

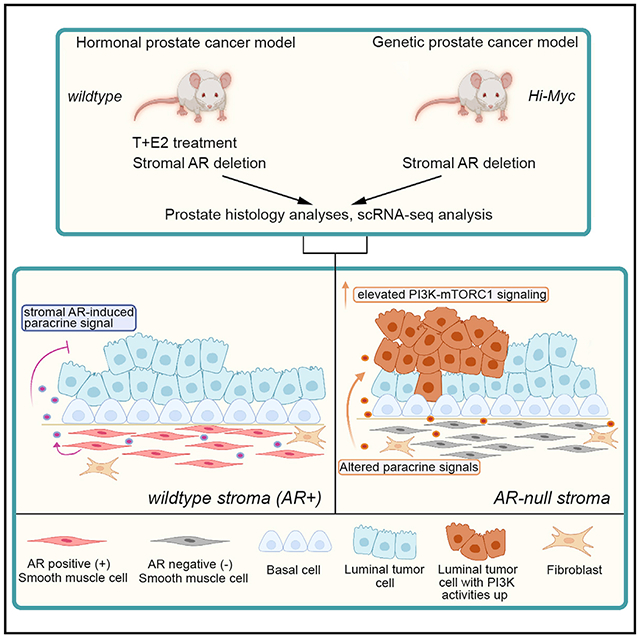
SUMMARY
Androgen receptor (AR) is expressed in both the prostate epithelium and the prostate stroma and plays diverse roles in prostate physiology. Although low expression of stromal AR is clinically associated with advanced cancer stage and worse outcome, whether stromal AR inhibits or promotes prostate cancer progression remains controversial. Here, we specifically delete AR in smooth muscle cells of the adult mouse prostate under two tumorigenic conditions, namely, the Hi-Myc genetic model and the T + E2 hormonal carcinogenesis model. Histology analyses show that stromal AR deletion exacerbates tumor progression phenotypes in both models. Furthermore, single-cell analyses of the tumor samples reveal that secretory luminal cells are the cell population particularly affected by stromal AR deletion, as they transition to a cellular state of potentiated PI3K-mTORC1 activities. Our results suggest that stromal AR normally inhibits prostate cancer progression by restraining secretory luminal cells and imply possible unintended negative effects of androgen deprivation therapy.
In brief
Whether stromal androgen receptor (AR) promotes or inhibits prostate cancer progression is controversial. Liu et al. report that AR loss in smooth muscle cells exacerbates tumor phenotypes by potentiating the PI3K pathway activity in a subset of luminal epithelial cells, suggesting a tumor-suppressing role for AR in the prostate stroma.
Graphical Abstract
INTRODUCTION
Most prostate cancers (PCas) are adenocarcinomas that originate from the epithelial cells of the gland (Shen and Abate-Shen, 2010). Both basal and luminal cells, the two major cell types of the prostate epithelium, can serve as cells of origin for PCa (Choi et al., 2012; Wang et al., 2013). On the other hand, the stromal compartment, which is composed of smooth muscle cells (SMCs), fibroblasts, endothelial cells, immune cells, and neurons, provides the crucial microenvironment for prostate development and cancer progression. Classic tissue recombination experiments have demonstrated the essential role of stromal-epithelial interactions in prostate organogenesis (Cunha et al., 1987; Hayward, 2002). In PCa, the altered stromal microenvironment, named reactive stroma, can promote cancer progression (Tuxhorn et al., 2001). For example, cancer-associated fibroblasts (CAFs) send paracrine signals, including growth factors and interleukins, to promote epithelial transformation (Sasaki et al., 2017).
Androgen is one of the key signals regulating prostate development and cancer (Murashima et al., 2014; Watson et al., 2015). The androgen receptor (AR) is expressed in both the prostate epithelium and the prostate stroma. In the epithelium, recent lineage-tracing analyses showed that ARs in basal and luminal cells play distinct roles in maintaining adult prostate homeostasis (Xie et al., 2017; Chua et al., 2018). In the stroma, classic experiments showed that AR-deficient urogenital sinus mesenchyme (UGSM) was unable to induce prostate budding when recombined with wild-type epithelium, suggesting that the stromal AR is essential for prostate organogenesis (Cunha and Lung, 1978). Consistent with this finding, genetic deletion of stromal AR in the developing mouse prostate inhibited epithelial cell growth and the differentiation of prostatic ducts and glandular acini (Welsh et al., 2011; Yu et al., 2011, 2012; Lai et al., 2012b; Lee et al., 2021).
How stromal cells and stromal AR affect PCa has been controversial (Singh et al., 2014; Wen et al., 2015; Leach and Buchanan, 2017). Early studies using epithelial-stromal cell co-culture conflicted regarding the role of stromal cells in epithelial cancer cell growth, with some favoring a negative role (Konig et al., 1987; Degeorges et al., 1996; Kooistra et al., 1997) and others a positive one (Kabalin et al., 1989; Lang et al., 2000; Castellon et al., 2005). Clinically, lower expression of stromal AR is associated with advanced PCa and worse patient outcome (Olapade-Olaopa et al., 1999; Henshall et al., 2001; Ricciardelli et al., 2005; Wikstrom et al., 2009; Leach et al., 2015). Paradoxically, AR-deficient mouse stroma was less capable of promoting tumor formation than wild-type stroma in epithelial co-culture assays and tumor cell renal grafts (Lai et al., 2012b; Ricke et al., 2012; Yu et al., 2012). In genetic mouse models, a study comparing AR deletion in the epithelium versus the entire organ in the TRAMP PCa model suggested a tumor-promoting role for stromal AR (Niu et al., 2008). Similarly, stromal AR deletion by FSP1-Cre and Tgln-Cre in the Pten−/+ cancer model was reported to diminish prostatic intraepithelial neoplasia (PIN) development (Lai et al., 2012a). In contrast, Welsh and colleagues used SMH-Cre to ablate AR in SMCs and found stromal AR to play a tumor-suppressing role in a hormonal carcinogenesis model (Welsh et al., 2011).
A possible cause of the discrepancy may be the different approaches used and their associated caveats. Renal grafting assay, and stromal-epithelial co-culture in particular, may not faithfully recapitulate actual cancer progression due to a lack of orthotopic microenvironment. On the other hand, previous studies of conditional stromal AR knockout utilized Cre lines that were not inducible, potentially complicating data interpretation by having AR loss during development. To date, no stromal AR ablation experiment has been rigorously performed in adult prostate carcinogenesis in vivo. Here, we used an inducible SMC-specific CreER line to delete AR at the adult stage in two distinct PCa mouse models. We show that stromal AR deletion accelerated PCa progression for both, and such effect mostly worked through secretory luminal cells by altering their molecular program to a PI3K-activated state.
RESULTS
Stromal AR maintains normal prostate epithelial homeostasis and morphology
To delete AR in the adult prostate stroma, we obtained the Myh11-CreERT2 transgenic mouse, in which the inducible Cre is driven by the promoter of the gene smooth muscle myosin heavy polypeptide 11 (Wirth et al., 2008). To test its specificity, we generated Myh11-CreERT2; R26R-CAG-EYFP/+ (denoted wt) mice, induced them with tamoxifen at 7 weeks of age, and analyzed the prostate 2 weeks later (Figure 1A). Immunofluorescence (IF) staining showed that YFP+ cells were in the stromal layer and did not express basal marker CK5 or luminal marker CK18 (Figure 1B). Instead, staining with a smooth muscle actin (SMA) antibody showed that over 99% of the SMCs were labeled by YFP (Figure 1C). The vast majority of the YFP+ cells were AR positive, with slightly varying proportions in different lobes (anterior lobe [AP] 92.7%; ventral lobe [VP] 88.4%; dorsal-lateral lobe [DLP] 83.3%) (Figures 1D and 1F). We then tested the efficiency of stromal AR deletion by tamoxifen induction of Myh11-CreERT2; ARflox/Y; R26R-CAG-EYFP/+ (denoted strAR−) mice at 7 weeks of age (Figure 1A). Two weeks later, the proportions of AR+ cells in the YFP+ population (SMCs) significantly decreased for all the lobes (AP 16.3%; VP 13.6%; DLP 13.0%) (Figures 1E and 1F). In contrast, epithelial AR expression was not affected (Figure 1E). Therefore, we achieved AR deletion in SMCs with high efficiency and specificity.
Figure 1. AR deletion in SMCs of the normal adult prostate.
