

Abstract
The enzyme 17-β-hydroxysteroid dehydrogenase type 3 (17β-HSD3) catalyzes the biosynthesis of testosterone (T) from Δ4- androstenedione, and plays an important role in the final steps of androgen synthesis. 17β-HSD3 deficiency originates from mutations in the HSD17B gene, causing an autosomal recessive 46,XY sex developmental disorder (DSD). Patients with 46,XY karyotype can exhibit a wide phenotypic spectrum, varying from complete external female genitalia to male genitalia with hypospadias. Here we report a case of 17β-HSD3 deficiency diagnosed in the infantile period who was later found to have a novel HSD17B3 gene variation. The 14-month old patient, who exhibited a female phenotype, presented with a bilateral lump in the inguinal area. Imaging revealed bilateral testicular gonads in the inguinal area. Hormonal evaluation showed low levels of basal and stimulated serum T, a high level of androstenedione (A), and a low T/A ratio. Chromosomal analysis showed 46,XY karyotype. Sequence analysis of the HSD17B3 gene revealed a c.673_1G>C homozygous class 2 (splice site) variation in intron 9. The consanguineous parents were sequenced, and both were heterozygous for the same mutation. This variation has not been previously reported in the literature. In conclusion, a 46,XY DSD should be considered in patients with a female phenotype who exhibit gonad(s) in the inguinal area at an early age. Furthermore, in patients with insufficient T synthesis and high levels of androstenedione, 17β-HSD3 should be considered, and molecular analysis should be done for a definitive diagnosis and subsequent genetic counseling.
Keywords: 17 beta-hydroxysteroid dehydrogenase type 3; 46,XY disorders of sex development; HSD17B3 gene
What is already known on this topic?
The 17-beta hydroxysteroid dehydrogenase type 3 (17β-HSD3) enzyme is primarily found in the testes and is involved in transforming Δ4-androstenedione, a weak androgen, to the more biologically active form, testosterone. Defects in the HSD17B3 gene, which encodes this enzyme, cause 17β-HSD3 deficiency.
What this study adds?
In this article, we describe a previously unreported c.673_1G>C homozygous variation that was identified in the HSD17B3 gene of a 46,XY patient.
Introduction
The 17-beta hydroxysteroid dehydrogenase (17b-HSD) enzyme family includes at least 14 isoenzymes identified to date. These contribute to the development of reproductive organs by taking part in the final steps of androgen and estrogen synthesis. The 17-beta hydroxysteroid dehydrogenase type 3 (17β-HSD3) enzyme is primarily found in the testes and is involved in transforming Δ4-androstenedione (A), a weak androgen, to the more biologically active form testosterone (T) (1). Defects in the HSD17B3 gene, which encodes this enzyme, causes 17β-HSD3 deficiency.
17β-HSD3 enzyme deficiency was initially described in 1971 and shows an autosomal recessive inheritance (2,3). Although not known exactly, the incidence rate has been reported to be 1/147,000 live births and the rate of heterozygosity to be 1/135 (4). Furthermore, in populations with a high rate of consanguineous marriage, such as the Gaza Strip Arabs, the incidence has been reported to be as high as 1/100-300 (5,6). Problems in T synthesis during fetal development result in insufficient development of male external genital organs. Although T synthesis is insufficient, the production of anti-Müllerian hormone (AMH) continues normally and prevents the development of Mullerian structures (7).
These patients with a 46,XY karyotype can exhibit a wide phenotypic spectrum varying from female external genitalia to male external genitalia with hypospadias, or ambiguous genitalia with microphallus (8). During puberty, the increase in gonadotropin levels lead to an increase in A levels and extra-testicular conversion of A to T, which ultimately leads to evident virilization. The degree of virilization can vary depending on 17β-HSD3 isoenzyme residue in the testes and the activity of other isoenzymes, such as 17b-HSD5 (9,10,11). Individuals with a 46,XX karyotype generally have normal female genitalia and are asymptomatic, making the condition difficult to diagnose (5,12).
