

Abstract
A Bayesian phase I-II design is presented that optimizes the dose of a new agent within predefined prognostic subgroups. The design is motivated by a trial to evaluate targeted agents for treating metastatic clear cell renal carcinoma, where a prognostic risk score defined by clinical variables and biomarkers is well established. Two clinical outcomes are used for dose-finding, time-to-toxicity during a prespecified follow-up period, and efficacy characterized by ordinal disease status evaluated at the end of follow-up. A joint probability model is constructed for these outcomes as functions of dose and subgroup. The model performs adaptive clustering of adjacent subgroups having similar dose-outcome distributions to facilitate borrowing information across subgroups. To quantify toxicity-efficacy risk-benefit trade-offs that may differ between subgroups, the objective function is based on outcome utilities elicited separately for each subgroup. In the context of the renal cancer trial, a design is constructed and a simulation study is presented to evaluate the design’s reliability, safety, and robustness, and to compare it to designs that either ignore subgroups or run a separate trial within each subgroup.
Keywords: adaptive randomization, Bayesian phase I-II clinical trial design, clustering, dose finding, patient prognostic subgroups
1 |. INTRODUCTION
In many medical settings, multiple prognostic factors are used to define patient prognostic risk subgroups that are expected to have different treatment effects. Such risk subgroups often are used by physicians to guide their therapeutic decisions. As a motivating example, we consider a phase I-II trial in metastatic clear cell renal cell carcinoma (mRCC). Patients with mRCC often are classified by their International Metastatic Renal-Cell Carcinoma Database Consortium (IMDC) prognostic risk scores. IMDC scores are defined based on a combination of clinical variables and biomarkers, including anemia, thrombocytosis, neutrophilia, hypercalcemia, Karnofsky performance status, and time from diagnosis to treatment.1 It is well established that IMDC risk scores are informative in predicting treatment outcomes for patients with mRCC. IMDC risk score is recommended by the National Comprehensive Cancer Network (NCCN) guidelines to guide treatment selection for mRCC, and it is used very commonly by oncologists. For example, Heng et al1 formed three prognostic subgroups of mRCC patients using IMDC risk scores: favorable (IMDC = 0), intermediate (IMDC = 1 or 2), and poor (IMDC ≥ 3), and reported that these subgroups had substantially different overall survival time distributions following first-line VEGF-targeted treatment. Motzer et al2 combined the intermediate and poor subgroups and compared the combination of immune checkpoint inhibitors nivolumab plus ipilimumab (N+I) to sunitinib within each of the favorable and intermediate/poor subgroups.
In this article, we develop a Bayesian phase I-II clinical trial design that optimizes prognostic subgroup-specific doses based on time-to-toxicity and ordinal efficacy outcomes. Our motivating application is a trial of sitravatinib, a tyrosine kinase inhibitor, combined with fixed doses of N+I, in patients with mRCC. The five doses of sitravatinib considered are 20, 40, 60, 80, and 120 mg, hereafter denoted by d1, … , d5. While targeted treatments can achieve durable responses in mRCC patients, the magnitudes of their effects often vary due to substantial prognostic heterogeneity, as shown in Heng et al1 and Motzer et al.2 The study reported by Motzer et al2 found that the effects of N+I on objective response, defined by RECIST, vary with IMDC risk subgroups. Consequently, it may be anticipated that the effects of sitravatinib dose, combined with N+I, also may vary with IMDC subgroups. Despite the established prognostic value of IMDC prognostic risk score, however, to date it has not been used explicitly in a dose-finding clinical trial design. The goal of our proposed design is to make decisions, including optimal dose selection and identification of excessively toxic or inefficacious doses, that may differ between prognostic risk subgroups.
Currently, there is a limited literature on phase I-II designs for finding subgroup-specific doses. For phase I trials based on toxicity, O’Quigley and Conaway,3 Iasonos and O’Quigley,4 and Horton et al5 considered the problem of finding the maximum-tolerated doses (MTD) for heterogeneous patient populations. Horton et al5 developed a two-stage design where a rule-based allocation under a shifted version of the continual reassessment model is used in each stage to find subgroup specific MTDs based on a binary toxicity outcome. However, there is a need for dose-finding designs that can handle more complex studies involving complex clinical outcomes and heterogeneous groups of patients, as in our motivating trial.
To provide a basis for subgroup-specific decision making, we build a Bayesian hierarchical model for regression of clinical outcomes on dose d and subgroup g. Our motivating trial has two co-primary patient outcomes, the time YT to severe toxicity over a 12-week follow-up period after the initiation of treatment, and disease severity, YE, evaluated as an ordinal categorical variable at the end of this follow up period. The possible values of YE are progressive disease (PD), stable disease (SD), partial response (PR), and complete response (CR), a very common ordinal response categorization in oncology. The regression model for [YT, YE |d, g] assumes additive dose and subgroup effects for each outcome. In general, we denote ordinal risk subgroups by {1, 2, … , G}, with g = 1 the lowest and g = G the highest risk. The mRCC trial accounts for three IMDC risk subgroups: favorable, intermediate, and poor, denoted by g = 1, 2, and 3. Our model exploits the prognostic subgroups established in previous studies, and avoids the task of attempting to identify new patient subgroups from baseline covariates, since this would be very difficult to do reliably given the limited sample size and sequentially adaptive decision making in a phase I-II trial. To improve the accuracy of subgroup-specific decisions, the model and design allow adaptive clustering of adjacent subgroups based on intermediate data during a trial. This may be especially useful in settings with sparse subgroups. While we will develop the design in the context of the mRCC trial, the model and method can be applied generally to design phase I-II trials with time-to-toxicity and ordinal efficacy outcomes, and ordinal prognostic subgroups.
Many clinical trial designs that base decisions on elicited utilities of multiple clinical outcomes have been proposed, including Thall and Nguyen6 for phase I-II dose-finding, Lee et al7 for two-cycle phase I-II dose-finding, Murray et al8 and Murray et al9 for randomized treatment comparisons, and Lin et al10 for a basket trial to jointly optimize dose and schedule. While these designs provide coherent decision making frameworks for homogeneous patient populations, assuming that one utility function U(y) is appropriate for all patients may be less useful for prognostic subgroup-specific decision making. For example, in mRCC subjective risk-benefit trade-offs of clinical outcomes can vary substantially with IMDC score, and even produce opposite conclusions.11 mRCC patients with favorable risk often prefer less intensive treatments with low risks of toxicity, while mRCC patients with poor risk may prefer more potent treatment combinations with a higher risk of toxicity,12 in order to increase the probability of a substantive anti-disease effect. This implies that the mRCC subgroups should have different outcome utility functions. This likely is also the case in other diseases where prognostic level affects risk-benefit trade-offs. To address this, we extend the usual utility-based approach by eliciting subgroup-specific utility functions and using these as a basis for making subgroup-specific decisions.
Section 2 presents the probability model. Section 3 describes and illustrates how the subgroup-specific utility function is constructed, and Section 4 presents the design. In Section 5, a simulation study is presented to evaluate the design’s operating characteristics (OCs) and robustness, and compare it to the two designs that either make the same decisions for all subgroups or run a separate trial for each subgroup. The simulations show that the proposed design produces much more accurate subgroup-specific decisions compared to these two more conventional approaches. We close with a brief discussion in Section 6.
2 |. PROBABILITY MODEL
2.1 |. Sampling and frailty models
For interim sample size n(t) ≤ Nmax at trial time t in days, index patients in order of enrollment by i = 1, … , n(t), with trial entry times 0 ≤ e1 ≤ e2 ≤ · · · ≤ en(t). Subgroups are predefined using prognostic scores, and treatment effects of a study drug in adjacent subgroups may be either similar or significantly different. At the time of each patient’s enrollment, their prognostic risk score is evaluated and the patient is classified into one of the G ordinal risk subgroups. Let gi ∈ {1, … , G} denote the subgroup of the ith patient. In practice, the prevalences of the subgroup will differ from 1∕G, and the numbers of patients enrolled in the subgroups will differ accordingly.
Given fixed follow-up time C, for patient i severe toxicity is monitored continuously until ei + C. The toxicity event occurs at trial time ei + Yi,T, with Yi,T < C if it is observed. Let denote the observed time from ei to toxicity Yi,T or right-censoring, with binary indicator δi,T = 1 if and δi,T = 0 otherwise. At trial time t > ei, if Yi,T ≤ min(t − ei, C), then is the observed time of toxicity, and δi,T = 1. If Yi,T > min(t − ei, C), then is the time of independent right censoring with δi,T = 0. In particular, the event of and δi,T = 0 implies that no toxicity has occurred up to time t. Ordinal disease status Yi,E is observed at time ei + C, and we define δi,E = 1 at any t ≥ ei + C. For t < ei + C, Yi,E is not observed and δi,E = 0. Denote the ordinal disease status outcome of patient i at time ei + C by Yi,E, taking on values in {0, 1, … , K − 1}. For example, disease severity levels {PD, SD, PR, CR}, are represented by Yi,E = 0, 1, 2, 3 and K = 4. Disease status is evaluated independently of (Yi,T, δi,T).
For the dose-outcome model, we denote the doses standardized to have mean 0 and variance 1 by {d1, … , dM}. In the renal cancer trial, given the raw doses {20, 40, 60, 80, 120} mg/day of oral sitravatinib, the standardized doses are {−1.144, −0.624, −0.104, 0.416, 1.456}, with M = 5. For each patient i = 1, … , n(t), denote the dose by d[i], subgroup by gi, and the latent frailty vector by . We assume conditional independence of Yi,T and Yi,E given γi, specify marginal models, and obtain a joint distribution by averaging over the distribution of γi.
