (20 nM) was incubated for 1 h with 40 nM Keap1 protein in 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol, and 0.01% Triton-X. Serial dilutions of competing peptide were prepared in the same buffer. Aliquots of the equilibrated peptide probe-protein mixture were mixed with each peptide dilution and incubated for 1 h. The samples were transferred to a 384-black microplate (Greiner) and FP values were measured on a Tecan Infinite M1000 Pro plate reader. The data were analyzed by GraphPad Prism with log [inhibitor] vs. response (four parameters).
Luciferase Complementation Assay.
HEK293T cells (400,000 cells per well) were seeded in 2 mL of a seeding medium (DMEM, 10% FBS, and 1% penicillin-streptomycin sulfate) in a 6-well plate and cultured overnight. The next day, cells were transfected with 0.5 μg of LgBit expression vector (Promega, WI) using Lipofectamine 2000 transfection reagent. After overnight incubation, cells were reseeded (20,000 cells per well) in a white 96-well plate in the seeding medium and cultured overnight. The cells were treated with varying concentrations of CPP12-HiBit, CPP12-2-HiBit, or HiBit (0-10 μM) in an assay medium (DMEM, 1% FBS, and 1% penicillin-streptomycin sulfate) for 4 h at 37 °C in the presence of 5% CO2. After incubation and removal of the medium, the cells were gently washed with DPBS and Nano-Glo® Live Cell Assay System reagent (25 μL, Promega, WI) was added to cells in OptiMEM medium immediately before measurement. The luminescence was measured using a Tecan Infinite M1000 Pro microplate reader. P values (<0.05) were calculated by one-way ANOVA using GraphPad.
Luciferase Reporter Assay.
ARE reporter-HepG2 cells (1000 cells per well) were seeded in 100 μL of assay medium (MEM, 0% FBS, and 1% penicillin-streptomycin sulfate) in an opaque 96-well plate and cultured overnight. Cells were treated with varying concentrations of P1 and CPP-P1 conjugates (0-5 μM) for 18 h at 37 °C in the presence of 5% CO2. After incubation, 100 μL of One-Step luciferase assay reagent was added to each well and after 15 min of shaking, the luminescence was measured using a Tecan Infinite M1000 Pro microplate reader. P values (<0.05) were calculated by one-way ANOVA using GraphPad.
RESULTS AND DISCUSSION
Discovery of CPP12 Analogs of Improved Cytosolic Delivery Efficiency.
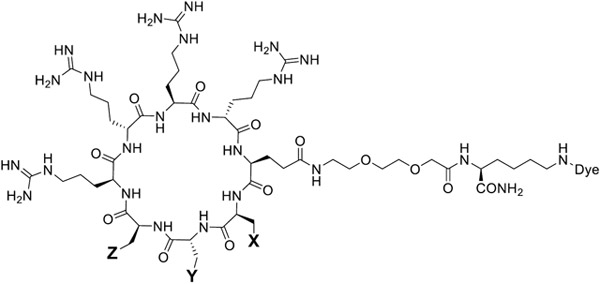
To reduce the protein binding of CPP12 and potentially improve its cellular entry efficiency at high serum concentrations, we synthesized a series of CPP12 analogs by replacing its hydrophobic residues with other amino acids of varying hydrophobicity (peptides CPP12-1 to CPP12-10, Table 1). Each peptide was labeled with naphthofluorescein (NF) at the Glu side chain through a long, flexible linker, miniPEG-Lys. NF is a pH-sensitive dye with a pKa of ~7.8, which is protonated and non-fluorescent inside the acidic environments of endosomes and lysosomes (pH 4.5-6.5) but becomes fluorescent upon entering the cytosol and nucleus (pH 7.4).41 HeLa cells were incubated with 5 μM NF-labeled peptides for 2 h in the presence of 10% FBS and washed extensively to remove any extracellular peptide. The cells were analyzed by flow cytometry and the cytosolic entry efficiencies of the CPP12 analogs (relative to that of CPP12, which is defined as 100%) were calculated from the MFICPPNF/MFICPP12 ratios.41 Replacement of 2-Nal with L-1-naphthylalanine (1-Nal; Table 1, CPP12-1) decreased its CPP activity by 1.4-fold, whereas substitution of L-3-benzothienylalanine (Bta; Table 1, CPP12-2) resulted in a 3.8-fold improvement in the cytosolic entry efficiency over CPP12 (Figure S2 and S3). Substitution of D-4-fluorophenylalanine (D-Fpa) for D-Phe (CPP12-3) or L-4-fluorophenylalanine (Fpa) for Phe (CPP12-7) decreased the CPP activity by 1.7- and 1.4-fold, respectively (Table 1 and Figure S2). Replacement of Phe or D-Phe with more hydrophilic residues [L-/D-Tyr or L-/D-2- or 4-pyridylalanine (Pya)] decreased the CPP activity by more than 3-fold (CPP12-4, -6, -8, -9, -10), except for CPP12-5, which showed slightly improved cytosolic entry efficiency.