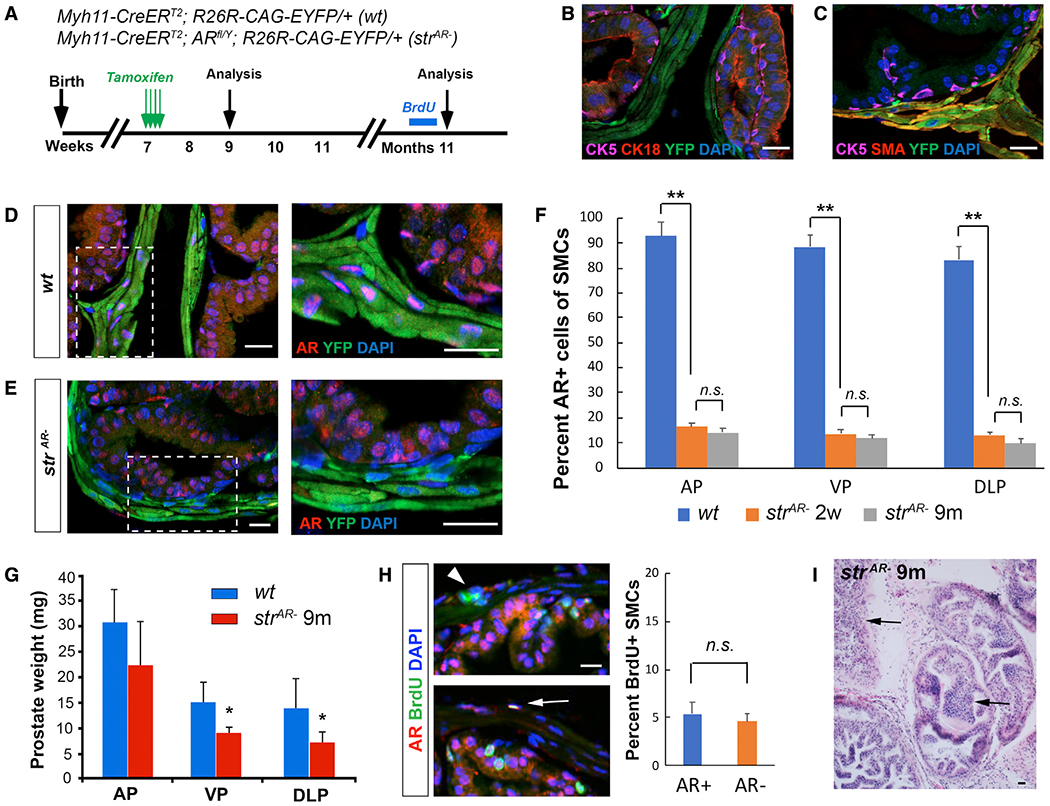
(A) Timeline of experiments for the wt and strAR− mice.
(B and C) IF staining showing YFP-marked cells present only in the stroma (B) and positive for SMA (C).
(D and E) IF staining showing loss of AR expression in YFP+ cells from wt (D) to strAR− (E). Zoom-in views of the boxed areas are shown on the right.
(F) Quantitation of the proportions of AR+ SMCs in different prostate lobes before and after AR deletion through time.
(G) Quantitation of prostate lobe weights in wt and strAR− mice 9 months after induction.
(H) Representative IF staining of BrdU (left) and quantitation of the proportions of BrdU+ cells in AR+ and AR− SMCs (right). Arrowhead points to an AR−BrdU+ SMC and arrow to an AR+BrdU+ SMC.
(I) H&E staining showing prostate morphology of strAR− mice 9 months after induction. Arrows point to regions of hyperplasia. *p < 0.05 and **p < 0.001 by Student’s t test. Scale bars, 20 μm.
We then characterized the long-term effects of stromal AR deletion in the strAR− mice by analyzing the prostate phenotypes 9 months after tamoxifen induction (11 months of age) (Figure 1A). Prostate weight was reduced in the strAR− mice compared with age-matched wt mice for all lobes (Figure 1G), consistent with the notion that stromal AR mediates androgen-dependent prostate epithelial growth (Yu et al., 2011). Size reduction was also observed for seminal vesicles and testes, since AR was deleted in SMCs of other urogenital organs as well (Figure S1). However, no difference in overall body weight or reproductive capability was noticed between strAR− and wt mice. The proportions of AR+ SMCs in the prostate remained constant from 2 weeks to 9 months after induction (Figure 1F), and BrdU incorporation assay at 9 months after induction showed that AR+ and AR− SMCs proliferated at similar rates (Figure 1H). No significant differences in luminal and basal cell proliferation were detected between strAR− and wt mice at this stage, either (Figure S2). Notably, hematoxylin and eosin (H&E) staining showed that strAR− prostate at 9 month after induction contained smaller lumens compared to wt and displayed hypertrophy and foci of epithelial hyperplasia (Figure 1I), suggesting that stromal AR regulates the normal morphology and epithelial cell integrity in the adult prostate.
Loss of stromal AR enhances Hi-Myc tumor progression
To test the role of stromal AR in PCa progression, we first utilized the ARR2/probasin-Myc (Hi-Myc) model (Ellwood-Yen et al., 2003), in which Myc overexpression mimics one of the frequent oncogenic events in human PCa (Fraser et al., 2017). Myh11-CreERT2; R26R-CAG-EYFP/+; Hi-Myc (denoted Myc) and Myh11-CreERT2; ARflox/Y; R26R-CAG-EYFP/+; Hi-Myc (denoted strAR− Myc) male mice were generated, induced with tamoxifen at 7 weeks of age, and analyzed at different time points later (Figure 2A). One month after induction, IF staining confirmed that AR was absent in over 90% of SMCs in the strAR− Myc group (Figure 2B). H&E staining at 3 and 9 months post induction revealed frequent hyperplasia and PIN lesions in both groups. However, the strAR− Myc group contained more high-grade PINs compared with the Myc group, and often displayed dilated ducts, cribriform patterns, and inflammation (Figure 2C and Table S1). To rigorously compare the histology between the two groups, we quantified tumor grades by evaluating each sample and assigning a histology score using a predefined formula in a double-blind setting (see STAR Methods). Our results (Figure 2D and Table S1) showed that stromal AR deletion promoted PCa progression from the Hi-Myc model. Indeed, BrdU incorporation assay showed that luminal cells were more proliferative in the strAR− Myc group compared with the Myc group (Figures 2E and 2F), whereas no significant difference was found for basal or stromal cells between the two groups (Figure 2F). These data demonstrate that AR in the prostate SMCs plays a tumor-suppressing role in the Hi-Myc model.
Figure 2. Stromal AR loss promotes tumor progression in the Hi-Myc and T + E models.
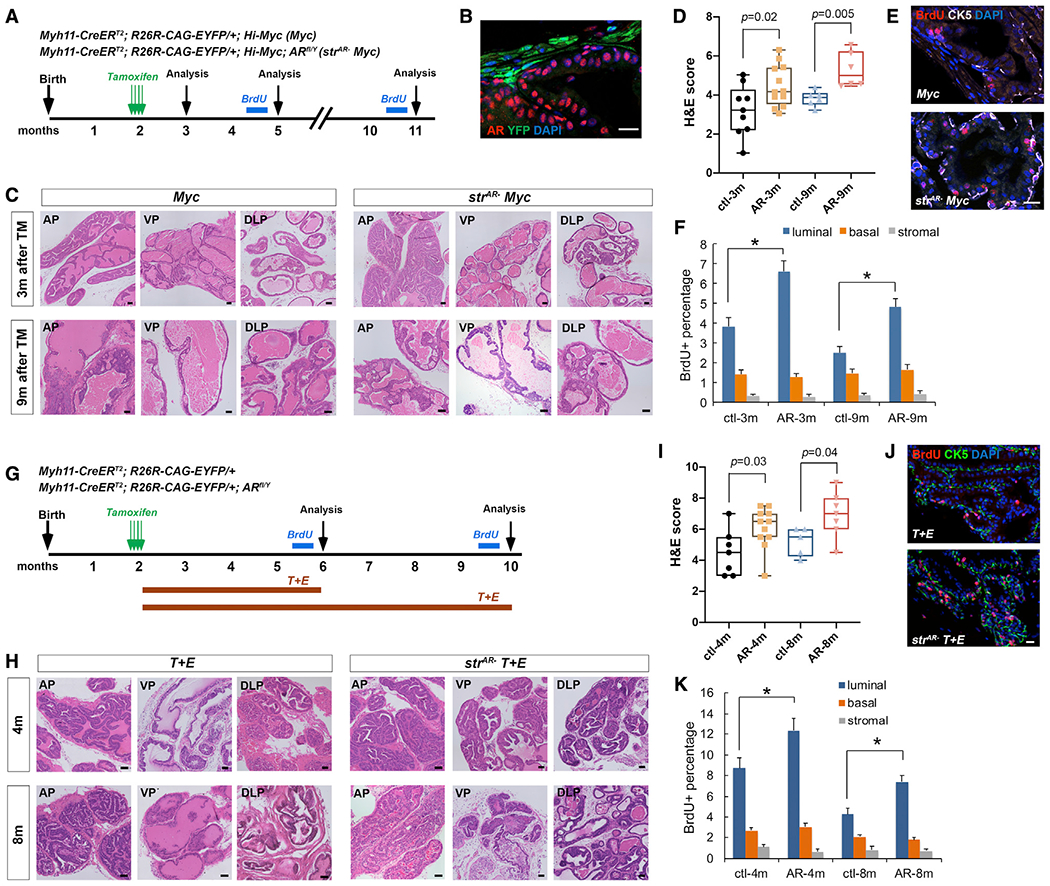
(A) Timeline of experiments for the Myc and strAR− Myc mice.
(B) IF showing efficient stromal AR deletion in strAR− Myc mice.
(C) Representative H&E images of different prostate lobes of Myc and strAR− Myc mice 3 and 9 months post induction.
