

Abstract
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive disease, affecting mainly patients of Slavic origin. It is caused by a defect in the NBN gene, resulting in defective nibrin protein formation. This leads to chromosomal instability, which predisposes to cancer, with lymphoid malignancies predominating. Nibrin is also involved in gonadal development and its disfunction in females with NBS frequently results in a pure gonadal dysgenesis (PGD) causing hypergonadotropic hypogonadism. However, only a few ovarian tumors in NBS patients have been reported to date. We describe the first case of a girl with NBS with PGD, who developed metachronous bilateral ovarian germ cell tumors (dysgerminoma and gonadoblastoma). Pathogenesis of PGD, neoplastic transformation and therapeutic approach in females with NBS are discussed.
Keywords: Pure gonadal dysgenesis, solid tumor, gonadoblastoma, dysgerminoma, children, cancer predisposition
What is already known on this topic?
Nijmegen breakage syndrome (NBS) is an autosomal recessive disease characterized by chromosomal instability and increased risk of various malignancies, including solid tumors. Pure gonadal dysgenesis (PGD) resulting in hypergonadotropic hypogonadism is frequently encountered in females with NBS. Disorders of sex development, including PGD, are associated with an increased risk of germ cell tumors. The risk of ovarian germ cell tumors seems not to be significantly increased in NBS females with PGD therefore a routine prophylactic oophorectomy is not recommended in these patients.
What this study adds?
In 46,XX NBS females diagnosed with ovarian germ cell tumor, an early prophylactic oophorectomy of the remaining nonfunctional streak ovary should be considered to exclude or avoid second germ cell tumor. Early detection and complete resection of ovarian malignancy in female NBS patients may enable successful treatment without the need for adjuvant chemotherapy, which should be avoided in patients with NBS as it significantly increases the risk of development of subsequent cancers.
Introduction
Nijmegen breakage syndrome (NBS; OMIM#251260, ORPHA:647) is a rare autosomal recessive disease characterized by chromosomal instability, similar to that of ataxia-telangiectasia, with preferential involvement of chromosomes 7 and/or 14 in a variable proportion of analyzed metaphases. The majority of patients are of Slavic origin with founder effect for the most common pathogenic variant, namely NM_002485.5(NBN):c.657_661delACAAA (p.Lys219Asnfs*16; rs587776650) (previously referred to as c.657_661del5) in exon 6 of the NBN gene. The NBN gene encodes nibrin, the crucial component of the of the Mre11/Rad50/NBN (MRN) complex, responsible for repair of DNA-double-strand breaks. A genetic defect in this gene causes defective nibrin protein formation, which in turn impairs G2/M checkpoint control during the cell cycle. This results in spontaneous chromosomal instability and hypersensitivity to ionizing radiation (1,2).
The hallmark features of NBS patients include microcephaly, distinct dysmorphic facial features (sloping forehead, prominent midface and receding mandible), growth retardation in the first years of life, café au lait spots and some minor developmental defects (3). While microcephaly is a dominating feature, mental capabilities of NBS patients may not be affected in infancy and early childhood, but cognitive functions usually worsen with age (1). Combined immunodeficiency of both the cellular and humoral arms of the immune system is an essential feature of NBS (4) and increased susceptibility to severe infections, particularly of the respiratory tract, is commonly observed (5).
Malignancies are the most frequent cause of mortality in NBS patients (6). The risk of malignancy is approximately 50-fold higher, with risk for lymphomas 250-fold higher than in general population (5,7,8). In NBS, lymphoid malignancies also occur significantly more frequently than in other chromosome-breakage disorders, such as ataxia-telangiectasia, Bloom syndrome, and Fanconi Anemia (5,6,7). Malignant solid tumors are uncommon, constituting less than 10% of cases. Among them, medulloblastoma and perianal rhabdomyosarcoma predominate (6,9,10), while breast cancer, prostate cancer, thyroid cancer, neuroblastoma and melanoma have been reported occasionally (6,11).