For the toxicity event time Yi,T, if patient i in prognostic subgroup gi is treated with dose d[i], we assume a proportional hazards (PH) model with conditional hazard function
| (1) |
where h0T(t) is a baseline hazard function and αT = (αT,1, … , αT,G) are the prognostic subgroup effects on the risk of toxicity. In (1), the regression function ηT(d[i]) accounts for dose effects on toxicity, and is the additive effect of subgroup gi. We will give a parametric model for ηT(dm) below. We fix αT,1 = 0 to avoid identifiability problems with h0T. To ensure that the toxicity hazard is non-decreasing in the ordinal risk subgroups, we impose the ordering constraint αT,1 ≤ · · · ≤ αT,G. The survival function of [Yi, T|gi, d[i]] is
We assume a multinomial probit model for ordinal disease status Yi,E. Denote subgroup-specific cutoffs ug,0 < ug,1 < … < ug,K for each g. The conditional marginals are
| (2) |
for k = 0, … , K − 1, where αE = (αE,1, … , αE,G) is the vector of prognostic subgroup effects on Yi,E and is a fixed hyperparameter. In (2), Φ(·|a, b2) denotes the cumulative distribution function of a normal distribution with mean a and variance b2. We let ug,k, k = 2, … , K − 1 be random, while fixing ug,1 = 0 for all g to ensure identifiablilty. We set ug,0 = −∞ and ug,K = ∞ for all g, so . The function ηE quantifies the dose effect on YE. Subgroup effects are accounted for through ug,k and αE,g, and various patterns in the relationships between subgroup and the efficacy outcome can be accommodated flexibly. We assume αE,1 ≥ · · · ≥ αE,G to ensure that the probability of PD is non-decreasing in subgroups. No restriction by g is imposed on ug,k for k = 2, … , K − 1, and under the model P(Yi,E ≤ k|gi, d[i],𝛾i,T) is not necessarily stochastically increasing in subgroups for k > 0. If desired, stochastic ordering of the distribution of YE in g can be imposed by requiring u1,k − αE,1 ≥ · · · ≥ uG,k − αE,G for all k.
We model the relationship between Yi,j and d[i] through the regression function
| (3) |
that appears in the linear component of each marginal, j = E, T. Dose is multiplied by 10 to stabilize numerical computations. We assume 𝛽j,1, and , so that ηj(d), P(YT ≤ CT|g, d, γ), and P(YE ≥ k|g, d, γ) each increases in d. The functional form in (3) may take many different shapes, including an “S” shape that allows the probabilities to plateau at some dose, with ηj → 0 as d → −∞, while ηj → βj3 as d → ∞.
Let θ denote the vector of all model parameters and the vector of all fixed hyperparameters, which we will specify below. The joint likelihood of all observable outcomes of patient i, conditional on the latent frailty vector γi, subgroup gi, dose d[i], and model parameters, is the product likelihood given by
| (4) |
We assume with random Ω. We include γi,1, … , γi,n(t) in the model to account for between-patient heterogeneity not explained by d[i] and gi, with correlations among the γi,j’s inducing dependence between Yi,E and Yi,T. Frailty models have been used widely for multivariate failure time data in a wide variety of settings, including competing risks,13 and a phase I-II design with a complex 5-variate time-to-event outcome.14 The joint distribution of Yi = (Yi,T, Yi,E) is obtained by integrating over the frailty distribution,
2.2 |. Subgroup clustering and posterior computation
To reduce notation, temporarily suppress patient index, i. We allow the possibility of adaptively combining prognostic subgroups that have similar subgroup effects on Y, based on the observed trial data, by introducing latent cluster membership indicators for the subgroups, z = (z1, … , zG). To account for risk subgroup ordinality, the model allows only adjacent subgroups to be combined. Each set of one or more combined subgroups is a cluster, and the R ≤ G clusters are indexed consecutively by r = 1, … , R. For example, if G = 3, the four possible configurations of z = (z1, z2, z3) are (1, 1, 1), (1, 1, 2), (1, 2, 2), and (1, 2, 3) which define, respectively, R = 1, 2, 2, and 3 clusters. The case with z = (1, 1, 2) is that where subgroups 1 and 2 are combined and subgroup 3 is distinct, and the clusters are {1,2} indexed by r = 1 and the singleton {3} indexed by r = 2. To implement the clustering, we set z1 = 1 and assume P(zg = zg−1|zg−1) = ξg and P(zg = zg−1 + 1|zg−1) = 1 − ξg for subgroups g = 2, … ,G, so the prior of z is
| (5) |
where I(A) = 1 if A is true and 0 otherwise. Under this model, subgroup g may join the cluster to which subgroup g − 1 belongs with probability ξg, so zg = zg−1, and with the remaining probability, 1 − ξg, subgroup g may start a new cluster, zg = zg−1 + 1. This ensures non-decreasing ordering of cluster membership indicators, zg ≤ zg+1, and subgroup g cannot be in the cluster to which subgroup g − 2 belongs if subgroup g − 1 is not in the cluster. The cluster membership indicator zG of the highest risk subgroup is R, and both R and z are random. If the subgroups are not ordinal, the ordering constraint on zg is not imposed and any model-based clustering approach, such as using a Gaussian mixture model, can be assumed as a prior for z, e.g., Chapple and Thall (2018).15
We assume that subgroup effects are identical within each cluster. To formalize this, we introduce cluster-specific parameters and for clusters r = 1, … , R, denoting for j = T or E, and let . A cluster-specific cutoff vector is defined similarly, with each the same for all subgroups in cluster r. The probabilities and do not change with g in each cluster.
We next specify priors for and u⋆. Given z, we assume normal distributions with order constrains on , given by
| (6) |
where and , and and are fixed hyperparameters. Due to the ordering on , higher risk subgroups have larger probabilities of toxicity and PD. For k = 2, … , K − 1 and r = 1, … , R, we let , and assume with prior mean ρr−1,k and prior variance ρr−1,k/κr,k, and fix ρ0,k and κr,k. The distributions of z in (5), in (6), and u⋆ jointly define the distributions of αj and u. In the posterior computations, z, and u⋆ are included as a parameter subvector of θ instead of αj and u.
Clustering is done to borrow strength by combining subgroups having similar effect distributions. However, optimal doses still are chosen for subgroups using their respective utilities and the distributions of αj,g and ug. Doses are not chosen for clusters. In clinical practice, physicians often cluster prognostic subgroups in an ad hoc manner to simplify their decision making. In contrast, our adaptive clustering algorithm is a Bayesian statistical methodology that forms subgroup clusters based on data.
We specify priors for the model parameters h0T(t), β = {βj,ℓ,j = T or E, and ℓ = 1, 2, 3}, and Ω as follows. For h0T, we assume a constant hazard over (0, C) and let , since previous studies indicate that it is reasonable to assume a constant hazard during the follow-up period.2,16,17 If a hazard that varies over time is more appropriate, other models such as a Weibull or piecewise exponential distribution can be used. To ensure that relationships of doses with the outcomes are monotonically increasing, we assume normal or truncated normal distributions for βj,ℓ, j = T,E and ℓ = 1, 2, 3, accordingly. We assume βj,1 and βj,3 have normal distributions truncated below at 0, for βj,ℓ > 0, ℓ = 1,3. We assume normal prior distributions for βj,2, j = E, T. To complete the prior specification of γi, we let Ω|ν, Ω0 ~ inv-Wishart(ν, Ω0) for fixed ν > 1 and 2 × 2 positive definite hyperparameter matrix Ω0.
Collecting terms, θ = (hT0, β, α⋆, z, ρ, Ω) denotes the vector of all model parameters, where β = {βj,ℓ,j = T,E,ℓ = 1, 2, 3}, , and ρ = {ρr,k, r = 1, … , R, k = 2, … , K − 1}. For j = E, T, ℓ = 1, 2, 3, r = 1, … , G, and k = 2, … , K − 1, we denote , , ξ = (ξ2, … , ξG), , , ρ0 = {ρ0,k}, and κ = {κr,k}, so the vector of all fixed hyperparameters is . For the mRCC trial design, we established by using data from the phase 3 CheckMate 214 trial described by Motzer et al,2 and elicited prior probabilities from the clinical investigators. In the CheckMate 214 trial, patients were categorized into favorable, intermediate, and poor risk subgroups by their IMDC risk status, and randomly assigned to one of two treatments including a combination of nivolumab and ipilimumab. Table 1 of Motzer et al2 reports P(YE = k) for g = 1 and g > 1 when no sitravatinib is given, that is, ηj = 0 under our model in (3). Using the historical data and elicited probabilities, we fit our model to pseudo data simulated from the elicited probabilities or the historical data, and used posterior means to determine location hyper-parameters. We calibrated dispersion hyper-parameters to reflect prior uncertainty, and examined the calibrated with pseudo data simulated under various settings. Details of prior calibration are given in Supplementary Section 2.
TABLE 1.