(D) Quantitation of H&E scores.
(E) Representative IF staining of BrdU in Myc and strAR− Myc mice 9 months post induction.
(F) Quantitation of the proportions of BrdU+ cells in the basal, luminal, and stromal layers of Myc and strAR− Myc mice 3 and 9 months post induction.
(G) Timeline of experiments for the T + E and strAR− T + E mice.
(H) Representative H&E images of different prostate lobes of T + E and strAR− T + E mice 4 and 8 months post treatment.
(I) Quantitation of H&E scores.
(J) Representative IF staining of BrdU in T + E and strAR− T + E mice 8 months post treatment.
(K) Quantitation of the proportions of BrdU+ cells in the basal, luminal, and stromal layers of T + E and strAR−T+E mice 4 and 8 months post treatment. Each dot in (D) and (I) represents a mouse in the group. *p < 0.05 by Student’s t test. Scale bars in (B), (E), and (J), 20 μm, and in (C) and (H), 100 μm.
Stromal AR inhibits PCa progression during hormonal carcinogenesis
Given that the Hi-Myc model relies on the probasin promoter, which is turned on at the neonatal stage, we next set out to test whether our conclusion could be extended to other PCa models where tumor initiates strictly at the adult stage. In rodents, combined treatment of testosterone and estradiol-17β (named the T + E2 model) has been used to mimic the hormonal change in aging men (Ricke et al., 2008; Wang et al., 2014), whose relative increase in estrogen and decrease in androgen levels are associated with increased PCa incidence (Wilson, 1980). We therefore induced Myh11-CreERT2; R26R-EYFP/+ and Myh11-CreERT2; ARfl/Y; R26R-EYFP/+ mice with tamoxifen at 7 weeks of age, implanted T and E2 tubes subcutaneously to induce tumor initiation (denoted T + E and strAR− T + E, respectively), and analyzed prostate histology 4 and 8 months later (Figure 2G). ELISA confirmed that serum estrogen levels were significantly enhanced after T + E2 treatment for both groups of mice (Figure S3). We found that, compared with the T + E group, the strAR− T + E group contained more prominent high-grade PIN lesions, with some regions displaying cribriform patterns and epithelial cells filling into the lumen (Figure 2H and Table S2). Double-blind quantification of the histology scores showed that strAR− T + E tumors were significantly more aggressive than the T + E tumors (Figure 2I and Table S2), in agreement with the previous report using SMH-Cre (Welsh et al., 2011). BrdU incorporation assay again revealed higher proliferation of luminal cells, but not basal and stromal cells, in the strAR− T + E group compared with the T + E group (Figures 2J and 2K). Taken together, we conclude that stromal AR deletion induced more aggressive PCa phenotypes in both the genetic and the hormonal carcinogenesis models.
Loss of stromal AR primarily affects secretory luminal tumor cells
To understand how stromal AR deletion promotes PCa progression, we next performed single-cell RNA sequencing (scRNA-seq) for the 8-month T + E and strAR− T + E tumors by 10× Genomics Drop-seq. We profiled 3,373 T + E and 2,689 strAR− T + E prostate cells that passed quality control, and ~1,500 genes were detected per cell. Based on known marker expression, we assigned cell-type identities in the t-distributed stochastic neighbor embedding (t-SNE) plots (Figure 3A), including basal cells, secretory luminal cells, the recently identified proximal luminal cells (Karthaus et al., 2020; Kwon et al., 2020), SMCs, fibroblasts, leukocytes, endothelial cells, erythrocytes, and seminal vesicle cell contaminants. Notably, in both samples, secretory luminal cells (identified by markers Krt8, Krt18, Nkx3.1, and Pbsn) contained multiple cell clusters, indicating that they became highly heterogeneous during tumor progression. The two samples were then integrated with single-cell variational inference (scVI) tools (Gayoso et al., 2021) and re-clustered (Figure 3B). Differentially expressed genes in each cluster are listed in Table S3. This analysis showed that the two samples were similar on a global level, as most clusters aligned well between them, except for the secretory luminal cell clusters (clusters 1, 3, 4, and 6) (Figure 3C). Specifically, in uniform manifold approximation and projection (UMAP), clusters 1 and 3 mostly belong to the T + E sample, whereas clusters 4 and 6 belong to the strAR− T + E sample (Figures 3B and 3C). We hypothesized that these four secretory luminal cell clusters were responsible for the phenotypic differences observed between the two groups of mice, and therefore focused on them for further analyses.
Figure 3. scRNA-seq analysis of T + E and strAR− T + E prostate.
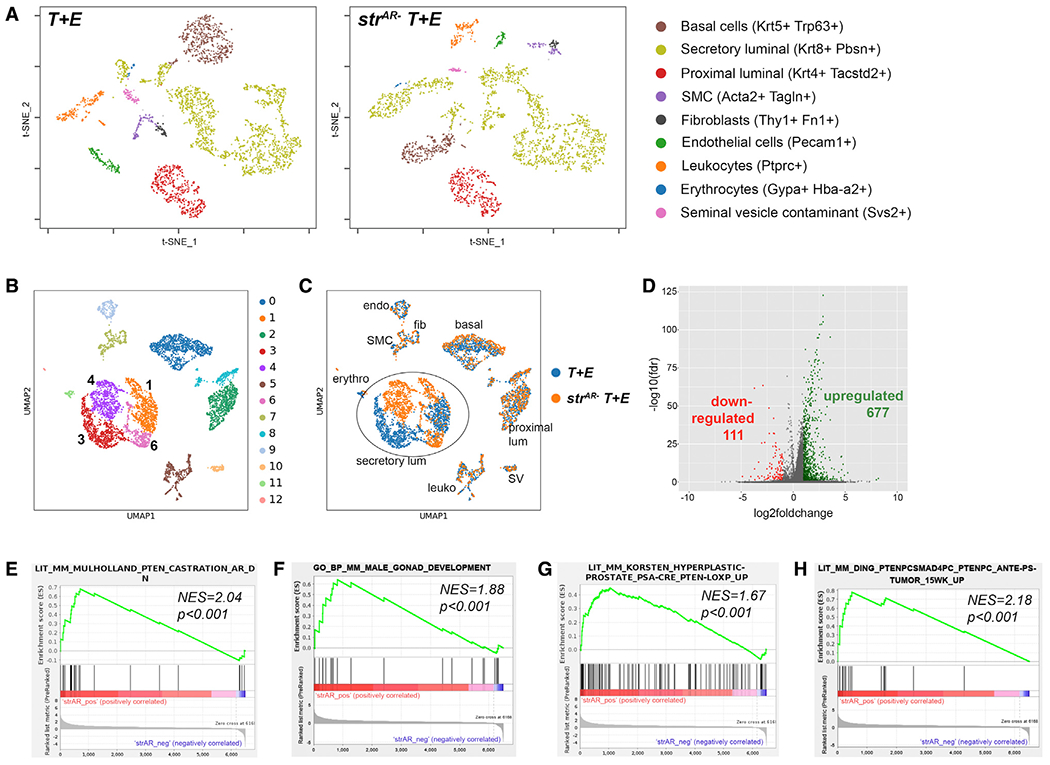
(A) t-SNE plots showing various cell populations (identifiable by markers listed on the right) in the T + E and strAR− T + E prostate.
(B) UMAP showing color-coded cell clusters of integrated samples using scVI tools.
(C) UMAP showing alignment of cell types between the T + E and the strAR− T + E samples.
(D) Volcano plot showing up- and downregulated genes in strAR− T + E versus T + E.
(E–H) GSEA comparing differentially expressed genes in secretory luminal cells of strAR− T + E versus T + E against GO pathways and published gene signatures.
Gene expression differential analysis comparing all secretory luminal cells (clusters 1, 3, 4, and 6) between the two samples showed that 677 genes were upregulated and 111 downregulated in strAR− T + E relative to T + E (false discovery rate [FDR] < 0.05 and log fold change > 1 or < −1) (Figure 3D, Tables S4 and S5). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) showed that genes overexpressed in the strAR− T + E sample were enriched in a previously published AR-repressed-gene signature obtained via mouse castration (Mulholland et al., 2011) (Figure 3E), indicating that stromal AR deletion has effects similar to those of castration. GSEA further showed that genes overexpressed in strAR− T + E were enriched in the Gene Ontology (GO) male gonad development (Figure 3F), in genes upregulated in a PSA-Cre; Ptenfl/fl model relative to wild type (Korsten et al., 2016) (Figure 3G), as well as genes upregulated in a Smad4-Pten-null model relative to Pten-null alone (Ding et al., 2011) (Figure 3H). GSEA of downregulated genes in the strAR− T + E sample showed that the reverse trend was also true (Figure S4). These data demonstrate that the strAR− T + E tumor is molecularly more aggressive than the age-matched T + E tumor.
