

Abstract
Tatton-Brown-Rahman syndrome is an autosomal dominant overgrowth syndrome caused by pathogenic DNMT3A variants in the germline. Clinical findings of tall stature due to postnatal overgrowth, intellectual disability, and characteristic facial features are the most consistent findings observed in patients with Tatton-Brown-Rahman syndrome (TBRS). Since the syndrome was first described in 2014, an expanding spectrum of neuropsychiatric, musculoskeletal, neurological, and cardiovascular manifestations have been reported. However, most TBRS cases described in the literature are children with de novo DNMT3A variants, signaling a need to better characterize the phenotypes in adults. In this report, we describe a 34 year old referred to genetics for possible Marfan syndrome with aortic root dilatation, mitral valve prolapse and dilated cardiomyopathy, who was diagnosed with TBRS due to a heterozygous de novo DNMT3A variant. This represents the third reported TBRS case with aortic root dilation and the second with cardiomyopathy. Collectively, these data provide evidence for an association with aortic disease and cardiomyopathy, highlight the clinical overlap with Marfan syndrome, and suggest that cardiovascular surveillance into adulthood is indicated.
Keywords: Aortic aneurysm, DNMT3A, overgrowth syndrome, cardiomyopathy, Tatton-Brown-Rahman syndrome
INTRODUCTION
Tatton-Brown-Rahman syndrome is an autosomal dominant overgrowth condition caused by pathogenic germline variants in DNMT3A, which encodes a DNA methyltransferase responsible for de novo methylation during embryogenesis (Okano, Bell, Haber, & Li, 1999). Delineation of the Tatton-Brown-Rahman syndrome (TBRS) phenotype continues to evolve, but consistently includes intellectual disability, postnatal overgrowth manifesting as tall stature and increased head circumference, neuropsychiatric conditions, and coarse facial features consisting of a round or long face, thick horizontal eyebrows, deeply set eyes, and narrow palpebral fissures (Balci et al., 2020; Tatton-Brown et al., 2014). Other clinical findings include joint hypermobility, scoliosis, afebrile seizures, hypotonia, umbilical hernia and less frequently, cryptorchidism, ventriculomegaly, Chiari malformations and malignancies (Balci et al., 2020; Hollink et al., 2017; Sweeney et al., 2019; Tatton-Brown et al., 2018; Tenorio et al., 2019). Additionally, a spectrum of cardiovascular abnormalities have been described including septal defects, patent ductus arteriosus, patent foramen ovale, mitral and tricuspid valve dysfunction, aortic root dilatation, and cardiomyopathy (Balci et al., 2020; Shen et al., 2017; Tatton-Brown et al., 2018; Tenorio et al., 2019; Xin et al., 2017).
Fewer than 100 TBRS cases have been reported in the literature and the majority are children, signifying a need to further characterize the phenotype in adults (Balci et al., 2020; Tatton-Brown et al., 2018). Diagnosis of TBRS is critical to inform long-term management as some of the cardiovascular and neuropsychiatric manifestations, and malignancies, are age-dependent with variable onset. This report describes a 34 year old with dilated cardiomyopathy and aortic root dilatation diagnosed with TBRS in adulthood after referral to genetics for possible Marfan syndrome.
CLINICAL REPORT
This 34-year-old male patient was referred to our aortic and vascular genetics clinic for evaluation of Marfan syndrome due to aortic root dilatation, tall stature, and long fingers. He was the product of an uncomplicated full-term vaginal delivery, born to a 39-year-old gravida 2 mother and 49-year-old father. The pregnancy was unremarkable, including an amniocentesis that demonstrated a normal male karyotype. His birthweight was 3.94 kg (88th centile), length was 50.8 cm (68th centile) and head circumference was unknown. No medical complications were reported within the first year of life. Early developmental milestones were achieved on time until age 4 when psychostimulant medications were initiated for attention deficit hyperactivity symptoms; however, developmental delays, and behavioral and learning difficulties persisted. Subsequent brain MRI, EEG, karyotype, serum amino acids, urine organic acids, lactate, and thyroid function testing, did not reveal a diagnosis. At age 7, he was prescribed antiepileptic medication for suspicion of petit mal seizures.
