

Abstract
Pseudohypoaldosteronism type 1 (PHA1) is an autosomal-recessive disorder characterized by defective regulation of body sodium (Na) levels. The abnormality results from mutations in the genes encoding subunits of the epithelial Na channel. Patients with PHA1 present in infancy as being in adrenal crisis. A 41-day-old female who presented with recurrent adrenal crisis did not adequately respond to hydrocortisone and required mineralocorticoid therapy. The patient’s demographic data and clinical features were recorded. Blood samples were collected and tested for endocrine and metabolic characteristics and for use in genetic studies. Bidirectional Sanger sequencing of SCNN1A was conducted. The entire coding region of 12 exons and 20 bp of flanking intron were sequenced. Genetic analyses revealed a new mutation - c.729_730delAG (p.Val245Glyfs*65) - in SCNN1A exon four. Adrenal crisis during the neonatal period highlights the importance of early screening for PHA1. Genetic testing could help to anticipate the prognosis, severity, onset of the disease, and the mode of inheritance, especially given its extensive phenotype.
Keywords: Pseudohypoaldosteronism, hyperkalemia, hyponatremia, adrenal crisis, congenital adrenal hyperplasia
What is already known on this topic?
Autosomal-recessive pseudohypoaldosteronism type 1 (PHA1) is a rare genetic disorder caused by different variations in the epithelial sodium channel (ENaC) subunit genes. Most of these variations appear in SCNN1A, mainly in exon eight, which encodes for the alpha subunit of the ENaC. Variations are nonsense, single-base deletions or insertions, or splice site variations, leading to mRNA and proteins of abnormal length. In addition, a few new missense variations have been reported.
What this study adds?
We report a novel mutation [c.729_730delAG (p.Val245Glyfs*65)] in exon 4 of the SCNN1A gene in a case of autosomal recessive PHA1. A patient with PHA1 requires early recognition, proper treatment, and close follow-up. Parents are advised to seek genetic counseling and plan future pregnancies.
Introduction
Pseudohypoaldosteronism type 1 (PHA1) is a rare disorder of mineralocorticoid resistance that is characterized by defective regulation of body sodium (Na) levels because of the inability of aldosterone to exert its effect on target tissues, aldosterone resistance. PHA1 is a life-threatening disease that presents during the neonatal period with dehydration, severe salt wasting, and failure to thrive, accompanied by hyponatremia, hyperkalemia, metabolic acidosis, and increased plasma renin, with an elevated aldosterone level that is consistent with aldosterone resistance despite normal renal and adrenal functions (1,2). PHA1 can be divided into renal (PHA1A, OMIM #600983) and systemic (PHA1B, OMIM #264350) types based on its physiologic and genetic characteristics. Renal type PHA1A is an autosomal-dominant disorder with heterogeneous inactivating mutations in the NR3C2 gene that codes for the aldosterone receptor (3,4). The renal autosomal-dominant type is characterized by Na loss that is restricted to the kidneys. It is usually less severe with gradual clinical improvement during the first several years of life, thus allowing Na supplementation to be terminated at some point. In contrast, the systemic PHA1 type has an autosomal-recessive inheritance pattern caused by inactivating mutations in any subunits of the epithelial Na channel (ENaC) encoded by SCNN1A, -1B, -1G (5). The systemic autosomal-recessive type typically presents shortly after birth. It is characterized by Na loss from the kidneys and other mineralocorticoid target tissues, such as the colon, lungs, salivary glands, and sweat glands, which increases the frequency of lower respiratory tract infections. The disorder is more common and severe in infancy, persists into adulthood, and requires lifelong therapy using Na supplementation (6,7).
There are no reports of the frequency of PHA1 in Arab populations or the Middle East. However, Al-Shaikh reported two Omani children who presented with PHA1 during their first week of life, both of whom came from a consanguineous family. The first child (a male) had a severe clinical course with skin manifestations, recurrent episodes of severe chest infections, and electrolyte imbalances. Feeding difficulty and treating the electrolyte imbalances were challenges for the treating physician. A genetic analysis was not conducted in this case. The second case (a female) had a milder clinical course with fewer episodes of respiratory infections. Her genetic analysis revealed a new mutation in the SCNN1A ENaC subunit (8).