Subpopulation of luminal tumor cells shows enhanced PI3K-mTORC1 activity
We next examined the heterogeneity within the secretory luminal cells. Clusters 3 and 4 corresponded to the subpopulation of secretory luminal cells with lower luminal marker expression than clusters 1 and 6 in the T + E and strAR− T + E samples, respectively (Figures 4A and S5). They also appeared to represent more advanced-stage tumor cells compared with clusters 1 and 6. For example, GSEA showed that genes downregulated in cluster 6 relative to 4 were highly enriched in the KEGG pathway chemical carcinogenesis (Figure 4B). Furthermore, the progression of these luminal tumor cells was similar between the strAR− T + E and the T + E samples, since GSEA showed that upregulated genes in cluster 4 versus 6 were significantly enriched in the top 300 upregulated genes in cluster 3 versus 1 (Figure 4C). When we compared the advanced-stage tumor cells between the two samples (cluster 4 in strAR− T + E versus cluster 3 in T + E), we found that genes downregulated in cluster 4 versus 3 were enriched in two published signatures of genes downregulated in Pten-null tumors versus wild-type tissues (Ding et al., 2011; Korsten et al., 2016) (Figures 4D and 4E). These data suggest that the advanced-stage luminal tumor cells in the strAR− T + E prostate were more aggressive than their counterparts in the T + E prostate and that stromal AR deletion elicited a tumor progression program resembling PI3K pathway activation in the secretory luminal cells. Consistent with this notion, we found that, after excluding cells below a minimal detection threshold, Pten expression in clusters 3 and 4 was lower than in clusters 1 and 6 (Figures 4F and 4G). Between clusters 3 and 4, cluster 3 also contained more cells with relatively high Pten expression (Figure 4G), suggesting that the loss of stromal AR may downregulate Pten in luminal tumor cells to accelerate their progression.
Figure 4. Luminal tumor cell heterogeneity in the T + E and strAR− T + E prostate.
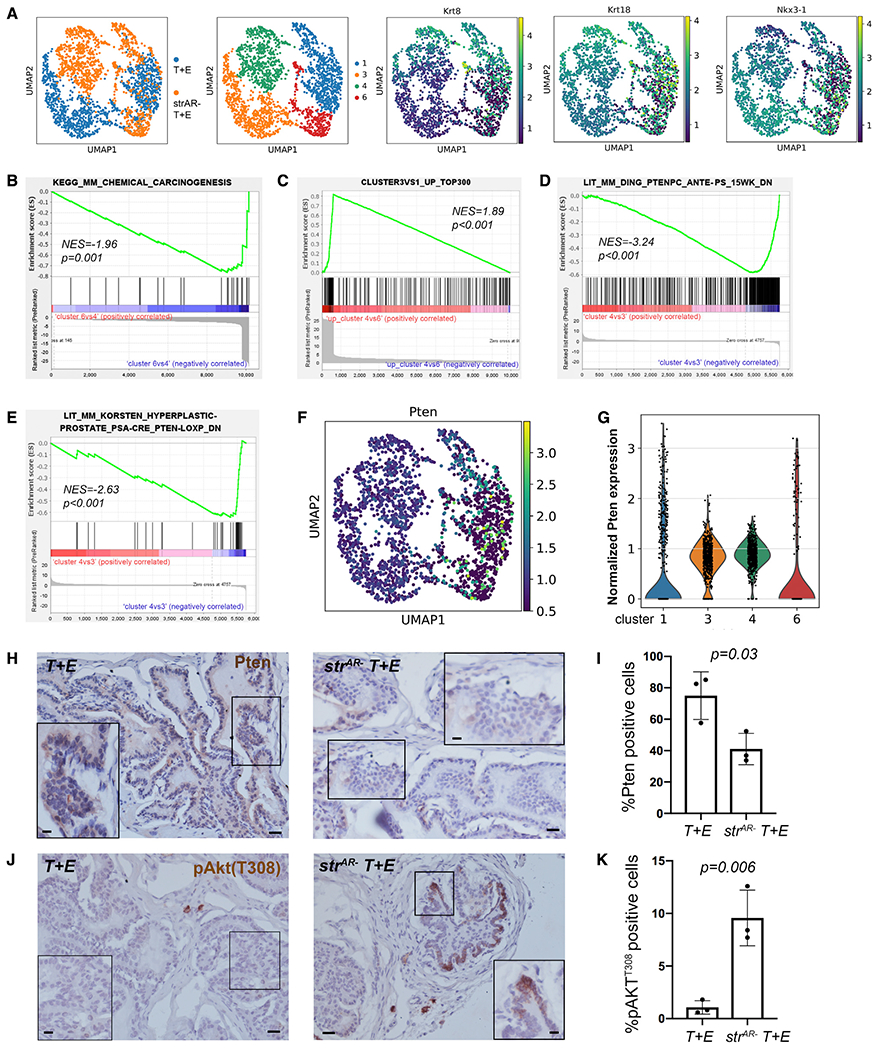
(A) UMAP plots showing the expression of luminal marker genes in secretory luminal clusters 1, 3, 4, and 6.
(B and C) GSEA showing that upregulated genes in cluster 4 versus 6 are enriched in KEGG pathway chemical carcinogenesis (B) and in the top 300 upregulated genes in cluster 3 versus 1 (C).
(D and E) GSEA showing that downregulated genes in cluster 4 versus 3 are enriched in two downregulated gene signatures of Pten-null tumors.
(F and G) UMAP (F) and violin plots (G) showing Pten expression in clusters 1, 3, 4, and 6.
(H and J) Representative IHC images showing Pten (H) and pAkt-T308 (J) staining in T + E (left) and strAR− T + E (right) tissues. Scale bars, 50 μm. Scale bars in insets, 10 μm.
(I and K) Quantitation of percentages of Pten-positive (I) and pAkt-T308-positive (K) epithelial cells. n = 3 animals per group. The p values were calculated by Student’s t test.
To further validate this finding, we performed immunohistochemistry (IHC) to detect Pten in T + E and strAR− T + E tissues (n = 3 animals each). While the vast majority of the T + E epithelial cells showed positive signals, Pten staining was significantly decreased in many PIN foci of the strAR− T + E epithelium (Figures 4H and 4I). To evaluate the PI3K signaling downstream effectors, we performed IHC to detect phospho-Akt Ser473 (pAkt-S473), an mTORC2 target, as well as mTORC1 signaling components phospho-Akt Thr308 (pAkt-T308), ribosomal protein S6 phosphorylated at Ser235/236 (pRPS6), and 4e-binding protein 1 phosphorylated at Thr37/46 (p4E-BP1) (Pearson et al., 2018). We did not detect pAkt-S473-positive epithelial cells in either group (Figure S6A), suggesting no activation of the mTORC2 pathway. However, clusters of pAkt-T308-positive epithelial cells were present in all the samples, and its percentage significantly increased in the strAR− T + E tissues compared with T + E (Figures 4J and 4K). Moreover, strAR− T + E epithelium contained significantly higher percentages of p4E-BP1-positive (Figures S6B and S6C) and pRPS6-positive cells (Figures S6D and S6E). Taken together, these results are consistent with the scRNA-seq finding, and indicate that stromal AR loss stimulated the PI3K-mTORC1 signaling activity in a subpopulation of luminal tumor cells.
Enhanced PI3K activity in a luminal subpopulation of the strAR− Myc tumor
Since the T + E model tends to capture an earlier window of tumorigenesis, while the Hi-Myc model offers a wider spectrum of pathological stages, we next asked whether similar mechanisms could also explain the more aggressive phenotypes in the strAR− Myc tumors. To this end, we performed scRNA-seq comparing Myc and strAR− Myc tumors that had progressed for 12 months, with 1,749 cells and 1,713 cells profiled at ~1,200 genes per cell, respectively. Integration of the two samples and re-clustering by scVI showed that, while most of the cell clusters aligned well between Myc and strAR− Myc samples, differences again existed in the secretory luminal cell populations (identified by Pbsn+ Krt8+) (Figures 5A and 5B). We assigned cell types to different clusters based on marker expression (Figure 5C and Table S6) and noticed that various stromal cell populations, including leukocytes, were more prominently represented compared with the previous single-cell analysis of the T + E model. Importantly, we found that, while cluster 1 contained secretory luminal cells that overlapped between the two samples, clusters 6 and 11 contained secretory luminal cells specific to strAR− Myc (Figures 5A and 5C). GSEA showed that genes upregulated in cluster 6 relative to cluster 1 were highly enriched in two aforementioned upregulated gene signatures of Pten-null prostate tumors (Ding et al., 2011; Korsten et al., 2016) (Figures 5D and 5E). Conversely, genes downregulated in cluster 6 versus 1 were significantly enriched in the downregulated Pten-null gene signature (Ding et al., 2011) (Figure 5F). These data suggest that, similar to the T + E model, strAR− Myc tumors also harbored more aggressive luminal tumor cells with enhanced PI3K activity. In further support, scRNA-seq data showed lower Pten expression in cluster 6 versus cluster 1 (Figures 5G) and IHC showed significantly higher proportions of pAkt-T308-positive epithelial cells in the strAR− Myc tumors compared with the Myc tumors (Figures 5H and 5I). On the other hand, no Pten-related gene signature was significantly enriched in a GSEA comparison between clusters 11 and 1. Instead, genes upregulated in cluster 11 versus 1 were highly enriched in a gene signature induced by interferon γ (IFNγ) (Jehl et al., 2012) (Figure 5J), as well as several upregulated gene signatures of lipopolysaccharide (LPS) treatment (Shell et al., 2005; Kitamura et al., 2008; Bonow et al., 2009) (Figures S7A–S7C). Given the high frequency of inflammatory foci in strAR− Myc tumors (Table S1), cluster 11 likely represented luminal cells receiving immune cell signals in an inflammatory microenvironment. Overall, upregulation of PI3K activity in luminal tumor cells appears to be a shared feature between the T + E and the Hi-Myc models after stromal AR deletion.