This patient was first referred to a medical genetics clinic at 14 years old (75–90th centile weight; 95th centile height, 98th centile head circumference) for suspected Marfan syndrome due to joint hypermobility, tall stature, and echocardiographic evidence of mild aortic root dilatation (3.5 cm, Z=2.24) and mitral valve prolapse (Sluysmans & Colan, 2005). Plasma homocysteine values were within normal limits. Molecular genetic testing was not performed and he did not meet clinical diagnostic criteria for Marfan syndrome based on Ghent-1 nosology (Paepe et al., 1996). Echocardiograms at 16 and 18 years demonstrated stable mitral valve prolapse and an unchanged aortic root diameter of 3.5 cm, which was within normal limits for his age, sex and body surface area (Z=1.84) (Sluysmans & Colan, 2005). The echocardiogram at age 18 did not show signs of cardiomyopathy based on the cardiologist’s interpretation and limited data reported (left ventricular ejection fraction of 59% and fractional shortening of 26%). His electrocardiogram was unremarkable
He was suspected to have neurodevelopmental impairment in childhood, but was not formally evaluated until adulthood. He was enrolled in special education classes from adolescence through high school and attended a higher education institution for individuals with learning disabilities. Neuropsychiatric symptoms, including hallucinations, delusions, paranoia, sleeplessness, and lower extremity numbness, began at age 27, approximately three months after a closed head traumatic brain injury. CT and MRI imaging of the brain after the injury ruled out acute trauma. These symptoms of psychosis led to hospitalization and raised concern for schizophrenia, but resolved within one year and he was never formally diagnosed. Psychological testing at 31 years old demonstrated a full-scale intelligence quotient (FSIQ) of 75 (5th centile). Adaptive behavior testing suggested mildly impaired functioning. He was diagnosed with autism spectrum disorder (borderline normal FSIQ without language impairment), impulse control disorder, and major depressive disorder.
A transthoracic echocardiogram at 34 years old incidentally revealed cardiomegaly during a hospitalization for an appendectomy. On that study, his aortic root measured 4.4 cm at the sinuses of Valsalva (Z=2.39–3.17) (Figure 1a) (Campens et al., 2014; Devereux et al., 2012). The diameter of his thoracic aorta was otherwise within normal limits. Formal cardiology evaluation and cardiac MRI findings were consistent with dilated cardiomyopathy: the left ventricle (LV) was severely dilated with a maximum end-diastolic diameter of 7.0 cm, and LV wall motion was globally depressed. The LV ejection fraction had decreased to 34% (Figure 1b). Three months later, he received an implantable cardioverter defibrillator.