Table 1.
Structures and Cytosolic Entry Efficiencies of CPP12 Analogs
|
|||||
|---|---|---|---|---|---|
| Peptide No. | Peptide Sequencea | Cytosolic Entry at 10% FBS (MFINF, %)b |
Cytosolic Entry at 1% FBS (MFINF, %)b |
||
| X = | Y = | Z = | |||
| CPP12 | Phe | D-Phe | 2-Nal | 100 | 100 |
| CPP12-1 | Phe | D-Phe | 1-Nal | 69 ± 9 | 105 ± 16 |
| CPP12-2 | Phe | D-Phe | Bta | 375 ± 48 | 85 ± 7 |
| CPP12-3 | Phe | D-Fpa | 2-Nal | 59 ± 12 | 83 ± 2 |
| CPP12-4 | Phe | D-2-Pya | 2-Nal | 29 ± 9 | 35 ± 5 |
| CPP12-5 | Phe | D-4-Pya | 2-Nal | 115 ± 18 | 36 ± 5 |
| CPP12-6 | Phe | D-Tyr | 2-Nal | 32 ± 15 | 30 ± 12 |
| CPP12-7 | Fpa | D-Phe | 2-Nal | 74 ± 9 | 86 ± 15 |
| CPP12-8 | 2-Pya | D-Phe | 2-Nal | 27 ± 8 | 41 ± 16 |
| CPP12-9 | 4-Pya | D-Phe | 2-Nal | 22 ± 9 | 57 ± 18 |
| CPP12-10 | Tyr | D-Phe | 2-Nal | 25 ± 15 | 44 ± 10 |
Bta, L-3-benzothienylalanine; Fpa, L-4-fluorophenylalanine; D-Fpa, D-4-fluorophenylalanine; 1-Nal, L-1-naphthylalanine; 2-Nal, L-2-naphthylalanine; 2-Pya, L-2-pyridylalanine; D-2-Pya, D-2-pyridylalanine; 4-Pya, L-4-pyridylalanine; D-4-Pya, D-4-pyridylalanine.
All values are relative to that of CPP12 (100%) and represent the mean ± SD of three independent experiments.
We next assessed the “intrinsic” cytosolic entry efficiencies of the CPP12 analogs by repeating the flow cytometry experiments in the presence of reduced serum concentration (1% FBS). Replacement of 2-Nal with 1-Nal (CPP12-1) did not significantly change the intrinsic cytosolic entry efficiency, while substitution of Bta for 2-Nal (CPP12-2) or D-/L-Fpa for D-/L-Phe (CPP12-3 and CPP12-7) slightly reduced the CPP activity (by 14% to 17%) (Table 1). Again, substitution of more hydrophilic residues for Phe or D-Phe substantially reduced the intrinsic CPP activity of the CPP12 (by 2- to 3-fold). Interestingly, contrary to our original hypothesis, most of the analogs were more sensitive to serum proteins (performed worse at higher FBS concentration) than CPP12, whereas CPP12-2 and CPP12-5 were notable exceptions. To ascertain that the increased fluorescence was not due to peptide binding to the outer surface of the cell, we repeated the flow cytometry experiment at a lower pH for CPP12 and CPP12-2. The peptide-treated cells were suspended in an acidic solution (pH 6.0) to quench the fluorescence of any cell surface-associated CPPs immediately before the flow cytometry analysis. Similar results were obtained, indicating that the observed fluorescence was not significantly impacted by cell surface-associated peptides (Figure S4). We selected CPP12-2 for further evaluation, as it showed substantially improved performance over CPP12 at 10% FBS.