Case Report
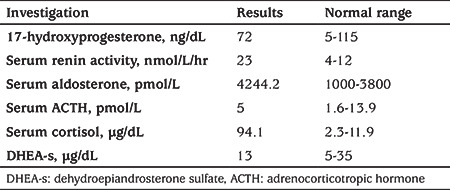
A 41-day-old female was referred to our hospital as a case of possible congenital adrenal hyperplasia. She was a product of a consanguineous marriage and an uneventful pregnancy. The patient was born at a gestational age of 37 weeks by normal spontaneous vaginal delivery with a birth weight of 3 kg without complications. At the age of six days, she began to have difficulty feeding, lethargy, and frequent vomiting. She was taken to the emergency room with severe dehydration and acidosis (arterial pH=7.20; serum bicarbonate=11 mmol/L), high potassium (K) of 10 mmol/L, and low Na of 119 mmol/L. Cardiopulmonary resuscitation was initiated and the patient was intubated. The treating team considered the possibility of an adrenal crisis and conducted initial hormonal studies (Table 1). The patient was treated with a stress dose of hydrocortisone, along with a fluid bolus and Na bicarbonate (NaHCO3). Hyperkalemia was managed with calcium gluconate, insulin, and glucose. The patient was admitted to the neonatal intensive care unit and tested for sepsis and metabolic issues, both of which were negative. She was later discharged from the hospital and treated with hydrocortisone and fludrocortisone with no Na chloride (NaCl).
Table 1. Results of initial hormonal studies at presentation.
This patient had an elder sibling and a cousin who died with similar presentations. At the age of 20 days, the newborn was readmitted to the hospital with episodes of vomiting, poor feeding, and lethargy and was found to have high K and low Na levels. The conditions were managed using a stress dose of hydrocortisone, a fluid bolus, and NaHCO3. Hyperkalemia was managed using calcium gluconate, insulin, glucose, and emergency peritoneal dialysis to lower her K levels. After the patient was stabilized, she was referred to our hospital for further investigation and management. On examination, the patient was conscious, well hydrated and was not dysmorphic. She had normal female genitalia and showed no systemic abnormalities.
Our initial lab results revealed Na: 138 mmol/L, K: 5 mmol/L, and bicarbonate (HCO–3) 23 mmol/L. The urinalysis and urine cultures were normal. A fluorescence in situ hybridization study was sent from the primary hospital, which was negative for the sex-determining region gene, and chromosomal analysis revealed an XX female karyotype. The results of abdominal and pelvic ultrasound examinations were normal. The results of hormonal estimations are provided in Table 1.
Initially, the patient’s symptoms were managed with hydrocortisone and fludrocortisone treatment for possible congenital adrenal hyperplasia. However, because the initial lab results for hormone levels (Table 1) were not consistent with congenital adrenal hyperplasia, and the patient had presented with recurrent adrenal crisis since the neonatal period, she did not adequately respond to hydrocortisone and required mineralocorticoid therapy. In addition, follow-up examinations revealed papular skin rashes and recurrent chest findings, which suggested a diagnosis of pseudohypoaldosteronism. A sweat-chloride test showed elevated levels (128 mmol/L; normal range, 0-39 mmol/L for patients >4 months).
The present study was approved by the research and ethics committee of King Fahad Medical City, Riyadh, Kingdom of Saudi Arabia (IRB log number: 18-609).
The patient’s demographic data and clinical symptoms were recorded. Blood samples were collected and tested for endocrine and metabolic characteristics and for use in genetic studies. Molecular genetic testing was conducted initially for NR3C2 sequencing for autosomal-dominant pseudohypoaldosteronism. Bidirectional Sanger sequencing of SCNN1A was then conducted. The entire coding region of 12 exons and 20 bp of flanking intron were sequenced. Informed consent for all tests was obtained from the patient’s parents.
The results of the hormone tests on the patient are presented in Table 1. Molecular genetic testing for NR3C2 sequencing for autosomal-dominant pseudohypoaldosteronism wereperformed and did not show any sequence variant that may have caused the disease. Based on the majority of reported mutations, Bidirectional Sanger sequencing of SCNN1A was then conducted. The results revealed that the patient was homozygous for a novel mutation at c.729_730delAG in exon 4 of SCNN1A (Figure 1),which was predicted to result in a frameshift and premature protein termination (p. Val245Glyfs*65).
Figure 1.