Figure 1.
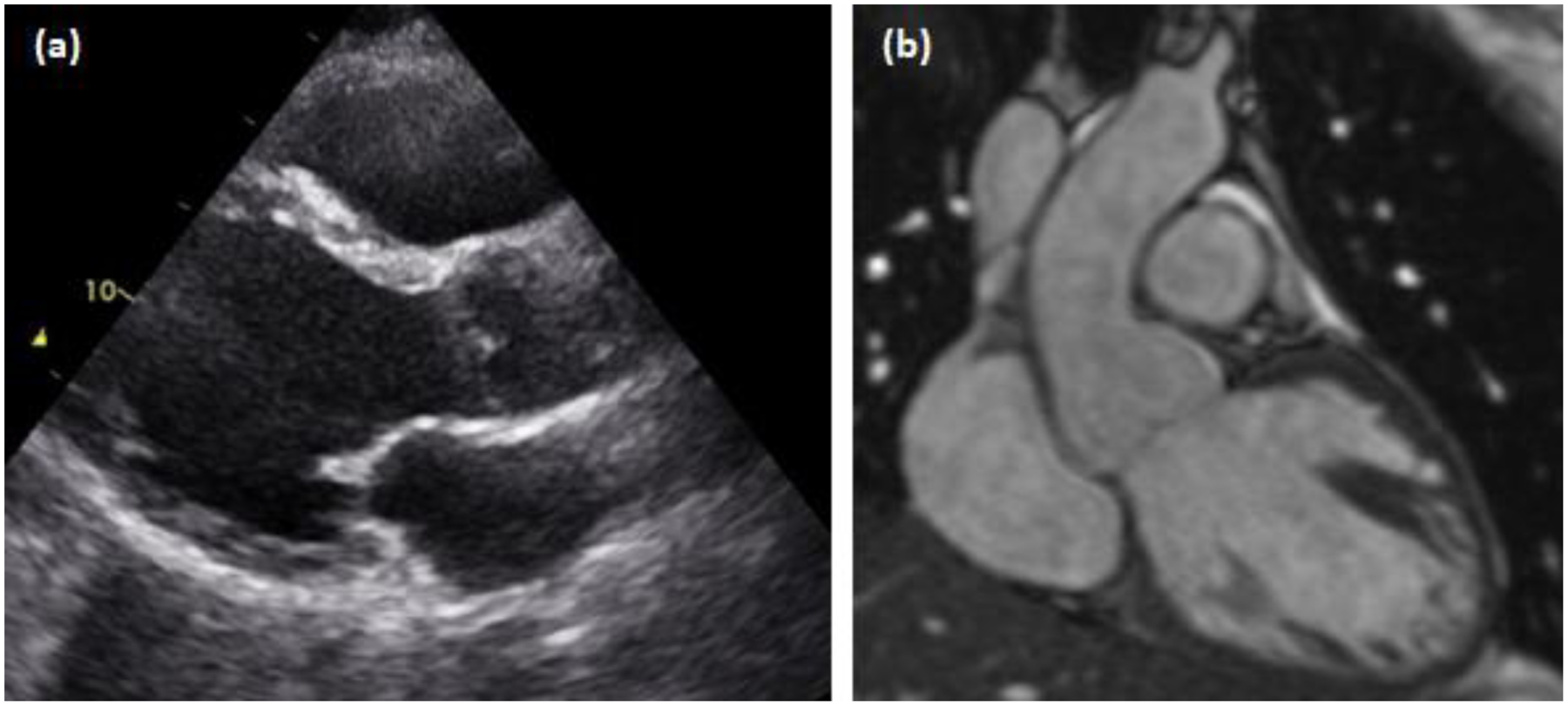
Transthoracic echocardiogram and cardiac MRI. (a) Dilated aortic root and left ventricle via parasternal long axis view. (b) Cardiac MRI demonstrating a dilated aortic root and left ventricle with normal ascending aortic dimensions distally.
These new cardiovascular findings prompted referral to our genetics clinic. At 34 years old, he was 6’7” (203.2 cm) and weighed 224 pounds (101.61 kg); notably, his mother and father were 5’6” and 5’11”, respectively. Clinical examination revealed a long face, tall forehead, deeply set eyes, prominent nasal bridge, thick horizontal eyebrows, and synophrys. The musculoskeletal exam was remarkable for long fingers with a positive wrist sign, long toes, long feet, pes planus, mild scoliosis, and joint hypermobility (Figure 2). He did not have ectopia lentis, although he had severe myopia with astigmatism. His systemic Marfan syndrome score was 5 based on revised Ghent-2 criteria (Loeys et al., 2010). The remainder of the physical exam was unremarkable for cutaneous findings, pectus deformities, easy bruising, high arched palate, and bifid uvula. He did not have a history of hypertension. His family history was negative for arterial aneurysms and dissections, cardiomyopathy, premature sudden death, congenital anomalies, neurodevelopmental disability, known genetic conditions, and consanguinity.
Figure 2.
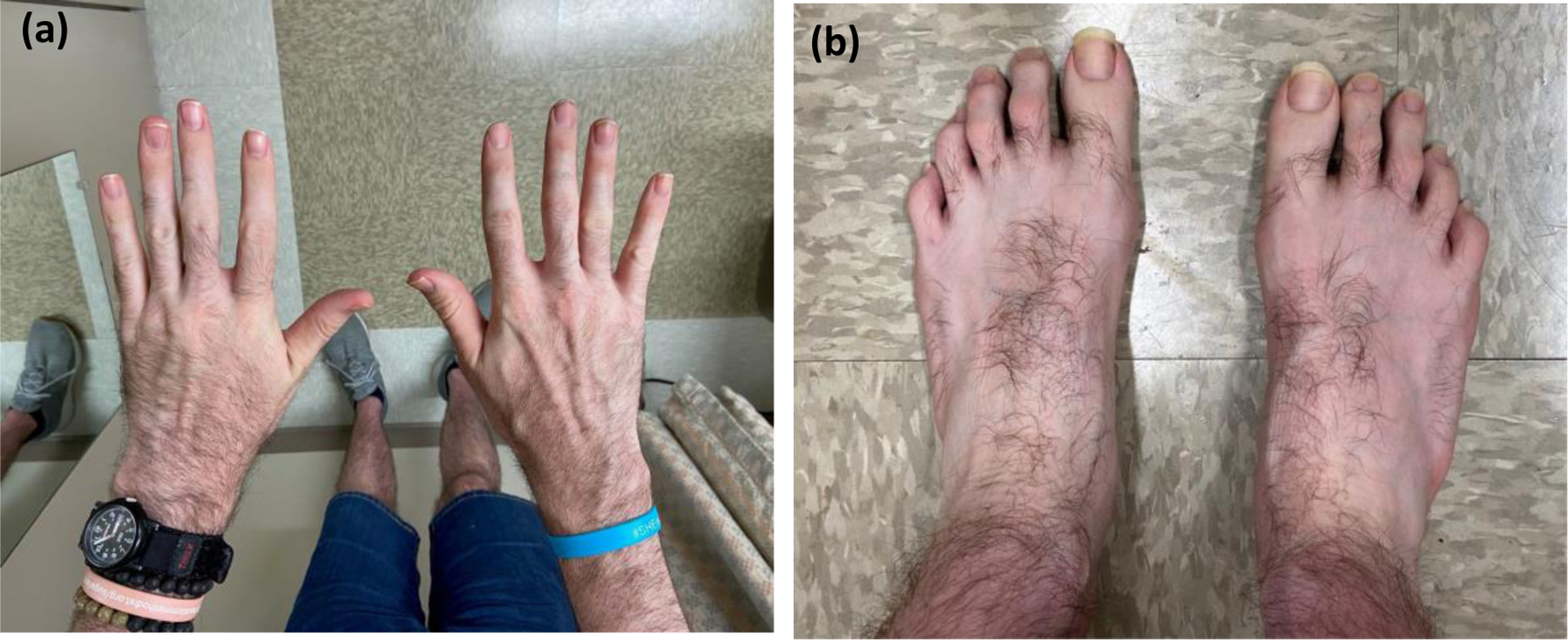
Phenotypic features. (a) Long slender fingers; more impressive on the left. (b) Long toes with mild flexion contractures; more prominent on the left.
GENETIC TESTING
Clinical exome sequencing was ordered for the patient and his parents. A heterozygous DNMT3A de novo variant was identified in the proband, c.1904G>A, p. Arg635Gln (NM_175629.2) and met criteria for a likely pathogenic classification by ACMG-AMP guidelines (PM6_strong, PS4_moderate [incorporated PP4], PP3, *PM2, *PP2) (Richards et al., 2015). The variant was observed in gnomAD v2.1.1 (2/188,710 alleles), but did not pass gnomAD quality filters. Because genes linked to hematopoietic clonal expansion, such as DNMT3A, have increased allele frequencies in population databases due to somatic mosaicism and the gnomAD variant calls were not deemed high quality, allele frequency data were not considered as evidence for or against pathogenicity (Carlston et al., 2017). The p.Arg635Gln somatic variant was observed in 