CPP12-2 Has Improved Endosomal Escape Efficiency.
The improved cytosolic entry efficiency of CPP12-2 at high serum concentrations can potentially be caused by higher endocytic uptake, more efficient endosomal escape, or both. To differentiate these possibilities, we labeled CPP12 and CPP12-2 with a pH-insensitive dye, tetramethylrhodamine (TMR), treated HeLa cells with 5 μM TMR-labeled peptide for 2 h, and quantitated the total cellular uptake of CPP12TMR and CPP12-2TMR by flow cytometry. CPP12-2 showed similar total cellular uptake to CPP12 in the presence of 10% FBS (98% relative to that of CPP12) but slightly lower uptake in the presence of 1% FBS (79%) (Table 2 and Figure S5). The relative endosomal escape efficiency of CPP12-2 was then calculated from the MFINF/MFITMR ratio.41 CPP12-2 exhibited relative endosomal escape efficiencies of 108% and 383% in the presence of 1% and 10% FBS, respectively [relative to that of CPP12 (100%)]. These results reveal that the higher cytosolic entry efficiency of CPP12-2 than CPP12 at high serum concentrations is primarily the result of improved endosomal escape efficiency. However, its different endosomal escape efficiencies in the presence of 1% vs 10% FBS was intriguing.
Table 2.
Total Cellular Uptake, Cytosolic Entry, and Endosomal Escape efficiencies of CPP12-2a
| [CPP12-2] (μM) |
[FBS] | Total Cellular Uptake (MFITMR, %) |
Cytosolic Entry (MFINF, %) |
Apparent Endosomal Escape Efficiency (γ, %) |
|---|---|---|---|---|
| CPP 12 | 10% or 1% | 100 | 100 | 100 |
| 5.0 | 10% | 98 ± 11 | 375 ± 48 | 383 ± 49 |
| 5.0 | 1% | 79 ± 13 | 85 ± 7 | 108 ± 9 |
| 2.0 | 1% | 84 ± 10 | 116 ± 5 | 138 ± 6 |
| 1.0 | 1% | 88 ± 8 | 145 ± 21 | 164 ± 24 |
| 0.5 | 1% | 94 ± 10 | 186 ± 7 | 198 ± 7 |
All values represent the mean ± SD of at least three independent experiments and are relative to that of CPP12 (100%).
The above results can potentially be rationalized by the improved endosomal escape efficiency of CPP12-2 (relative to that of CPP12) at lower peptide concentrations, as high serum concentrations result in binding/sequestration of the CPP, reducing the free CPP concentration. To test this notion, we treated HeLa cells with different concentrations of CPP12-2TMR or CPP12-2NF (5.0, 2.0, 1.0, and 0.5 μM) in the presence of 1% FBS and quantitated the total cellular uptake and cytosolic entry efficiencies as a function of CPP concentration. Although both the total cellular uptake and cytosolic entry efficiencies of CPP12-2 (relative to CPP12) decreased as the CPP concentration increased, the magnitude of reduction was greater for cytosolic entry (from 186% at 0.5 μM to 85% at 5.0 μM) than total cellular uptake (from 94% to 79%) (Table 2 and Figure S6). Again, we calculated the apparent endosomal escape efficiencies of CPP12-2 (relative to that of CPP12) at different CPP concentrations. In agreement with our hypothesis, CPP12-2 showed the highest relative endosomal escape efficiency (198% of that of CPP12) at 0.5 μM CPP12-2, which progressively decreased as the CPP concentration increased (to 108% at 5 μM). Thus, CPP12-2 has higher endosomal escape efficiency than CPP12 under all CPP concentrations tested, but especially at lower CPP concentrations.