In the laboratory analysis of 17β-HSD3 deficiency, low serum T levels and high serum A levels are observed. A human chorionic gonadotropin (hCG) stimulation test generally results in a serum T/A ratio of below 0.8 (13). A final diagnosis is made through molecular genetic testing.
In this article, we describe a case who presented with a history of bilateral lumps in the inguinal area during the infantile period, the patient’s physical examination revealed external genitalia of female phenotype and palpable gonads. The evaluations were consistent with 17β-HSD3 deficiency, and a previously unreported c.673_1G>C homozygous variation was identified in the HSD17B3 gene of the 46,XY patient.
Case Report
A 14-month old female patient presented with a history of a bilateral lump in the inguinal area. Her mother and father were first-degree cousins. Physical examination showed female-appearing external genitalia with the absence of clitoromegaly. Gonads were palpable in both inguinal regions. Ultrasound imaging revealed gonads that were compatible with testes in both inguinal areas, and no Mullerian structures were observed in the pelvis. It was speculated that the patient may have 46,XY disorder of sex development (DSD). Laboratory testing was used to assess gonad functions, showing that the serum follicle stimulating hormone level was 2.29 IU/L (0.26-3.0), luteinizing hormone (LH) was 1.19 IU/L (0.02-0.3), and T level was <0.693 nmol/L. Her serum AMH level was >73 ng/ML (maximum 3.9 for female, 9.9-444.1 for male) and inhibin B level was 388 ng/L (91-400). Evaluation of basal hormone levels revealed that T synthesis was insufficient. In order to precisely evaluate T synthesis ability, an hCG stimulation test was conducted. Both basal and stimulated serum T levels were found to be low and the serum T level did not increase following stimulation. The patient’s T synthesis defect was confirmed. Serum T/A ratio was 0.14 (Table 1). A standard dose (250 µg) synacthen test was done to exclude disorders in which T synthesis defect and adrenal insufficiency may be seen together (such as 17-alpha hydroxylase deficiency). The test showed an adequate level of stimulated cortisol, and normal basal and stimulated serum progesterone and dehydroepiandrosterone sulphate levels. 17-alpha hydroxylase deficiency was ruled out. The karyotype was 46,XY, and fluorescence in situ hybridization analysis showed the absence of SRY gene variations. Pelvic magnetic resonance imaging (MRI) was used to comprehensively examine internal gonadal structures. The MRI revealed structures proximal to both inguinal canals that were compatible with testes, and a 30x4 mm structure that was compatible with vaginal tissue between the bladder and rectum. Diagnostic cystoscopy and laparoscopy showed the presence of gonads identical to testes in both inguinal canals. The vagina was 2 cm long. Since AMH levels were high and imaging showed the absence of Mullerian structures, gonadal dysgenesis was excluded.
Table 1. Serum androgen concentrations before and after human chorionic gonadotropin stimulation.

A gonad biopsy was made for pathological evaluation. The pathology report stated “Bilateral testes containing seminiferous tubules and surrounded by the tunica albuginea were observed. Spermatogonia were not observed in the seminiferous tubules. Leydig cells were not present with hematoxylin and eosin staining. Of the immune markers used, Inhibin led to a strong positive immunoreaction in Sertoli cells and a mild positive immunoreaction in a small number of Leydig cells; calretinin led to a mild, positive reaction in Leydig cells; mild staining with PLAP; CD138 negative; OCT3/4 negative; LH receptor showed a negative reaction.” As the pathology report stated that the Leydig cells were insufficient and LH receptors were absent with specific staining, Leydig cell hypoplasia was considered but LH/choriogonadotropin receptor (LHCGR) gene analysis revealed no mutation. The patient, who had a T synthesis defect as well as a low serum T/A ratio, and did not have adrenal insufficiency or gonadal dysgenesis, was considered to have 17β-HSD3 deficiency. A sequence analysis of the patient’s HSD17B3 gene revealed a c.673_1G>C homozygous class 2 (splice site) variation on intron 9 (Figure 1). Both parents exhibited an identical heterozygous variation. This variation has not been reported in the literature previously, and was most likely pathological according to in silico analyses (Table 2). It was reported that in 46,XY patients with 17β-HSD3 deficiency who exhibit a total female phenotype, it is possible to achieve a penis size within normal limits through treatment with 25-50 mg/dose of intramuscular T for 3-9 months during the infantile period (14). Accordingly, our patient was treated with 50 mg/month of intramuscular T and gender determination was made based on response to treatment.