6 samples in the COSMIC database; 5 were hematopoietic neoplasms (Tate et al., 2019). Although the exome assay was not validated to detect mosaicism, the DNMT3A variant was observed in approximately 50% of the reads sequenced in our proband from a saliva specimen; additional tissues were not analyzed. No additional pathogenic, likely pathogenic, or variants of uncertain significance were identified in other genes, including established aortopathy and dilated cardiomyopathy genes (Jordan et al., 2021; Renard et al., 2018). Neither parent had TBRS features, and the chest CT of his father did not show evidence of cardiomyopathy or aortic disease. His two older paternal half-siblings were not reported to have TBRS features either.
DISCUSSION
This report describes a 34-year-old male with TBRS due to a de novo DNMT3A variant, p.Arg635Gln, who presented with dilated cardiomyopathy and aortic root dilatation, which ultimately led to his diagnosis. The p.Arg635Gln variant in the MTase domain was previously identified in another patient diagnosed with TBRS at 3 years old, but no cardiovascular findings were reported (Yokoi, Enomoto, Naruto, Kurosawa, & Higurashi, 2020). Another de novo variant disrupting the same amino acid (p.Arg635Trp) was observed in a patient with autism spectrum disorder, but limited clinical features were reported (C Yuen et al., 2017; Jiang et al., 2013).
Although the number of adults with TBRS reported in the literature has exceeded 20, minimal data describing phenotypic features through adulthood limits clinical recommendations to the pediatric population, particularly with respect to age-dependent manifestations (Tatton-Brown et al., 2018). Cardiovascular pathology has been described in more than 15 patients with TBRS, ranging from structural disease diagnosed early in life including atrial and ventricular septal defects, patent ductus arteriosus, patent foramen ovale, mitral and tricuspid valve dysfunction (with and without supraventricular tachycardia), to aortic root dilatation in two cases at 16 and 30 years and cardiomyopathy in a 41 year old (Hage et al., 2020; Kosaki, Terashima, Kubota, & Kosaki, 2017; Shen et al., 2017; Tatton-Brown et al., 2018; Tenorio et al., 2019; Tlemsani et al., 2016; Xin et al., 2017). Before TBRS was described in 2014, a 14 year old with a complex phenotype that included TBRS features and dilated cardiomyopathy was found to harbor an 8.97 Mb de novo deletion of 2p23.3–24.3; although this region included more than 60 genes, DNMT3A was among them, suggesting that haploinsufficiency likely contributed to his phenotype (Shoukier et al., 2012).
