

Abstract
Introduction:
Patients with a confirmed germline mutation in the von Hippel-Lindau (VHL) tumor suppressor gene have been followed at the National Cancer Institute since the 1980s. In this study, we identify VHL patients with pheochromocytoma and long-term follow-up to determine the best candidates for active surveillance and surgical resection.
Methods:
A prospectively collected database of patients with a confirmed germline VHL mutation was reviewed to identify patients with a history of pheochromocytoma and at least 10 years of follow up. The presence of symptoms was assessed at the time of resection. Imaging data obtained at each clinic visit was reviewed to evaluate mass size and annual growth rate. Catecholamine data were reviewed to evaluate for data above the upper limit of the reference range. Masses that underwent imaging at least 3 months apart were considered in our surveillance cohort.
Results:
Median follow up was 16.7 years. There was a size-dependent increase in catecholamine production (P<0.05). For 36 masses on active surveillance, growth rate increased exponentially from 0.03 cm/y when masses were <1 cm to 0.32 cm/y when masses were greater than 2 cm. Approximately 1/3 of patients developed another pheochromocytoma after initial resection with a median time of 7.9 years. Partial adrenalectomy was associated with no metastatic events and a steroid-free rate of 97%.
Conclusion:
Active surveillance is a safe strategy for management of VHL associated pheochromocytoma in masses less than 2 cm.
Keywords: Pheochromocytoma, von Hippel Lindau, Active surveillance, Catecholamines, Adrenalectomy, Partial adrenalectomy, Metastatic pheochromocytoma
Introduction
Individuals affected with von Hippel-Lindau (VHL) are at risk for the development tumors in multiple organs, including spine and/or brain and/or retinal hemangioblastomas, pancreatic neuroendocrine tumors, endolymphatic sac tumors, bilateral clear cell renal cell carcinoma, papillary cystadenoma of the epididymis and broad ligament, and pheochromocytomas [1, 2]. Approximately 15% to 30% of patients with VHL will develop a pheochromocytoma with long term follow-up, although in VHL patients with missense mutations the rates of development of pheochromocytoma are as high as 59% [3–5]. While pheochromocytomas can secrete catecholamines resulting in symptoms (headaches, hypertension, mood changes, diaphoresis), they are malignant in only 5% to 10% of VHL patients [6]. Management strategies for patients with VHL-associated pheochromocytomas focus on minimizing the morbidity of treatment by preserving adrenal tissue whenever possible in order to reduce the potential need for steroid replacement. Active surveillance strategies were implemented at the National Institutes of Health (NIH) for small, asymptomatic pheochromocytomas in the 1980s in order to retain organ function and to reduce the morbidity of long-term steroid replacement and the need for multiple surgeries in these patients. In patients who required adrenal surgery, partial adrenalectomy was performed when deemed feasible [7]. In the current study we evaluate the long term natural history of VHL patients with pheochromocytoma who have been followed for at least 10 years at the NIH in order to elucidate the role of active surveillance and timing for intervention in this population.
Methods
Patients enrolled between 1987 and 2005 in a prospectively maintained National Cancer Institute Institutional Review Board-approved protocol evaluating urologic manifestations of patients with hereditary diseases were evaluated. Patients were included in this analysis if they met the following criteria: [1] Confirmed individual or family germline pathogenic variation of the VHL gene, [2] history of surgery for either pheochromocytoma or paraganglioma, [3] at least 10 years of follow-up at the NIH. For patients with VHL, our practice has been to perform cross sectional abdominal imaging every 1 to 3 years (depending on the phenotype) and annual testing for catecholamines and catecholamine byproducts.
Assessment of growth rates for patients on active surveillance
The active surveillance cohort consisted of patients managed with a period of observation if at least 3 months between cross sectional imaging. The surveillance cohort was split into 3 subsets based on mass size: less than 1 cm (smallest masses), 1 to 2 cm (intermediate size masses), and greater than 2 cm (larger masses) and the growth rate per year was calculated using the following formula: size (end of surveillance period)—size (beginning of surveillance period)/ time (years). If a mass progressed from one size category to another, the growth rate was calculated for each size category; consequently, one mass could have multiple growth rates in our analysis. Although there has been no absolute indication for surgical resection when patients were on active surveillance, the general indications for surgery were mass size approaching 2 cm, catecholamine values twice the upper limit of normal, and/or symptomatic patients. These indications were developed after years of observation that masses greater than 2 cm had a higher propensity to become more functionally active, and patient tended to be more symptomatic when their catecholamine levels were more than 2x above the upper limited of the laboratory reference range. Additionally, when concomitant abdominal or retroperitoneal surgery was being performed (i.e. pancreatic resection or partial nephrectomy), pheochromocytomas stable on surveillance were often resected simultaneously to reduce the need for future procedures.
Assessment of symptoms and abnormal catecholamines and/or catecholamine byproducts
As symptoms were assessed most reliably at the time of surgery for pheochromocytoma, presence, or absence of symptoms was taken from this time point. If the presence or absence of symptoms was not noted in the chart at this point, the patient’s symptoms were deemed “not assessable.” The presence of specific symptoms (i.e., headaches, anxiety, palpitations) was also ascertained at the time of surgery. In addition, patient records were reviewed for a history of hospitalization for hypertensive crisis or history of stroke-related to hypertension in the setting of active pheochromocytoma.
