

Abstract
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) are clonal myeloid malignancies that are characterized by dysplasia resulting in cytopenias as well as proliferative features such as thrombocytosis or splenomegaly. Recent studies have better defined the genetics underlying this diverse group of disorders. Trisomy 8, monosomy 7, and loss of Y chromosome are the most common cytogenetic abnormalities seen. Chronic myelomonocytic leukemia (CMML) likely develops from early clones with TET2 mutations that drive granulomonocytic differentiation. Mutations in SRSF2 are common and those in the RAS‐MAPK pathway are typically implicated in disease with a proliferative phenotype. Several prognostic systems have incorporated genetic features, with ASXL1 most consistently demonstrating worse prognosis. Atypical chronic myeloid leukemia (aCML) is most known for granulocytosis with marked dysplasia and often harbors ASXL1 mutations, but SETBP1 and ETNK1 are more specific to this disease. MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN‐RS‐T) most commonly involves spliceosome mutations (namely SF3B1) and mutations in the JAK‐STAT pathway. Finally, MDS/MPN‐unclassifiable (MDS/MPN‐U) is least characterized but a significant fraction carries mutations in TP53. The remaining patients have clinical and/or genetic features similar to the other MDS/MPNs, suggesting there is room to better characterize this entity. Evolution from age‐related clonal hematopoiesis to MDS/MPN likely depends on the order of mutation acquisition and interactions between various biologic factors. Genetics will continue to play a critical role in our understanding of these illnesses and advancing patient care.
Keywords: clonal architecture, disease classification, genomic landscapes, MDS/MPN overlap syndromes, myeloid malignancies
1. INTRODUCTION
Myelodysplastic/myeloproliferative‐overlap neoplasms (MDS/MPN) are a group of clonal myeloid disorders with characteristics of dysplasia, resulting in dysfunctional hematopoiesis and cytopenias, as well as proliferative features causing increased cell counts, organ infiltration, and constitutional symptoms. They include chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML), MDS/MPN with ring sideroblasts (RSs) and thrombocytosis (MDS/MPN‐RS‐T), and MDS/MPN‐unclassifiable (MDS/MPN‐U) in adults, while juvenile myelomonocytic leukemia (JMML) is exclusively seen in pediatric populations.
With the advent of next generation sequencing technologies, the genetics and clonal architecture of MDS/MPNs have been increasingly elucidated. Much like the myeloid disorders that is this group's namesake, the mutations underlying the MDS/MPNs affect DNA methylation, chromatin modification, RNA splicing, transcription regulation, cytokine receptors, and proliferative signaling pathways.
Cytogenetically, these syndromes are most defined by the absence of specific chromosomal abnormalities so as to exclude more prevalent diagnoses. For example, all diagnoses must exclude the presence of the BCR‐ABL fusion gene seen with t(9;22) [1]. Testing must also be done to ensure there are no rearrangements involving platelet‐derived growth factor A (PDGFRA), PDGFRB, fibroblast growth factor receptor 1 (FGFR1), or pericentriolar material 1 (PCM1)‐Janus kinase 2 (JAK2) fusions in patients with eosinophilia to exclude this group of myeloid neoplasms [1]. Finally, MDS/MPN‐RS‐T cannot have isolated del(5q), t(3;3)(q21;q26), or inv(3)(q21q26) [1]. Cytogenetic abnormalities are more prevalent in MDS/MPN‐U and aCML than CMML and MDS/MPN‐RS‐T, the most common of which are trisomy 8, monosomy 7/deletion 7q, and loss of Y chromosome [2, 3, 4].
Much of our initial understanding of these diseases was extrapolated from studies of the relatively common MDS and MPNs. Subsequent studies, including both large collaborative and small single center ones, further advanced the knowledge of genetic pathways and their pathological consequences. In this review, we will discuss these findings and their impact on disease prognosis and pathophysiology, then discuss implications for disease characterization.
1.1. CMML
The most common of the MDS/MPNs, CMML is defined as a myeloid stem cell neoplasm characterized by presence of greater than 1000 monocytes/μl in the peripheral blood (PB), with monocytes comprising at least 10% of PB white blood cells (WBCs), evidence of dysplasia in at least one lineage, and less than 20% blasts [1]. Monocyte partitioning by flow cytometry may distinguish reactive from classical monocytes and aid in diagnosis [5]. It is often diagnosed incidentally or in the context of cytopenias or antecedent autoimmune illness [6, 7]. Patients may have a dysplastic phenotype characterized by low counts or a proliferative one characterized by leukocytosis (≥13,000 WBC/μl) and hepatosplenomegaly. A notable characteristic observed in vitro is hypersensitivity to granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) [8], prompting study of the anti‐GM‐CSF monoclonal antibody lenzilumab [9].