Figure 1.
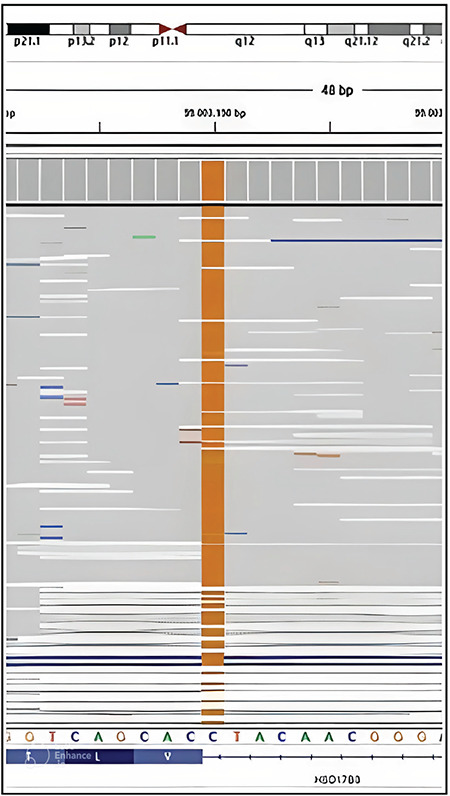
HSD17B3 gene, c.673_1G>C homozygous class 2 (splice site) variation on intron 9
Table 2. The in silico analysis, revealing the genetic variation in the HSD17B3 gene of our patient.

The parents gave their written consent for sharing the patient’s examination, laboratory, imaging, and genetic results in scientific publications, on the condition that the child remains anonymous.
Discussion
The clinical signs of 17β-HSD3 deficiency may vary due to its wide phenotypic spectrum. These 46,XY patients may have differing external genital appearances depending on the residual activity of enzymes. Patients most frequently have a complete external female genital structure, usually with separate urethral and vaginal openings. However, some patients have been reported to only have a short urogenital sinus (3,11,15). Patients with complete external female genitalia are usually diagnosed late, and are often raised as female individuals. These patients usually present during puberty with primary amenorrhea and varying degrees of virilization. In patients with evident lumps in the inguinal canals or labioscrotal folds, the palpation of gonads may lead to an early diagnosis, similar to our patient (4,11,16). 46,XY patients may less frequently present with micropenis and hypospadias, in which case the patient is generally raised as a male individual (5).
Due to their female phenotype and evident virilization during puberty, 46,XY patients with 17β-HSD3 deficiency are clinically similar to other conditions, such as androgen insensitivity syndrome (AIS), partial 5-α-reductase type 2 deficiency, or steroidogenic factor 1 deficiency (17). Boehmer and colleagues reported that 19 patients who were initially believed to have AIS were diagnosed with 17β-HSD3 deficiency after further investigation (4). Leydig cell aplasia is also included in the differential diagnosis of 46,XY female patients who have been diagnosed at an early age.