Simulation results
| d 1 | d 2 | d 3 | d 4 | d 5 | d 1 | d 2 | d 3 | d 4 | d 5 | ||
|
|
|
||||||||||
| ztrue = (1, 2, 3) | Scenario 1 | Scenario 2 | |||||||||
| U true | g = 1 | 56.89 | 51.38 | 47.69 | 43.81 | 39.46 | 34.34 | 35.57 | 34.21 | 32.38 | 32.45 |
| g = 2 | 58.17 | 53.25 | 51.91 | 49.83 | 47.67 | 35.85 | 38.94 | 40.73 | 40.27 | 41.70 | |
| g = 3 | 39.85 | 35.41 | 33.63 | 32.13 | 30.50 | 34.59 | 38.40 | 41.17 | 41.94 | 43.86 | |
| g = 1 | 0.05 | 0.20 | 0.35 | 0.50 | 0.65 | 0.25 | 0.35 | 0.50 | 0.60 | 0.65 | |
| g = 2 | 0.07 | 0.28 | 0.47 | 0.64 | 0.79 | 0.32 | 0.44 | 0.61 | 0.71 | 0.76 | |
| g = 3 | 0.11 | 0.39 | 0.61 | 0.78 | 0.90 | 0.38 | 0.51 | 0.68 | 0.78 | 0.82 | |
| g = 1 | 0.16 | 0.16 | 0.12 | 0.09 | 0.07 | 0.50 | 0.37 | 0.25 | 0.20 | 0.16 | |
| g = 2 | 0.25 | 0.25 | 0.20 | 0.16 | 0.12 | 0.57 | 0.43 | 0.31 | 0.25 | 0.20 | |
| g = 3 | 0.57 | 0.56 | 0.50 | 0.43 | 0.37 | 0.63 | 0.50 | 0.37 | 0.31 | 0.25 | |
| D-Sub | g = 1 | 0.84 | 0.06 | 0.03 | 0.01 | 0.03 | 0.00 | 0.02 | 0.03 | 0.00 | 0.02 |
| g = 2 | 0.85 | 0.07 | 0.04 | 0.01 | 0.02 | 0.01 | 0.02 | 0.01 | 0.00 | 0.00 | |
| g = 3 | 0.13 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | |
| D-Comb | all g | 0.75 | 0.04 | 0.02 | 0.00 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D-Sep | g = 1 | 0.62 | 0.14 | 0.10 | 0.02 | 0.10 | 0.05 | 0.02 | 0.06 | 0.03 | 0.07 |
| g = 2 | 0.83 | 0.06 | 0.06 | 0.01 | 0.02 | 0.02 | 0.02 | 0.01 | 0.00 | 0.00 | |
| g = 3 | 0.10 | 0.03 | 0.04 | 0.02 | 0.02 | 0.08 | 0.03 | 0.05 | 0.01 | 0.01 | |
| D-Sub | g = 1 | 0.06 | 0.07 | 0.32 | 0.55 | 0.62 | 0.99 | 0.97 | 0.95 | 0.97 | 0.97 |
| g = 2 | 0.02 | 0.05 | 0.54 | 0.92 | 0.95 | 0.98 | 0.96 | 0.99 | 1.00 | 1.00 | |
| g = 3 | 0.86 | 0.91 | 0.99 | 1.00 | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 | 1.00 | |
| D-Comb | all g | 0.18 | 0.23 | 0.74 | 0.96 | 0.97 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| D-Sep | g = 1 | 0.06 | 0.06 | 0.20 | 0.43 | 0.51 | 0.94 | 0.92 | 0.86 | 0.89 | 0.89 |
| g = 2 | 0.03 | 0.07 | 0.57 | 0.91 | 0.94 | 0.97 | 0.96 | 0.99 | 1.00 | 1.00 | |
| g = 3 | 0.86 | 0.89 | 0.92 | 0.97 | 0.98 | 0.88 | 0.90 | 0.94 | 0.99 | 0.99 | |
| D-Sub | g = 1 | 7.35 | 7.19 | 5.31 | 3.80 | 3.43 | 1.87 | 2.66 | 2.90 | 2.16 | 2.04 |
| g = 2 | 25.71 | 23.94 | 13.13 | 4.55 | 3.31 | 8.90 | 8.53 | 4.60 | 1.76 | 1.16 | |
| g = 3 | 3.88 | 3.06 | 1.30 | 0.30 | 0.08 | 1.67 | 1.81 | 0.66 | 0.16 | 0.05 | |
| D-Comb | g = 1 | 9.68 | 8.43 | 3.90 | 1.41 | 1.07 | 2.44 | 2.08 | 1.27 | 0.54 | 0.45 |
| g = 2 | 24.76 | 22.25 | 10.07 | 3.69 | 2.72 | 6.21 | 5.63 | 3.35 | 1.50 | 1.16 | |
| g = 3 | 7.06 | 6.25 | 2.80 | 1.05 | 0.78 | 1.66 | 1.52 | 0.99 | 0.44 | 0.34 | |
| D-Sep | g = 1 | 6.96 | 6.93 | 5.82 | 4.36 | 3.89 | 7.49 | 7.11 | 5.14 | 3.31 | 2.88 |
| g = 2 | 25.85 | 23.47 | 12.53 | 5.22 | 3.81 | 11.74 | 10.01 | 5.66 | 2.49 | 1.86 | |
| g = 3 | 6.96 | 6.09 | 3.66 | 1.75 | 1.34 | 7.09 | 5.52 | 2.94 | 1.53 | 1.15 | |
| d 1 | d 2 | d 3 | d 4 | d 5 | d 1 | d 2 | d 3 | d 4 | d 5 | ||
|
|
|
||||||||||
| ztrue = (1, 2, 3) | Scenario 3 | Scenario 4 | |||||||||
| U true | g = 1 | 59.95 | 64.84 | 72.19 | 64.57 | 58.12 | 54.07 | 48.75 | 48.63 | 40.37 | 36.04 |
| g = 2 | 64.96 | 70.83 | 79.05 | 74.63 | 70.18 | 56.47 | 54.15 | 55.52 | 49.64 | 46.55 | |
| g = 3 | 56.53 | 63.35 | 73.50 | 65.89 | 60.18 | 51.47 | 52.05 | 54.98 | 50.88 | 48.50 | |
| g = 1 | 0.05 | 0.08 | 0.10 | 0.35 | 0.50 | 0.05 | 0.25 | 0.30 | 0.50 | 0.60 | |
| g = 2 | 0.05 | 0.08 | 0.10 | 0.35 | 0.50 | 0.05 | 0.25 | 0.30 | 0.50 | 0.60 | |
| g = 3 | 0.13 | 0.20 | 0.25 | 0.69 | 0.85 | 0.05 | 0.25 | 0.30 | 0.50 | 0.60 | |
| g = 1 | 0.16 | 0.09 | 0.03 | 0.02 | 0.02 | 0.16 | 0.12 | 0.09 | 0.09 | 0.09 | |
| g = 2 | 0.16 | 0.09 | 0.03 | 0.02 | 0.02 | 0.16 | 0.12 | 0.09 | 0.09 | 0.09 | |
| g = 3 | 0.31 | 0.20 | 0.09 | 0.07 | 0.07 | 0.41 | 0.34 | 0.29 | 0.29 | 0.29 | |
| D-Sub | g = 1 | 0.01 | 0.04 | 0.87 | 0.05 | 0.03 | 0.74 | 0.13 | 0.13 | 0.00 | 0.00 |
| g = 2 | 0.00 | 0.02 | 0.85 | 0.08 | 0.05 | 0.60 | 0.16 | 0.19 | 0.01 | 0.04 | |
| g = 3 | 0.02 | 0.04 | 0.85 | 0.04 | 0.01 | 0.44 | 0.14 | 0.22 | 0.01 | 0.05 | |
| D-Comb | all g | 0.01 | 0.03 | 0.86 | 0.07 | 0.04 | 0.65 | 0.13 | 0.17 | 0.02 | 0.03 |
| D-Sep | g = 1 | 0.06 | 0.11 | 0.47 | 0.14 | 0.21 | 0.64 | 0.16 | 0.12 | 0.02 | 0.05 |
| g = 2 | 0.01 | 0.04 | 0.70 | 0.14 | 0.12 | 0.64 | 0.12 | 0.16 | 0.03 | 0.05 | |
| g = 3 | 0.09 | 0.13 | 0.59 | 0.11 | 0.08 | 0.14 | 0.09 | 0.20 | 0.09 | 0.41 | |
| D-Sub | g = 1 | 0.01 | 0.00 | 0.00 | 0.10 | 0.26 | 0.01 | 0.01 | 0.07 | 0.32 | 0.47 |
| g = 2 | 0.00 | 0.00 | 0.00 | 0.15 | 0.37 | 0.01 | 0.01 | 0.08 | 0.40 | 0.57 | |
| g = 3 | 0.08 | 0.05 | 0.06 | 0.74 | 0.92 | 0.17 | 0.20 | 0.34 | 0.73 | 0.82 | |
| D-Comb | all g | 0.00 | 0.00 | 0.01 | 0.31 | 0.60 | 0.01 | 0.02 | 0.11 | 0.39 | 0.55 |
| D-Sep | g = 1 | 0.02 | 0.01 | 0.01 | 0.12 | 0.20 | 0.05 | 0.05 | 0.14 | 0.36 | 0.46 |
| g = 2 | 0.00 | 0.00 | 0.01 | 0.14 | 0.28 | 0.00 | 0.03 | 0.11 | 0.37 | 0.52 | |
| g = 3 | 0.03 | 0.03 | 0.07 | 0.65 | 0.81 | 0.23 | 0.22 | 0.23 | 0.38 | 0.46 | |
| D-Sub | g = 1 | 5.83 | 6.02 | 5.86 | 5.22 | 4.73 | 6.49 | 6.49 | 5.91 | 4.64 | 4.10 |
| g = 2 | 15.86 | 15.87 | 15.86 | 13.17 | 11.08 | 18.29 | 17.82 | 15.72 | 10.87 | 8.97 | |
| g = 3 | 5.14 | 5.45 | 5.40 | 2.33 | 1.32 | 5.49 | 5.20 | 4.02 | 1.95 | 1.43 | |
| D-Comb | g = 1 | 6.66 | 6.56 | 6.27 | 4.59 | 3.60 | 7.17 | 6.74 | 5.98 | 4.11 | 3.51 |
| g = 2 | 16.93 | 16.97 | 16.61 | 12.33 | 9.10 | 18.88 | 17.62 | 15.17 | 11.15 | 9.16 | |
| g = 3 | 4.74 | 4.80 | 4.60 | 3.52 | 2.62 | 5.28 | 5.04 | 4.20 | 3.03 | 2.64 | |
| D-Sep | g = 1 | 5.95 | 6.01 | 5.88 | 5.27 | 4.90 | 6.87 | 6.74 | 5.80 | 4.50 | 4.04 |
| g = 2 | 15.97 | 15.63 | 15.28 | 13.26 | 11.79 | 18.55 | 17.23 | 15.28 | 11.19 | 9.63 | |
| g = 3 | 5.14 | 5.19 | 4.79 | 2.78 | 2.04 | 4.71 | 4.70 | 4.21 | 3.33 | 3.01 | |
| d 1 | d 2 | d 3 | d 4 | d 5 | d 1 | d 2 | d 3 | d 4 | d 5 | ||
|
|
|
||||||||||
| ztrue = (1, 2, 3) | Scenario 5 | Scenario 6 | |||||||||