Evidence of hypergonadotropic hypogonadism affecting females with NBS has been reported, based on a longitudinal observation of a large cohort of Polish patients and anecdotal case-reports (12,13). Interestingly, homozygous and compound heterozygous pathogenic variants in NBN gene have also been found in patients with isolated infertility, lacking other typical NBS features. The infertility was caused either by premature ovarian insufficiency (14,15,16) or by oligo-terato-asthenozoospermia (15). These reports suggest that nibrin may play a role in normal development and function of gonads, and explain the cause and pathomechanism of gonadal insufficiency in NBS patients lacking function of this protein (12).
Various specific forms of disorders of sex development, in which pure gonadal dysgenesis (PGD) is one of examples, are prone to development of germ-cell tumors. In NBS, only one case of ovarian gonadoblastoma and one of dysgerminoma have been previously reported (6,11). However, heterozygous carriers of c.657_661del5 variant have been shown to be at increased risk of developing rare types of ovarian cancers and other malignancies (17,18,19).
In this report, we document a girl with NBS who developed bilateral ovarian germ cell tumor. The first clinically appeared as a dysgerminoma and, two years later, gonadoblastoma was detected contralaterally in the prophylactic ovariectomy.
Case Report
A 13-year-old girl with NBS was admitted to the Department of Pediatrics, Hematology and Oncology of the University Clinical Center in Gdańsk, Poland, in 2007 due to an asymptomatic tumor of the left ovary, detected by screening ultrasonography (US) imaging. At the age of eight years, the patient had been diagnosed with NBS, based on typical clinical features, including: microcephaly; typical facial features; growth retardation; immunodeficiency; and recurrent respiratory tract infections. The diagnosis of NBS was confirmed by molecular testing, detecting a homozygous five base-pair deletion NM_002485.5(NBN):c.657_661delACAAA in exon 6. The family history of the patient was remarkable. Among five siblings she had an older sister with very similar clinical features, suggestive of NBS, but never confirmed molecularly. In 1999, at the age of 17 years, her sister died of rapidly progressing, abdominal, diffuse large B-cell lymphoma. Parents are both Polish and not related.
On admission, our patient was in a good condition, assessed as Tanner stage I. Her height was 146 cm, weight 35 kg, head circumference 49 cm and chest circumference 60 cm - all measurements were below the 3rd percentiles. US examination showed a solid and well demarcated tumor of the left ovary, measuring 35x30x26 mm. There was an abundant pathological vasculature invading from the periphery of the lesion and a very low resistivity index of 0.51. The presence of a localized ovarian tumor was confirmed on magnetic resonance imaging (MRI) (Figure 1a). No lymph nodes and distant metastases were detected. The serum levels of lactate dehydrogenase, uric acid, C-reactive protein, beta-chorionic gonadotropin alpha-fetoprotein and carcinoembryonic antigen were within normal ranges for age. The suspicion of ovarian non-Hodgkin lymphoma was raised, and a laparoscopic left oophorectomy was performed at the Department of Pediatric Surgery, Gdańsk. Histopathology revealed total ovarian involvement by dysgerminoma with a typical immunophenotype. The ovarian capsule was not infiltrated and no angioinvasion was detected (Figure 2a). The right ovary was found to be normal on intraoperative macroscopic evaluation. According to the International Federation of Gynecologists and Obstetricians the tumor was classified as stage 1A, which did not require adjuvant oncologic therapy.
Figure 1.