The typical hormonal findings for 17β-HSD3 deficiency includes reduced T and increased A levels. While it is possible to diagnose patients through basal hormone levels during adulthood, puberty or minipuberty, an hCG stimulation test must be performed in the other age periods, or the diagnosis may be missed (11,13). In our patient, T levels did not increase with the hCG stimulation test and the T/A ratio was found to be low, suggesting 17β-HSD3 deficiency. Through imaging techniques, the observation of Wolffian structures and the absence of Mullerian structures are supportive in diagnosing 17β-HSD3 deficiency. However, since these findings are also present in both 5a-reductase deficiency and androgen receptor mutations, they are inadequate for a definitive diagnosis. In individuals with 17β-HSD3 deficiency, while histological examination can reveal near normal testicular structure at early ages, patients who reach adulthood with undescended testes usually display characteristics of testicular atrophy (exaggerated thickening of the basement membrane, evident decrease in the seminiferous tubule germinative epithelium, interstitial fibrosis, increased Leydig cells) (18). According to the literature, in the pathological examination of gonads that were removed for prophylactic measures, 2-3% of cases had germ cell tumors (19). In 40 patients diagnosed with 17β-HSD3 deficiency histological examination of testicular tissue stained with hematoxylin and eosin revealed that 5% of cases had germ cell tumors (18). On medical imaging, our patient had gonads in both inguinal canals that were compatible with testes and Müllerian structures were absent; moreover on the pathology report, the gonad biopsy was described as testicular tissue.
17β-HSD3 deficiency arises from the compound heterozygous or homozygous mutation of the HSD17B3 gene. 17β-HSD3 deficiency shows an autosomal recessive inheritance pattern and is a frequent cause of 46,XY DSD among populations with high rates of consanguineous marriage. The HSD17B3 gene consists of 11 exons and is located on chromosome 9q22. To date, more than 30 mutations have been identified in this gene, including insertion, exonic deletion, missense, and nonsense mutations (8,20,21,22,23). Most of these mutations have been identified in the Arab population of the Gaza strip. The most widespread mutation in the Arab population is the p.Arg80Gln mutation on exon 3 (4). In the Turkish population, c655-1;G-A, p.Ala188Val, and c.777-783del_GATAACC mutations have previously been identified (24). In a study by Özen et al (25), 20 patients being followed-up for 46,XY DSD, who did not have mutations in genes SRD5A2 and AR, were analyzed using targeted next generation sequence (TNGS) analysis for 56 potential genes which may be involved in the etiology of 46,XY DSD. Mutations were identified in the HSD17B3 gene in 30% of patients. It was reported that two patients had a homozygous p. Y287X variation, one patient had combined heterozygous p.R80Q and p.E93K variations, and three patients each had one homozygous p.T54A, p.R175T, or p.R80Q variation (25). The literature has reported no genotype-phenotype correlation in 17β-HSD3 deficiency (26). Our patient exhibited a c.673_1G>C homozygous class 2 (splice site) variation in intron 9 of the HSD17B3 gene. This variant has not been previously reported in the Human Gene Mutation Database. In silico analyses (Human Splicing Finder; VarSome; Mutation- Taster; https://varsome.com) predicted this variant as likely to be pathogenic.Similar to other DSDs, gender selection proves to be a difficult decision in individuals with 17β-HSD3 deficiency, especially in cases diagnosed at an early age. The 2006 report of the Chicago Consensus Meeting recommends discussing both the fertility potential (unclear) and the development of sexual identity (mostly male) while determining sex in patients with 17β-HSD3 deficiency diagnosed during infancy (27). Male individuals with cryptorchidism and 17β-HSD3 deficiency show regression in spermatogenesis over time. These patients have an uncertain fertility potential, and a fertile 46,XY patient has not been previously identified. Gonads that are preserved should be lowered into the scrotum and routinely checked for malignancy (18,27). However, prepubescent gonadectomy is recommended for patients that are raised female because of the potential risks of germ cell tumors and virilization caused by a pubertal increase in androgens.
A significant proportion of female individuals (39-64%) who did not undergo gonadectomy and experienced virilization during their adolescence, later transitioned to the male sex (28). However, females who did undergo gonadectomy during their childhood were usually satisfied and very few individuals exhibited a desire for future sex change (4,8,29). It was reported that no individual with male-dominant phenotype who was raised as a male desired a change in sex (28).