Most genetic abnormalities in CMML involve mutations of a select few genes involved in DNA methylation, splicing, and proliferative signaling. Ten‐eleven translocation‐2 (TET2) is the most commonly mutated gene, seen in 50%–70% of patients, [3, 10, 11] followed by SRSF2 (∼50%) and additional sex combs‐like 1 gene (ASXL1; ∼40%). Biallelic mutation of TET2 has been frequently recognized [3, 11]. Mutations in the RAS pathway (e.g., NRAS, KRAS, casitas B‐lineage lymphoma [CBL], PTPN11) are also frequently observed, in up to 40% of patients [12]. Karyotype abnormalities exist in less than a quarter of CMML patients [3,4] and typically involve chromosomes 7 and 8. Patients with therapy‐related CMML may have more frequent cytogenetic abnormalities but carry a similar mutation burden [13].
TET2 plays an important role in DNA methylation balance by performing hydroxylation of methylated cytosine residues, facilitating demethylation of these nucleotide residues. Mutations cause a loss of function, resulting in aberrant DNA methylation, dysmyelopoiesis, and clonal expansion [14, 15, 16]. One model of CMML pathogenesis suggests that early clonal dominance of TET2‐mutant (TET MT) clones promotes granulomonocytic differentiation of immature progenitors [17], whereas without this early dominance, additional mutations or other stochastic factors cause evolution toward other phenotypes.
Given the increased presence of mutations associated with age‐related clonal hematopoiesis in CMML [18], notably TET2 and ASXL1, CMML appears to stem from hematopoietic cells that stochastically acquire culprit mutations over time. Ancestral TET2 mutations are most common [19] and are highly associated with biallelic TET2 mutation and TET2‐SRSF2 co‐mutation [3]. Additionally, ASXL1 has been found to be a common ancestral event [3, 19]. The sequence of driver mutation acquisition and gene interactions likely skews disease toward either a more proliferative or dysplastic phenotype [20] with RAS pathway mutations responsible for the former [12] and SF3B1 occasionally associated with the latter [21].
Genetic features have been incorporated into multiple prognostic models. A large Spanish registry categorized cytogenetic risk based on overall survival (OS) and progression to acute myeloid leukemia (AML): low risk (normal karyotype or isolated loss of Y chromosome), high risk (trisomy 8, abnormalities of chromosome 7, or complex karyotype [CK]), and intermediate (all other abnormalities) [22]. These risk groups were later incorporated into the CMML‐specific prognostic scoring system (CPSS) [23]. Published the same year, the Groupe Français des Myélodysplasies prognostic score included ASXL1 nonsense and frameshift mutations because they conferred poor prognosis in multivariate analysis [10], confirmed later in a larger study [24]. Subsequently, ASXL1, NRAS, RUNX1, and SET‐binding protein‐1 (SETBP1) were independently linked to worse OS and increased risk of leukemic transformation. These mutations were integrated with the established cytogenetic risk categories into a CMML‐specific genetic score, which was incorporated into the clinical/molecular CPSS (CPSS‐Mol) [25].
Although a smaller study found that patients with TET2 MT CMML‐1 had adverse prognosis [11], this has not been consistently observed. In one large, multi‐institutional study, not only was TET2 MT disease associated with a significant survival advantage, but truncating or multiple TET2 mutations were particularly advantageous [26]. Additionally, the impact of ASXL1 was found to be ameliorated by co‐mutation with TET2 in this study [26]. Finally, another study found mutations in STAG2 and U2AF1 correlated with worse survival [3].
There have been multiple attempts to use genetic and epigenetic signatures to predict response to therapy. While somatic mutations did not predict response to hypomethylating agent (HMA) therapy in one international study, methylation profiling of mostly nonpromoter regions distinguished responders from non‐responders [27]. Gene profiling of the responders found that expression of cell cycle‐related genes were upregulated. In other evaluations, mutations in RUNX1 and CBL predicted worse OS in patients undergoing HMA therapy, but TET2 MTand ASXL1‐wildtype (ASXL1 WT) genotypes had better outcomes [28, 29]. Interestingly, HMA therapy does not appear to alter disease mutation burden or be cytotoxic to cancer cells but instead alters gene expression through epigenetic changes [30]. Finally, genetics impact response to hematopoietic stem cell transplantation: patients with high overall mutation burden (>10 mutations) were found to be at higher risk of relapse [31]. In a recently developed prognostic model, ASXL1 and NRAS mutations independently worsened survival in CMML patients post‐transplant [32].