The ability of CPP12-2 to enter the cytosol of mammalian cells was confirmed by live-cell confocal microscopy of HeLa cells after treatment with 5 μM CPP12-2TMR for 2 h in the presence of 1% FBS. All treated cells showed intense and diffuse fluorescence throughout the entire cell volume (Figure 1a). The presence of strong TMR fluorescence inside the nucleus demonstrates that at least a fraction of the internalized CPP12-2 reached the cytosol, prior to nuclear localization. In some of the treated cells, punctate fluorescence was also visible in the cytoplasmic region and may represent the fraction of CPP12-2 that had not yet escaped from the endosomes and/or cytosolic CPP12-2 that became associated with intracellular organelles (e.g., microfilaments and the cytoplasmic leaflet of the endosomal membrane). CPP12TMR exhibited very similar intracellular distribution (Figure 1b). Similar results were obtained after treatment of HeLa cells with 5 μM CPP12TMR and CPP12-2TMR for 2 h in the presence of 10% FBS (Figure S7). Note that under the assay condition (5 μM CPP and 1% or 10% FBS), CPP12 and CPP12-2 are expected to have similar total cellular uptake (Table 2). HeLa cells treated with lower concentrations of CPP12TMR or CPP12-2TMR (0.2 and 1.0 μM) for 2 h produced predominantly punctate fluorescence in the cytoplasmic region (Figure S8), consistent with less efficient endosomal escape at lower CPP concentrations (Figure 1). Treatment of HeLa cells with 5 μM fluorescein-labeled CPP12 and CPP12-2 for 2 h in the presence of 1% FBS resulted in similar observations (Figure S9).
Figure 1.

Confocal microscopic images of HeLa cells after treatment with 5 μM TMR-labeled CPP12-2 (a) or CPP12 (b) for 2 h in the presence of 1% FBS. I, TMR fluorescence; II, DIC; III, Merge of I and II. Scale bars, 20 μm.
The cytosolic entry efficiencies of CPP12-2 and CPP12 were also evaluated by direct fluorescence imaging. HeLa cells were treated with 5 μM NF-labeled CPP12 or CPP12-2 for 2 h in the presence of 10% FBS and imaged by confocal microscopy. Diffuse fluorescence throughout the cell volume was observed for both CPPs, although the fluorescence intensity was visibly lower for CPP12 (Figure 2a, b). Intense punctate fluorescence was also present in the cytoplasmic region, which may represent cytosolic CPPs associated with intracellular structures (e.g., microfilaments). To avoid the punctate fluorescence in cytosolic regions, we estimated the intracellular CPP concentration by quantifying the fluorescence intensity in nuclear regions with diffuse and evenly distributed fluorescence (Figure S10). Comparison of the average values (MFINF) from a large population of cells (145 and 469 cells for CPP12 and CPP12-2, respectively) revealed a 3.2-fold higher cytosolic/nuclear concentration for CPP12-2 than CPP12 (Figure 2c).
Figure 2.

Quantitation of cytosolic/nuclear CPP concentration by fluorescence imaging. (a,b) Representative confocal microscopic images of HeLa cells after treatment with 5 μM CPP12-2NF (a) or CPP12NF (b) for 2 h in the presence of 10% FBS. I, NF fluorescence; II, DIC. Scale bars, 10 μm. (c) Comparison of cytosolic/nuclear NF fluorescence intensities of HeLa cells treated with 5 μM CPP12NF (145 cells) or CPP12-2NF (469 cells). ****, p ≤ 0.0001.
Finally, to test whether the differential sensitivity of CPP12 and CPP12-2 to serum concentration is caused by difference in binding to serum proteins, we determined the binding affinity of FITC-, NF-, or TMR-labeled CPP12 and CPP12-2 to serum proteins by fluorescence polarization (FP). CPP12FITC and CPP12-2FITC exhibited apparent KD values of 29 ± 3 μM and 30 ± 5 μM, respectively, to proteins in FBS (Figure S11). CPP12TMR and CPP12-2TMR showed apparent KD values of 27 ± 9 μM and 68 ± 9 μM, while CPP12NF and CPP12-2NF showed apparent KD values of 1.3 ± 0.3 μM and 1.2 ± 0.4 μM, respectively. Thus, although the dye structure significantly affects the binding affinity of CPP12 and CPP12-2 to serum proteins, the two CPPs bind to serum proteins with very similar affinities, excluding protein binding per se as a significant contributor to the differential sensitivity of CPP12 and CPP12-2 to serum concentration. Rather, binding to serum proteins results in lower free CPP concentration, under which CPP12-2 is more active than CPP12.