Figure 5. Single-cell analysis comparing Myc and strAR− Myc prostate.
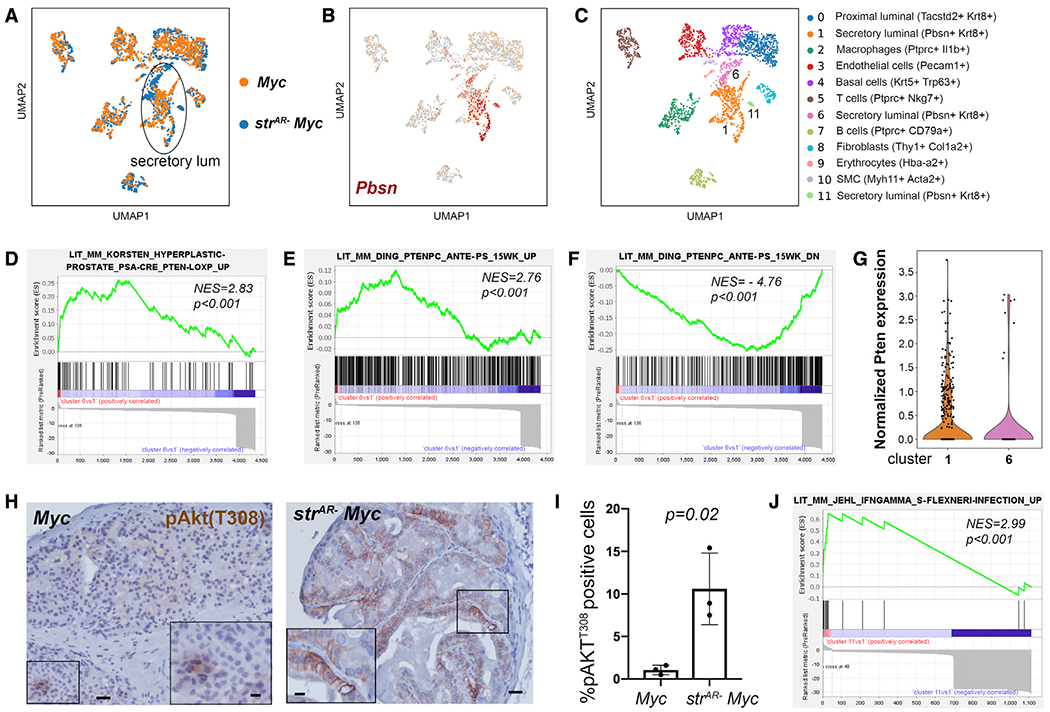
(A) UMAP showing alignment of cell types between the Myc and the strAR− Myc prostate.
(B) Identification of secretory luminal clusters based on Pbsn expression.
(C) UMAP showing color-coded cell clusters of integrated samples using scVI tools and assignment of cell types based on marker expression.
(D–F) GSEA comparing differentially expressed genes in cluster 6 versus 1 against published Pten-null tumor gene signatures.
(G) Violin plot showing Pten expression in clusters 1 and 6.
(H) Representative IHC images showing pAkt-T308 staining in Myc (left) and strAR− Myc (right) tissues. Scale bars, 50 μm. Scale bars in insets, 10 μm.
(I) Quantitation of percentages of pAkt-T308-positive epithelial cells. n = 3 animals per group. The p value was calculated by Student’s t test.
(J) GSEA showing enrichment of upregulated genes in cluster 11 versus 1 in a published IFNγ-induced gene signature.
DISCUSSION
The role of stromal AR in PCa has been controversial. Experiments recombining AR-deficient mouse stromal cells with transformed prostate epithelial cells in renal grafts suggested that stromal AR promotes PCa progression (Ricke et al., 2012). However, the renal grafting assay mimics prostate organogenesis (Wang and Shen, 2011; Wang et al., 2014), and therefore the requirement of stromal AR for epithelial cell growth in prostate development (Cunha and Lung, 1978; Yu et al., 2011) may have contributed to the outcomes of those studies. Using genetic approaches, AR has been deleted in the stroma, epithelium, or whole prostate via different Cre drivers, and the findings regarding the role of the stromal AR were still contradictory (Niu et al., 2008; Welsh et al., 2011; Lai et al., 2012a). One possibility underlying the discrepancy is the timing of AR deletion, since various Cre drivers may be turned on at different developmental stages. The inducible Myh11-CreERT2 driver used here ensures stromal AR deletion in the adult prostate. Our results obtained from both genetic and chemical-induced models pointed to a role for stromal ARs in inhibiting PCa progression, explaining the clinical observation that lower stromal AR expression is associated with advanced PCa stages and worse outcomes (Olapade-Olaopa et al., 1999; Henshall et al., 2001; Ricciardelli et al., 2005; Wikstrom et al., 2009; Leach et al., 2015).
SMCs and fibroblasts are the two major stromal cell types in the prostate. In scRNA-seq analysis of the T + E model, SMCs and fibroblasts were clustered together (Figure 3C) and shared marker gene Myh11, Acta2, and Tagln expression to variable degrees, consistent with a recent study (Kwon et al., 2019). Presumably, AR could also have been deleted in some fibroblasts due to their low Myh11 expression. Therefore, our conclusion drawn from SMC AR deletion is likely extendable to the whole stroma. For the Hi-Myc model, although graph-based clustering assigned SMCs and fibroblasts into one cluster, we could distinguish them into clusters 10 and 8 in Figure 5C, respectively, based on their UMAP spatial distance and distinct expression levels of Myh11 and Col1a2 (Figure S7D). In this analysis, the difference in AR expression was evident between Myc and strAR− Myc SMCs, but hardly detectable between Myc and strAR− Myc fibroblasts (Figure S7E), suggesting that accelerated tumor progression was mainly due to AR loss in SMCs. Future research deleting AR using a fibroblast-specific CreER driver should help assess the relative contribution of these two stromal cell types.
We note that AR deletion in SMCs resulted in subtle changes in the SMCs themselves, as reflected by no difference in SMC proliferation as well as the overall alignment of strAR− and control SMCs in the UMAP. Such cell overlap suggests that the changes in SMCs were masked by the greater differences among different cell types on the global level. Indeed, secretory luminal cells appeared to be the cell population most affected by stromal AR deletion under tumorigenic conditions, suggesting that AR-null SMCs modulate paracrine signals to promote luminal tumor cell progression. A spectrum of growth factors and cytokines has been implicated as under the regulation of stromal ARs in the prostate (Konig et al., 1987; Li et al., 2008; Wen et al., 2015). We observed the expression of factors such as IGF1, HGF, VEGFb, and FGF2 in both SMCs and fibroblasts in scRNA-seq. However, changes in their expression after AR deletion may not be discernable due to low data resolution. Nevertheless, TGFβ2 appeared to be an interesting candidate, since it was downregulated in the strAR− SMCs of both tumor models (Figure S7F), and TGFβ signaling was shown to play a cytostatic role in normal prostate epithelial cells (Salm et al., 2005; Valdez et al., 2012) and serves as a barrier to PCa progression (Ding et al., 2011). We caution that our scRNA-seq samples were acquired from progressed tumors, and the stromal cells may have changed since the onset of AR deletion. Future research with an SMC-enriched single-cell analysis at different time points may clarify key paracrine signals.
The finding that stromal AR deletion has distinct impacts on normal and malignant prostate luminal cells is intriguing. One possibility is that the oncogenic stimuli altered the properties of SMCs. Another model we speculate is that stromal AR deletion may result in changes in multiple paracrine signals with opposing roles. In the normal prostate, the tumor-promoting signals may not be sufficient to overcome the growth-inhibitory signals, resulting in overall reduced prostate size but sporadic foci of hyperplasia as seen in Figure 1I. Under tumorigenic conditions, the oncogenic stimuli may cooperate with the tumor-promoting signals to become the dominant force. Regardless of the cause, the combinatory effects appear to potentiate PI3K-mTORC1 activity in at least a subset of secretory luminal cells, as revealed by our bioinformatics and IHC data. This mechanism is conserved in the two PCa models we tested, but additional mechanisms may exist. For example, we discovered a cluster of strAR− Myc-specific luminal cells under the influence of an inflammatory microenvironment, and tumor cells are known to be capable of taking advantage of cytokine signals such as IFNγ to acquire immune evasion and/or high invasiveness (Jorgovanovic et al., 2020).