It is crucial that every case is evaluated individually while trying to determine sex. Since our patient was diagnosed early and evaluations showed that the patient and family embraced the individual’s male identity, T-based treatments were given and the patient awaited the response of the external genital structures to androgen therapy. We plan to determine the sex based on the patient’s response to the treatment.
Conclusion
17β-HSD3 deficiency is an autosomal recessive form of 46,XY DSD. Although the diagnosis can be made with the appropriate endocrinological evaluations, it is confirmed by molecular genetic analysis. Our case showed a novel variation (c.673_1G> C homozygous) in the HSD17B3 gene. 46,XY DSD should be considered in females who present with inguinal lumps and/or mild clitoromegaly during infancy or childhood, and in adolescent females who experience virilization during puberty. For a definitive diagnosis and subsequent genetic counseling, molecular analysis should be performed in cases with insufficient T synthesis and high androstenedione levels who are suspected of 17β-HSD3 deficiency. An early and accurate diagnosis is important for determining sex, patient management, and genetic counseling.
Footnotes
Ethics
Informed Consent: Consent form was filled out by all participants.
Peer-review: Externally peer-reviewed
Authorship Contributions
Surgical and Medical Practices: Nurdan Çiftci, Leman Kayaş, Emine Çamtosun, Ayşehan Akıncı, Concept: Nurdan Çiftci, Emine Çamtosun, Ayşehan Akıncı, Design: Nurdan Çiftci, Emine Çamtosun, Ayşehan Akıncı, Data Collection or Processing: Nurdan Çiftci, Analysis or Interpretation: Nurdan Çiftci, Emine Çamtosun, Ayşehan Akıncı, Literature Search: Nurdan Çiftci, Emine Çamtosun, Writing: Nurdan Çiftci, Emine Çamtosun, Ayşehan Akıncı.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Moeller G, Adamski J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2009;301:7–19. doi: 10.1016/j.mce.2008.10.040. [DOI] [PubMed] [Google Scholar]
- 2.Saez JM, De Peretti E, Morera AM, David M, Bertrand J. Familial male pseudohermaphroditism with gynecomastia due to a testicular 17-ketosteroid reductase defect. Studies in vivo. J Clin Endocrinol Metab. 1971;32:604–610. doi: 10.1210/jcem-32-5-604. [DOI] [PubMed] [Google Scholar]
- 3.Saez JM, Morera AM, De Peretti E, Bertrand J. Further in vivo studies in male pseudohermaphroditism with gynecomastia due to a testicular 17-ketosteroid reductase defect (compared to a case of testicular feminization) J Clin Endocrinol Metab. 1972;34:598–600. doi: 10.1210/jcem-34-3-598. [DOI] [PubMed] [Google Scholar]
- 4.Boehmer AL, Brinkmann AO, Sandkuijl LA, Halley DJ, Niermeijer MF, Andersson S, de Jong FH, Kayserili H, de Vroede MA, Otten BJ, Rouwé CW, Mendonça BB, Rodrigues C, Bode HH, de Ruiter PE, Delemarre-van de Waal HA, Drop SL. 17β-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics and world wide distribution of ancient and de novo mutations. J Clin Endocrinol Metab. 1999;84:4713–4721. doi: 10.1210/jcem.84.12.6174. [DOI] [PubMed] [Google Scholar]