Improved Cytosolic Delivery of Peptidyl Cargos.
We further assessed the ability of CPP12 and CPP12-2 to deliver functional cargos into the cytosol of mammalian cells. To avoid any complication from serum proteins and simplify the data interpretation, we conducted the cargo delivery experiments in the presence of minimal FBS (0 or 1%). We first adapted a previously established luciferase complementation assay42 and conjugated HiBit, an 11-residue peptide derived from NanoLuc (VSGWRLFKKIS), to CPP12 or CPP12-2 through a disulfide bond (Figure S1; CPP12-HiBit and CPP12-2-HiBit). HEK293T cells were transfected with plasmid DNA coding for an 18-kDa subunit of NanoLuc, LgBit. When successfully delivered into the cytosol of the transfected HEK293T cells, HiBit binds to LgBit with high affinity (KD = 700 pM) to form a catalytically active luciferase, NanoLuc. Treatment of the HEK293T cells with the CPP-HiBit conjugates resulted in dose-dependent increases in luminescence, with 200- and 240-fold luciferase induction at 10 μM CPP12-HiBit and CPP12-2-HiBit, respectively (Figure 3). While CPP12-2 delivered HiBit more efficiently than CPP12 at all concentrations tested, greater differences were observed at lower peptide concentrations. In the concentration range of 0.04-0.16 μM, CPP12-2-HiBit produced ~2-fold higher luminescence than CPP12-HiBit, whereas the difference was much smaller at higher concentrations. The unconjugated HiBit resulted in only a small increase in luminescence (20-fold at 10 μM); this residual activity was likely caused by the intrinsic cell permeability of HiBit (which contains both positively charged and hydrophobic residues) and/or secretion of LgBit into the cell medium.43 We also attempted to perform the HiBit assay in the presence of 10% FBS. However, for reasons that are not clear to us, the induction of luminescence was very low (~5-fold induction at 10 uM in 10% FBS).
Figure 3.

Cytosolic delivery of HiBit by CPP12 and CPP12-2 and complementation with transiently expressed LgBit in HEK293T cells in the presence of 1% FBS. Data shown represent the mean ± SD of multiple independent experiments (n = 3 for HiBit and n = 7 for CPP12-HiBit and CPP12-2-HiBit. *, p ≤ 0.05.
We next tested the capacity of CPP12-2 to deliver a peptidyl cargo of potential therapeutic utility. A previously reported cyclic peptidyl inhibitor against the Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid-2 (Nrf2) interaction, Cyclo(GQLDPETGEFL) (P1; KD = 18 nM),44 was conjugated to CPP12 or CPP12-2 through a flexible miniPEG linker (CPP12-P1 and CPP12-2-P1; Figure 4a).
Figure 4.

Intracellular delivery of peptidyl inhibitor of Keap1. (a) Structures of CPP12-P1 and CPP12-2-P1. (b) Binding of P1, CPP12-P1 and CPP12-2-P1 to Keap1 as monitored by fluorescence polarization (FP). Keap1 (40 nM), fluorescein-labeled peptide 2 (20 nM), and increasing concentrations of P1, CPP12-P1 and CPP12-2-P1 were incubated for 1 h and FP values were measured and plotted as a function of peptide concentration. Data shown represent the mean ± SD of three independent experiments. (c) Induction of luciferase expression in HepG2-ARE (Luc) cells by P1, CPP12-P1, and CPP12-2-P1. Data shown represent the mean ± SD of n = 5 independent experiments. *, p ≤ 0.05; **, p ≤ 0.01.