| U true | g = 1 | 57.64 | 48.14 | 47.08 | 49.66 | 52.30 | 57.62 | 55.12 | 53.76 | 63.17 | 56.94 |
| g = 2 | 65.44 | 59.25 | 58.92 | 63.74 | 67.67 | 52.93 | 46.64 | 44.45 | 52.85 | 42.68 | |
| g = 3 | 70.05 | 65.27 | 64.67 | 70.81 | 75.03 | 55.65 | 50.80 | 48.84 | 59.80 | 51.27 | |
| g = 1 | 0.03 | 0.28 | 0.30 | 0.35 | 0.38 | 0.03 | 0.10 | 0.13 | 0.15 | 0.30 | |
| g = 2 | 0.03 | 0.28 | 0.30 | 0.35 | 0.38 | 0.13 | 0.38 | 0.46 | 0.52 | 0.80 | |
| g = 3 | 0.03 | 0.28 | 0.30 | 0.35 | 0.38 | 0.13 | 0.37 | 0.46 | 0.52 | 0.80 | |
| g = 1 | 0.12 | 0.12 | 0.12 | 0.07 | 0.04 | 0.16 | 0.16 | 0.16 | 0.05 | 0.05 | |
| g = 2 | 0.16 | 0.16 | 0.16 | 0.09 | 0.06 | 0.20 | 0.20 | 0.20 | 0.07 | 0.07 | |
| g = 3 | 0.16 | 0.16 | 0.16 | 0.09 | 0.05 | 0.20 | 0.20 | 0.20 | 0.07 | 0.07 | |
| D-Sub | g = 1 | 0.64 | 0.01 | 0.00 | 0.01 | 0.34 | 0.21 | 0.02 | 0.03 | 0.26 | 0.49 |
| g = 2 | 0.28 | 0.01 | 0.00 | 0.02 | 0.68 | 0.77 | 0.03 | 0.03 | 0.14 | 0.03 | |
| g = 3 | 0.15 | 0.01 | 0.00 | 0.02 | 0.81 | 0.69 | 0.02 | 0.04 | 0.12 | 0.02 | |
| D-Comb | all g | 0.27 | 0.01 | 0.01 | 0.02 | 0.70 | 0.60 | 0.01 | 0.03 | 0.22 | 0.11 |
| D-Sep | g = 1 | 0.50 | 0.04 | 0.03 | 0.03 | 0.39 | 0.09 | 0.05 | 0.06 | 0.22 | 0.58 |
| g = 2 | 0.24 | 0.02 | 0.01 | 0.03 | 0.70 | 0.77 | 0.03 | 0.03 | 0.12 | 0.03 | |
| g = 3 | 0.10 | 0.04 | 0.04 | 0.05 | 0.77 | 0.45 | 0.09 | 0.11 | 0.17 | 0.17 | |
| D-Sub | g = 1 | 0.00 | 0.01 | 0.02 | 0.02 | 0.03 | 0.00 | 0.00 | 0.01 | 0.02 | 0.03 |
| g = 2 | 0.00 | 0.01 | 0.03 | 0.04 | 0.05 | 0.01 | 0.10 | 0.46 | 0.75 | 0.92 | |
| g = 3 | 0.02 | 0.04 | 0.06 | 0.08 | 0.09 | 0.11 | 0.23 | 0.59 | 0.84 | 0.96 | |
| D-Comb | all g | 0.00 | 0.02 | 0.03 | 0.04 | 0.05 | 0.03 | 0.08 | 0.28 | 0.54 | 0.77 |
| D-Sep | g = 1 | 0.02 | 0.04 | 0.10 | 0.13 | 0.16 | 0.06 | 0.05 | 0.03 | 0.03 | 0.05 |
| g = 2 | 0.00 | 0.02 | 0.04 | 0.06 | 0.07 | 0.03 | 0.14 | 0.49 | 0.77 | 0.91 | |
| g = 3 | 0.00 | 0.02 | 0.07 | 0.12 | 0.15 | 0.04 | 0.11 | 0.33 | 0.59 | 0.77 | |
| D-Sub | g = 1 | 5.63 | 5.64 | 5.48 | 5.40 | 5.41 | 5.59 | 5.58 | 5.53 | 5.40 | 5.29 |
| g = 2 | 15.55 | 15.00 | 14.28 | 13.50 | 13.44 | 24.17 | 20.77 | 13.62 | 7.98 | 4.87 | |
| g = 3 | 4.58 | 4.31 | 3.93 | 3.63 | 3.56 | 6.91 | 5.73 | 3.22 | 1.49 | 0.65 | |
| D-Comb | g = 1 | 6.15 | 5.66 | 5.33 | 5.23 | 5.22 | 8.31 | 7.16 | 5.32 | 3.69 | 2.54 |
| g = 2 | 15.97 | 15.02 | 13.89 | 13.59 | 13.50 | 21.86 | 18.73 | 13.79 | 9.32 | 6.48 | |
| g = 3 | 4.41 | 4.27 | 4.06 | 3.96 | 3.75 | 6.29 | 5.34 | 3.87 | 2.72 | 1.86 | |
| D-Sep | g = 1 | 6.36 | 6.19 | 5.50 | 5.05 | 4.87 | 5.73 | 5.79 | 5.54 | 5.52 | 5.42 |
| g = 2 | 16.10 | 15.14 | 14.17 | 13.37 | 13.22 | 25.84 | 20.59 | 12.58 | 7.08 | 4.46 | |
| g = 3 | 4.47 | 4.47 | 4.00 | 3.55 | 3.51 | 5.64 | 5.23 | 4.00 | 2.72 | 2.20 | |
| d 1 | d 2 | d 3 | d 4 | d 5 | d 1 | d 2 | d 3 | d 4 | d 5 | ||
|
|
|
||||||||||
| ztrue = (1, 2, 3) | Scenario 7 | Scenario 8 | |||||||||
| U true | g = 1 | 39.68 | 42.13 | 46.41 | 50.73 | 54.11 | 46.93 | 53.95 | 46.36 | 40.27 | 36.10 |
| g = 2 | 40.33 | 43.08 | 48.81 | 55.18 | 60.06 | 48.04 | 55.63 | 52.02 | 47.72 | 44.74 | |
| g = 3 | 40.83 | 43.79 | 50.06 | 57.99 | 63.55 | 48.90 | 57.16 | 55.02 | 51.73 | 49.26 | |
| g = 1 | 0.03 | 0.05 | 0.10 | 0.20 | 0.25 | 0.05 | 0.05 | 0.30 | 0.45 | 0.55 | |
| g = 2 | 0.03 | 0.05 | 0.10 | 0.20 | 0.25 | 0.05 | 0.05 | 0.30 | 0.45 | 0.55 | |
| g = 3 | 0.03 | 0.05 | 0.10 | 0.20 | 0.25 | 0.05 | 0.05 | 0.30 | 0.45 | 0.55 | |
| g = 1 | 0.54 | 0.46 | 0.31 | 0.16 | 0.09 | 0.31 | 0.16 | 0.12 | 0.12 | 0.12 | |
| g = 2 | 0.54 | 0.46 | 0.31 | 0.16 | 0.09 | 0.31 | 0.16 | 0.12 | 0.12 | 0.12 | |
| g = 3 | 0.54 | 0.46 | 0.31 | 0.16 | 0.09 | 0.31 | 0.16 | 0.12 | 0.12 | 0.12 | |
| D-Sub | g = 1 | 0.00 | 0.00 | 0.05 | 0.17 | 0.76 | 0.08 | 0.82 | 0.09 | 0.00 | 0.00 |
| g = 2 | 0.00 | 0.00 | 0.04 | 0.11 | 0.84 | 0.04 | 0.76 | 0.17 | 0.00 | 0.03 | |
| g = 3 | 0.00 | 0.00 | 0.02 | 0.10 | 0.82 | 0.03 | 0.68 | 0.21 | 0.01 | 0.05 | |
| D-Comb | all g | 0.00 | 0.00 | 0.03 | 0.12 | 0.82 | 0.06 | 0.74 | 0.15 | 0.00 | 0.03 |
| D-Sep | g = 1 | 0.01 | 0.01 | 0.07 | 0.14 | 0.72 | 0.26 | 0.40 | 0.17 | 0.02 | 0.08 |
| g = 2 | 0.00 | 0.00 | 0.05 | 0.12 | 0.82 | 0.08 | 0.64 | 0.17 | 0.02 | 0.09 | |
| g = 3 | 0.00 | 0.00 | 0.04 | 0.09 | 0.86 | 0.05 | 0.25 | 0.20 | 0.09 | 0.41 | |
| D-Sub | g = 1 | 0.98 | 0.97 | 0.58 | 0.03 | 0.02 | 0.27 | 0.02 | 0.05 | 0.29 | 0.38 |
| g = 2 | 0.66 | 0.54 | 0.15 | 0.02 | 0.02 | 0.04 | 0.00 | 0.07 | 0.38 | 0.49 | |
| g = 3 | 0.77 | 0.68 | 0.23 | 0.06 | 0.05 | 0.08 | 0.02 | 0.12 | 0.51 | 0.60 | |
| D-Comb | all g | 0.90 | 0.85 | 0.34 | 0.03 | 0.03 | 0.10 | 0.02 | 0.12 | 0.41 | 0.51 |
| D-Sep | g = 1 | 0.93 | 0.88 | 0.52 | 0.13 | 0.07 | 0.28 | 0.18 | 0.20 | 0.37 | 0.42 |
| g = 2 | 0.62 | 0.54 | 0.17 | 0.02 | 0.03 | 0.04 | 0.01 | 0.10 | 0.32 | 0.42 | |
| g = 3 | 0.44 | 0.35 | 0.11 | 0.04 | 0.04 | 0.04 | 0.02 | 0.09 | 0.26 | 0.33 | |
| D-Sub | g = 1 | 1.46 | 2.27 | 5.18 | 8.93 | 9.25 | 5.43 | 6.53 | 6.20 | 4.92 | 4.49 |
| g = 2 | 8.96 | 10.19 | 15.49 | 18.37 | 18.43 | 17.46 | 18.00 | 15.96 | 11.21 | 9.69 | |
| g = 3 | 1.65 | 2.47 | 4.21 | 5.49 | 5.49 | 4.87 | 5.50 | 4.49 | 2.55 | 2.07 | |
| D-Comb | g = 1 | 2.32 | 2.64 | 5.72 | 8.36 | 8.19 | 6.46 | 7.05 | 5.78 | 4.23 | 3.62 |
| g = 2 | 6.03 | 7.24 | 14.84 | 21.34 | 21.28 | 17.20 | 18.49 | 15.36 | 10.72 | 9.57 | |
| g = 3 | 1.70 | 2.05 | 4.23 | 5.95 | 6.04 | 4.95 | 5.09 | 4.32 | 3.10 | 2.69 | |
| D-Sep | g = 1 | 5.18 | 5.36 | 5.56 | 5.92 | 5.88 | 6.35 | 6.61 | 5.79 | 4.71 | 4.41 |
| g = 2 | 9.97 | 10.90 | 15.42 | 17.65 | 17.53 | 17.23 | 17.40 | 15.29 | 11.42 | 10.39 | |
| g = 3 | 3.96 | 4.12 | 4.10 | 3.98 | 3.84 | 4.51 | 4.60 | 4.17 | 3.52 | 3.21 | |
Note: punacc(g, dm) = P(declare dose dm unacceptable for subgroup g), psel(g, dm) = P(select dose dm as optimal for subgroup g), and = mean number of patients in subgroup g treated at dose dm. Values for true unacceptable and true optimal doses are given in italics and bold, respectively. = 0.20,0.35, and 0.35 for g− = 1, 2, 3 and for all subgroups.