1.2. JMML
Unlike CMML, JMML is a disease of infancy and early childhood characterized by organ infiltration of malignant cells resulting in lymphadenopathy, hepatosplenomegaly, cutaneous lesions, as well as constitutional symptoms and infectious sequelae. Its canonical feature is hypersensitivity to GM‐CSF [33], as seen in CMML, thought to be related to its nature as a RASopathy. Indeed, most cases arise from somatic or germline mutations in genes involving the RAS‐MAPK pathway: PTPN11, N/KRAS, NF1, or CBL are seen in about 90% of cases [34]. Mutations involving other signal transduction pathways, splicing, transcription, and polycomb repressive complex 2 have also been described [35]. Two‐thirds of patients will have normal karyotype, but the remaining will have monosomy 7 (26%) and 10% with other aberrancies [36].
1.3. aCML
This entity is characterized by neutrophilia with severe dysgranulopoiesis, along with bone marrow findings of hypercellularity and granulocytic dysplasia [37]. Perhaps due to significant clinical, morphologic, and genetic overlap with the MPN chronic neutrophilic leukemia (CNL) [38], WHO guidelines advise that granulocyte precursors (promyelocytes, myelocytes, and metamyelocytes) should comprise at least 10% of PB WBCs [1]. To exclude CML and CMML, there must be no or minimal absolute basophilia or monocytosis as well as no evidence of BCR‐ABL [1]. This disease is considered relatively aggressive, with a median OS of about 11–14 months [37,39,40] and a known propensity for leukemic transformation.
aCML is most specifically associated with mutations in SETBP1 and ethanolamine kinase 1 (ETNK1), which typically are found in 23%–38% and 3%–9% of patients, respectively [3, 4, 41, 42, 43]. As SETBP1 has a lower prevalence in other MDS/MPN except CMML or CNL, and ETNK1 is only otherwise seen in CMML and systemic mastocytosis [43], these genetic markers can be useful to distinguish aCML from other myeloid neoplasms. Less specific but far more prevalent is ASXL1, the most frequently mutated gene (60%–90%) [3, 4]. SETBP1 was found to be equally codominant or secondary to ASXL1 in aCML as ASXL1 mutations were found in a high percentage (>90%) of ancestral clones [3]. Other common mutations in aCML are TET2 (43%), SRSF2 (34%), NRAS (31%) [4], EZH2 (>30%) [3], RUNX1 (20%) [3, 4], and CBL (8%) [44]. Although mutations in the granulocyte‐colony stimulating factor 3 receptor (CSF3R) were previously thought to be closely linked to both aCML and CNL [45], it is now recognized to be more common in CNL [1, 46, 47]. About 40% of aCML patients have an abnormal karyotype [3, 4, 40].
Evaluation of the most aCML‐specific mutations has yielded insight into the pathophysiology of this disease. For instance, SETBP1, found on the long arm of chromosome 18, plays a role in cell proliferation by producing a nuclear protein that binds SET and inhibits the tumor suppressor gene PP2A [42, 48]. Overexpression of this gene is associated with poor outcome in AML, and it may represent a unique mechanism in leukemogenesis [48, 49].
In 2015, heterozygous mutation of ETNK1 was recognized as an abnormality largely unique to aCML [43]. As this enzyme is the first in a specific phospholipid biosynthesis pathway, its loss of catalytic function may impact cell membrane architecture, cytokinesis during cell division [50], and optimal respiratory chain performance in the mitochondrial inner membrane [51]. In fact, a recent study found that the metabolic product of ETNK1 attenuates mitochondrial activity, with loss of function driving increased production of reactive oxygen species (ROS) and mutagenesis. In vitro experiments suggested a normal phenotype can be restored by administering the underproduced enzyme product or the antibiotic tigecycline [52], which is known to inhibit synthesis of mitochondrial proteins that enable ROS production.
Despite evidence that SETBP1 confers worse prognosis in myeloid malignancies in general [53, 54] and aCML specifically [42], its impact has not been found deleterious in all studies [3, 40]. The frequent coexistence of mutations in ASXL1 and CBL with SETBP1 suggests an association with worse outcomes; [41, 42] CBL mutation alone suggests more aggressive disease [44]. Notably, TET2 mutation is independently associated with worse OS in aCML [40], although may be mutually exclusive with mutations in SETBP1 [41]. Finally, SRSF2 mutations may correlate with better OS while those in RUNX1, NRAS, and CUX1 suggest poor outcomes [3].
1.4. MDS/MPN‐RS‐T
Only upgraded from a provisional entity in the 2016 WHO classification of myeloid neoplasms [1], MDS/MPN‐RS‐T is essentially the epitome of overlap between these two disease spectrums. As its name suggests, greater than 15% RSs are found on marrow iron stain [1]. These erythroid precursors have a perinuclear ring of blue granules on Prussian blue stain, reflective of mitochondrial iron overload resulting in dyserythropoiesis [55]. Proliferative mutations drive elevated platelet counts, arterial and venous thrombosis [56, 57], vasomotor symptoms, and bleeding from acquired von Willebrand syndrome [58]. Relative to the other MDS/MPNs, prognosis is better (median OS 76 months) [57].