Catecholamine data has been collected routinely since the 1980s during every clinic visit for patients with VHL. Plasma catecholamines and catecholamine byproducts were assessed via phlebotomy performed after the patient has been resting in the in the supine position; urine catecholamines and catecholamine byproducts were collected utilizing a 24-hour urine collection. The initial analysis of abnormalities in catecholamines and catecholamine byproducts evaluated each separately and placed each laboratory value in 3 categories: below upper limit of the reference range, above the upper limit of the reference range, and 2x above the upper limit of the reference range. For each lab value, the highest value at any point during the evaluation was used to determine abnormality. For example, if a patient had a plasma normetanephrine 2x the upper limit of normal on just one measurement with the remaining measurements being lower, this patient was placed in the 2x abnormal category for plasma normetanephrines. Abnormal laboratory datapoint values were then matched to imaging dates for patients on active surveillance to determine the impact of mass size on active surveillance on the presence of abnormal catecholamines and catecholamine byproducts.
Assessment of recurrences
Recurrences were defined as the presence of a pheochromocytoma in an adrenal gland that had previously undergone a partial adrenalectomy. Recurrences were divided into 3 groups. Ipsilateral recurrences were defined as recurrences in an adrenal gland in which a prior adrenal surgery had been performed. Contralateral recurrences were defined as the presence of an adrenal mass after resection on the contralateral adrenal gland. Extra-adrenal recurrences were defined as all masses outside the adrenal gland after a resection of any pheochromocytoma or paraganglioma. Metastatic disease was defined as the presence of lesions in multiple organs or lesions in areas not known to form pheochromocytoma or paraganglioma (i.e., bone, lung, and liver). Time to recurrence was defined by the amount of time (in years) from the initial resection. Recurrences were further sub-divided by the extent of initial resection: a unilateral partial adrenalectomy was deemed any adrenal resection where adrenal gland was purposefully left behind. A total adrenalectomy was defined as complete resection of an adrenal gland. If bilateral adrenal surgery was performed concomitantly, a partial resection was considered if any adrenal tissue was preserved on one side.
Statistical analysis
For continuous variables, t-test was used to evaluate the mean difference in 2 continuous variables when there were 2 conditions. ANOVA was used for continuous variables when there were more than 2 conditions. Chi squared tests were used to evaluate for differences in categorical variables. For all statistical comparisons, P <0.05 was considered to be statistically significant. All analyses were performed in python 3.6 utilizing the numpy, pandas, and statistics packages. For data visualization, matplotlib package was used.
Results
Patient demographics
One hundred and eighty-three patients were identified with VHL with at least 1 surgery for pheochromocytoma, of which 72 had at least 10 years of follow up. The only criteria for exclusion from this initial cohort of 183 was follow up less than 10 years. Seventy-two patients from 54 VHL families met all inclusion criteria. Of these patients, 42 were included in a prior report of patients monitored on a pancreatic protocol at the NIH [8], and 30 patients are unique to this study. The median age at which patients in this cohort were enrolled in protocol at the NIH was 31 (range 6–63). In 37 patients (51%), pheochromocytoma was the initial VHL manifestation. The median length of follow up was 16.7 years. There were 50 (69%) males and 22 (31%) females; 71 (98%) were Caucasian, 1 patient was Hispanic.
Surgical data
One hundred and fifteen adrenal surgical procedures were performed on 72 patients. A total of 70 adrenal operations (61%) were performed by 7 senior surgeons at the NIH. The remaining 45 operations (39%) were performed at other institutions. For the masses resected at the NIH, the average mass size was 2.5 cm (0.7 cm–6.5 cm). Partial adrenal resection was performed in 45 surgical procedures; complete adrenal resection was performed in 25 operations (Table 2). The average mass size resection via partial adrenalectomy was 2.5 cm (range 0.7–5.0 cm); the average mass size resected via total adrenalectomy was 3.4 cm (range 0.8–6.5 cm), P<0.01. Table 1 summarizes the size of masses resected by partial and total adrenalectomy.
Table 2.
Summary of surgical data for patient undergoing surgery at the NIH vs. outside NIH
| Category | Sub–Category | NIH (N = 70) N,% |
Outside NIH (N = 45) N,% |
Total (N = 115) N,% |
P value |
|---|---|---|---|---|---|
| Adrenal Mass Location | Right Side | 22 (31) | 13 (29) | 35 (30) | 0.9 |
| Left Side | 24 (34) | 16 (36) | 40 (35) | 0.9 | |
| Bilateral | 13 (19) | 12 (27) | 25 (22) | 0.4 | |
| Extra Adrenal | 11 (16) | 4 (9) | 15 (13) | 0.4 | |
| Partial versus Total Adrenalectomy | Partial Adrenalectomy | 45 (64) | 11 (24) | 55 (48) | < 0.001 |
| Total Adrenalectomy | 25 (37) | 32 (71) | 58 (50) | < 0.001 | |
| Unknown | 0 (0) | 2 (4) | 2 (2) | 0.3 | |
| Laparoscopic vs. Open | Laparoscopic (including robotic) | 39 (56) | 8 (18) | 47 (41) | < 0.001 |
| Open | 31 (44) | 36 (80) | 67 (58) | < 0.001 | |
| Unknown | 0 (0) | 1 (2) | 2 (1) | 0.4 |
Abbreviation: NIH = National institute of health
Table 1.