Careful phenotyping with consideration of variable expressivity and phenocopies will be important for evaluating cardiomyopathy and aortic disease associated with TBRS. Tenorio et. al. reported a case similar to our patient: a 30 year-old male with a 5.7 cm aortic root aneurysm referred to genetics for Marfan syndrome who was found to carry a DNMT3A de novo nonsense variant identified by exome sequencing (Tenorio et al., 2019). This patient’s father also had a dilated aortic root; thus, the authors posited that the proband’s aortic disease was unlikely related to the DNMT3A variant. However, thoracic aortic aneurysm and cardiomyopathy phenotypes are driven by a variety of genetic, clinical, and environmental factors. Nuances that can only be ascertained by clinical history and diagnostic imaging review may be necessary to differentiate phenocopies. For example, aneurysms involving different segments of the aorta often signify distinct etiologies. Acquisition of post-zygotic mutations at various developmental stages and gonadal mosaicism may contribute to variable expressivity and impact recurrence risk (Lemire, Gauthier, Soucy, & Delrue, 2017; Xin et al., 2017). Our patient’s variant appeared to be de novo in the germline, but analyses of other tissues were not pursued as results were unlikely to alter his medical management and recurrence risk was not a concern.
In our patient, mild aortic root dilatation (Z=2.24) and mitral valve prolapse were initially reported at 14 years old, but subsequent echocardiograms up to 18 years revealed normal aortic root dimensions when adjusted for body surface area (Z=1.84), which is particularly relevant for individuals with overgrowth syndromes. Imaging at 34 years incidentally led to the diagnosis of dilated cardiomyopathy and confirmed aortic root enlargement to 4.4 cm (Z=2.39–3.17) (Figure 1). Notably, our patient exhibited several classic TBRS features, including neurodevelopmental disability (with borderline normal IQ), autism spectrum disorder, impulse control disorder, major depressive disorder, postnatal overgrowth evidenced by tall stature, and characteristic facial features. However, his tall stature, long fingers, aortic root aneurysm, mitral valve prolapse, scoliosis, pes planus and joint hypermobility, triggered clinical suspicion for Marfan syndrome, contributing to diagnostic confusion throughout his teen and young adult years.
Aberrant epigenetic regulation affecting DNA methylation and histone modifications drives pathogenesis in three overgrowth syndromes associated with aortopathy and cardiomyopathy: TBRS (DNMT3A), Sotos syndrome (NSD1) and Malan syndrome (NFIX). Aortic root aneurysms and Marfan-like musculoskeletal features have been reported in patients with all three syndromes and may be explained by crosstalk among the associated genes and the transforming growth factor β (TGF-β) signaling pathway, which plays a role in aortic aneurysm formation (Hood et al., 2016; Nimmakayalu et al., 2013; Oshima et al., 2017; Pezzani et al., 2020; Priolo et al., 2018; Tatton-Brown et al., 2018; Tenorio et al., 2019). Histone modifications mediated by histone methyltransferase NSD1 are required for DNMT3A localization and maintenance of DNA methylation (Weinberg et al., 2019). Nuclear factor-I transcription factors, including NFIX, were shown to form a nuclear complex with SMAD4 to regulate gene expression and are known to interact with SKI which regulates SMAD-dependent TGF-β signaling (Kretova et al., 2014; Tarapore et al., 1997). Further, TGF-β was shown to upregulate DNMT3A through SMAD3 signaling in dermal fibroblasts from patients with fibrotic disease (Dees et al., 2020). Conversely, knockdown of DNMT3A in trophoblasts activated TGF-β/Smad signaling and increased TGFBR1 expression (Jia, Xie, Zhang, & Ying, 2020). In addition to aortic disease, cardiomyopathy has been reported in patients with TBRS and Sotos syndrome, and is linked to epigenetic dysregulation (Martinez, Belmont, Craigen, Taylor, & Jefferies, 2011; Saccucci et al., 2011). Patients with dilated cardiomyopathy have altered DNA methylation patterns and other studies demonstrated a role for DNMT3A in cardiac remodeling and fibrosis (De Pauw et al., 2017; Gu et al., 2018; Meder et al., 2017). Finally, knockout of DNMT3A in human induced pluripotent stem cell derived cardiomyocytes led to decreased expression of contractile proteins and altered cell morphology (Madsen et al., 2020).