Given and interim data at trial time t, the joint posterior of θ and the patient specific random effects γ = {γi, i = 1, … , n(t)} is
| (7) |
Where is specified in (4). We use Markov chain Monte Carlo (MCMC) simulation to generate posterior samples of (θ, γ). Since z is a subvector of θ, its posterior is obtained as a marginal of the posterior of (θ, γ), which in turn provides a posterior on R, the clusters, and the cluster-specific subgroup effects α⋆. Computational details are given in Supplementary Section 1. A computer program “Dose-finding-subgroup” for implementing the proposed design is available from https://users.soe.ucsc.edu/~juheelee/.
3 |. SUBGROUP-SPECIFIC UTILITY FUNCTION
Our utility function is constructed to accommodate the possibility that clinicians may be more willing to accept a higher risk of toxicity if disease status is likely to be improved for patients in a poor risk subgroup, and they also consider an early occurrence of toxicity less desirable for patients in more favorable risk subgroups. To construct a utility function that allows risk-benefit preferences between YT and YE to differ between subgroups in this way, we elicited G subgroup-specific utility functions, Ug(Y), g = 1, … , G. Denote the utility of Yj in subgroup g by Uj,g(Yj), for j = E, T. For each g, we first establish UT,g(yT) for 0 ≤ yT, then establish UE,g(yE) for yE = 0, 1, … , K − 1, scaled to have values on the same numerical domain as those of UT,g(yT), and finally define Ug(Y) = UT,g(YT) + UE,g(YE).
We begin by eliciting the numerical utility, UT,max, of not observing toxicity during the follow up period [0, C], and define
The shape parameter ag > 0 is determined during the utility elicitation process by showing several plots of UT,g(y) as a function of y on the interval [0, C] to the physician(s) providing the utility for a set of candidate ag values. The function UT,g(y) increases continuously with y over the follow-up period [0, C] to its maximum value UT,max for all YT ≥ C.
For the motivating mRCC trial, based on the clinicians’ experiences and preferences, we elicited UT,max = 140 and UT,g(YT) = 70 at YT = 70 days for g = 1 (good prognosis), at YT = 42 days for g = 2 (intermediate prognosis), and at YT = 28 days for g = 3 (poor prognosis). The resulting shape parameters are a1 = 3.80, a2 = 1.00, and a3 = 0.63, and the functions {UT,g, g = 1, 2, 3} are illustrated in Figure 1A. The figure shows that an early occurrence of toxicity has a lower utility compared to a later occurrence within each subgroup. Also, toxicity-vs-no toxicity utility differences are largest for subgroup 1. Thus, a dose with a high toxicity probability is less likely to be optimal for subgroup 1 than for the other subgroups, even when the dose has good efficacy.
FIGURE 1.
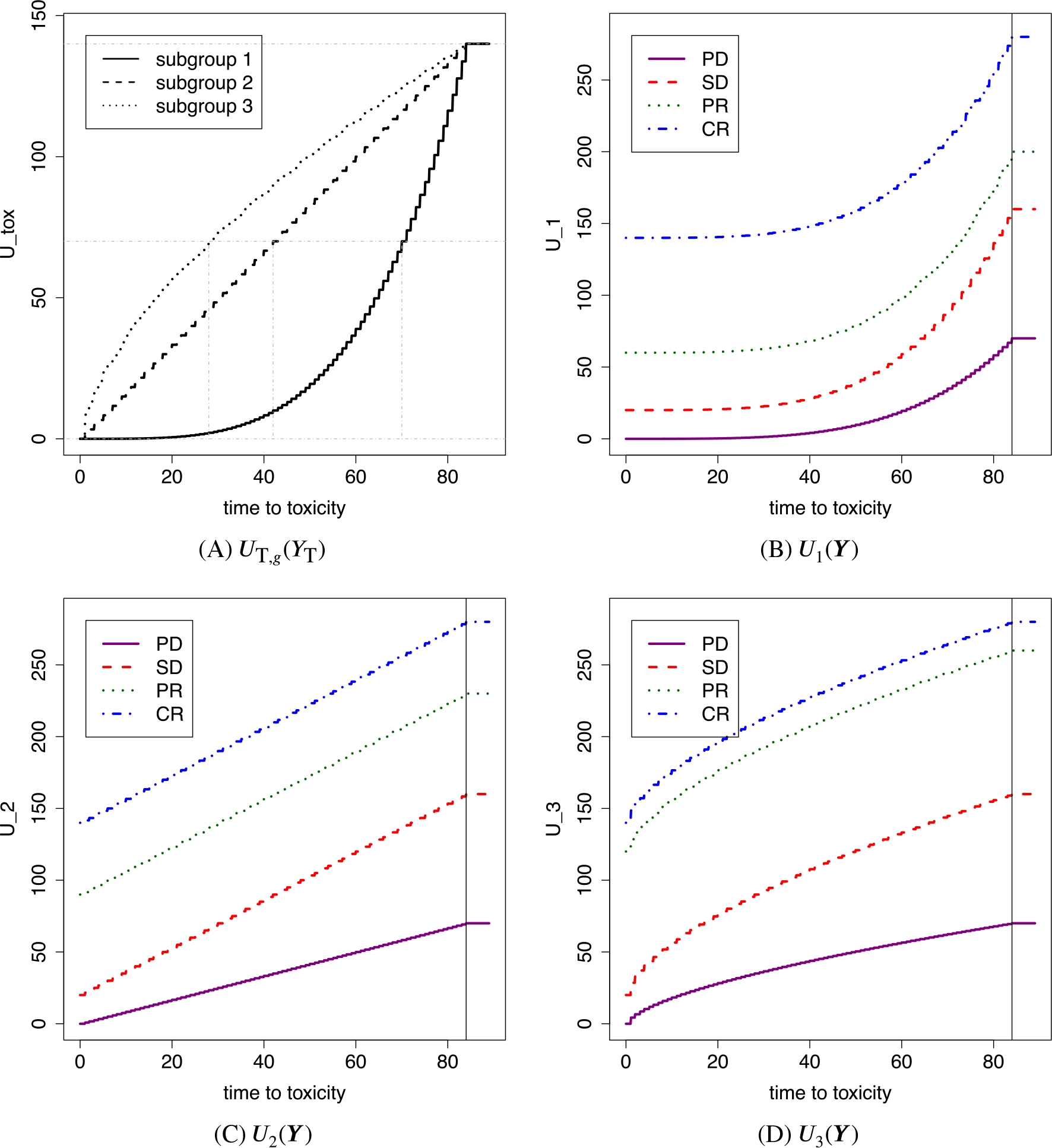
Illustration of subgroup-specific utilities Ug. A, The utility of toxicity for subgroup g, UT,g(YT), . B-D, The utilities of a bivariate outcome, Ug(Y) with Y = (YT, YE), for subgroups 1, 2, and 3, respectively. YE = 0, 1, 2, and 3 represent PD, SD, PR, and CR, respectively
We incorporate YE into the total utility Ug as follows: If YE = 0 (PD), we define Ug(Y) = UT,g(YT)∕2, and for YE = 1, 2, or 3, we define Ug(Y) = UT,g(YT) + UE,g(YE). Thus, having PD reduces the total utility by half of UT,g, while the better efficacy outcomes, SD, PR, and CR, each increase the utility additively. We fix UE,g(1) = 20 and UE,g(3) = 140 for all g and set UE,1(2) = 60, UE,2(2) = 90, and UE,3(2) = 120. The numerical value of UE,g(2) is the preference of PR relative to SD and CR in subgroup g, and a larger value of UE,g(2) makes a higher dose more desirable. The maximum utility 280 is achieved if a toxicity event does not occur during the follow-up period and CR is observed, that is, YT > C and YE = 3. Outcomes with YT < 1 and YE = 0 are assigned the minimum utility, 0. Figure 1B-D illustrates the utility functions. The relative preferences of the outcomes in subgroup g is determined jointly by ag and UE,g(2). Absolute numerical values of Ug are of little importance, whereas the relative sizes of Ug(Y) over different Y values within each subgroup are most relevant for optimal dose selection in that subgroup. The elicited utility should reflect experts’ experience treating the disease of the study. For example, whereas more potent therapies are preferable in IMDC poor risk mRCC, patients with poor prognosis metastatic triple negative breast cancer may be treated with less intensive therapies to preserve quality of life.18 Thus, elicited subgroup-specific utilities for breast cancer patients can be very different from those for mRCC patients. A detailed discussion of how subgroup-specific utilities may be elicited is given in Supplementary Section 2.
The design uses posterior predictive (PP) mean utilities as optimality criteria for dose selection. Given data at trial time t, the PP mean utility of giving dose dm to a future patient in subgroup g is
| (8) |
We compute using the empirical posterior sample mean of θ values simulated from . Computational details are given in Supplementary Section 1.
4 |. TRIAL DESIGN
Since lower doses carry a higher risk of PD, and higher doses carry a higher risk of toxicity, we identify acceptable doses by imposing the following subgroup-specific safety conditions. In each subgroup, we will restrict optimal dose selection and treatment assignment to the set of acceptable doses. The first safety constraint is that an untried dose may not be skipped when escalating, with this rule applied separately in each subgroup. If is the highest dose that has been administered by trial time t in subgroup g, then the search for the optimal dose and the treatment assignment are constrained to . We monitor both toxicity and efficacy as follows. Recalling that (YE = 0) = PD, for each pair (g, dm), we denote the probabilities of observing PD or severe toxicity during the full 9-week (84-day) follow-up period in subgroup g by
For each outcome and subgroup g, let be an elicited fixed upper limit on ζj(g, dm, θ), and let p⋆ be a fixed cut-off probability. A dose dm is unacceptable for subgroup g if
| (9) |
Expression (9) says that the probability of PD or toxicity in subgroup g is unacceptably high with dm. In this case, dm is not administered to any new patients enrolled in that subgroup. The set of acceptable doses at time t for subgroup g that do not satisfy (9) is denoted by . For the mRCC trial, elicited values were for all g, and and 0.35 for the three subgroups. The upper limit for the probability of PD was set based on the historical data on nivolumab and ipilimuma in Motzer et al,2 so adding sitravatinib to the combination should not be permitted to have worse safety. To obtain a design with high probabilities of stopping a truly unsafe or inefficacious dose, while still having high probabilities of selecting the best safe and efficacious dose, we investigated decision probability cutoffs 0.80 and 0.85 by simulation, and chose p⋆ = 0.85.