Summary of Pheochromocytoma size partial vs. total adrenalectomy
| Size | Partial Resection N (%) |
Total Resection N (%) |
|---|---|---|
| <1cm | 1 (2%) | 2 (8%) |
| 1–2cm | 8 (18%) | 2 (8%) |
| 2–3cm | 24 (53%) | 5 (25%) |
| >3cm | 12 (27%) | 16 (64%) |
| Total | 45 | 25 |
| Median Size in cm | 2.5 | 3.4 |
Unilateral surgery was performed as the initial surgery in 48 patients. In 4 patients, the initial surgery performed was for an extra-adrenal mass. Bilateral surgery was performed as the initial surgery in 20 patients—in 12 of these mostly earlier patients a total bilateral adrenalectomy was performed. In 8 of these patients, a partial adrenalectomy was performed on at least 1 side. Of the surgeries performed at the NIH, 17 of the surgeries were performed between 1987 and 1997, 39 surgeries were performed between 1998 and 2007, and 12 surgeries were performed between 2008 and 2014. Patients undergoing surgery at the NIH historically had higher rates of undergoing partial adrenalectomy and higher rates of undergoing laparoscopic resection than patients undergoing surgery at outside facilities (Table 2). Of note, among the 70 surgeries performed at the NIH, 19 (27%) were performed with concomitant procedures—5 (7%) were performed at the same time as a pancreatic resection, 13 (19%) were performed with a concomitant renal procedure, and 1 (1.4%) was performed in the same setting as a neurosurgery procedure. There was no difference in the mean size of the lesion removed in the setting of concomitant surgery versus adrenal-only surgery (2.6 cm vs. 2.7 cm respectively, P = 0.5). Surgical data for patients undergoing surgery at the NIH versus outside the NIH are summarized in Table 2.
Recurrence
A total of 21 patients (29%) had 28 new pheochromocytomas detected after complete resection of the initial lesion (via either partial or total adrenalectomy). There were 9 ipsilateral recurrences, 13 contralateral new pheochromocytomas, and 6 extra-adrenal recurrences Table 3. The average time to the development of any new pheochromocytoma or paraganglioma was 7.9 years. Average time to ipsilateral recurrence was 8.7 years (range 0.4–34) and average time to development of contralateral pheochromocytoma was 8.0 years. There was no difference in time to recurrence when stratified by partial vs. total adrenalectomy(P = 0.81) (Fig. 4). The majority of patients with recurrence (18/21) presented with initial mass in the adrenal, but of the 6 patients who initially presented with a mass in an extra-adrenal location, 3 had a recurrence within the adrenal gland. A summary of recurrences based on initial mass location and extent of first surgery can be seen in Table 2.
Table 3.
Recurrences and functional outcomes for patients with unilateral and bilateral masses
| Unilateral Adrenal Mass (N = 45) |
Bilateral Adrenal Mass (N = 21) |
|||
|---|---|---|---|---|
| Partial Adrenalectomy (N = 22) |
Total Adrenalectomy (N = 23) |
Partial Adrenalectomy (N = 13) |
Total Adrenalectomy (N = 8) |
|
| Evaluation of Patients with Recurrences | ||||
| Patients with at least one additional pheo: N, (%) | 7(32) | 6 (26) | 4 (31) | 0 (0) |
| Years to first additional pheo: mean, (range) | 5.6 (0.2–15.0) | 5.4 (0.9–14.3) | 13.2 (0.4–34.6) | – |
| Patients with multiple additional pheo: N, (%) | 3 (14) | 4 (17) | 0 (0) | 0 |
| Evaluation of Total Number of Recurrences | ||||
| Total ipsilateral recurrences: N, (%) | 3 (15) | 1 (4) | 3 (38) | 0 (0) |
| Years to ipsilateral recurrence: mean (range) | 3.8 (0.4–8.0) | 3.3 (3.3–3.3) | 13.2 (0.4–34.6) | – |
| Total contralateral pheo: N, (%) | 6 (23) | 6 (26) | – | – |
| Years to contralateral pheo: mean (range) | 6.0 (0.2–15.0) | 6.1 (1.2–12.7) | – | – |
| Total extra adrenal pheo: N (%) | 2 (9) | 4 (17) | 0 (0) | 0 |
| Years to extra adrenal pheo: mean (range) | 7.2 (0.4–8.5) | 4.7 (0.9–14.3) | – | – |
| Functional Outcomes | ||||
| Require steroid replacement: N, (%) | 0 (0) | 2 (8) | 1 (13) | 8 (100) |
Figure 4.