Similar to its phenotype, the genotype of this disease is a melding of stereotypical dysplastic and proliferative genetic aberrations. Founder mutations are frequently in genes involved in RNA splicing: SF3B1 is found in about 90% of patients [4], and SF3B1 WT patients typically harbor other spliceosome mutations [59]. SF3B1 encodes subunit 1 of the splicing factor 3b complex and may cause RS formation via downregulation of key mitochondrial gene networks [60]. Curiously, in vitro experiments showed hotspot SF3B1 mutations induce cellular metabolic changes by aberrant mRNA splicing, causing defects in the synthesis of the non‐essential amino acid serine and thereby inducing a vulnerability to serine deprivation [61]. Proliferative features are attributed to mutations involving the JAK‐STAT pathway, with about half of patients having JAK2 V617F(59) or less frequently (<10%) mutations in CALR or MPL [1]. One model suggests that disease develops from an SF3B1 MT clone with RSs, which then develops thrombocytosis following somatic mutation in the JAK‐STAT pathway [62].
Mutation in DNA (cytosine‐5)‐methyltransferase 3 alpha (DNMT3A), a gene involved in epigenetic regulation via DNA methylation, is associated with both MDS/MPN‐RS‐T and myelodysplasia with RSs (MDS‐RS) [63]. Palomo et al found that DNMT3A was always the founder mutation in MDS/MPN‐RS‐T patients who had it, whereas ASXL1 and TET2 never originated prior to SF3B1 [3]. Perhaps DNMT3A mutation is well‐suited to promoting a RS phenotype; patients with a DNMT3A MT/SF3B1 MT genotype were found to have a higher percentage of RS than DNMT3A WT/SF3B1 MT cases [59]. Although JAK2 mutation is classically associated with thrombosis in MPNs, there is evidence that SF3B1 also increases the thrombosis risk in both MDS‐RS and MDS‐RS‐T [56, 63]. Unlike CMML, the RAS‐MAPK pathway is not commonly affected in MDS/MPN‐RS‐T [3].
Like in CMML, cytogenetic abnormalities are uncommon and found in about only 10%–20% of patients [3, 4, 55]. Abnormal karyotype and mutations in ASXL1 and SETBP1 are associated with worse OS in this population [3, 64]. These mutations were found in 29% and 13% of patients in one cohort, respectively [64]. EZH2 also correlates with worse OS [3].
1.5. MDS/MPN‐U
The least well‐defined of the chronic myeloid neoplasms, MDS/MPN‐U is defined as a malignancy with dysplastic features in at least one cell lineage, prominent myeloproliferative features (either platelet count ≥450,000/μl or WBC ≥13,000/μl), and <20% blasts in blood and bone marrow. Splenomegaly may be seen. Patients must not have a history of either MDS or MPN nor recent cytotoxic growth factor therapy. The median OS is 12.4 months with thrombocytosis being an independent positive prognostic factor [65].
The genetic abnormalities found in MDS/MPN‐U are broad. ASXL1 is the most frequent mutation, occurring in about 30%–50% of patients. Others include TET2, JAK2, EZH2, SRSF2, NRAS, SETBP1, RUNX1, STAG2, U2AF1, and TP53 [2, 3, 4, 66]. Diploid cytogenetics are seen in 49%–71% of patients [2–4,65], and about 12% of patients have CK [2, 3, 65]. CBL and TP53 are independent risk factors for worse prognosis [2], whereas ASXL1, EZH2, and STAG2 mutations are associated with worse survival [3].
In a large (n = 106) study of MDS/MPN‐U clonal architecture, unclassifiable cases actually segregated into subgroups that fit the other MDS/MPNs based on genetic and clinical features [3]: 17% had a CMML‐like signature, 33% were aCML‐like, and 11% had MDS/MPN‐RS‐T‐like disease. The CMML‐like disease, for instance, included patients with either biallelic TET2 mutations, TET2 and SRSF2 co‐mutation, or RUNX1 and SRSF2 co‐mutation; survival curves were similar between this group and WHO‐classified CMML patients. Patients with mono‐ or biallelic mutations in TP53 segregated into a fourth group (13%), characterized by worse anemia, higher bone marrow blast percentage, and worse prognosis [3]. Finally, the remaining 26% of patients more frequently had mutations in JAK2, U2AF1, and ASXL1 and were more likely to have thrombocytosis. Survival in these “other” patients trended worse than CMML‐like patients but was better than aCML‐like patients.