- 5.Rösler A, Silverstein S, Abeliovich D. A (R80Q) mutation in 17 betahydroxysteroid dehydrogenase type 3 gene among Arabs of Israel is associated with pseudohermaphroditism in males and normal asymptomatic females. J Clin Endocrinol Metab. 1996;81:1827–1831. doi: 10.1210/jcem.81.5.8626842. [DOI] [PubMed] [Google Scholar]
- 6.Rösler A. 17 beta-hydroxysteroid dehydrogenase 3 deficiency in the Mediterranean population. Pediatr Endocrinol Rev. 2006;3:455–461. [PubMed] [Google Scholar]
- 7.Cools M, Köhler B. Disorders of Sex Development. Brook’s Clinical Pediatric Endocrinology 7th Edition. 2020;114. [Google Scholar]
- 8.George MM, New MI, Ten S, Sultan C, Bhangoo A. The clinical and molecular heterogeneity of 17βHSD-3 enzyme deficiency. Horm Res Paediatr. 2010;74:229–240. doi: 10.1159/000318004. [DOI] [PubMed] [Google Scholar]
- 9.Andersson S, Russell DW, Wilson JD. 17 beta-Hydroxysteroid dehydrogenase 3 deficiency. Trends Endocrinol Metab. 1996;7:121–126. doi: 10.1016/1043-2760(96)00034-3. [DOI] [PubMed] [Google Scholar]
- 10.Eckstein B, Cohen S, Farkas A, Rösler A. The nature of the defect in familial male pseudohermaphroditism in Arabs of Gaza. J Clin Endocrinol Metab. 1989;68:477–485. doi: 10.1210/jcem-68-2-477. [DOI] [PubMed] [Google Scholar]
- 11.Lee YS, Kirk JM, Stanhope RG, Johnston DI, Harland S, Auchus RJ, Andersson S, Hughes IA. Phenotypicvariability in 17beta-hydroxysteroid dehydrogenase-3 deficiency and diagnostic pitfalls. Clin Endocrinol (Oxf) 2007;67:20–28. doi: 10.1111/j.1365-2265.2007.02829.x. [DOI] [PubMed] [Google Scholar]
- 12.Mendonca BB, Arnhold IJ, Bloise W, Andersson S, Russell DW, Wilson JD. 17Beta-hydroxysteroid dehydrogenase 3 deficiency in women. J Clin Endocrinol Metab. 1999;84:802–804. doi: 10.1210/jcem.84.2.5477. [DOI] [PubMed] [Google Scholar]
- 13.Faisal Ahmed S, Iqbal A, Hughes IA. The testosterone: androstenedione ratio in male under masculinization. Clin Endocrinol (Oxf) 2000;53:697–702. doi: 10.1046/j.1365-2265.2000.01166.x. [DOI] [PubMed] [Google Scholar]
- 14.Gross DJ, Landau H, Kohn G, Farkas A, Elrayyes E, el-Shawwa R, Lasch EE, Rösler A. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase deficiency: gender reassignment in early infancy. Acta Endocrinol (Copenh) 1986;112:238–246. doi: 10.1530/acta.0.1120238. [DOI] [PubMed] [Google Scholar]
- 15.Bertelloni S, Maggio MC, Federico G, Baroncelli G, Hiort O. 17beta-hydroxysteroid dehydrogenase-3 deficiency: a rare endocrine cause of male-to-female sex reversal. Gynecol Endocrinol. 2006;22:488–494. doi: 10.1080/09513590600921358. [DOI] [PubMed] [Google Scholar]
- 16.Andersson S, Geissler WM, Wu L, Davis DL, Grumbach MM, New MI, Schwarz HP, Blethen SL, Mendonca BB, Bloise W, Witchel SF, Cutler GB Jr, Griffin JE, Wilson JD, Russel DW. Molecular genetics and pathophysiology of 17 beta-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrinol Metab. 1996;81:130–136. doi: 10.1210/jcem.81.1.8550739. [DOI] [PubMed] [Google Scholar]
- 17.Cools M, Hoebeke P, Wolffenbuttel KP, Stoop H, Hersmus R, Barbaro M, Wedell A, Brüggenwirth H, Looijenga LH, Drop SL. Pubertal androgenization and gonadal histology in two 46, XY adolescents with NR5A1 mutations and predominantly female phenotype at birth. Eur J Endocrinol. 2012;166:341–349. doi: 10.1530/EJE-11-0392. [DOI] [PubMed] [Google Scholar]