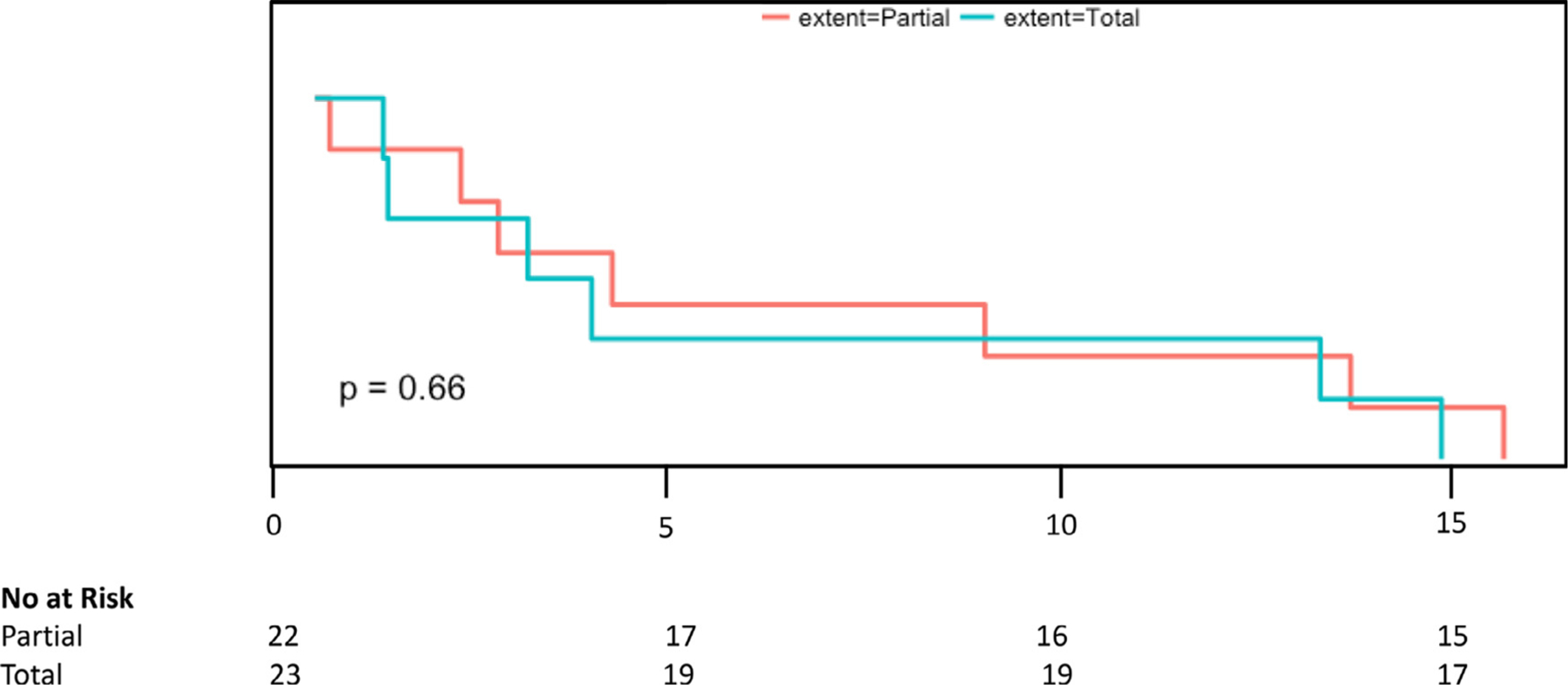
Survival curve analysis after partial vs. total adrenalectomy. There was no difference in recurrence rates between partial and total adrenalectomy over time.
Surveillance cohort
There were 38 masses that underwent a period of surveillance in 26 patients. The median age at which surveillance for beginning surveillance was 30 (range 7–61). The median time on surveillance was 4.3 years (range 0.3–20.1). The median size of mass when starting surveillance was 1.0 cm (range 0.3–2.5 cm) and the median size when the mass was resected was 2.0 (range 0.3–4.5). The median growth rate for all masses was 0.1 cm per year (range −0.02–0.6).
Relationship between mass size and growth rate, symptoms, and catecholamine abnormalities
Growth rate by size
When masses on surveillance were split into 3 groups based on size, 10 of the 38 masses progressed through multiple size groups resulting in 48 evaluable growth rates. Growth rates were evaluated for 10 masses under 1.0 cm (small), 24 masses between 1.0 and 2.0 cm (medium), and 14 masses greater than 2.0 cm (large). The median growth rate for masses under 1.0 cm was 0.03 cm per year. The median growth rate for masses between 1.0 and 2.0 cm was 0.12 cm per year. The median growth rate for masses larger than 2.0 cm was 0.32 cm per year (P = 0.018), Fig. 1. This exponential increase in growth rate is well-demonstrated by 1 mass that was followed for over 10 years in Fig. 2.
Fig. 1.