Our findings could have important implications for PCa treatment. Androgen deprivation therapy (ADT) remains a widely used method for treating advanced PCa. However, despite initial tumor shrinkage after ADT, virtually all patients relapse. Multiple molecular alteration mechanisms have been discovered to explain the castration-resistant PCa, including AR amplification and hypersensitivity, gain-of-function mutations in the AR gene, AR coactivator/corepressor mutations, and intratumoral androgen production (Chandrasekar et al., 2015). These discoveries highlight the importance of continued AR signaling within cancer cells, which are of epithelial origin. Our study suggests that stromal AR signaling plays an opposite role in PCa progression, at least in the early stages. If such mechanism persists in late-stage tumors, ADT may thereby have unintended negative effects in promoting epithelial cancer cell progression through decreased stromal AR signaling. If so, therapies that target epithelial ARs locally or enhance stromal AR downstream signals during ADT may achieve more favorable clinical outcomes.
Limitations of the study
We report the antitumor effects of stromal AR signaling using two distinct mouse PCa models. While our findings suggest elevation of the PI3K signaling pathway in secretory luminal cells as the key mechanism, other mechanisms cannot be ruled out, especially under different oncogenic conditions. More models are needed to assess the conservation of such mechanism. Due to the limited scope of the scRNA-seq experiments, we were not able to pinpoint the key paracrine factors under stromal AR regulation, and ARs in SMCs and fibroblasts could have different roles. Finally, although low stromal AR is associated with poorer clinical outcome, findings in mouse models may not be fully transferrable to humans, and we do not know whether the mechanism discovered here persists in advanced-stage human PCa.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhu A. Wang (zwang36@ucsc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Tumor scRNA-seq data are deposited in the Gene Expression Omnibus database under GSE186114.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Adult male mice (7–60 weeks old) were used in this study in accordance with protocols approved by the Institutional Animal Care and Use Committee at UCSC. The mice were housed in an animal facility with a regular 12-h light/dark cycle with ad libitum access to food and water. The ARflox (De Gendt et al., 2004), Hi-Myc (Ellwood-Yen et al., 2003), and R26R-CAG-EYFP (Madisen et al., 2010) lines were used previously. The Myh11-CreERT2 line (Wirth et al., 2008) was obtained from JAX. All animals used were maintained in C57BL/6N or mixed background, including the Hi-Myc allele (originally FVB background), which has been continuously backcrossed to C57BL/6N mice in the lab for over 15 generations. Genotyping was performed by PCR using tail genomic DNA, with the primer sequences listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-AR (1:500) | Sigma-Aldrich | Cat#A9853; RRID: AB_262132 |
| rat anti-BrdU (1:500) | Bio-Rad | Cat# MCA2060; RRID: AB_323427 |
| rabbit anti-CK5 (1:1000) | Covance | Cat# PRB-160P-100; RRID: AB_291581 |
| mouse anti-CK18 (1:100) | Abcam | Cat# ab668; RRID: AB_305647 |
| mouse anti-SMA (1:500) | Sigma-Aldrich | Cat# A2547; RRID: AB_476701 |
| chick anti-YFP (1:2000) | Abcam | Cat# ab13970; RRID: AB_300798 |
| rabbit anti-Pten (1:100) | Cell Signaling Technology | Cat# 9559; RRID: AB_390810 |
| rabbit anti-pAkt(Ser473) (1:50) | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| rabbit anti-pAkt(Thr308) (1:100) | Cell Signaling Technology | Cat# 13038; RRID: AB_2629447 |
| rabbit anti-pRPS6(Ser235/236) (1:400) | Cell Signaling Technology | Cat# 2211; RRID: AB_331679 |
| rabbit anti-p4E-BP1(Thr37/46) (1:1600) | Cell Signaling Technology | Cat# 2855; RRID: AB_560835 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat#T5648-5G |
| BrdU | Sigma-Aldrich | Cat#5002-5G |
| Testosterone | Sigma-Aldrich | Cat#T1500-1G |
| Estradiol-17β | Sigma-Aldrich | Cat#E8875-1G |
| Critical commercial assays | ||
| DetectX Serum 17β-Estradiol Enzyme Immunoassay Kit | Arbor Assays | Cat#KB30-H1 |
| Vectastain Elite ABC HRP Kit | Vector Labs | Cat#PK-6101 |
| Vector NovaRED Substrate Kit | Vector Labs | Cat#SK-4800 |
| Chromium Single Cell 3′ Reagent Kits (v3.1 Chemistry) | 10x Genomics | Cat#1000121, 1000127 |
| Deposited data | ||
| scRNA-seq raw data | This paper | GEO: GSE186114 |
| Experimental models: Organisms/strains | ||
| Tg(Myh11-icre/ERT2)1Soff | Jackson Laboratory (Wirth et al., 2008) | Strain #019079 |
| B6.Cg-Gt(ROSA)26Sortm3(CAG–EYFP)Hze/J | Jackson Laboratory (Madisen et al., 2010) | Strain #007903 |
| ARflox/Y | De Gendt et al. (2004) | Kindly provided by Dr. Michael Shen |
| Hi-Myc | Ellwood-Yen et al. (2003) | Kindly provided by Dr. Michael Shen |
| Oligonucleotides | ||
| Genotyping primer for Myh11-CreERT2 forward: TGACCCCATCTCTTCACTCC | This paper | N/A |
| Genotyping primer for Myh11-CreERT2 reverse: AGTCCCTCACATCCTCAGGTT | This paper | N/A |
| Genotyping primer for ARflox forward: GTTGATACCTTAACCTCTGC | This paper | N/A |
| Genotyping primer for ARflox reverse: CTTCAGCGGCTCTTTTGAAG | This paper | N/A |
| Genotyping primer for Hi-Myc forward: AAACATGATGACTACCAAGCTTGGC | This paper | N/A |
| Genotyping primer for Hi-Myc reverse: ATGATAGCATCTTGTTCTTAGTCTTTTTCTTAATAGGG | This paper | N/A |
| Genotyping primer for R26R-CAG-EYFP wt forward: AAGGGAGCTGCAGTGGAGTA | This paper | N/A |
| Genotyping primer for R26R-CAG-EYFP wt reverse: CCGAAAATCTGTGGGAAGTC | This paper | N/A |
| Genotyping primer for R26R-CAG-EYFP mutated forward: ACATGGTCCTGCTGGAGTTC | This paper | N/A |
| Genotyping primer for R26R-CAG-EYFP mutated reverse: GGCATTAAAGCAGCGTATCC | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 8.0 | GraphPad | RRID: SCR_002798 |
| 10x Genomics Cell Ranger pipeline v5.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Scanpy v1.7.1 | Wolf et al. (2018) | RRID: SCR_018139 |
| Scrublet v0.2.1 | Wolock et al. (2019) | RRID: SCR_018098 |
| scVI-tools v0.7.0 | Gayoso et al. (2021) | https://scvi-tools.org |
| GSEA v4.1.0 | Subramanian et al. (2005) | RRID: SCR_003199 |
METHOD DETAILS
Tamoxifen induction
Mice were administered 9 mg per 40 g body weight tamoxifen (Sigma) suspended in corn oil by oral gavage once daily for 4 consecutive days.
BrdU incorporation assay
BrdU (Sigma) was dissolved in PBS (10 mg/mL) and administered by intraperitoneal injection twice daily (0.1 mL per dose) for 7 consecutive days to label proliferating cells.
T + E2 treatment
A 1.0 cm Silastic capsule (No. 602–305 Silastic tubing; 1.54 mm inside diameter, 3.18 mm outside diameter; Dow-Corning #2415569) filled with testosterone (Sigma) and a 0.4 cm Silastic capsule filled with estradiol-17β (Sigma) were implanted subcutaneously. Mice were treated with hormones for 4 months, and tubes were replenished at that time if the treatment would be extended to 8 months.
Serum estradiol ELISA
Blood sampling was performed by venipuncture from the facial vein and allowed to clot for at least 30 min at 4°C in a BD Microtainer MAP microtube (BD cat #363706). The blood was then centrifuged for 15 min at 2000g, and the serum was collected and stored at −80°C. Serum levels of estradiol were determined using the DetectX serum 17β-estradiol enzyme immunoassay kit, as per the manufacturer’s instructions (Arbor assays). Values for the samples were derived via interpolation using standards provided with the kit.
Tissue collection and dissociation
Mouse prostate tissues were dissected and fixed in 4% paraformaldehyde for subsequent cryo-embedding in OCT compound (Sakura), or fixed in 10% formalin followed by paraffin embedding. For dissociation, prostate tissues were dissected and minced to small clumps, followed by enzymatic dissociation with 0.2% Collagenase/Hyaluronidase (StemCell Technologies) in DMEM/F12 media with 5% FBS for 3 h at 37°C. Tissues were digested with 0.25% Trypsin-EDTA (StemCell Technologies) for 1 h at 4°C, passed through 21- to 26-gauge syringes and filtered through a 40-mm cell strainer to obtain single-cell suspensions. Dissociated prostate cells were suspended in Hanks’ Balanced Salt Solution Modified/2% FBS.
Immunofluorescence staining
Immunofluorescence staining was performed using 6 mm cryo sections. Samples were incubated with 10% normal goat serum (NGS) and primary antibodies diluted in 10% NGS overnight at 4°C. Samples were then incubated with secondary antibodies (diluted 1:500 in PBST) labeled with Alexa Fluor 488, 555, or 647 (Invitrogen/Molecular Probes). Slides were mounted with VectaShield mounting medium with DAPI (Vector Labs), and images were taken on a Leica TCS SP5 spectral confocal microscope in the UCSC Microscopy Shared Facility. Primary antibodies and dilutions used are listed in the key resources table.