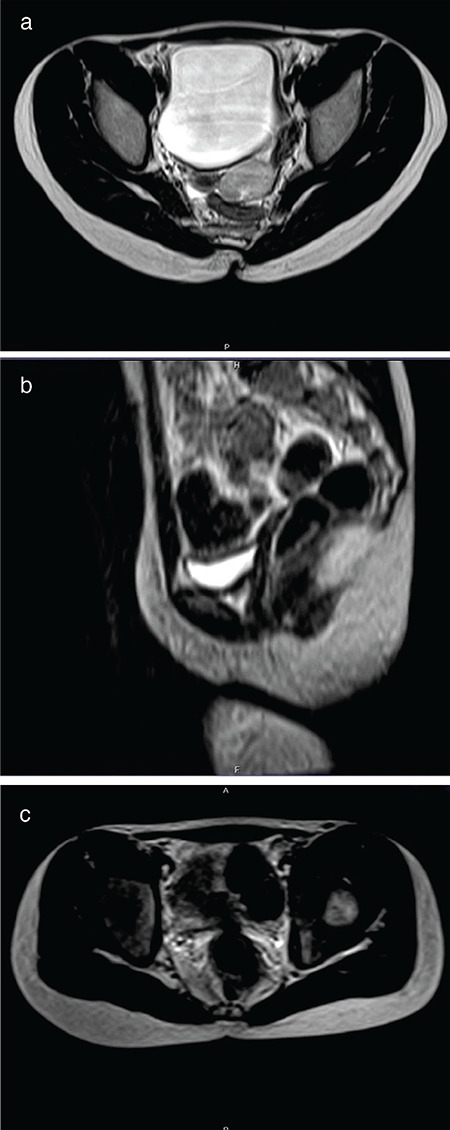
a) Axial T2-weighted magnetic resonance (MR) scan shows well demarcated hyperintense pathological mass in the left ovary, which does not infiltrate the ovarian capsule. The tumor is oval and homogeneous. b) Sagittal T2-weighted MR image shows a small hypoplastic uterine remnant in the anatomic location of the uterus. c) Axial T2-weighted MR image shows a hypoplastic ovary on the right side (the longest diameter of the ovary is 13 mm). The morphology of the right ovary is normal without any visible pathological structure inside
Figure 2.
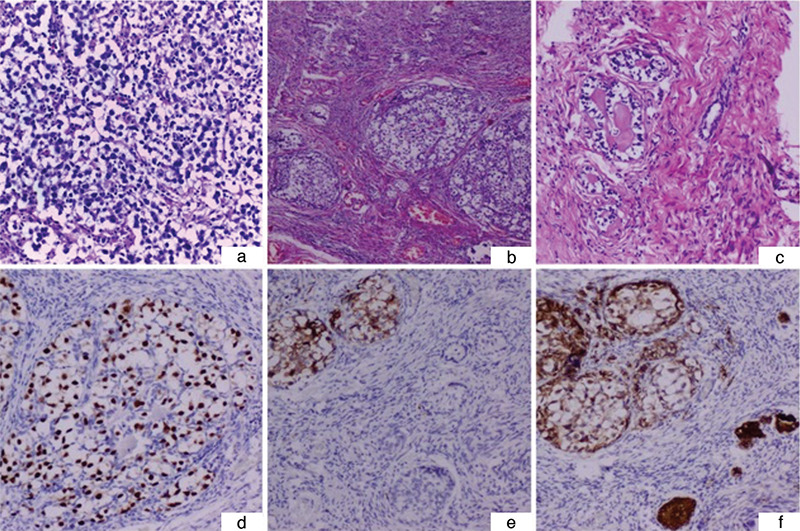
a) Histology of the dysgerminoma from the left ovary [hematoxylin-eosin (HE) 200x]. b) Dysgenetic gonad containing gonadoblastoma foci within the ovarian type stroma (HE 100x). c) A small group of tubular structures of sex cord cells arranged around the hyaline cores, scattered in the stroma of the dysgenetic gonad (HE 400x). d) Oct 3-positive germ cells within gonadoblastoma structures (Oct 3, 400x). e) CD117 expression within the germ cell component of gonadoblastoma, and immunonegative sex cord elements within the surrounding gonad (CD117, 200x). f) Inhibin alpha staining present within the sex cord element of gonadoblastoma and stromal sex cord structures (inhibin, 200x)
After the surgery, the patient remained under regular follow-up by a pediatric oncologist, with screening US of the abdomen and pelvis performed every 3 months and MRI every 6 months. No recurrence of the left ovarian tumor was found. However, both a small uterus and a streaked hypoplastic right ovary were recorded in subsequent examinations (Figure 1b, c). Finally, at the age of 15 years, primary amenorrhea with no secondary signs of puberty was diagnosed in the girl. Based on serum hormone levels, including follicle-stimulating hormone (FSH) - 131.32 mIU/L, luteinizing hormone - 35.86 U/L, and estradiol <37 pmol/L, hypergonadotropic hypogonadism was diagnosed and right ovary dysplasia was suspected. Cytogenetic studies revealed female karyotype with a number of distinct, nonclonal aberrations involving chromosome 7 and 14 in a significant proportion (13%) of the cells analyzed. A paracentric inversion of chromosome 14 was also present in all metaphases studied. No SRY signal was detected by fluorescence in situ hybridization.