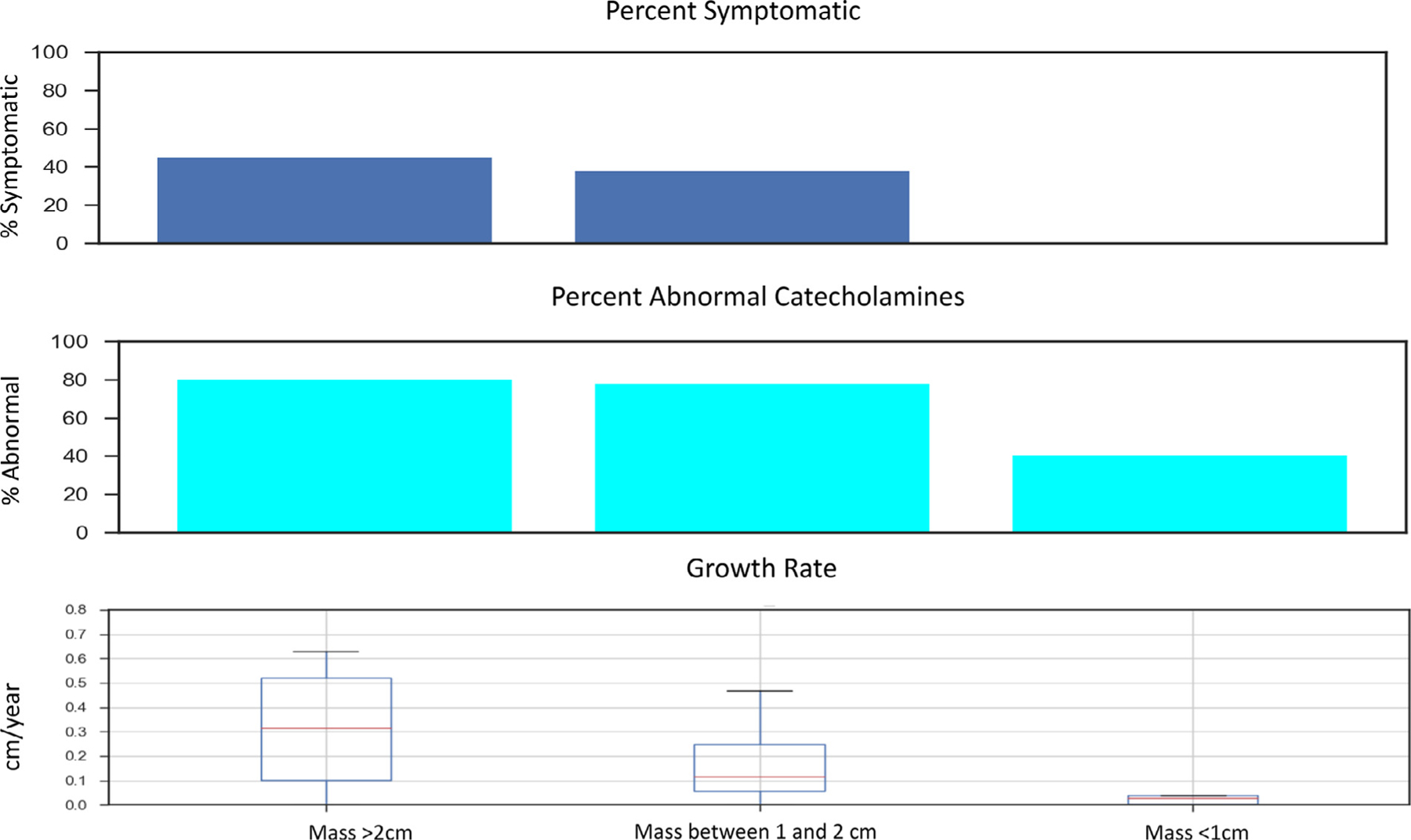
Symptoms, abnormal catecholamines, and growth rate by adrenal mass size. The percentage of patients who were symptomatic and the percentage of patients with at least one abnormal catecholamine lab value abnormality increase with increasing mass size. Average growth rate also increased from less than 0.1cm/y for masses less than 1cm to over 0.3cm/y in patients with masses >2cm.
Fig. 2.
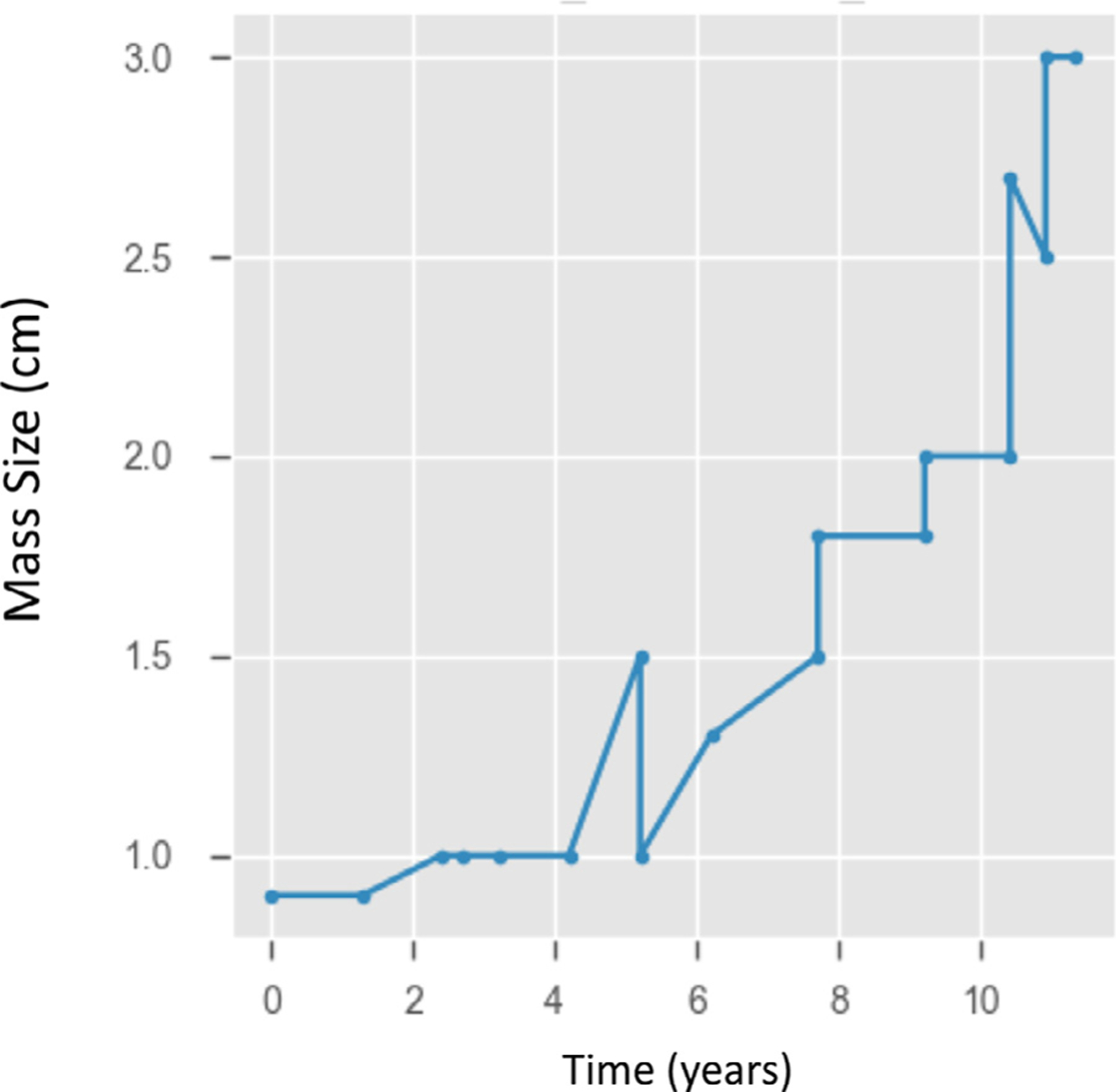
Nonlinear growth kinetics of an adrenal mass. In a patient with an adrenal mass followed for 12 years at the National Institutes of Health, the growth rate there was an acceleration of the growth curve over time. For the first 8 years, the mass increased in size 0.8cm. In the next 4 years, the mass grew an additional 1.5 cm.
Catecholamine abnormalities by size
A total of 4 patients (40%) had at least 1 lab outside the reference range while under surveillance with a smaller (<1 cm) mass. For patients with an intermediate size masses (1–2 cm), 21 patients (78%) had at least 1 lab outside the reference range and for patients with larger masses (>2 cm), 20 (80%) had at least 1 lab outside the reference range (P = 0.04). Although the rates of any abnormal catecholamine elevation were high, the majority of these abnormalities were mild with very few patients having elevation more than double the upper limit of normal (Fig. 1). Both plasma and urine normetanephrine levels showed a size-dependent increase with plasma normetanephrines increasing from a 10% abnormal rate with masses less than 1 cm to a 64% abnormal rate in masses greater than 2 cm and urine normetanephrines increasing from 10% abnormal in masses less than 1 cm to 44% in masses greater than 2 cm (P = 0.01)
Symptom prevalence and relationship to mass size
Of the 70 adrenal surgeries performed at the NIH, there were a total of 30 patients (43%) who were symptomatic at the time of surgery. The most common symptoms were headaches (40%), palpitations (36%), diaphoresis (37%), anxiety (13%). Agitation and mood change were much less common, each of which was documented in 1 patient (3%). Thirty seven percent of patient reported a history of hypertension. There was an increase in the prevalence of symptoms as mass size increased from 0% in the small mass group (<1 cm), 37.5% in the medium mass group (1–2 cm) to 46.5% in the large mass group (>2 cm), but this difference was not statistically significant (P= 0.18). The relationship between mass size, growth rate, and symptom prevalence is shown in Fig. 3. There were no patients who had a history of hospitalization for hypertensive crisis and no patients with a history of stroke attributed to hypertension.
Fig. 3.