For the mRCC trial, the first patient enrolled in each subgroup is treated at d2, chosen by physicians. For all subsequent patients in each subgroup, the dose acceptability rules first are applied. At trial time t, the safety and efficacy constraints together define the set of acceptable doses based on interim data . Since the efficacy outcomes are observed only at ei + C, we define based on toxicity only to produce reliable decisions of acceptability until at least 20 patients have been fully followed up to 84 days. Subject to the acceptability rules during trial conduct, patients are adaptively randomized among . A patient in subgroup g is assigned to dose with probability proportional to 1∕{ng,m(t) + 1}, where ng,m(t) is the number of patients in subgroup g treated at dose dm up to time t. This implies that a new patient tends to be treated at a dose that is acceptable but less explored. If , then no patients in subgroup g are enrolled. If for all g, then the trial is terminated and no dose is selected, denoted by dsel(g) = None for all g. If the trial is not terminated early, subgroup-specific final optimal actions are taken at time . We first identify , and let dsel(g) = None if . Otherwise, the optimal dose selected for subgroup g is
5 |. SIMULATION STUDY
5.1 |. Simulation design
To evaluate the design’s performance, we simulated the mRCC trial under eight scenarios. For all scenarios, three subgroups, five doses, and four categorical efficacy outcomes were assumed, that is, G = 3, M = 5, and K = 4. We simulated each gi from a multinomial distribution with probability vector pg = (0.23, 0.60, 0.17), to reflect the historical data in Motzer et al.2 With Nmax = 120, we expect 27.6, 72.0, and 20.4 patients, on average, for the three subgroups. Thus, learning about the dose-outcome distributions for subgroups 1 and 3 may be challenging due to their relatively small sample sizes. For each scenario, we first specified the true latent clustering of the three predefined subgroups, . We specified the covariance matrix Ωtrue for the frailty vectors, and simulated frailties . To simulate Yi,T, we specified marginal probabilities of toxicity occurring during the follow-up period , for gi, d[i] ∈ {d1, … ,d5} and . We simulated Yi,E from {0, … , K − 1} with probabilities conditional on gi, d[i] and . Additional details are given in Supplementary Section 4. Table 1 gives the assumed true probabilities of observing severe toxicity during the follow-up period and of observing PD, given the frailty equals zero,
with k = 0 for each scenario. Also, the table gives the expected utility, Utrue(g, dm), under the truth after scaling to have maximum utility 100 for the best outcome. Truly unacceptable doses are given in italics, and truly optimal doses are given in bold. Recall that the elicited thresholds are for all g, and for g = 1, 2, 3. Supplementary Table 5 illustrates for all k, dm, and g. The simulation truth is different from the assumed model, since the assumed true dose-outcome relationships are arbitrarily specified, and do not follow the regression model assumed for the design. Either or may remain unchanged for multiple doses, as in Scenarios 3, 4, and 8. For some scenarios, the subgroups in different clusters have different distributions for one outcome, but the same distribution for the other outcome, as in Scenarios 4 and 5, while the model assumes that both distributions vary with subgroup clusters. The simulation truth also is very different from the historical data used to calibrate the prior’s location hyperparameters in Supplementary Section 2. Possibly due to the weakly informative prior in Section 2.2, as will be shown below, the design performs well in a range of different simulation scenarios.
With G = 3, four configurations of ztrue are possible due to the constraint of subgroup ordinality. Scenarios 1 and 2 have ztrue = (1, 2, 3), Scenarios 3 and 4 have ztrue = (1, 1, 2), Scenarios 5 and 6 have ztrue = (1, 2, 2), and Scenarios 7 and 8 have ztrue = (1, 1, 1). Due to potential subgroup effects and the subgroup-specific utility function, the pattern of the true expected utilities in doses varies with subgroups in all scenarios, including Scenarios 7 and 8. In Scenario 1, dose 1 is optimal for subgroups 1 and 2, but no dose is acceptable for subgroup 3 due to excessive probabilities of toxicity or PD. In Scenario 2, no dose is acceptable for any subgroup. In Scenarios 3, 7, and 8, the optimal dose is the same for all subgroups, but in Scenario 3 the true dose acceptability changes with subgroup. In Scenarios 4 to 6, the optimal dose varies with subgroup. For example, in Scenario 4, dose 1 is optimal for subgroups 1 and 2, whereas dose 3 is optimal for subgroup 3. In Scenarios 4 and 5, a higher risk subgroup has a higher optimal dose. In Scenario 6, subgroup 1 has dose 4 as optimal, but dose 1 is optimal in subgroups 2 and 3 due to a large increase in the severe toxicity probabilities.
We call the proposed design “D-Sub”, and considered two comparators, “D-Comb,” which ignores patient subgroups, and “D-Sep,” which runs a separate trial for each subgroup. Both D-comb and D-Sep still are more sophisticated than most phase I-II designs used in practice, such as designs based on two binary outcomes, which would be very impractical for conduct of the mRCC trial due to the 84-day evaluation period.
For D-Comb, we assumed the same model developed for D-Sub, but removed all subgroup-specific factors from the model, so the hazard function was hT(t|d[i], 𝛾i,T) = h0T(t) exp{ηT(d[i]) + 𝛾i,T} and the disease status distribution was . For dose acceptability, we used the upper limits ζT = 0.40 and ζE = 0.30, elicited for D-Comb. We used U2(Y) as the common utility function since subgroup 2 has the highest prevalence. Under D-Comb, if a dose is identified as unacceptable, no patient will be treated at that dose regardless of the patient’s subgroup, and if all doses are identified as unacceptable, the trial is terminated. A dose selected as optimal is recommended for all subgroups.
For the D-Sep design, we removed all subgroup specific-factors and kept the remaining parts of the model unchanged, as done for D-Comb. Under D-Sep, trials are run separately in the three subgroups, and no information is borrowed across trials. For each g, we used the same upper limits and Ug(Y) used for D-Sub, and let Nmax = 28, 72, and 20 for the three subgroups.
We evaluated each of the three designs using the following criteria. In subgroup g,
punacc(g, dm) = probability of identifying an unacceptable dose dm with a truly excessive probability of either severe toxicity or PD.
psel(g, dm) = probability of selecting the truly optimal dose dm.
nptrt(g, dm) = mean number of patients in subgroup g treated at dm.
Thus, punacc(g, dm) and psel(g, dm) vary with g under D-Sub and D-Sep, but they are the same for all g under D-Comb. For each simulated trial, b = 1, … , B under each design, we denote by dsel,b(g) the dose selected as optimal for subgroup g. We let wb,m(g) = 1 if dose dm is identified as unacceptable for subgroup g in simulated trial b, or 0 if not, and the number of patients treated in trial b is denoted by Nb. For each scenario and design, we summarized simulation results by the following simulation sample proportions:
5.2 |. Simulation results
Simulation results are summarized in Table 1. A total of R = 1000 trials with Nmax = 120 were simulated under each scenario. Overall, for each subgroup, the D-Sub design reliably identifies doses that are either unsafe or have low efficacy, and selects subgroup-specific optimal acceptable doses. Large punacc(g, dm) is achieved for dm with unacceptably large or . When either of or is clearly greater than its threshold , punacc(g, dm) is particularly high. We also observe that punacc(g, dm) tends to be larger for higher risk subgroups even with the same or , possibly due to the order constraint on . For example, in Scenario 4, in all g, and punacc(g, d5) = 0.45, 0.57, and 0.81 for the three subgroups, respectively. When all doses are unacceptable, for example, subgroup 3 in Scenario 1 or all subgroups in Scenario 2, punacc(g, dm) is especially high for all m, possibly because the model borrows information across doses through ηj and across subgroups through and thus improves reliability. The D-Sub design is likely to selectively identify unacceptable doses even for scenarios where only some of doses are unacceptable, as in Scenarios 3, 4, and 6 to 8. When all doses are truly unacceptable for all subgroups, as in Scenario 2, trials are stopped and it is concluded that there is no optimal dose with high probability. In Scenario 2, the design identifies all doses as unacceptable, and stops accrual of patients or selects no dose 98%, 96%, and 99% of the time for the three subgroups, respectively. If all doses are truly unacceptable for some subgroups only, trials are continued but accrual is likely to be stopped in those subgroups. For example, in Scenario 1, accrual in subgroup 3 is stopped or no dose is selected for that subgroup 86% of the time, but accrual of patients in subgroups 1 and 2 is continued and dose 1 is correctly selected as the optimal 84% and 85% of the time, respectively. For cases where at least one dose is acceptable for a subgroup, psel(g, dm) tends to be large for the true optimal doses. In most cases, psel(g, dm) is at least 0.60 for the true optimal doses. This indicates that the design performs very well in subgroup-specific optimal dose selection. For example, psel(g, dm) = 0.86, 0.83, and 0.85 for the subgroups, respectively, for the true optimal dose d2 in Scenario 3. In Scenario 5 where the true optimal dose varies greatly by subgroup, psel(g, dm) corresponding to the true optimal dose is 0.65, 0.66, and 0.80 for the subgroups, respectively. In some other cases where the optimal dose varies by subgroups, psel(g, dm) is not very large, especially for a sparse subgroup. Specifically, subgroup 1 in Scenario 6 has psel(1, d4) = 0.26 for its true optimal dose d4. Rather, the design more often selects d5 as optimal, psel(1, d5) = 0.47. However, given that the difference in the true expected utility between doses 4 and 5 is small, 6.23, the design often tends to select the largest dose as optimal. On the other hand, psel(g, dm) = 0.76 and 0.68 are obtained for subgroups 2 and 3, respectively, for their true optimal dose d1. Subgroup 3 in Scenario 4 is a similar case.
The mean numbers nptrt(g, dm) of patients in subgroup g treated at dm show that the design is very safe in that it reliably identifies unacceptable doses during the trial and assigns fewer patients to truly unacceptable doses. In Scenarios 3, 4, and 6 to 8, fewer patients were treated at unacceptable doses, and more patients were treated at truly acceptable doses. Recall that the true probabilities of observing events during the follow-up period, , and , are arbitrarily specified for the doses, whereas the design assumes regression models for the dose-outcome relationships. Thus, in terms of all criteria, D-Sub is robust in that it performs well in a variety of scenarios not based on the underlying model.