Findings of aortic root dilatation, mitral valve prolapse, and dilated cardiomyopathy in a 34 year old with TBRS, contribute to the emerging cardiovascular phenotype. This report is the second in the literature to describe cardiomyopathy in a patient with TBRS due to a single nucleotide DNMT3A variant. Another patient with a complex phenotype that included TBRS features and dilated cardiomyopathy was found to have a multigene deletion encompassing DNMT3A. Aortic root dilation has been described in three patients with TBRS including ours. This emphasizes a need to consider TBRS in the differential diagnosis for patients with thoracic aortic disease, Marfan-associated musculoskeletal findings, and cardiomyopathy. Accordingly, modifications to multigene diagnostic panels or exome sequencing may be indicated as syndromic overgrowth genes are not included on standard aortopathy and cardiomyopathy multigene panels. This report highlights a possible role for DNMT3A and altered epigenetic regulation as drivers of aortic root dilatation and cardiomyopathy. Periodic surveillance should be considered after a baseline echocardiogram is obtained at the time of diagnosis as these phenotypes may present later in childhood or adulthood.
ACKNOWLEDGEMENTS
This work was supported by NHLBI R01HL109942 (D.M.M.). We are grateful to this family for their collaboration in this report. Informed consent was obtained to publish photographs and enroll in this study approved by the Institutional Review Board at the University of Texas Health Science Center at Houston.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DATA AVAILABILITY STATEMENT
The variant described in this report can be accessed in ClinVar (accession VCV000872724.6). No datasets were generated or analyzed. Additional data supporting the findings in this report may be requested from the corresponding author.
REFERENCES
- Balci TB, Strong A, Kalish JM, Zackai E, Maris JM, Reilly A, … Carter MT (2020). Tatton-Brown-Rahman syndrome: Six individuals with novel features. American Journal of Medical Genetics. Part A, 182(4), 673–680. doi: 10.1002/ajmg.a.61475 [DOI] [PubMed] [Google Scholar]
- Campens L, Demulier L, De Groote K, Vandekerckhove K, De Wolf D, Roman MJ, … De Backer J (2014). Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. The American Journal of Cardiology, 114(6), 914–920. doi: 10.1016/j.amjcard.2014.06.024 [DOI] [PubMed] [Google Scholar]
- Carlston CM, O’Donnell-Luria AH, Underhill HR, Cummings BB, Weisburd B, Minikel EV, … Exome Aggregation Consortium. (2017). Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Human Mutation, 38, 517–523. doi: 10.1002/humu.23203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- C Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, Patel RV, … Scherer SW (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature Neuroscience, 20(4), 602–611. doi: 10.1038/nn.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees C, Pötter S, Zhang Y, Bergmann C, Zhou X, Luber M, … Distler JHW (2020). TGF-β–induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. The Journal of Clinical Investigation, 130(5), 2347–2363. doi: 10.1172/JCI122462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pauw A, Andre E, Sekkali B, Bouzin C, Esfahani H, Barbier N, … Balligand J-L (2017). Dnmt3a-mediated inhibition of Wnt in cardiac progenitor cells improves differentiation and remote remodeling after infarction. JCI Insight, 2(12). doi: 10.1172/jci.insight.91810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux RB, de Simone G, Arnett DK, Best LG, Boerwinkle E, Howard BV, … Roman MJ (2012). Normal Limits in Relation to Age, Body Size and Gender of Two-Dimensional Echocardiographic Aortic Root Dimensions in Persons ≥15 Years of Age. The American Journal of Cardiology. doi: 10.1016/j.amjcard.2012.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T, Lin X, Cullen SM, Luo M, Jeong M, Estecio M, … Goodell MA (2018). DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biology, 19(1), 88. doi: 10.1186/s13059-018-1464-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage C, Sabini E, Alsharhan H, Fahrner JA, Beckers A, Daly A, & Salvatori R (2020). Acromegaly in the setting of Tatton-Brown-Rahman Syndrome. Pituitary, 23(2), 167–170. doi: 10.1007/s11102-019-01019-w [DOI] [PubMed] [Google Scholar]