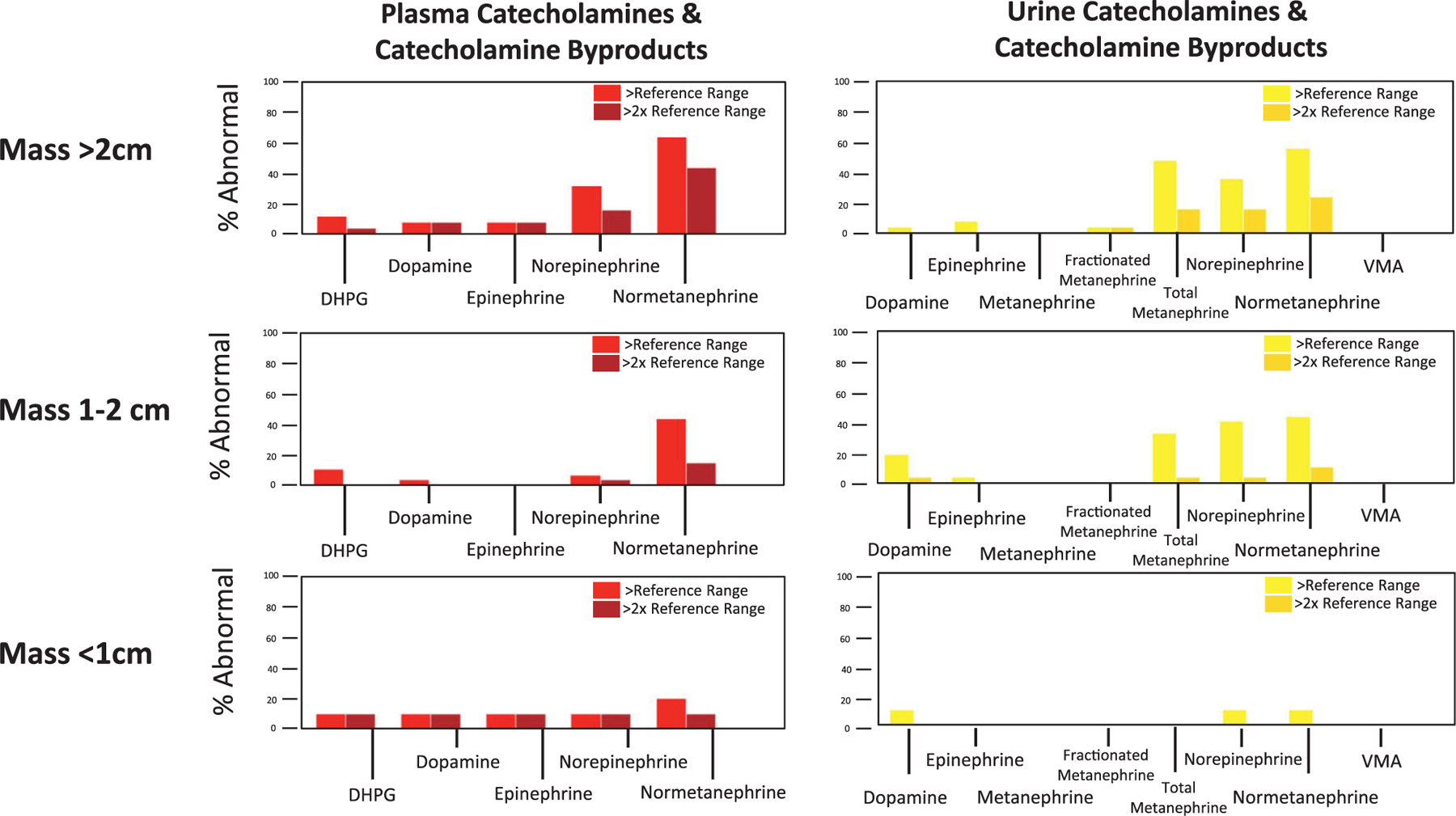
Abnormalities of catecholamines and catecholamine byproducts by adrenal mass size. Patient were more likely to have catecholamine abnormalities as the mass increased in size. It is notable that the majority of adrenal masses less than 1 cm were not metabolically active.
Functional outcomes
A total of 11 patients (15%) in the surgical cohort required long-term steroid replacement and of these patients, 73% (8/11) had a history of total bilateral adrenalectomy. Six of the 8 patients who underwent a total bilateral adrenalectomy underwent this procedure at a facility outside the NIH. The 2 total bilateral adrenalectomies performed at the NIH performed were both in the 1990s, and one of these patients had pan-hypopituitarism and was already on hormone replacement. Of the patients who were initially managed by unilateral partial adrenalectomy, no patients subsequently required long term steroid replacement. Of the patients who underwent unilateral total adrenalectomy, 1 patient underwent a contralateral total adrenalectomy and another underwent a contralateral partial adrenalectomy. The latter patient was able to avoid replacement steroid therapy for 8 years, but eventually became steroid dependent. The only patient who required steroid replacement who had bilateral adrenal masses managed initially with bilateral partial adrenalectomy eventually required repeat resection for a 4 cm adrenal lesion. Although attempts were made to spare adrenal tissue, this patient eventually required steroid replacement. Of the 35 patients (22 unilateral partial, 13 bilateral partial) who underwent a partial resection as the initial management strategy, 34 of 35 (97%) were able to stay off long-term steroids.
Metastatic disease
Two patients developed metastatic disease during follow-up. One patient underwent a total adrenalectomy surgery as a teenager for a 3.2 cm mass. This patient subsequently developed an extra adrenal recurrence, which was resected. The patient then developed ipsilateral recurrence in the adrenal fossa which was resected. The patient then developed a solitary lung mass which was resected. The patient was subsequently found to have multiple small spinal lesions and as well as lesions in his liver. The largest spinal cord lesion was biopsied and the pathological findings were consistent with metastatic pheochromocytoma. The diagnosis of metastatic disease was made 20 years after his first adrenal resection. The other patient who developed metastatic disease underwent a resection at a facility outside the NIH and the largest size tumor size was not known. This patient has been followed at our institution for 18 years. He developed metastatic disease 12 years after the resection of his primary adrenal mass. His metastatic disease has been managed with multiple resections. Both patients are still alive and are asymptomatic from their metastasis without need for alpha blockade.