2. DISCUSSION
It was not until 2001 that the WHO re‐classified CMML from a subset of MDS to the newly created MDS/MPN category [67]. Since then, there have been significant advancements in understanding this group of neoplasms (Table 1) and an increase in clinical trials targeting CMML patients in particular. There remains a need to better characterize MDS/MPNs by genetic features—to more accurately understand the natural history of the disease and ensure patient eligibility for clinical trials and FDA‐approved treatment options [68]. Additionally, genetics plays a critical role in disease prognostication; for instance, the worse outcomes seen among men with MDS/MPN as compared to women may be explained by gender‐related differences in somatic mutation profiles [69]. Our review highlights the advances in our understanding of the genetics of these diseases.
TABLE 1.
Overview of diagnostic, clinical, and genetic features of the MDS/MPN overlap syndromes. MDS/MPNs all must have <20% blasts in PB and BM, with the RS‐T subtype only allowing for <1% PB and <5% BM blasts. No evidence of the BCR‐ABL1 fusion gene, PDGFR, FGFR1, and PCM1‐JAK2 is permitted. No history of cytotoxic or growth factor therapy is permitted in the RS‐T or U subtypes
| Disease | 2016 WHO diagnostic criteria | Clinical features | Cytogenetics | Molecular genetics | Other |
|---|---|---|---|---|---|
| CMML |
|
|
|
|
|
| JMML |
|
|
|
|
|
| aCML |
|
|
|
|
|
| MDS/MPN‐RS‐T |
|
|
|
|
|
| MDS/MPN‐U |
|
|
|
|
|
Abbreviations: aCML, atypical chronic myeloid leukemia; aVWS, acquired von Willebrand syndrome; BM, bone marrow; CK, complex karyotype; CMML, chronic myelomonocytic leukemia; CNL, chronic neutrophilic leukemia; HCT, hematopoietic cell transplantation; HMA, hypomethylating agents; JMML, juvenile myelomonocytic leukemia; LOH, loss of heterozygosity; MDS, myelodysplasia; MPN, myeloproliferative neoplasm; NK, normal karyotype; OS, overall survival; PB, peripheral blood; RS‐T, ring sideroblasts with thrombocytosis; U, unclassifiable.
As observers of natural phenomena, our inclination is to categorize disease based on features such as cell count abnormalities, morphology, and genetic signatures. If the cutoffs for these characteristics are not specific enough, then disease classifications become less meaningful. However, if the prescribed cutoffs are too restrictive, meaningful data about cases that fall outside rigid parameters may be lost into an exclusionary “wastebasket” category. Thoughtfully liberalizing criteria can notably help encompass cases that may be at a more immature stage, as done in 2016: The criteria for MDS‐RS were adjusted from requiring ≥15% RS on marrow exam to needing ≥5% RS in the presence of SF3B1 mutation [1]. Conceivably, these cases represent disease that may eventually progress to ≥15% RS or simply have prognoses that are not meaningfully different than those defined by the pre‐2016 classifications.
Evidence suggests that such disease exists in the MDS/MPNs and is frequently labeled under alternative categories (Figure 1). For example, “oligomonocytic” CMML shares genetic and clinical features with CMML but does not meet the WHO criteria due to absolute monocytosis < 1000/μl [70, 71], likely representing early‐phase dysplastic‐type CMML. A similar example is found within a group of MDS/MPN‐U patients with ≥15% RS on bone marrow and enriched for JAK2 and SF3B1 mutations. Although they did not meet criteria for MDS/MPN‐RS‐T due to lack of thrombocytosis or other features, they had prognoses similar to an independent cohort of patients with MDS/MPN‐RS‐T [2].
FIGURE 1.
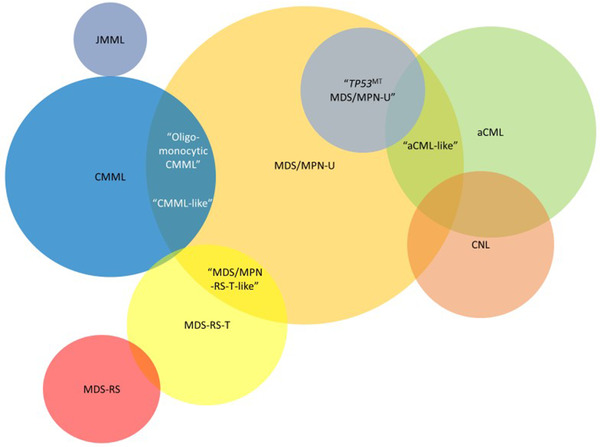
Diagram depicting the MDS/MPN as defined by genetic and clinical features, which may overlap themselves due to genetic heterogeneity or early‐stage disease. Within these are several entities (in quotation marks) that represent diseases with similar genetics and clinical characteristics, but not meeting WHO criteria
Abbreviations: aCML, atypical chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; CNL, chronic neutrophilic leukemia; JMML, juvenile myelomonocytic leukemia; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; MDS‐RS, myelodysplasia with ring sideroblasts; MDS/MPN‐RS‐T, ring sideroblast with thrombocytosis; MDS/MPN‐U, unclassifiable.