- Hollink IHIM, van den Ouweland AMW, Beverloo HB, Arentsen-Peters STCJM, Zwaan CM, & Wagner A (2017). Acute myeloid leukaemia in a case with Tatton-Brown-Rahman syndrome: the peculiar DNMT3A R882 mutation. Journal of Medical Genetics, 54(12), 805–808. doi: 10.1136/jmedgenet-2017-104574 [DOI] [PubMed] [Google Scholar]
- Hood RL, McGillivray G, Hunter MF, Roberston SP, Bulman DE, Boycott KM, … Care4Rare Canada Consortium. (2016). Severe connective tissue laxity including aortic dilatation in Sotos syndrome. American Journal of Medical Genetics. Part A, 170A(2), 531–535. doi: 10.1002/ajmg.a.37402 [DOI] [PubMed] [Google Scholar]
- Jiang Y-H, Yuen RKC, Jin X, Wang M, Chen N, Wu X, … Scherer SW (2013). Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. American Journal of Human Genetics, 93(2), 249–263. doi: 10.1016/j.ajhg.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Xie H, Zhang J, & Ying H (2020). Induction of TGF-β receptor I expression in a DNA methylation-independent manner mediated by DNMT3A downregulation is involved in early-onset severe preeclampsia. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 34(10), 13224–13238. doi: 10.1096/fj.202000253RR [DOI] [PubMed] [Google Scholar]
- Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, … Hershberger RE (2021). Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation, 144(1), 7–19. doi: 10.1161/CIRCULATIONAHA.120.053033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaki R, Terashima H, Kubota M, & Kosaki K (2017). Acute myeloid leukemia-associated DNMT3A p.Arg882His mutation in a patient with Tatton-Brown-Rahman overgrowth syndrome as a constitutional mutation. American Journal of Medical Genetics. Part A, 173(1), 250–253. doi: 10.1002/ajmg.a.37995 [DOI] [PubMed] [Google Scholar]
- Kretova M, Sabova L, Hodny Z, Bartek J, Kollarovic G, Nelson BD, … Luciakova K (2014). TGF-β/NF1/Smad4-mediated suppression of ANT2 contributes to oxidative stress in cellular senescence. Cellular Signalling, 26(12), 2903–2911. doi: 10.1016/j.cellsig.2014.08.029 [DOI] [PubMed] [Google Scholar]
- Lemire G, Gauthier J, Soucy J-F, & Delrue M-A (2017). A case of familial transmission of the newly described DNMT3A-Overgrowth Syndrome. American Journal of Medical Genetics. Part A, 173(7), 1887–1890. doi: 10.1002/ajmg.a.38119 [DOI] [PubMed] [Google Scholar]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, … De Paepe AM (2010). The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics, 47(7), 476–485. doi: 10.1136/jmg.2009.072785 [DOI] [PubMed] [Google Scholar]
- Madsen A, Höppner G, Krause J, Hirt MN, Laufer SD, Schweizer M, … Stenzig J (2020). An Important Role for DNMT3A-Mediated DNA Methylation in Cardiomyocyte Metabolism and Contractility. Circulation, 142(16), 1562–1578. doi: 10.1161/CIRCULATIONAHA.119.044444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez HR, Belmont JW, Craigen WJ, Taylor MD, & Jefferies JL (2011). Left ventricular noncompaction in Sotos syndrome. American Journal of Medical Genetics. Part A, 155A(5), 1115–1118. doi: 10.1002/ajmg.a.33838 [DOI] [PubMed] [Google Scholar]
- Meder B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Frese K, Lai A, … Posch AE (2017). Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation, 136(16), 1528–1544. doi: 10.1161/CIRCULATIONAHA.117.027355 [DOI] [PubMed] [Google Scholar]
- Nimmakayalu M, Horton VK, Darbro B, Patil SR, Alsayouf H, Keppler-Noreuil K, & Shchelochkov OA (2013). Apparent germline mosaicism for a novel 19p13.13 deletion disrupting NFIX and CACNA1A. American Journal of Medical Genetics. Part A, 161A(5), 1105–1109. doi: 10.1002/ajmg.a.35790 [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, & Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99(3), 247–257. doi: 10.1016/s0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- Oshima T, Hara H, Takeda N, Hasumi E, Kuroda Y, Taniguchi G, … Komuro I (2017). A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection. Human Genome Variation, 4, 17022. doi: 10.1038/hgv.2017.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paepe AD, De Paepe A, Devereux RB, Dietz HC, Hennekam RCM, & Pyeritz RE (1996). Revised diagnostic criteria for the Marfan syndrome. American Journal of Medical Genetics. doi: [DOI] [PubMed] [Google Scholar]
- Pezzani L, Mauri L, Selicorni A, Peron A, Grasso M, Codazzi AC, … Milani D (2020). Aortic dilation in Sotos syndrome: An underestimated feature? American Journal of Medical Genetics. Part A, 182(7), 1819–1823. doi: 10.1002/ajmg.a.61591 [DOI] [PubMed] [Google Scholar]