- 18.Mendonca BB, Gomes NL, Costa EM, Inacio M, Martin RM, Nishi MY, Carvalho FM, Tibor FD, Domenice S. 46,XY disorder of sex development(DSD) due to 17β-hydroxysteroid dehydrogenasetype 3 deficiency. J Steroid Biochem Mol Biol. 2017;165:79–85. doi: 10.1016/j.jsbmb.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27:468–484. doi: 10.1210/er.2006-0005. [DOI] [PubMed] [Google Scholar]
- 20.Massanyi EZ, Gearhart JP, Kolp LA, Migeon CJ. Novel mutation among two sisters with 17β hydroxysteroid dehydrogenase type 3 deficiency. Urology. 2013;81:1069–1071. doi: 10.1016/j.urology.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Hassan HA, Mazen I, Gad YZ, Ali OS, Mekkawy M, Essawi ML. A nove lnonsense mutation in exon 1 of HSD17B3 gene in an Egyptian 46, XY adult female presenting with primary amenorrhea. Sex Dev. 2013;7:277–281. doi: 10.1159/000351822. [DOI] [PubMed] [Google Scholar]
- 22.Ben Rhouma B, Belguith N, Mnif MF, Kamoun T, Charfi N, Kamoun M, Abdelhedi F, Hachicha M, Kamoun H, Abid M, Fakhfakh F. A novel nonsense mutation in HSD17B3 gene in a Tunisian patient with sexual ambiguity. J Sex Med. 2013;10:2586–2589. doi: 10.1111/j.1743-6109.2012.02763.x. [DOI] [PubMed] [Google Scholar]
- 23.Neocleous V, Sismani C, Shammas C, Efstathiou E, Alexandrou A, Ioannides M, Argyrou M, Patsalis PC, Phylactou LA, Skordis N. Duplication of exons 3-10 of the HSD17B3 gene: a noveltype of genetic defect underlying 17β-HSD-3 deficiency. Gene. 2012;499:250–255. doi: 10.1016/j.gene.2012.03.031. [DOI] [PubMed] [Google Scholar]
- 24.Alikasifoglu A, Hiort O, Gonc N, Demirbilek H, Isik E, Kandemir N. 17beta-hydroxysteroid dehydrogenase type 3 deficiency as a result of a homozygous 7 base pair deletion in 17betaHSD3 gene. J Pediatr Endocrinol Metab. 2012;25:561–563. doi: 10.1515/jpem-2012-0009. [DOI] [PubMed] [Google Scholar]
- 25.Özen S, Onay H, Atik T, Solmaz AE, Özkınay F, Gökşen D, Darcan Ş. Rapid molecular genetic diagnosis with next-generation sequencing in 46, XY disorders of sex development cases: efficiency and cost assessment. Horm Res Pediatr. 2017;87:81–87. doi: 10.1159/000452995. [DOI] [PubMed] [Google Scholar]
- 26.Farkas A, Rosler A. Ten years experience with masculinizing genitoplasty in male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase deficiency. Eur J Pediatr. 1993;152(Suppl 2):88–90. doi: 10.1007/BF02125448. [DOI] [PubMed] [Google Scholar]
- 27.Hughes IA, Houk C, Ahmed SF, Lee PA LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554–563. doi: 10.1136/adc.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen-Kettenis PT. Gender Change in 46,XY Persons with 5α-Reductase-2 Deficiency and 17β-Hydroxysteroid Dehydrogenase-3 Deficiency. Arch Sex Behav. 2005;34:399–410. doi: 10.1007/s10508-005-4339-4. [DOI] [PubMed] [Google Scholar]
- 29.Mendonca BB, Inacio M, Arnhold IJ, Costa EM, Bloise W, Martin RM, Denes FT, Silva FA, Andersson S, Lindqvist A, Wilson JD. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase 3 deficiency.Diagnosis, psychologic evaluation, and management. Medicine (Baltimore) 2000;79:299–309. doi: 10.1097/00005792-200009000-00003. [DOI] [PubMed] [Google Scholar]