Our current classifications likely do not allow for enough nuance, too. Although there is evidence that the less well‐defined chronic myeloid malignancies are distinct entities based on cytogenetic risk stratification and leukemia free survival [39], a more recent study found significant heterogeneity among patients with CNL, aCML, MPN‐U, MDS/MPN‐U, and CMML [15]. Based on whole exome and RNA sequencing, researchers identified at least 15 groups of patients with different combinatorial mutations patterns, suggesting these diseases represent a continuum as opposed to distinct pathologies. Perhaps disease classification for these more genetically diverse entities should be more granular by specifying aberrations in their names, as done with some AML subtypes [1].
3. SUMMARY AND FUTURE DIRECTIONS
The MDS/MPNs are rare and heterogeneous, making systematic study more difficult relative to other myeloid malignancies. Advances in gene sequencing technology along with large multi‐institution collaborations have shed light on these illnesses: We now know that most have signaling gene mutations responsible for proliferative phenotypes, and many have gene mutations affecting epigenetic regulation, causing dysplastic features. These syndromes likely often evolve out of preexisting clones that mature into neoplasms with phenotypes based on the order of mutation acquisition and interaction between epigenetic and genetic factors. Genetic studies will likely aid in revising WHO criteria so as to more holistically classify the overlap syndromes.
CONFLICT OF INTEREST
Tania Jain serves as a consultant for Targeted Healthcare Communications and has served on the advisory board for CareDx and Bristol Myers Squibb. The authors Michael J. Hochman and Bipin Savani have no disclosures.
AUTHOR CONTRIBUTIONS
Michael J. Hochman did the literature review and wrote the manuscript. Bipin Savani did the literature search and reviewed the manuscript. Tania Jain did the literature search, reviewed the manuscript and supervised the project. All authors approve the final draft of the manuscript.
Hochman MJ, Savani B, Jain T. Examining disease boundaries: Genetics of myelodysplastic/myeloproliferative neoplasms. eJHaem. 2021;2:607–615. 10.1002/jha2.264
REFERENCES
- 1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405. [DOI] [PubMed] [Google Scholar]
- 2. Mangaonkar AA, Swoboda DM, Coltro G, Lasho TL, Novotny PJ, Pophali P, et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN‐U): Mayo Clinic‐Moffitt Cancer Center study of 135 consecutive patients. Leukemia. 2020;34(2):656–61. [DOI] [PubMed] [Google Scholar]
- 3. Palomo L, Meggendorfer M, Hutter S, Twardziok S, Ademà V, Fuhrmann I, et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood. 2020;136(16):1851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meggendorfer M, Jeromin S, Haferlach C, Kern W, Haferlach T. The mutational landscape of 18 investigated genes clearly separates four subtypes of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2018;103(5):e192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shallis RM, Siddon AJ, Zeidan AM. Clinical and molecular approach to adult‐onset, neoplastic monocytosis. Curr Hematol Malig Rep. 2021;16(3):276–25. [DOI] [PubMed] [Google Scholar]
- 6. Grignano E, Mekinian A, Braun T, Liozon E, Hamidou M, Decaux O, et al. Autoimmune and inflammatory diseases associated with chronic myelomonocytic leukemia: A series of 26 cases and literature review. Leuk Res. 2016;47:136–41. [DOI] [PubMed] [Google Scholar]
- 7. Zahid MF, Barraco D, Lasho TL, Finke C, Ketterling RP, Gangat N, et al. Spectrum of autoimmune diseases and systemic inflammatory syndromes in patients with chronic myelomonocytic leukemia. Leuk Lymphoma. 2017;58(6):1488–93. [DOI] [PubMed] [Google Scholar]
- 8. Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, et al. GM‐CSF‐dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patnaik MM, Sallman DA, Mangaonkar AA, Heuer R, Hirvela J, Zblewski D, et al. Phase 1 study of lenzilumab, a recombinant anti‐human GM‐CSF antibody, for chronic myelomonocytic leukemia. Blood. 2020;136(7):909–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Itzykson R, Kosmider O, Renneville A, Gelsi‐Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36. [DOI] [PubMed] [Google Scholar]
- 11. Kosmider O, Gelsi‐Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94(12):1676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, et al. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16(8):2246–56. [DOI] [PubMed] [Google Scholar]
- 13. Patnaik MM, Vallapureddy R, Yalniz FF, Hanson CA, Ketterling RP, Lasho TL, et al. Therapy related‐chronic myelomonocytic leukemia (CMML): Molecular, cytogenetic, and clinical distinctions from de novo CMML. Am J Hematol. 2018;93(1):65–73. [DOI] [PubMed] [Google Scholar]