After multidisciplinary consultation, it was decided to remove the remaining non-functioning ovary and to start the patient on hormone replacement therapy. The small right (1.5x1.0 cm) gonad was resected by laparoscopy, histologically showing gonadal dysgenesis with gonadoblastoma. The gonad contained fibrous, focally evident ovarian type stroma with scattered granulosa cell groups and dispersed microcalcifications. Moreover, Sertoli cell nodules, as well as germ cells accompanied by Sertoli/granulosa cells in cord-like structures were present. Neither primordial follicles nor seminiferous tubules were present. Coexisting gonadoblastoma nests presented typical immunophenotype with Oct 3/4, placental alkaline phosphatase, CD117, and inhibin positivity (Figure 2b, c, d, e, f).
The patient was started on hormone replacement therapy, and in three months she developed secondary sexual characteristics (breast Tanner stage 3 and growth gain of 5 cm). On follow-up US examinationsa few months later, an enlargement of the uterus from 11x5.7x22 mm to 31x14x55 mm and prominent endometrium were detected. Currently, this 27-year-old patient is alive and free of any symptoms of cancer recurrence. She is under the care of a hematologist and on continuous immunoglobulin supplementation. However, after she turned 18 and was discharged from pediatric care, she discontinued hormone replacement therapy. In a recently performed US examination, her uterus size was 24.5x11.6x30.5 mm, with no visible endometrium. An informed consent from the patient and her parents to publish this case-report has been obtained.
Discussion
NBS is a rare and possibly still underdiagnosed disease. It has been estimated that approximately 20-30% of patients are diagnosed with neoplasms prior to the diagnosis of this syndrome. Our patient was diagnosed with NBS soon after her sister’s death from cancer, based on their specific phenotypic similarity. This led to discontinuation of repeated diagnostic X-rays of chest and paranasal sinuses during her recurrent infections. These procedures are now highly contraindicated in NBS patients, due to their hypersensitivity to ionizing radiation and the high risk of malignancy induction (20). Since then, our patient underwent regular prophylactic US examinations, leading to the discovery of an asymptomatic left ovarian dysgerminoma at an early stage.
Gonadal dysgenesis is defined as an incomplete or defective formation of gonads, resulting from a disturbed process of migration of the germ cells and the organization of the primitive gonads. It is caused by aberrations of sex chromosomes or defects in genes involved in the formation of the urogenital ridge and sex determination of the primarily bipotential gonad (21). Gonadal dysgenesis predisposes to the development of gonadoblastoma and dysgerminoma, particularly in patients with 46,XY karyotype (22). However, rare cases of these tumors in patients with female 46,XX karyotypes have been reported (23,24). Capito et al (25) suggested that a highly elevated serum FSH level might be an indirect marker of suspected ovarian tumor in patients with PGD. Gonadal dysgenesis resulting from defective nibrin function is a typical feature of females with NBS.
Dysgerminomas can be associated with many genetic syndromes, such as: Frasier; ataxia-telangiectasia; WAGR (Wilms tumor, aniridia, genitourinary anomalies and intellectual disability); and Apert syndromes (26,27,28,29). However, to the best of our knowledge, only one case of dysgerminoma in a patient with NBS has been reported to date (6). In our patient, the 46,XX karyotype was confirmed; however, numerous abnormalities involving chromosomes 7 and 14 were detected in a significant percentage of cells. These aberrations are characteristic for NBS patients, reflecting the flag mark of the disorder - chromosomal instability. In our patient, along with the typical abnormalities of chromosomes 7 and 14 in 13% of cells, all metaphases revealed the presence of pericentric inversion of chromosome 14. It is generally acknowledged that aberrations involving chromosomes 7 and 14 are present in 10-50% of the cells (30). To some extent, the rate observed in our patient might be age-related, resulting from simple accumulation of individual aberrations over time. This patient was cytogenetically evaluated at the age of 20 years, while most of the published NBS patients had karyotype performed in early childhood. We cannot exclude, however, the parental origin of chromosome 14 inversion, because the parents were unavailable for genetic testing at that time.