Probabilities of identifying unacceptable doses and of dose selection under the comparators, D-Comb and D-Sep, also are summarized in Table 1. D-Sub has greatly superior performance compared to the two comparators in most scenarios. D-Comb failed severely when optimal decisions should differ between subgroups. For example, in Scenario 1, D-Comb assigned patients in subgroup 3 to a dose although no dose is acceptable for subgroup 3, and selected d1 as optimal for subgroup 3, although d1 is clearly unacceptable for subgroup 3 due to its unacceptably large probability of PD. D-Comb also identified d1 and d2, which are acceptable for subgroups 1 and 2 but not acceptable for subgroup 3, as unacceptable only 18% and 22% of the time, respectively. The corresponding punacc(g, dm) values for d1 and d2 under D-Sub are 6% and 6% for subgroup 1, 2% and 4% for subgroup 2, and 87% and 92% for subgroup 3, respectively. In Scenario 5, d1 is the true optimal dose for subgroup 1 with Utrue(1, d1) = 57.64, followed by Utrue(1, d5) = 52.30. D-Sub selected d1 as optimal 65% of the time for subgroup 1, but D-Comb selected d1 only 27% of the time. In Scenario 6, D-Comb performs very poorly, selecting d1, which is the true optimal dose for subgroups 1 and 2, but not for subgroup 3, as an optimal dose for subgroup 3 more often than its true optimal dose d4. When there is no subgroup effect, as in Scenarios 7 and 8, D-Sub and D-Comb perform similarly. In Scenarios 7 and 8, due to the subgroup-specific utilities, the patterns of Utrue(g, dm) differ by subgroups. In Scenario 8, dopt(g) = 2 for all g, but the difference between Utrue(g, d2) and Utrue(g, d3) is greater for subgroup 1. Consequently, D-Sub selected d2 as optimal more often for subgroup 1 (83%) than for subgroups 2 (76%) and 3 (70%). In contrast, D-Comb selected d2 as optimal for all subgroups 74% of the time.
D-Sep has very poor performance for subgroups 1 and 3 for most scenarios, due to their low prevalences and the fact that D-Sep does not borrow strength between subgroups. In Scenario 2, where all doses clearly are unacceptable, D-Sep stopped trials with no dose was selected as optimal 78% and 82% of the time for subgroups 1 and 3, respectively, while D-Sub was much safer in that it correctly selected no dose 94% and 100% of the time for those subgroups. When the true optimal doses are middle doses, as in Scenarios 3 and 8, psel(g, dm) values for D-Sep are very small for the true optimal doses, especially in subgroups 1 and 3. For example, in Scenario 8, D-Sep obtained psel(g, dm) = 39% and 23% for the true optimal dose d2 for subgroups 1 and 3, respectively, compared to 83% and 70% for these subgroups with D-Sub. In Scenario 1, where subgroup-specific decision making is critical, D-Sep behaves more reasonably than D-Comb but is much less reliable than D-Sub. Under D-Sep, trials were terminated or no dose was selected as optimal for subgroup 3 79% of the time, but D-Sep selected d1 as optimal, for subgroups 1 and 2, 61% and 84% of the time, respectively. Figures 2 and 3 compare the performance metrics of D-Sub, D-Comb, and D-Sep. The figures show histograms of between-design differences in psel(g, dm) for the truly optimal doses, punacc(g, dm) for all doses, and nptrt(g, dm) for the truly unacceptable doses. Positive differences in psel(g, dm) and punacc(g, dm), and negative differences in nptrt(g, dm), correspond to superior performance by D-Sub. The figures show that, overall, D-Sub is greatly superior to both D-Comb and D-Sep in terms of both dose selection and safety.
FIGURE 2.
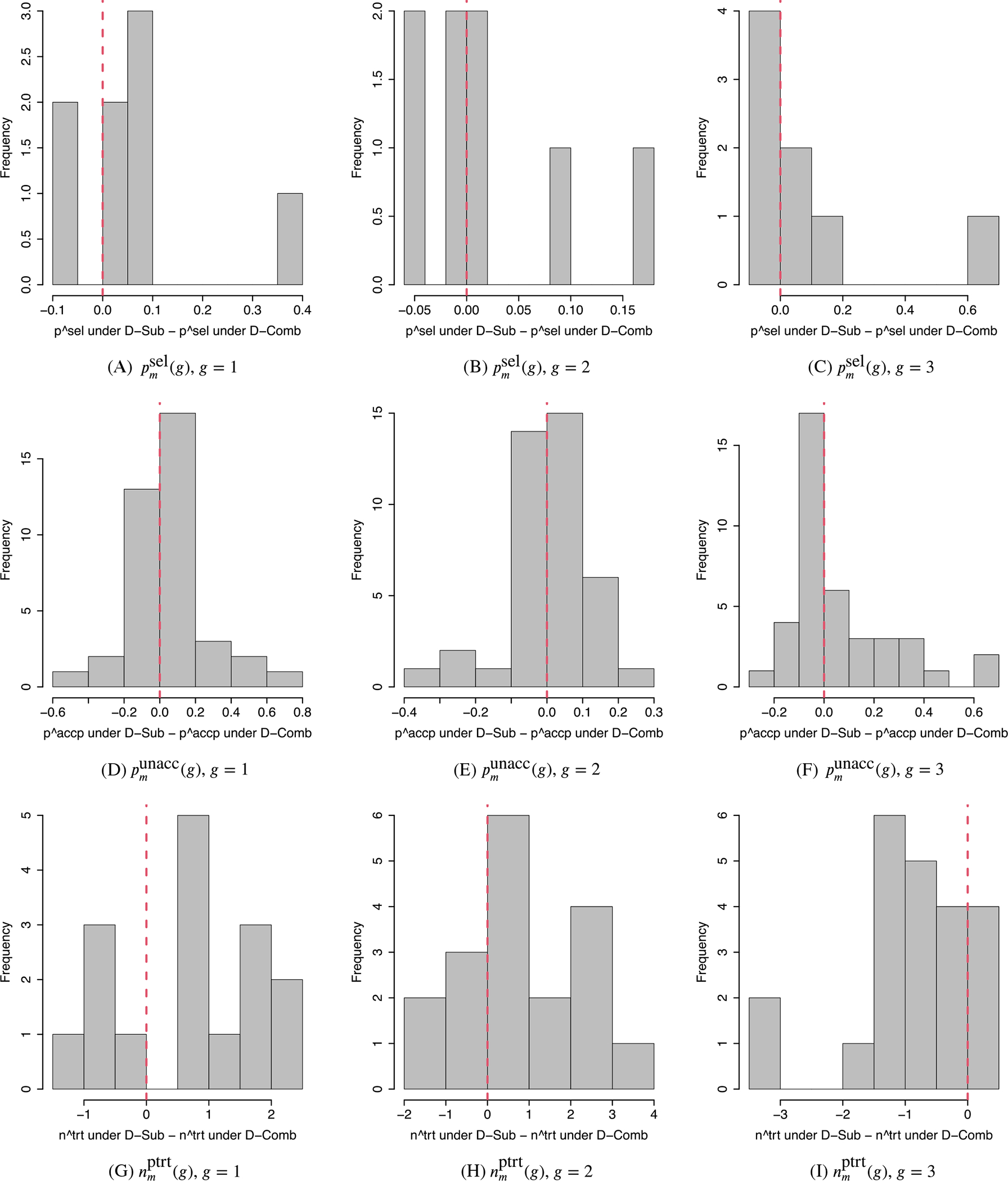
Comparison between D-Sub and D-Comb. A-C, Histograms of differences in between D-Sub and D-Comb. D-F, Histograms of differences in between D-Sub and D-Comb for the truly unacceptable doses and those between D-Comb and D-Sub for the truly acceptable doses. G-I, Histograms of differences in between D-Sub and D-Comb for the truly unacceptable doses. The left, middle, and right columns are for subgroups g = 1 (favorable), g = 2 (intermediate), and g = 3 (poor). A positive value indicates better performance of D-Sub than D-Comb in Panels A-F, and a worse performance in Panels G-I
FIGURE 3.