- 14. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5‐methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L, Padron E, Lancet J. The molecular basis and clinical significance of genetic mutations identified in myelodysplastic syndromes. Leuk Res. 2015;39(1):6–17. [DOI] [PubMed] [Google Scholar]
- 16. Guan Y, Hasipek M, Tiwari AD, Maciejewski JP, Jha BK. TET‐dioxygenase deficiency in oncogenesis and its targeting for tumor‐selective therapeutics. Semin Hematol. 2021;58(1):27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186–98. [DOI] [PubMed] [Google Scholar]
- 18. Mason C, Khorashad J, Tantravahi S, Kelley T, Zabriskie M, Yan D, et al. Age‐related mutations and chronic myelomonocytic leukemia. Leukemia. 2016;30(4):906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patel BJ, Przychodzen B, Thota S, Radivoyevitch T, Visconte V, Kuzmanovic T, et al. Genomic determinants of chronic myelomonocytic leukemia. Leukemia. 2017;31(12):2815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carr RM, Patnaik MM. Genetic and epigenetic factors interacting with clonal hematopoiesis resulting in chronic myelomonocytic leukemia. Curr Opin Hematol. 2020;27(1):2–10. [DOI] [PubMed] [Google Scholar]
- 21. Wudhikarn K, Loghavi S, Mangaonkar AA, Al‐Kali A, Binder M, Carr R, et al. SF3B1‐mutant CMML defines a predominantly dysplastic CMML subtype with a superior acute leukemia‐free survival. Blood Adv. 2020;4(22):5716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Such E, Cervera J, Costa D, Solé F, Vallespí T, Luño E, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005–15. [DOI] [PubMed] [Google Scholar]
- 24. Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two‐center study of 466 patients. Leukemia. 2014;28(11):2206–12. [DOI] [PubMed] [Google Scholar]
- 25. Elena C, Gallì A, Such E, Meggendorfer M, Germing U, Rizzo E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128(10):1408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coltro G, Mangaonkar AA, Lasho TL, Finke CM, Pophali P, Carr R, et al. Clinical, molecular, and prognostic correlates of number, type, and functional localization of TET2 mutations in chronic myelomonocytic leukemia (CMML)—a study of 1084 patients. Leukemia. 2020;34(5):1407–21. [DOI] [PubMed] [Google Scholar]
- 27. Meldi K, Qin T, Buchi F, Droin N, Sotzen J, Micol J‐B, et al. Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J Clin Invest. 2015;125(5):1857–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duchmann M, Yalniz FF, Sanna A, Sallman D, Coombs CC, Renneville A, et al. Prognostic role of gene mutations in chronic myelomonocytic leukemia patients treated with hypomethylating agents. EBioMedicine. 2018;31:174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bejar R, Lord A, Stevenson K, Bar‐Natan M, Pérez‐Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124(17):2705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M, et al. Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat Commun. 2016;7(1):10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woo J, Choi DR, Storer BE, Yeung C, Halpern AB, Salit RB, et al. Impact of clinical, cytogenetic, and molecular profiles on long‐term survival after transplantation in patients with chronic myelomonocytic leukemia. Haematologica. 2020;105(3):652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gagelmann N, Badbaran A, Beelen DW, Salit RB, Stölzel F, Rautenberg C, et al. A prognostic score including mutation profile and clinical features for patients with CMML undergoing stem cell transplantation. Blood Adv. 2021;5(6):1760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte‐macrophage colony‐stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925–9. [PubMed] [Google Scholar]
- 34. Sakashita K, Matsuda K, Koike K. Diagnosis and treatment of juvenile myelomonocytic leukemia. Pediatr Int. 2016;58(8):681–90. [DOI] [PubMed] [Google Scholar]
- 35. Stieglitz E, Taylor‐Weiner AN, Chang TY, Gelston LC, Wang Y‐D, Mazor T, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. 2015;47(11):1326–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG‐MDS). Blood. 1997;89(10):3534–43. [PubMed] [Google Scholar]
- 37. Hernández JM, del Cañizo MC, Cuneo A, García JL, Gutiérrez NC, González M, et al. Clinical, hematological and cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000;11(4):441–4. [DOI] [PubMed] [Google Scholar]
- 38. Dao K‐HT, Tyner JW. What's different about atypical CML and chronic neutrophilic leukemia? Hematology. 2015;2015(1):264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot‐Richards D, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014;123(17):2645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patnaik MM, Barraco D, Lasho TL, Finke CM, Reichard K, Hoversten KP, et al. Targeted next generation sequencing and identification of risk factors in World Health Organization defined atypical chronic myeloid leukemia. Am J Hematol. 2017;92(6):542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti‐Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852–60. [DOI] [PubMed] [Google Scholar]