The prognosis and treatment of dysgerminomas depend on their pathological and clinical stage. Patients with disease limited to one ovary may be treated by unilateral oophorectomy alone, especially when fertility is to be maintained. However, in patients with PGD, early bilateral gonadectomy is recommended to avoid the risk of subsequent neoplasm development. In our patient, no secondary signs of puberty with primary amenorrhea were observed at the age of 15. On US and MRI, performed repeatedly after left ovariectomy, the hypoplastic uterus and right ovary were described. With very low estradiol levels and significantly elevated FSH concentrations, hypergonadotropic hypogonadism was diagnosed and right ovarian dysplasia was suspected. Due to the history of dysgerminoma in the left ovary, a decision to perform prophylactic right oophorectomy was made, following the rules established for PGD XY females (Swyer syndrome) (25). Histopathological examination of the resected right ovary confirmed its dysgenetic features and revealed areas of gonadoblastoma. The surgery was complete so the patient did not require adjuvant oncological therapy and remains cancer-free. Hormone replacement therapy produced a satisfactory clinical effect with induction of secondary sexual characteristics.
Our patient is the first case with metachronous ovarian gonadoblastoma and dysgerminoma developing within the dysgenetic ovaries, occurring in a patient with NBS and 46,XX karyotype. Although it seems that nibrin may play a role in normal development and function of the ovary, only one case of gonadoblastoma and one of dysgerminoma in NBS patients have been previously reported to date (6,11).
The optimal management of NBS females with hypergonadotropic hypogonadism suggesting PGD is controversial. In 2010 Chrzanowska et al (12) published results of a longitudinal study showing that primary ovarian insufficiency with associated hypergonadotropic hypogonadism are common and typical features in females with NBS. The authors suggested that careful, but minimally invasive diagnostics, including auxiological follow-up, US examinations and basic hormonal assays are sufficient in these patients. Accordingly, Huang et al (24) demonstrated within a group of 33 patients with XX PGD, that the risk of malignant transformation in case of absence of the Y chromosome is low. Therefore, in XX PGD patients prophylactic gonadectomy is not recommended and only careful follow-up should be provided. However, in patients in whom ovarian germ cell tumor has already been diagnosed within one gonad, we advocate for early prophylactic oophorectomy of the remaining nonfunctional streak ovary to exclude or avoid a second germ cell tumor. This recommendation is particularly meaningful for XX, PGD females with NBS because early detection and complete resection of malignancy in these patients gives a chance for successful outcome without need of adjuvant chemotherapy, which is extremely risky in terms of development of subsequent cancers. Such a strategy for clinical management was adopted in our patient, resulting in a long-lasting clinical remission of metachronous bilateral ovarian germ cell tumors.
Acknowledgments
We would like to express our appreciation to professor Krystyna H. Chrzanowska from the Department of Medical Genetics, the Children’s Memorial Health Institute in Warsaw, Poland for providing valuable meritorical support during the analysis of this patient. We would like to thank the students of The English Division Pediatric Oncology Scientific Circle, Medical University of Gdańsk: Marta Kedrzycki and Sylwia Mackiewicz for their help in preparing the case report.