Their binding affinity to the Kelch domain of Keap1 was determined by a FP-based competition assay. Peptides P1, CPP12-P1, and CPP12-2-P1 showed IC50 values of 14 ± 1, 18 ± 1, and 22 ± 1 nM, respectively (Figure 4b). These data indicate that conjugation of P1 to the cyclic CPPs does not significantly change its Keap1-binding affinity. We next examined CPP12-P1 and CPP12-2-P1 for their inhibition of the intracellular Keap1-Nrf2 interaction by using an ARE reporter-HepG2 cell line, which contains a firefly luciferase gene under the transcriptional control of Nrf2.45 Under basal conditions, Nrf2 interacts with Keap1 and is retained in the cytosol or degraded by the proteasome. However, upon blocking the Keap1-Nrf2 interaction, Nrf2 accumulates and translocates into the nucleus, inducing the expression of luciferase. As reported previously,28 P1 did not result in significant increase in luciferase activity up to 5 μM concentration (Figure 4c). On the other hand, both CPP12-P1 and CPP12-2-P1 increased the luciferase expression in a dose-dependent manner, with a maximal induction of three- to four-fold at the highest concentration tested (5 μM). Again, CPP12-2-P1 was more active than CPP12-P1 at all concentrations tested, especially at lower concentrations. For example, at 0.31 μM concentration, CPP12-2-P1 increased the luciferase activity by ~2-fold, whereas CPP12-P1 did not have significant effect at this concentration (Figure 4c).
CPP12-2 Is Non-cytotoxic.
CPP12-2 was assessed for potential cytotoxicity against HeLa cells by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Like CPP12, CPP12-2 did not reduce the viability of HeLa cells at up to 25 μM concentration (Figure S12). However, loss of viability (~40%) was observed at ⩾550 μM concentration for both CPPs.
CONCLUSION
In this study, we have discovered a new cyclic CPP, CPP12-2, which has up to ~4-fold greater cytosolic delivery efficiency than CPP12, one of the most active CPPs known to date. The improved cytosolic delivery efficiency of CPP12-2 is primarily the result of more efficient endosomal escape. Since CPPs generally have poor cytosolic entry efficiencies at low concentrations, the improved endosomal escape and cytosolic entry efficiencies of CPP12-2 at low concentrations renders it an excellent choice for delivering highly potent cargos to achieve robust biological activities at low concentrations.
Supplementary Material
Funding Sources
This work was supported by the National Institutes of Health (GM122459) and Entrada Therapeutics. R.Y. was supported by a visiting professorship from the China Scholarship Council. Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. This facility is supported in part by grant P30 CA016058, National Cancer Institute, Bethesda, MD.
ABBREVIATIONS
- Bta
L-3-benzothienylalanine
- CPP
cell-penetrating peptide
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization
- Nal
naphthylalanine
- NF
naphthofluorescein
- TMR
tetramethylrhodamine
Footnotes
Supporting Information
Experimental details and additional data are available free of charge online at http://pubs.acs.org.
The authors declare the following competing financial interests: A patent application has been filed on the findings of this work. D.P. is a co-founder and owns equity of Entrada Therapeutics, Inc.
REFERENCES
- (1).Futaki S Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Deliv. Rev 2005, 57 (4), 547–558. [DOI] [PubMed] [Google Scholar]
- (2).Stanzl EG; Trantow BM; Vargas JR; Wender PA Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc. Chem. Res 2013, 46 (12), 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bechara C; Sagan S Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587 (12), 1693–1702. [DOI] [PubMed] [Google Scholar]
- (4).Rothbard JB; Garlington S; Lin Q; Kirschberg T; Kreider E; McGrane PL; Wender PA; Khavari PA Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med 2000, 6, 1253–1257. [DOI] [PubMed] [Google Scholar]
- (5).Gupta B; Levchenko T; Torchilin V Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev 2005, 57 (4), 637–651. [DOI] [PubMed] [Google Scholar]
- (6).Wadia J; Dowdy S Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Deliv. Rev 2005, 57 (4), 579–596. [DOI] [PubMed] [Google Scholar]