- 42. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gambacorti‐Passerini CB, Donadoni C, Parmiani A, Pirola A, Redaelli S, Signore G, et al. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood. 2015;125(3):499–503. [DOI] [PubMed] [Google Scholar]
- 44. Grand FH, Hidalgo‐Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009;113(24):6182–92. [DOI] [PubMed] [Google Scholar]
- 45. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. N Engl J Med. 2013;368(19):1781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27(9):1870–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014;99(12):e244–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cristóbal I, Blanco FJ, Garcia‐Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115(3):615–25. [DOI] [PubMed] [Google Scholar]
- 49. Cristóbal I, Garcia‐Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti‐leukemic effect. Leukemia. 2011;25(4):606–14. [DOI] [PubMed] [Google Scholar]
- 50. Mileykovskaya E, Dowhan W. Role of membrane lipids in bacterial division‐site selection. Curr Opin Microbiol. 2005;8(2):135–42. [DOI] [PubMed] [Google Scholar]
- 51. Furt F, Moreau P. Importance of lipid metabolism for intracellular and mitochondrial membrane fusion/fission processes. Int J Biochem Cell Biol. 2009;41(10):1828–36. [DOI] [PubMed] [Google Scholar]
- 52. Fontana D, Mauri M, Renso R, Docci M, Crespiatico I, Røst LM, et al. ETNK1 mutations induce a mutator phenotype that can be reverted with phosphoethanolamine. Nat Commun. 2020;11(1):5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27(6):1401–3. [DOI] [PubMed] [Google Scholar]
- 55. Patnaik MM, Tefferi A. Refractory anemia with ring sideroblasts (RARS) and RARS with thrombocytosis (rars ‐ t): 2017 update on diagnosis, risk‐stratification, and management. Am J Hematol. 2017;92(3):297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Vascular events and risk factors for thrombosis in refractory anemia with ring sideroblasts and thrombocytosis. Leukemia. 2016;30(11):2273–5. [DOI] [PubMed] [Google Scholar]
- 57. Broseus J, Florensa L, Zipperer E, Schnittger S, Malcovati L, Richebourg S, et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica. 2012;97(7):1036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Michiels JJ. Acquired von Willebrand disease due to increasing platelet count can readily explain the paradox of thrombosis and bleeding in thrombocythemia. Clin Appl Thromb Hemost. 1999;5(3):147–51. [DOI] [PubMed] [Google Scholar]
- 59. Jeromin S, Haferlach T, Weissmann S, Meggendorfer M, Eder C, Nadarajah N, et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica. 2015;100(4):e125–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dalton WB, Helmenstine E, Walsh N, Gondek LP, Kelkar DS, Read A, et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J Clin Invest. 2019;129(11):4708–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cazzola M, Malcovati L, Invernizzi R. Myelodysplastic/myeloproliferative neoplasms. Hematology. 2011;2011(1):264–72. [DOI] [PubMed] [Google Scholar]
- 63. Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26(3):542–5. [DOI] [PubMed] [Google Scholar]
- 64. Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS‐T) and the role of next‐generation sequencing. Am J Hematol. 2016;91(5):492–8. [DOI] [PubMed] [Google Scholar]
- 65. DiNardo CD, Daver N, Jain N, Pemmaraju N, Bueso‐Ramos C, Yin CC, et al. Myelodysplastic/myeloproliferative neoplasms, unclassifiable (MDS/MPN, U): natural history and clinical outcome by treatment strategy. Leukemia. 2014;28(4):958–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bose P, Nazha A, Komrokji RS, Patel KP, Pierce SA, Al‐Ali N, et al. Mutational landscape of myelodysplastic/myeloproliferative neoplasm‐unclassifiable. Blood. 2018;132(19):2100–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–302. [DOI] [PubMed] [Google Scholar]
- 68. Padron E. Toward classifying the unclassifiable. Blood. 2020;136(16):1800–1. [DOI] [PubMed] [Google Scholar]
- 69. Karantanos T, Gondek LP, Varadhan R, Moliterno AR, DeZern AE, Jones RJ, et al. Gender‐related differences in the outcomes and genomic landscape of patients with myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes. Br J Haematol. 2021;193(6):1142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Geyer JT, Tam W, Liu Y‐C, Chen Z, Wang SA, Bueso‐Ramos C, et al. Oligomonocytic chronic myelomonocytic leukemia (chronic myelomonocytic leukemia without absolute monocytosis) displays a similar clinicopathologic and mutational profile to classical chronic myelomonocytic leukemia. Mod Pathol. 2017;30(9):1213–22. [DOI] [PubMed] [Google Scholar]
- 71. Valent P, Orazi A, Savona MR, Patnaik MM, Onida F, van de Loosdrecht AA, et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre‐CMML conditions. Haematologica. 2019;104(10):1935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]