Footnotes
Ethics
Informed Consent: Consent form was filled out by all participants.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Malgorzata A. Krawczyk, Dorota Birkholz-Walerzak, Mariola Iliszko, Beata S. Lipska-Zietkiewicz, Wojciech Kosiak, Ninela Irga-Jaworska, Ewa Bien, Concept: Malgorzata A. Krawczyk, Malgorzata Styczewska, Ewa Izycka-Swieszewska, Ewa Bien, Design: Malgorzata A. Krawczyk, Malgorzata Styczewska, Data Collection or Processing: Malgorzata A. Krawczyk, Malgorzata Styczewska, Wojciech Kosiak, Analysis or Interpretation: Malgorzata A. Krawczyk, Malgorzata Styczewska, Dorota Birkholz-Walerzak, Mariola Iliszko, Beata S. Lipska-Zietkiewicz, Wojciech Kosiak, Ninela Irga-Jaworska, Ewa Izycka-Swieszewska, Ewa Bien, Literature Search: Malgorzata A. Krawczyk, Malgorzata Styczewska, Dorota Birkholz-Walerzak, Mariola Iliszko, Beata S. Lipska-Zietkiewicz, Writing: Malgorzata A. Krawczyk, Malgorzata Styczewska, Dorota Birkholz-Walerzak, Mariola Iliszko, Beata S. Lipska-Zietkiewicz, Wojciech Kosiak, Ninela Irga-Jaworska, Ewa Izycka-Swieszewska, Ewa Bien.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M. Nijmegen breakage syndrom (NBS) Orphanet J Rare Dis. 2012;7:13. doi: 10.1186/1750-1172-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor AMR, Rothblum-Oviatt C, Ellis NA, Hickson ID, Meyer S, Crawford TO, Smogorzewska A, Pietrucha B, Weemaes C, Stewart GS. Chromosome instability syndromes. Nat Rev Dis Primers. 2019;5:64. doi: 10.1038/s41572-019-0113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seemanová E, Passarge E, Beneskova D, Houstĕk J, Kasal P, Sevcíková M. Familial microcephaly with normal intelligence, immunodeficiency, and risk for lymphoreticular malignancies: A new autosomal recessive disorder. Am J Med Genet. 1985;20:639–648. doi: 10.1002/ajmg.1320200410. [DOI] [PubMed] [Google Scholar]
- 4.Meijers RWJ, Dzierzanowska-Fangrat K, Zborowska M, Solarska I, Tielemans D, van Turnhout BAC, Driessen G, van der Burg M, van Dongen JJM, Chrzanowska KH, Langerak AW. Circulating T Cells of Patients with Nijmegen Breakage Syndrome Show Signs of Senescence. J Clin Immunol. 2017;37:133–142. doi: 10.1007/s10875-016-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chrzanowska KH, Kleijer WJ, Krajewska-Walasek M, Białecka M, Gutkowska A, Goryluk-Kozakiewicz B, Michałkiewicz J, Stachowski J, Gregorek H, Lysón-Wojciechowska G, et al. Eleven Polish patients with microcephaly, immunodeficiency, and chromosomal instability: The Nijmegen breakage syndrome. Am J Med Genet. 1995;57:462–471. doi: 10.1002/ajmg.1320570321. [DOI] [PubMed] [Google Scholar]
- 6.Wolska-Kuśnierz B, Gregorek H, Chrzanowska K, Piątosa B, Pietrucha B, Heropolitańska-Pliszka E, Pac M, Klaudel-Dreszler M, Kostyuchenko L, Pasic S, Marodi L, Belohradsky BH, Čižnár P, Shcherbina A, Kilic SS, Baumann U, Seidel MG, Gennery AR, Syczewska M, Mikołuć B, Kałwak K, Styczyński J, Pieczonka A, Drabko K, Wakulińska A, Gathmann B, Albert MH, Skarżyńska U, Bernatowska E Inborn Errors Working Party of the Society for European Blood and Marrow Transplantation and the European Society for Immune Deficiencies. Nijmegen Breakage Syndrome: Clinical and Immunological Features, Long-Term Outcome and Treatment Options - a Retrospective Analysis. J Clin Immunol. 2015;35:538–549. doi: 10.1007/s10875-015-0186-9. [DOI] [PubMed] [Google Scholar]