- (7).Fawell S; Seery J; Daikh Y; Moore C; Chen LL; Pepinsky B; Barsoum J Tat-mediated delivery of heterologous proteins into cells. Proc. Natl Acad. Sci. U. S. A 1994, 91 (2), 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schwarze SR; Ho A; Vocero-Akbani A; Dowdy SF In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999, 285 (5433), 1569–1572. [DOI] [PubMed] [Google Scholar]
- (9).Säälik P; Elmquist A; Hansen M; Padari K; Saar K; Viht K; Langel Ü; Pooga M Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjugate Chem. 2004, 15 (6), 1246–1253. [DOI] [PubMed] [Google Scholar]
- (10).Patel SG; Sayers EJ; He L; Narayan R; Williams TL; Mills EM; Allemann RK; Luk LYP; Jones AT; Tsai Y-H Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep 2019, 9 (1), 6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gaston J; Maestrali N; Lalle G; Gagnaire M; Masiero A; Dumas B; Dabdoubi T; Radošević K; Berne P-F Intracellular delivery of therapeutic antibodies into specific cells using antibody-peptide fusions. Sci. Rep 2019, 9 (1), 18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Morris MC; Depollier J; Mery J; Heitz F; Divita G A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001, 19 (12), 1173–1176. [DOI] [PubMed] [Google Scholar]
- (13).Akishiba M; Takeuchi T; Kawaguchi Y; Sakamoto K; Yu H-H; Nakase I; Takatani-Nakase T; Madani F; Gräslund A; Futaki S Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem 2017, 9 (8), 751–761. [DOI] [PubMed] [Google Scholar]
- (14).Erazo-Oliveras A; Najjar K; Dayani L; Wang T-Y; Johnson GA; Pellois J-P Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014, 11 (8), 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lehto T; Kurrikoff K; Langel Ü Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv 2012, 9 (7), 823–836. [DOI] [PubMed] [Google Scholar]
- (16).Verdurmen WPR; Mazlami M; Plückthun A A quantitative comparison of cytosolic delivery via different protein uptake systems. Sci. Rep 2017, 7, 13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sarko D; Beijer B; Garcia Boy R; Nothelfer EM; Leotta K; Eisenhut M; Altmann A; Haberkorn U; Mier W The pharmacokinetics of cell-penetrating peptides. Mol. Pharmaceutics 2010, 7 (6), 2224–2231. [DOI] [PubMed] [Google Scholar]
- (18).Bode SA; Löwik DWPM Constrained cell penetrating peptides. Drug Discov Today Technol. 2017, 26, 33–42. [DOI] [PubMed] [Google Scholar]
- (19).Lättig-Tünnemann G; Prinz M; Hoffmann D; Behlke J; Palm-Apergi C; Morano I; Herce HD; Cardoso MC Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun 2011, 2, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mandal D; Nasrolahi Shirazi A; Parang K Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed 2011, 50 (41), 9633–9637. [DOI] [PubMed] [Google Scholar]
- (21).Qian Z; Liu T; Liu Y-Y; Briesewitz R; Barrios AM; Jhiang SM; Pei D Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol 2013, 8 (2), 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Horn M; Reichart F; Natividad-Tietz S; Diaz D; Neundorf I Tuning the properties of a novel short cell-penetrating peptide by intramolecular cyclization with a triazole bridge. Chem. Commun 2016, 52, 2261–2264. [DOI] [PubMed] [Google Scholar]
- (23).Amoura M; Illien F; Joliot A; Guitot K; Offer J; Sagan S; Burlina F Head to tail cyclisation of cell-penetrating peptides: impact on GAG-dependent internalisation and direct translocation. Chem. Commun 2019, 55 (31), 4566–4569. [DOI] [PubMed] [Google Scholar]
- (24).Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 2016, 55 (18), 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Qian Z; LaRochelle JR; Jiang B; Lian W; Hard RL; Seiner NG; Luechapanichkul R; Barrios AM; Pei D Early endosomal escape of a cyclic cell-penetrating peptide allowseffective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dougherty PG; Karpurapu M; Koley A; Lukowski JK; Qian Z; Srinivas Nirujogi T; Rusu L; Chung S; Hummon AB; Li HW; Christman JW; Pei D A peptidyl inhibitor that blocks calcineurin–NFAT interaction and prevents acute lung injury. J. Med. Chem 2020, 63 (21), 12853–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dougherty PG; Wen J; Pan X; Koley A; Ren J; Sahni A; Basu R; Salim H; Appiah Kubi G; Qian Z; Pei D Enhancing the cell permeability of stapled peptides with a cyclic cell-penetrating peptide. J. Med. Chem 2019, 62 (22), 10098–10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Salim H; Song J; Sahni A; Pei D Development of a cell-permeable cyclic peptidyl inhibitor against the Keap1–Nrf2 interaction. J. Org. Chem 2020, 85 (3), 1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nischan N; Herce HD; Natale F; Bohlke N; Budisa N; Cardoso MC; Hackenberger CPR Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed 2015, 54 (6), 1950–1953. [DOI] [PubMed] [Google Scholar]