- Priolo M, Schanze D, Tatton-Brown K, Mulder PA, Tenorio J, Kooblall K, … Hennekam RC (2018). Further delineation of Malan syndrome. Human Mutation, 39(9), 1226–1237. doi: 10.1002/humu.23563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, … De Backer J (2018). Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. Journal of the American College of Cardiology, 72(6), 605–615. doi: 10.1016/j.jacc.2018.04.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccucci P, Papetti F, Martinoli R, Dofcaci A, Tuderti U, Marcantonio A, … Banci M (2011). Isolated left ventricular noncompaction in a case of sotos syndrome: a casual or causal link? Cardiology Research and Practice, 2011, 824095. doi: 10.4061/2011/824095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Heeley JM, Carlston CM, Acuna-Hidalgo R, Nillesen WM, Dent KM, … Carey JC (2017). The spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies. American Journal of Medical Genetics Part A. doi: 10.1002/ajmg.a.38485 [DOI] [PubMed] [Google Scholar]
- Shoukier M, Schröder J, Zoll B, Burfeind P, Freiberg C, Klinge L, … Brockmann K (2012). A de novo interstitial deletion of 2p23.3–24.3 in a boy presenting with intellectual disability, overgrowth, dysmorphic features, skeletal myopathy, dilated cardiomyopathy. American Journal of Medical Genetics Part A. doi: 10.1002/ajmg.a.34427 [DOI] [PubMed] [Google Scholar]
- Sluysmans T, & Colan SD (2005). Theoretical and empirical derivation of cardiovascular allometric relationships in children. Journal of Applied Physiology, 99(2), 445–457. doi: 10.1152/japplphysiol.01144.2004 [DOI] [PubMed] [Google Scholar]
- Sweeney KJ, Mottolese C, Belot A, Szathmari A, Frappaz D, Lesca G, … Di Rocco F (2019). The first case report of medulloblastoma associated with Tatton-Brown-Rahman syndrome. American Journal of Medical Genetics. Part A, 179(7), 1357–1361. doi: 10.1002/ajmg.a.61180 [DOI] [PubMed] [Google Scholar]
- Tarapore P, Richmond C, Zheng G, Cohen SB, Kelder B, Kopchick J, … Stavnezer E (1997). DNA binding and transcriptional activation by the Ski oncoprotein mediated by interaction with NFI. Nucleic Acids Research, 25(19), 3895–3903. doi: 10.1093/nar/25.19.3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, … Forbes SA (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Research, 47(D1), D941–D947. doi: 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, … Rahman N (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nature Genetics, 46(4), 385–388. doi: 10.1038/ng.2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, … Rahman N (2018). The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Research, 3, 46. doi: 10.12688/wellcomeopenres.14430.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenorio J, Alarcón P, Arias P, Dapía I, García-Miñaur S, Bralo MP, … Lapunzina P (2019). Further delineation of neuropsychiatric findings in Tatton-Brown-Rahman syndrome due to disease-causing variants in DNMT3A: seven new patients. European Journal of Human Genetics: EJHG, 28(4), 469–479. doi: 10.1038/s41431-019-0485-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlemsani C, Luscan A, Leulliot N, Bieth E, Afenjar A, Baujat G, … Burglen L (2016). SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. Journal of Medical Genetics, 53(11), 743–751. doi: 10.1136/jmedgenet-2015-103638 [DOI] [PubMed] [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, … Lu C (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature, 573(7773), 281–286. doi: 10.1038/s41586-019-1534-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin B, Cruz Marino T, Szekely J, Leblanc J, Cechner K, Sency V, … Wang H (2017). Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clinical Genetics, 91(4), 623–628. doi: 10.1111/cge.12878 [DOI] [PubMed] [Google Scholar]
- Yokoi T, Enomoto Y, Naruto T, Kurosawa K, & Higurashi N (2020). Tatton-Brown-Rahman syndrome with a novel DNMT3A mutation presented severe intellectual disability and autism spectrum disorder. Human Genome Variation, 7, 15. doi: 10.1038/s41439-020-0102-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The variant described in this report can be accessed in ClinVar (accession VCV000872724.6). No datasets were generated or analyzed. Additional data supporting the findings in this report may be requested from the corresponding author.