- 7.Filipovich AH, Mathur A, Kamat D, Shapiro RS. Primary immunodeficiencies: Genetic risk factors for lymphoma. Cancer Res. 1992;52(19 Suppl):5465–5467. [PubMed] [Google Scholar]
- 8.Pastorczak A, Szczepanski T, Mlynarski W International Berlin-Frankfurt-Munster (I-BFM) ALL host genetic variation working group. Clinical course and therapeutic implications for lymphoid malignancies in Nijmegen breakage syndrome. Eur J Med Genet. 2016;59:126–132. doi: 10.1016/j.ejmg.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Bakhshi S, Cerosaletti KM, Concannon P, Bawle EV, Fontanesi J, Gatti RA, Bhambhani K. Medulloblastoma with adverse reaction to radiation therapy in Nijmegen breakage syndrome. J Pediatr Hematol Oncol. 2003;25:248–251. doi: 10.1097/00043426-200303000-00013. [DOI] [PubMed] [Google Scholar]
- 10.Meyer S, Kingston H, Taylor AM, Byrd PJ, Last JI, Brennan BM, Trueman S, Kelsey A, Taylor GM, Eden OB. Rhabdomyosarcoma in Nijmegen breakage syndrome: Strong association with perianal primary site. Cancer Genet Cytogenet. 2004;154:169–174. doi: 10.1016/j.cancergencyto.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 11.Wegner R, German J, Chrzanowska K. Chromosomal instability syndromes other than ataxia-telangiectasia. In: Ochs H, Smit C, Puck J (eds). Prim. Immunodefic. Dis A Mol Genet approach. 2nd ed. New York, Oxford University Press. 2007:427–453. [Google Scholar]
- 12.Chrzanowska KH, Szarras-Czapnik M, Gajdulewicz M, Kalina MA, Gajtko-Metera M, Walewska-Wolf M, Szufladowicz-Wozniak J, Rysiewski H, Gregorek H, Cukrowska B, Syczewska M, Piekutowska-Abramczuk D, Janas R, Krajewska-Walasek M. High prevalence of primary ovarian insufficiency in girls and young women with Nijmegen breakage syndrome: Evidence from a longitudinal study. J Clin Endocrinol Metab. 2010;95:3133–3140. doi: 10.1210/jc.2009-2628. [DOI] [PubMed] [Google Scholar]
- 13.Szeliga A, Zysnarska A, Szklarska Z, Truszkowska E, Podfigurna A, Czyzyk A, Genazzani AR, Chrzanowska K, Meczekalski B. A case of premature ovarian insufficiency in Nijmegen breakage syndrome patient and review of literature From gene mutation to clinical management. Gynecol Endocrinol. 2019;35:999–1002. doi: 10.1080/09513590.2019.1626366. [DOI] [PubMed] [Google Scholar]
- 14.Tucker EJ, Grover SR, Robevska G, van den Bergen J, Hanna C, Sinclair AH. Identification of variants in pleiotropic genes causing “isolated” premature ovarian insufficiency: implications for medical practice. Eur J Hum Genet. 2018;26:1319–1328. doi: 10.1038/s41431-018-0140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warcoin M, Lespinasse J, Despouy G, Dubois d’Enghien C, Laugé A, Portnoï MF, Christin-Maitre S, Stoppa-Lyonnet D, Stern MH. Fertility defects revealing germline biallelic nonsense NBN mutations. Hum Mutat. 2009;30:424–430. doi: 10.1002/humu.20904. [DOI] [PubMed] [Google Scholar]
- 16.Fiévet A, Bellanger D, Zahed L, Burglen L, Derrien AC, Dubois d’Enghien C, Lespinasse J, Parfait B, Pedespan JM, Rieunier G, Stoppa-Lyonnet D, Stern MH. DNA repair functional analyses of NBN hypomorphic variants associated with NBN-related infertility. Hum Mutat. 2020;41:608–618. doi: 10.1002/humu.23955. [DOI] [PubMed] [Google Scholar]
- 17.Plisiecka-Hałasa J, Dansonka-Mieszkowska A, Rembiszewska A, Bidziński M, Steffen J, Kupryjańczyk J. Nijmegen breakage syndrome gene (NBS1) alterations and its protein (nibrin) expression in human ovarian tumours. Ann Hum Genet. 2002;66:353–359. doi: 10.1017/S0003480002001227. [DOI] [PubMed] [Google Scholar]