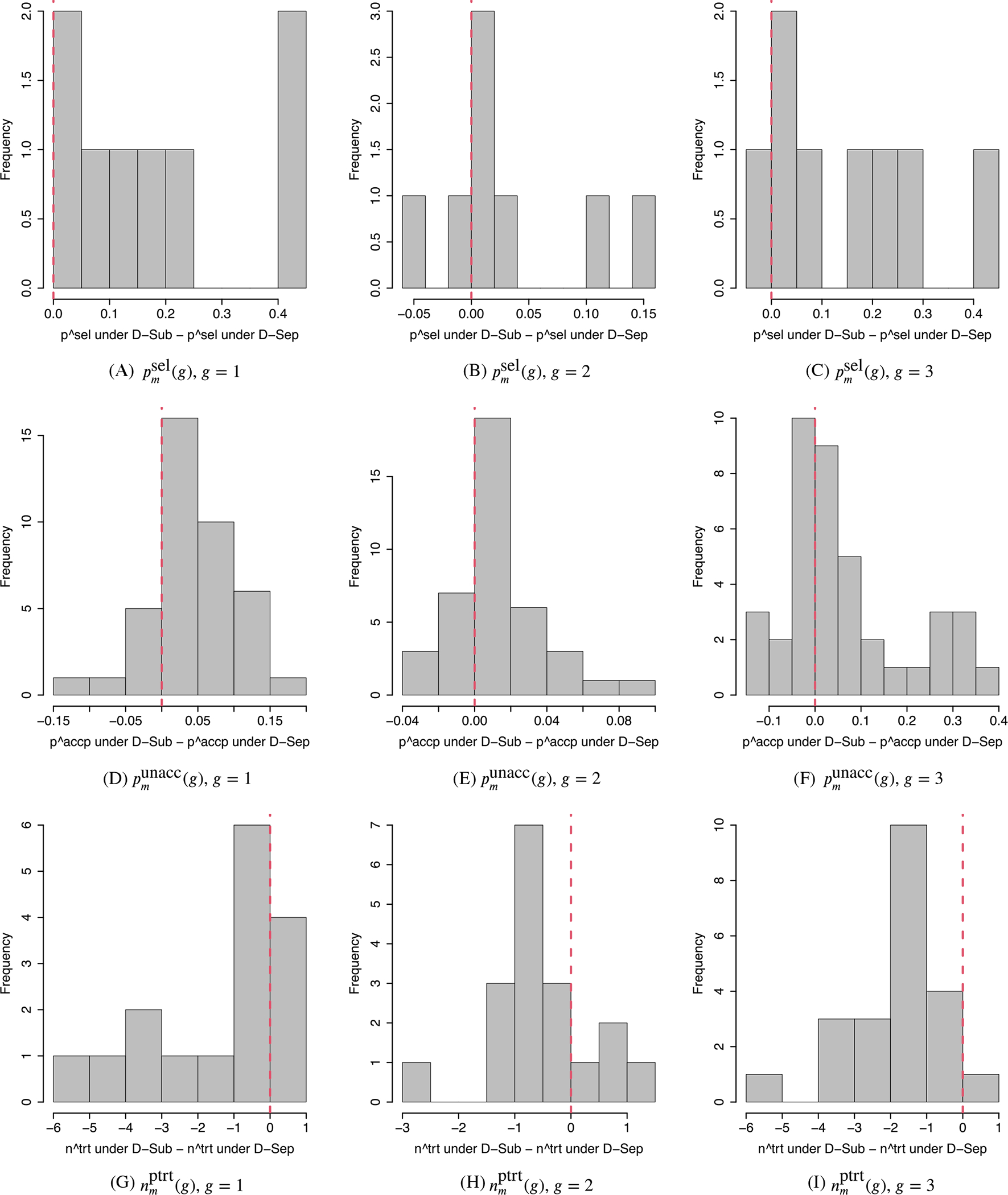
Comparison between D-Sub and D-Sep. A-C, Histograms of differences in between the D-Sub and D-Sep designs. D-F, Histograms of differences in between D-Sub and D-Sep for the truly unacceptable doses and those between D-Sep and D-Sub for the truly acceptable doses. G-I, Histograms of differences in between D-Sub and D-Sep for the truly unacceptable doses. The left, middle, and right columns are for subgroups g = 1 (favorable), g = 2 (intermediate), and g = 3 (poor). A positive value indicates better performance of D-Sub than D-Sep in Panels A-F, and a worse performance in Panels G-I
We performed additional simulations to examine the performance of D-Sub under several different scenarios and relative to the comparators. We also examined how D-Sub’s performance is affected by different specifications of certain design and model parameters, including pg, Ug(Y), and Nmax. We first examined the designs’ performances in the easier case where the subgroup proportions are equal, using pg = (1∕3, 1∕3, 1∕3) with Nmax = 120. Supplementary Table 6 summarizes the results under all scenarios for all three designs, D-Sub, D-Comb, and D-Sep. As expected, the performances of D-Sub and D-Sep are improved for subgroups 1 and 3 in most scenarios, since they have larger subsample sizes. D-Sub performs very similarly for subgroup 2, but D-Sep performs worse for this subgroup. D-Comb performs similarly, except in Scenarios 1 and 8 where decisions vary with subgroups. We also evaluated a more conventional utility based design by assuming that all subgroups have the same utility, using U2(Y) for all three subgroups. The results are summarized in Supplementary Table 7. Using four selected scenarios, Scenarios 1, 3, 5, and 7, we further examined the robustness of D-Sub by changing the assumption on and varying Nmax. We assumed to be either increasing or decreasing by using Weibull distributions to simulate Yi,T. Recall that D-Sub assumes a constant hazard. Supplementary Table 8 shows that D-Sub performs reasonably well under all the three evaluation criteria, even when the assumption on h0T is violated. We also studied D-Sub’s performance, assuming different maximum numbers of patients, Nmax = 60 and Nmax = 180, and compared it to those of D-Comb and D-Sep. The results are summarized in Supplementary Tables 9 and 10. For each Nmax, D-Sub has superior performance than the comparators by a large margin. As expected, the performance of D-Sub improves with larger Nmax over the range 60, 120, 180. In contrast, when truly optimal doses or dose acceptability vary between subgroups, as in Scenarios 1 and 5, the performance of D-Comb does not improve, even with Nmax = 120. This strongly suggests that ignoring subgroups is a very bad idea in settings where they have truly different dose-outcome distributions.
We compared D-Sub to four comparators, “D-w/o clustering,” “D-w/o frailty,” “D-diff AR,” and “D-linear tox,” where each simplifies the model or method used for D-Sub in a particular way. We used all scenarios for the comparison to D-w/o clustering, and four scenarios, Scenarios 1, 3, 5, and 7, for the comparison to the others. Comparison of D-Sub to these designs provides an empirical justification for using a complex model for D-Sub. A version of D-Sub that does not induce clustering of subgroups is “D-w/o clustering.” For D-w/o clustering, z = (1, 2, 3) is fixed and the subgroups have their own subgroup effects with the ordering constraint, αj,g < αj,g′, g < g′. Everything else is the same as in the model for D-Sub. Supplementary Table 11 provides a summary of the simulation results. To facilitate comparison, the table also includes the results for D-Sub. D-Sub and D-w/o clustering perform very similarly in many scenarios, but the performance of D-w/o clustering is sometimes much worse, especially for small subgroups, for example, subgroups 1 and 3 in Scenarios 4 and 5. For D-w/o frailty, the patient-specific random effects γ are removed, while the remaining model elements are the same as for D-Sub. The model for D-w/o frailty thus assumes that there is no between-patient heterogeneity beyond subgroups, and that the two outcomes of each patient are independent. Supplementary Table 12 shows that D-w/o frailty has much worse punacc(g, dm) and nptrt(g, dm) for dose that are truly unacceptable within some subgroups, while it is comparable to D-Sub for psel(g, dm). This may be due to the fact that variability between patients is not properly accommodated for by the model of D-w/o frailty. D-diff AR uses the same probability model as used for D-Sub, but it uses a different adaptive randomization method based on posterior expected utilities. Specifically, a patient in subgroup g is assigned to dose with probability proportional to . The results, summarized in Supplementary Table 14, show that the change in the design’s performance with this different AR method is very small. D-linear tox assumes a simpler linear regression model for the toxicity hazard, , while the remaining model components are the same as in the model of D-Sub. We assumed a normal distribution truncated below at 0 as a prior for . The results, summarized in Supplementary Table 14, show that the performance of D-linear tox is similar to that of D-Sub for Scenario 1, but D-linear tox shows an inferior performance in the other three scenarios.
6 |. DISCUSSION
We have presented a phase I-II clinical trial design in a setting where the patient population has multiple prognostic subgroups. The design was applied and evaluated for a trial in metastatic renal cancer. The design adaptively clusters patient risk subgroups with adjacent subgroups when there is no subgroup effect, and efficiently borrows information across subgroups and across doses. In a departure from established utility-based designs, subgroup-specific utilities are formulated to reflect risk-benefit trade-offs between efficacy and toxicity that vary with subgroups. Our simulations show that the design performs quite well under a wide variety of dose-outcome scenarios, and that incorporating all data while accounting for heterogeneity between patients significantly benefits correct decision making. The simulations also showed that the proposed design compares very favorably to both a design that ignores subgroups and a design that runs a separate trial within each subgroup.
Supplementary Material
ACKNOWLEDGEMENTS
Juhee Lee’s research was supported by NSF grant DMS-1662427. Pavlos Msaouel is supported by a Young Investigator Award from the Kidney Cancer Association, a Career Development Award from the American Society of Clinical Oncology/Conquer Cancer Foundation, a Concept Award from the United States Department of Defense, and by the MD Anderson Khalifa Scholar Award. Peter Thall’s research was supported by NIH/NCI grants 2 P30 CA016672 43 and 5R01CA061508.
Funding information
American Society of Clinical Oncology / Conquer Cancer Foundation, Grant/Award Number: Career Development Award; Kidney Cancer Association, Grant/Award Number: Young Investigator Award; National Science Foundation, Grant/Award Number: DMS-1662427; NIH/NCI, Grant/Award Number: 5R01CA061508; NIH/NCI, Grant/Award Number: 2 P30 CA016672 43; United States Department of Defense, Grant/Award Number: Concept Award; University of Texas MD Anderson Cancer Center, Grant/Award Number: Khalifa Scholar Award
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data used in this article are computer simulated. The codes that simulate datasets are available from one of the authors’ homepage.
REFERENCES
- 1.Heng DYC, Xie W, Regan MM, et al. External validation and comparison with other models of the international metastatic renal-cell carcinoma database consortium prognostic model: a population-based study. Lancet Oncol. 2013;14(2):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019;20(10):1370–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Quigley J, Conaway M. Continual reassessment and related dose-finding designs. Stat Sci Rev J Inst Math Stat. 2010;25(2):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iasonos A, O’Quigley J. Design considerations for dose-expansion cohorts in phase I trials. J Clin Oncol. 2013;31(31):4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horton BJ, Wages NA, Conaway MR. Shift models for dose-finding in partially ordered groups. Clin Trials. 2019;16(1):32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thall PF, Nguyen HQ. Adaptive randomization to improve utility-based dose-finding with bivariate ordinal outcomes. J Biopharm Stat. 2012;22(4):785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Thall PF, Ji Y, Müller P. Bayesian dose-finding in two treatment cycles based on the joint utility of efficacy and toxicity. J Am Stat Assoc. 2015;110(510):711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray TA, Thall PF, Yuan Y, McAvoy S, Gomez DR. Robust treatment comparison based on utilities of semi-competing risks in non-small-cell lung cancer. J Am Stat Assoc. 2017;112(517):11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murray TA, Yuan Y, Thall PF, Elizondo JH, Hofstetter WL. A utility-based design for randomized comparative trials with ordinal outcomes and prognostic subgroups. Biometrics. 2018;74(3):1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin R, Thall PF, Yuan Y. A Phase I–II basket trial design to optimize dose-schedule regimes based on delayed outcomes. Bayesian Anal. 2020;16(1):179–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snapinn S, Jiang Q. On the clinical meaningfulness of a treatment’s effect on a time-to-event variable. Stat Med 2011;30(19):2341–2348. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, George DJ. Choosing the best approach for patients with favorable-risk metastatic renal cell carcinoma. Clin Adv Hematol Oncol H&O. 2020;18(4):204–207. [PubMed] [Google Scholar]
- 13.Gorfine M, Hsu L. Frailty-based competing risks model for multivariate survival data. Biometrics. 2011;67(2):415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J, Thall PF, Rezvani K. Optimizing natural killer cell doses for heterogeneous cancer patients based on multiple event times. J R Stat Soc Ser C. 2019;68:809–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapple AG, Thall PF. Subgroup-specific dose finding in phase I clinical trials based on time to toxicity allowing adaptive subgroup combination. Pharmaceutical statistics. 2018;17(6):734–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1116–1127. [DOI] [PubMed] [Google Scholar]
- 17.Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isakoff SJ. Triple negative breast cancer: role of specific chemotherapy agents. Cancer J (Sudbury, Mass). 2010;16(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this article are computer simulated. The codes that simulate datasets are available from one of the authors’ homepage.