Discussion
This study summarizes the outcomes of patients with VHL-associated pheochromocytomas treated at the NIH over the course of 3 decades. While surgical approaches and practice patterns have changed over the course of the study period, the results of this study confirm early clinical insights noting small pheochromocytomas in patients with VHL are a largely indolent and non-functional entity [9]. This study adds several new observations. First, the adrenal pheochromocytomas grow very slowly (<0.1 cm/year) in masses less than 1 cm, but the growth rate accelerates as the masses increase in size with masses over 2 cm having a 10-fold higher growth rate when compared with masses less than 1 cm. We also demonstrate a size-dependent relationship in the development of both symptoms and elevation in catecholamines. Surveillance of VHL-associated pheochromocytomas until the mass reaches 2 cm or becomes metabolically active appears to be oncologically sound with the only 2 instances of development of metastatic disease in patients who did not undergo surveillance. In addition, no patient had a hypertensive crisis or stroke while on active surveillance. This study confirms findings in several prior studies demonstrating VHL-associated pheochromocytomas can be removed via partial adrenalectomy [10] in order to prevent the potential future need for steroid replacement, which is particularly important given approximately 1/3 of patients will develop a recurrence. The time course for recurrence is notable with most recurrences occurring an average of 8 years after the initial pheochromocytoma, supporting the need for continued high-quality surveillance that includes cross-sectional imaging that includes adequate visualization of the adrenal glands.
Given the data described here, our current practice consists of obtaining annual catecholamines in all patients with VHL. Imaging is obtained every 1–3 depending on the size and growth of adrenal and renal lesions, as previously described [11]. For patients with small adrenal lesions with imaging characteristics consistent with pheochromocytoma, our initial management strategy is active surveillance as long all of the following are true: no symptoms of catecholamine excess, serum and/or urine catecholamines are less than 2 times the upper limit of the references range, and there is no immediate need for ipsilateral kidney surgery. For patients where all of these conditions are negative, and the current size is less than 2 cm, we obtained follow-up imaging in 12–24 months, reflecting the modest growth potential of these lesions. In patients with adrenal lesion > 2 cm, follow up is generally 12 months or less, reflecting their increased growth potential.
Several limitations of this study should be noted. Although the data was collected prospectively, the study period was long enough that technology and practice changed significantly over time limiting uniformity in imaging evaluation and therapy during the study period. Additionally, this study reflects the input of 7 attending surgeons, each of whom had a unique practice pattern and approach to these patients. Additionally, 45 of 115 surgeries for pheochromocytoma (39%) were performed outside the NIH, and these procedures are not as well characterized. Also, symptoms were noted primarily around the time of surgery, and therefore the presence of symptoms was taken exclusively from this time point. Furthermore, we did not include quantitative data on hypertensive medication use or development of congestive heart failure.
Conclusions
VHL-associated pheochromocytomas can safely undergo active surveillance when less than 2 cm, which allows these patients to delay surgery and afford the opportunity to potentially combine adrenal resection with concomitant abdominal procedures. The growth rate of adrenal masses appears to be exponential with small (<1 cm) pheochromocytomas having a very slow growth rate, making these masses ideal for active surveillance. Active surveillance can be performed safely until the mass reaches 2 cm after which it should be recommended with caution. Partial adrenalectomy allows most patients to remain off steroid replacement and does not appear to increase the risk of recurrence.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Conflicts of Interest
No author has any conflict of interest to report.
References
- [1].Shuch B, Ricketts CJ, Metwalli AR, Pacak K, Linehan WM. The genetic basis of pheochromocytoma and paraganglioma: implications for management. Urology 2014;83(6):1225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet 2003;361(9374):2059–67. [DOI] [PubMed] [Google Scholar]
- [3].Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, et al. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996;33 (4):328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Feletti A, Anglani M, Scarpa B, Schiavi F, Boaretto F, Zovato S, et al. Von Hippel-Lindau disease: an evaluation of natural history and functional disability. Neuro Oncol 2016;18(7):1011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 1995;5(1):66–75. [DOI] [PubMed] [Google Scholar]
- [6].Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011;19(6):617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sanford TH, Storey BB, Linehan WM, Rogers CA, Pinto PA, Bratslavsky G. Outcomes and timing for intervention of partial adrenalectomy in patients with a solitary adrenal remnant and history of bilateral phaeochromocytomas. BJU Int 2011;107(4):571–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Aufforth RD, Ramakant P, Sadowski SM, Mehta A, Trebska-McGowan K, Nilubol N, et al. Pheochromocytoma screening initiation and frequency in von Hippel-Lindau Syndrome. J Clin Endocrinol Metab 2015;100(12):4498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aprill BS, Drake AJ, Lasseter DH 3rd, Shakir KM. Silent adrenal nodules in von Hippel-Lindau disease suggest pheochromocytoma. Ann Intern Med 1994;120(6):485–7. [DOI] [PubMed] [Google Scholar]
- [10].Gomella PT, Sanford TH, Pinto PA, Bratslavsky G, Metwalli AR, Linehan WM, et al. Long-term functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma. Urology 2020;140: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ball MW, An JY, Gomella PT, Gautam R, Ricketts CJ, Vocke CD, et al. Growth rates of genetically defined renal tumors: implications for active surveillance and intervention. J Clin Oncol 2020;38(11): 1146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]