- 18.Ciara E, Piekutowska-Abramczuk D, Popowska E, Grajkowska W, Barszcz S, Perek D, Dembowska-Bagińska B, Perek-Polnik M, Kowalewska E, Czajńska A, Syczewska M, Czornak K, Krajewska- Walasek M, Roszkowski M, Chrzanowska KH. Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients. Acta Neuropathol. 2010;119:325–334. doi: 10.1007/s00401-009-0608-y. [DOI] [PubMed] [Google Scholar]
- 19.Zhang G, Zeng Y, Liu Z, Wei W. Significant association between Nijmegen breakage syndrome 1 657del5 polymorphism and breast cancer risk. Tumor Biol. 2013;34:2753–2757. doi: 10.1007/s13277-013-0830-z. [DOI] [PubMed] [Google Scholar]
- 20.Walsh MF, Chang VY, Kohlmann WK, Scott HS, Cunniff C, Bourdeaut F, Molenaar JJ, Porter CC, Sandlund JT, Plon SE, Wang LL, Savage SA. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res. 2017;23:23–31. doi: 10.1158/1078-0432.CCR-17-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolffenbuttel KP, Hersmus R, Stoop H, Biermann K, Hoebeke P, Cools M, Looijenga LH. Gonadal dysgenesis in disorders of sex development: Diagnosis and surgical management. J Pediatr Urol. 2016;12:411–416. doi: 10.1016/j.jpurol.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Piazza MJ, Urbanetz AA. Germ Cell Tumors in Dysgenetic Gonads. Clinics (Sao Paulo) 2019;74:e408. doi: 10.6061/clinics/2019/e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth LM, Davis MM, Czernobilsky B. Classic and “dissecting” Gonadoblastoma in a Phenotypic Girl with a 46,XX Peripheral Karyotype and No Evidence of a Disorder of Sex Development. Int J Gynecol Pathol. 2019;38:581–587. doi: 10.1097/PGP.0000000000000551. [DOI] [PubMed] [Google Scholar]
- 24.Huang H, Wang CQ, Tian QJ. Clinical features and management of 33 patients with 46,XX pure gonadal dysgenesis. Gynecol Endocrinol. 2016;32:995–998. doi: 10.1080/09513590.2016.1190820. [DOI] [PubMed] [Google Scholar]
- 25.Capito C, Arnaud A, Hameury F, Fremond B, Lardy H, Leclair MD, Heloury Y. Dysgerminoma and gonadal dysgenesis: The need for a new diagnosis tree for suspected ovarian tumours. J Pediatr Urol. 2011;7:367–372. doi: 10.1016/j.jpurol.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Subbiah V, Huff V, Wolff JE, Ketonen L, Lang FF Jr, Stewart J, Langford L, Herzog CE. Bilateral gonadoblastoma with dysgerminoma and pilocytic astrocytoma with WT1 GT-IVS9 mutation: A 46 XY phenotypic female with Frasier syndrome. Pediatr Blood Cancer. 2009;53:1349–1351. doi: 10.1002/pbc.22152. [DOI] [PubMed] [Google Scholar]
- 27.Koksal Y, Caliskan U, Ucar C, Yurtcu M, Artac H, Ilerisoy-Yakut Z, Reisli I. Dysgerminoma in a child with ataxia-telangiectasia. Pediatr Hematol Oncol. 2007;24:431–436. doi: 10.1080/08880010701451434. [DOI] [PubMed] [Google Scholar]
- 28.Miura R, Yokoyama Y, Shigeto T, Futagami M, Mizunuma H, Kurose A, Tsuruga K, Sasaki S, Terui K, Ito E. Dysgerminoma developing from an ectopic ovary in a patient with WAGR syndrome: A case report. Mol Clin Oncol. 2016;5:503–506. doi: 10.3892/mco.2016.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouzier C, Soler C, Hofman P, Brennetot C, Bieth E, Pedeutour F. Ovarian dysgerminoma and Apert syndrome. Pediatr Blood Cancer. 2008;50:696–698. doi: 10.1002/pbc.21156. [DOI] [PubMed] [Google Scholar]
- 30.Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: Old paths, new directions, moving frontiers. Endocr Rev. 2006;27:468–484. doi: 10.1210/er.2006-0005. [DOI] [PubMed] [Google Scholar]