Tumor histology
For immunohistochemical staining, the procedures followed the instructions of the Vectastain Elite ABC Kit (PK-6101) and Vector NovaRED Substrate Kit (SK-4800). Briefly, 5 μm paraffin sections were deparaffinized and hydrated through xylenes and graded alcohol series, followed by antigen retrieval through boiling in antigen unmasking solution (Vector Labs). Slides were blocked in blocking serum (1.5% normal serum), and incubated with primary antibodies diluted in blocking serum overnight at 4°C. Biotinylated secondary anti-rabbit antibody from Vectastain Elite ABC Kit was diluted 1:200 in blocking serum. The signal was enhanced using the Vectastain Elite ABC system and visualized with the NovaRed Substrate Kit. The slides were counterstained with Harris Modified Hematoxylin (1:10 diluted in H2O) and mounted with Clearmount (American MasterTech). Primary antibodies and dilutions used are listed in the key resources table. H&E staining was performed using standard protocols as previously described. Histology slides were visualized using a Zeiss Axiolmager.
Single cell RNA-seq
Approximately 16,000 dissociated cells were obtained from each prostate sample. Library preparation was performed using Chromium Single Cell 3′ Solution with v3.1 chemistry following the manufacturer’s protocol (10x Genomics). The library purity, size, and quantity were validated by capillary electrophoresis using 2100 Bioanalyzer (Agilent Technologies). The libraries were sequenced at the UC Davis Genome Center with a NovaSeq 6000 S4 instrument (Illumina) to a depth of ~225k reads per cell.
Single cell data pre-processing and analyses
Raw sequencing data were processed using the 10x Genomics Cell Ranger pipeline (version 5.0.1) to generate FASTQ files and aligned to the mm10 genome (version 2020-A) to generate gene expression counts and quality control metrics. The count matrices were read into AnnData objects and concatenated using Scanpy (version 1.7.1) (Wolf et al., 2018). Genes that were not detected in any cells were removed and cells with genes<200 were removed. The quality of the scRNA-seq dataset was assessed by plotting the numbers of unique molecular identifiers (UMIs), genes, total counts, and mitochondrial gene percentage per cell. The cutoffs for the above metrics were manually determined. Specifically, cells with detected genes >10,000 or UMI counts >150,000 or mitochondrial UMI ratio >15% were excluded. After quality control filtering, the concatenated object was split into objects corresponding to each sample. Cell doublets were removed from the objects using Scrublet (version 0.2.1) (Wolock et al., 2019) with default parameters, while the doublet score thresholds were set by inspecting the simulated doublet score histograms. Then all the objects were concatenated and genes not detected in any cells were filtered again, normalized to 10,000 reads per cell, logarithmized, scaled and centered.
To integrate different samples, top3000 highly variable genes were selected. Sample batches were registered with scvi-tools (version 0.7.0) (Gayoso et al., 2021), and a deep generative modeling was trained (with n_latent set to 30 and n_layers set to 2). Then data imputation and integration were carried out to impute dropout events and correct batch effects. The latent variables obtained from the model were stored as annotations of observations (obsm). A nearest-neighbor graph using the 30 dimensions of the latent space was calculated using ‘pp.neighbors()’, followed by clustering using leiden algorithm ‘tl.leiden()’ with a resolution adopting from prior knowledge on prostate cell population composition. Then uniform manifold approximation and projection (UMAP) dimension reduction was performed for visualizing high-dimensional data.
Scanpy rank_genes_groups() and Wilcoxon rank-sum test were used to compute the statistical significance of differential genes expression between any groups and identify marker genes of each cluster.
Gene set enrichment analysis
The significantly differentially expressed genes (FDR <0.01) were ranked by their log-transformed fold change value. Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was conducted using GSEA software (Version 4.1.0). The pre-ranked gene list and MousePath_All_gmt-Format.gmt or MousePath_GO_gmt.gmt gene set (both were downloaded from http://ge-lab.org/gskb/) were used for running the tool “Run GSEA Preranked” with default parameters.
Comparing transition between samples
To investigate the molecular transition from cell cluster 1 to 3 in the T + E sample and cluster 6 to 4 in the strAR− T + E sample, we compared the cluster 3 vs cluster 1 signature with cluster 4 vs cluster 6 signature. Only significant differentially expressed genes between the clusters were kept (FDR<0.01). Then the query signature was defined as a list of top300 positive changed genes ranked by logfoldchange in cluster 3 compared to cluster 1 in sample T+E (higher in 3 than 1). The target signature was defined as a list of genes ranked by logfoldchange in cluster 4 compared to cluster 6 in sample strAR− T + E. Statistical significance of the enrichment between the query signature and the target signature was computed using GSEA.
QUANTIFICATION AND STATISTICAL ANALYSIS
For tumor H&E evaluation, samples were deidentified and scoring was performed in a double-blinded setting. Each sample was assigned a histology score (highest 10 and lowest 0) using the following formula: score = % area of low/moderate-grade PIN X 5 + % area of high-grade PIN X 10. PIN subtype classification was described previously (Berman-Booty et al., 2012). For BrdU and IHC quantification, the investigators were blinded to the ID/genotype of the mice before performing cell counting. Numbers of positive cells were counted manually across tissue sections.
Statistical analyses for IF and IHC staining images and H&E scores were performed using the two-sided student’s t-test. At least four animals for each IF staining quantification, three animals for each IHC staining quantification, and at least five animals per cohort for H&E scoring were used. The variances were similar between the groups that were being statistically compared. Specific details for these statistical tests are listed in figure legends.
Supplementary Material
Highlights.
AR loss in adult prostate stroma disrupts normal epithelial homeostasis and morphology
Deleting AR in adult smooth muscle cells accelerates Hi-Myc prostate cancer progression
A luminal tumor cell subset has enhanced PI3K-mTORC1 activity after stromal AR deletion
The mechanism is conserved between the Hi-Myc model and a hormonal carcinogenesis model
ACKNOWLEDGMENTS
We thank the Sofie Salama and David Haussler lab for sharing the 10× Genomics Chromium machine and the microscopy core facility at UCSC and DNA Technologies Core at UC Davis for technical support. We also thank Yvonne Gutierrez, Ysabelle-Jeah Habon, and Emily Person for helping with mouse genotyping and histology analysis. This work was supported by a TRDRP post-doctoral fellowship (Y.L.), a Santa Cruz Cancer Benefit Group award (Z.A.W.), ACS grant 134386-RSG-20-038-01-DDC (Z.A.W.), and NIH grant R01CA271452 (Z.A.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110848.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
REFERENCES
- Berman-Booty LD, Sargeant AM, Rosol TJ, Rengel RC, Clinton SK, Chen CS, and Kulp SK (2012). A review of the existing grading schemes and a proposal for a modified grading scheme for prostatic lesions in TRAMP mice. Toxicol. Pathol 40, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonow RH, AÔd S, Zhang Y, Becker KG, and Bosetti F (2009). The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccharide in mice. Pharmacogenomics J. 9, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellon E, Venegas K, S·enz L, Contreras H, and Huidobro C (2005). Secretion of prostatic specific antigen, proliferative activity and androgen response in epithelial-stromal co-cultures from human prostate carcinoma. Int. J. Androl 28, 39–46. [DOI] [PubMed] [Google Scholar]
- Chandrasekar T, Yang JC, Gao AC, and Evans CP (2015). Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol 4, 365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N, Zhang B, Zhang L, Ittmann M, and Xin L (2012). Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 21, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CW, Epsi NJ, Leung EY, Xuan S, Lei M, Li BI, Bergren SK, Hibshoosh H, Mitrofanova A, and Shen MM (2018). Differential requirements of androgen receptor in luminal progenitors during prostate regeneration and tumor initiation. Elife 7, e28768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR, and Lung B (1978). The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J. Exp. Zool 205, 181–193. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Donjacour AA, Cooke PS, Mee S, Bigsby RM, Higgins SJ, and Sugimura Y (1987). The endocrinology and developmental biology of the prostate. Endocr. Rev 8, 338–362. [DOI] [PubMed] [Google Scholar]
- De Gendt K, Swinnen JV, Saunders PT, Schoonjans L, Dewerchin M, Devos A, Tan K, Atanassova N, Claessens F, Lecureuil C, et al. (2004). A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc. Natl. Acad. Sci. U S A 101, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degeorges A, Tatoud R, Fauvel-Lafeve F, Podgorniak MP, Millot G, de Cremoux P, and Calvo F (1996). Stromal cells from human benign prostate hyperplasia produce a growth-inhibitory factor for LNCaP prostate cancer cells, identified as interleukin-6. Int. J. Cancer 68, 207–214. [DOI] [PubMed] [Google Scholar]
- Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, et al. (2011). SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, and Sawyers CL (2003). Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238. [DOI] [PubMed] [Google Scholar]
- Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, Shiah YJ, Yousif F, Lin X, Masella AP, et al. (2017). Genomic hallmarks of localized, non-indolent prostate cancer. Nature 541, 359–364. [DOI] [PubMed] [Google Scholar]
- Gayoso A, Lopez R, Xing G, Boyeau P, Wu K, Jayasuriya M, Melhman E, Langevin M, Liu Y, Samaran J, et al. (2021). scvi-tools: a library for deep probabilistic analysis of single-cell omics data. Preprint at bioRxiv. 10.1101/2021.04.28.441833. [DOI] [PubMed] [Google Scholar]
- Hayward SW (2002). Approaches to modeling stromal-epithelial interactions. J. Urol 168, 1165–1172. [DOI] [PubMed] [Google Scholar]
- Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, Delprado W, Stricker PD, Grygiel JJ, and Sutherland RL (2001). Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 61, 423–427. [PubMed] [Google Scholar]
- Jehl SP, Nogueira CV, Zhang X, and Starnbach MN (2012). IFNγ inhibits the cytosolic replication of Shigella flexneri via the cytoplasmic RNA sensor RIG-I. PLoS Pathog. 8, e1002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgovanovic D, Song M, Wang L, and Zhang Y (2020). Roles of IFN-γ in tumor progression and regression: a review. Biomark. Res 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabalin JN, Peehl DM, and Stamey TA (1989). Clonal growth of human prostatic epithelial cells is stimulated by fibroblasts. Prostate 14, 251–263. [DOI] [PubMed] [Google Scholar]
- Karthaus WR, Hofree M, Choi D, Linton EL, Turkekul M, Bejnood A, Carver B, Gopalan A, Abida W, Laudone V, et al. (2020). Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 368, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, Ito M, Yuasa T, Kikuguchi C, Hijikata A, Takayama M, Kimura Y, Yokoyama R, Kaji T, and Ohara O (2008). Genome-wide identification and characterization of transcripts translationally regulated by bacterial lipopolysaccharide in macrophage-like J774.1 cells. Physiol. Genomics 33, 121–132. [DOI] [PubMed] [Google Scholar]
- Konig JJ, Romijn JC, and Schr^der FH (1987). Prostatic epithelium inhibiting factor (PEIF): organ specificity and production by prostatic fibroblasts. Urol. Res 15, 145–149. [DOI] [PubMed] [Google Scholar]
- Kooistra A, Romijn JC, and Schr^der FH (1997). Stromal inhibition of epithelial cell growth in the prostate; overview of an experimental study. Urol. Res 25, S97–S105. [DOI] [PubMed] [Google Scholar]
- Korsten H, Ziel-van der Made AC, van Weerden WM, van der Kwast T, Trapman J, and Van Duijn PW (2016). Characterization of heterogeneous prostate tumors in targeted Pten knockout mice. PLoS One 11, e0147500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OJ, Zhang Y, Li Y, Wei X, Zhang L, Chen R, Creighton CJ, and Xin L (2019). Functional heterogeneity of mouse prostate stromal cells revealed by single-cell RNA-seq. iScience 13, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OJ, Choi JM, Zhang L, Jia D, Li Z, Zhang Y, Jung SY, Creighton CJ, and Xin L (2020). The Sca-1. Stem Cells 38, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KP, Yamashita S, Huang CK, Yeh S, and Chang C (2012a). Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol. Med 4, 791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KP, Yamashita S, Vitkus S, Shyr CR, Yeh S, and Chang C (2012b). Suppressed prostate epithelial development with impaired branching morphogenesis in mice lacking stromal fibromuscular androgen receptor. Mol. Endocrinol 26, 52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang SH, Stower M, and Maitland NJ (2000). In vitro modelling of epithelial and stromal interactions in non-malignant and malignant prostates. Br. J. Cancer 82, 990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DA, and Buchanan G (2017). Stromal androgen receptor in prostate cancer development and progression. Cancers (Basel) 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DA, Need EF, Toivanen R, Trotta AP, Palethorpe HM, Palenthorpe HM, Tamblyn DJ, Kopsaftis T, England GM, Smith E, et al. (2015). Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 6, 16135–16150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Olson AW, Wang J, Kim WK, Mi J, Zeng H, Le V, Aldahl J, Hiroto A, Wu X, et al. (2021). Androgen action in cell fate and communication during prostate development at single-cell resolution. Development 148, dev196048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li CX, Ye H, Chen F, Melamed J, Peng Y, Liu J, Wang Z, Tsou HC, Wei J, et al. (2008). Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J. Cell Mol. Med 12, 2790–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, et al. (2011). Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashima A, Kishigami S, Thomson A, and Yamada G (2014). Androgens and mammalian male reproductive tract development. Biochim. Biophys. Acta 1849, 163–170. [DOI] [PubMed] [Google Scholar]
- Niu Y, Altuwaijri S, Yeh S, Lai KP, Yu S, Chuang KH, Huang SP, Lardy H, and Chang C (2008). Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. U S A 105, 12188–12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olapade-Olaopa EO, MacKay EH, Taub NA, Sandhu DP, Terry TR, and Habib FK (1999). Malignant transformation of human prostatic epithelium is associated with the loss of androgen receptor immunoreactivity in the surrounding stroma. Clin. Cancer Res 5, 569–576. [PubMed] [Google Scholar]
- Pearson HB, Li J, Meniel VS, Fennell CM, Waring P, Montgomery KG, Rebello RJ, Macpherson AA, Koushyar S, Furic L, et al. (2018). Identification of Pik3ca mutation as a genetic driver of prostate cancer that co-operates with Pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 8, 764–779. [DOI] [PubMed] [Google Scholar]
- Ricciardelli C, Choong CS, Buchanan G, Vivekanandan S, Neufing P, Stahl J, Marshall VR, Horsfall DJ, and Tilley WD (2005). Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate 63, 19–28. [DOI] [PubMed] [Google Scholar]
- Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, and Risbridger GP (2008). Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 22, 1512–1520. [DOI] [PubMed] [Google Scholar]
- Ricke EA, Williams K, Lee YF, Couto S, Wang Y, Hayward SW, Cunha GR, and Ricke WA (2012).Androgen hormoneaction in prostatic-carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 33, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salm SN, Burger PE, Coetzee S, Goto K, Moscatelli D, and Wilson EL (2005). TGF-beta maintains dormancy of prostatic stem cells in the proximal region of ducts. J. Cell Biol 170, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Franco OE, and Hayward SW (2017). Interaction of prostate carcinoma-associated fibroblasts with human epithelial cell lines in vivo. Differ. Res. Biol. Divers 96, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell SA, Hesse C, Morris SM, and Milcarek C (2005). Elevated levels of the 64-kDa cleavage stimulatory factor (CstF-64) in lipopolysaccharide-stimulated macrophages influence gene expression and induce alternative poly(A) site selection. J. Biol. Chem 280, 39950–39961. [DOI] [PubMed] [Google Scholar]
- Shen MM, and Abate-Shen C (2010). Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 24, 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Jha R, Melamed J, Shapiro E, Hayward SW, and Lee P (2014). Stromal androgen receptor in prostate development and cancer. Am. J. Pathol 184, 2598–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuxhorn JA, Ayala GE, and Rowley DR (2001). Reactive stroma in prostate cancer progression. J. Urol 166, 2472–2483. [PubMed] [Google Scholar]
- Valdez JM, Zhang L, Su Q, Dakhova O, Zhang Y, Shahi P, Spencer DM, Creighton CJ, Ittmann MM, and Xin L (2012). Notch and TGFbeta form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell 11, 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, and Shen MM (2011). Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 30, 1261–1271. [DOI] [PubMed] [Google Scholar]
- Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, and Shen MM (2013). Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol 15, 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Toivanen R, Bergren SK, Chambon P, and Shen MM (2014). Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep. 8, 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M, Moffat L, McNeilly A, Brownstein D, Saunders PT, Sharpe RM, and Smith LB (2011). Smooth muscle cell-specific knockout of androgen receptor: a new model for prostatic disease. Endocrinology 152, 3541–3551. [DOI] [PubMed] [Google Scholar]
- Wen S, Chang HC, Tian J, Shang Z, Niu Y, and Chang C (2015). Stromal androgen receptor roles in the development of normal prostate, benign prostate hyperplasia, and prostate cancer. Am. J. Pathol 185, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom P, Marusic J, Stattin P, and Bergh A (2009). Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 69, 799–809. [DOI] [PubMed] [Google Scholar]
- Wilson JD (1980). The pathogenesis of benign prostatic hyperplasia. Am. J. Med 68, 745–756. [DOI] [PubMed] [Google Scholar]
- Wirth A, BenyÛ Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvth B, Maser-Gluth C, Greiner E, et al. (2008). G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med 14, 64–68. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Liu Y, Cai T, Horton C, Stefanson J, and Wang ZA (2017). Dissecting cell-type-specific roles of androgen receptor in prostate homeostasis and regeneration through lineage tracing. Nat. Commun 8, 14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Zhang C, Lin CC, Niu Y, Lai KP, Chang HC, Yeh SD, Chang C, and Yeh S (2011). Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 71, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Yeh CR, Niu Y, Chang HC, Tsai YC, Moses HL, Shyr CR, Chang C, and Yeh S (2012). Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 72, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Tumor scRNA-seq data are deposited in the Gene Expression Omnibus database under GSE186114.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.