- (30).Herce HD; Schumacher D; Schneider AFL; Ludwig AK; Mann FA; Fillies M; Kasper M-A; Reinke S; Krause E; Leonhardt H; Cardoso MC; Hackenberger CPR Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nature Chem. 2017, 9, 762–771. [DOI] [PubMed] [Google Scholar]
- (31).Schneider AFL; Wallabregue ALD; Franz L; Hackenberger CPR Targeted subcellular protein delivery using cleavable cyclic cell-penetrating peptides. Bioconjugate Chem. 2019, 30 (2), 400–404. [DOI] [PubMed] [Google Scholar]
- (32).Zhang W; Lin M; Yan Q; Budachetri K; Hou L; Sahni A; Liu H; Han NC; Lakritz J; Pei D; Rikihisa Y An intracellular nanobody targeting T4SS effector inhibits Ehrlichia infection. Proc. Natl Acad. Sci. U. S. A 2021, 118, e2024102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shintaku J; Lian W; Li X; Tadesse S; Theng Ting M; Xu X; Long NE; Ramirez-Paz J; Qian Z; Sethuraman N; Hirano M Thymidine phosphorylase intracellular enzyme replacement therapy in a murine model of mitochondrial neurogastrointestinal encephalopathy (MNGIE). Neurology 2020, 94, 3975. [Google Scholar]
- (34).Soudah T; Khawaled S; Aqeilan RI; Yavin E AntimiR-155 cyclic peptide–PNA conjugate: synthesis, cellular uptake, and biological activity. ACS Omega 2019, 4 (9), 13954–13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cai B; Kim D; Akhand S; Sun Y; Cassell RJ; Alpsoy A; Dykhuizen EC; Van Rijn RM; Wendt MK; Krusemark CJ Selection of DNA-encoded libraries to protein targets within and on Living Cells. J. Am. Chem. Soc 2019, 141 (43), 17057–17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sahni A; Qian Z; Pei D Cell-penetrating peptides escape the endosome by inducing vesicle budding and collapse. ACS Chem. Biol 2020, 15, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mäger I; Langel K; Lehto T; Eiríksdóttir E; Langel U The role of endocytosis on the uptake kinetics of luciferin-conjugated cell-penetrating peptides. Biochim Biophys Acta. 2012, 1818, 502–511. [DOI] [PubMed] [Google Scholar]
- (38).Lucchino M; Billet A; Bai SK; Dransart E; Hadjerci J; Schmidt F; Wunder C; Johannes L Absolute quantification of drug vector delivery to the cytosol. Angew. Chem. Int. Ed 2021, 60, 14824–14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lönn P; Kacsinta AD; Cui XS; Hamil AS; Kaulich M; Gogoi K; Dowdy SF Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci. Rep 2016, 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Song J; Qian Z; Sahni A; Chen K; Pei D Cyclic cell-penetrating peptides with single hydrophobic groups. ChemBioChem 2019, 20 (16), 2085–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Qian Z; Dougherty PG; Pei D Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluorophore. Chem. Commun 2015, 51 (11), 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schwinn MK; Machleidt T; Zimmerman K; Eggers CT; Dixon AS; Hurst R; Hall MP; Encell LP; Binkowski BF; Wood KV CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol 2018, 13 (2), 467–474. [DOI] [PubMed] [Google Scholar]
- (43).Teo SLY; Rennick JJ; Yuen D; Al-Wassiti H; Johnston APR; Pouton CW Unravelling cytosolic delivery of cell penetrating peptides with a quantitative endosomal escape assay. Nat Commun. 2021, 12 (1), 3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lu MC; Jiao Q; Liu T; Tan SJ; Zhou HS; You QD; Jiang ZY Discovery of a head-to-tail cyclic peptide as the Keap1-Nrf2 protein-protein interaction inhibitor with high cell potency. Eur. J. Med. Chem 2018, 143, 1578–1589. [DOI] [PubMed] [Google Scholar]
- (45).Lee JM; Johnson JA An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem Mol. Biol 2004, 37 (2), 139–143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.