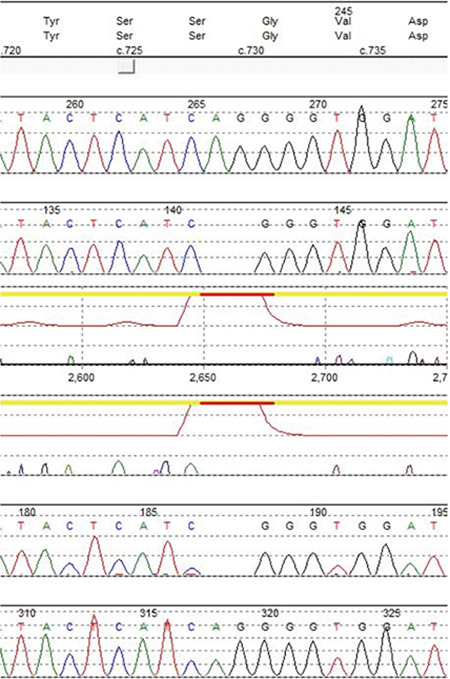
Electropherogram of the novel variant in SCNN1A: c.729_730delAG
Using in silico prediction tools, such as SIFT-indel and Mutation Taster, this variant is predicted to be deleterious (Table 2). Moreover, the amino acid valine in position 245 is highly conserved among different species (Figure 2). This variant is not found in gnomAD (PM2).
Table 2. In silico analysis of the SCNN1A gene mutation founded in our patient and ACMG classification.
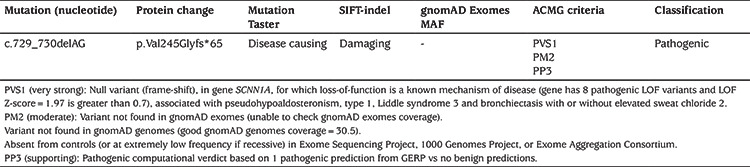
Figure 2.

Conservation of the amino acid (valine) at position 245 of the amino acid sequence among different species
NP_001153047.1: H. sapiens, XP_508948.4: P. troglodytes, XP_001103017.2: M. mulatta, XP_0ç05637310.1: C. lupus, NP_777023.1: B. taurus, NP_035454.2: M. musculus, NP_113736.1: R. norvegicus, NP_990476.2 :G. gallus, NP_001184124.1: X. tropicalis
Targeted sequencing for this variant was conducted in both parents, who were found to be carriers for the sequence variant c.729_730delAG (p.Val245Glyfs*65) in SCNN1A in heterozygous form.
Discussion
“Adrenal crisis” was the initial impression, which included adrenal hypoplasia and congenital adrenal hyperplasia, and was based on the patient’s clinical presentation and a history of poor feeding, lethargy, frequent vomiting, consanguinity, family history of sudden infant death, hyperkalemia, hyponatremia, and metabolic acidosis. The patient was accordingly treated with hydrocortisone. She had normal female genitalia, hyponatremia, and hyperkalemia with normal androgen precursor levels but a high level of plasma aldosterone, renin activity, and cortisol, all of which were more consistent with PHA1 than congenital adrenal hyperplasia.
Other differential diagnoses, based on the results of the initial electrolyte levels, included other causes of hyperkalemia and hyponatremia, including secondary PHA, which is a transient form of aldosterone resistance secondary to a urinary tract infection. A urinary tract malformation was also a possibility, but was unlikely because the results of the urine tests and ultrasound were normal. Renal tubular acidosis type 4 is characterized by low levels of ammonia in the urine and is associated with hyperkalemia, mild hyponatremia, and metabolic acidosis with hyperaldosteronism or aldosterone insensitivity.
A poor response of the patient to corticosteroids indicated aldosterone resistance and either renal or systemic PHA1. Patients with PHA1 usually present in the neonatal period with hyponatremia, hyperkalemia, and metabolic acidosis, and the condition can be diagnosed by significantly elevated plasma aldosterone levels and renin activity (9,10,11).
The clinical manifestations of renal PHA1 may vary in asymptomatic patients and can only be diagnosed using the presentation of hyperaldosteronism and elevated renin levels compared with patients presenting with a salt-wasting crisis. The clinical characteristics and severity of this condition can vary widely. Severe phenotype has been reported due to mutations in both alleles in the NR3C2 gene (12). Overall, PHA1 has a mild clinical course followed by remission over time (13,14,15,16,17). The systemic form of PHA1 leads to salt loss from organs that express ENaC, such as the kidneys, salivary and sweat glands, and the colon. Children typically present shortly after birth with electrolyte disturbances mimicking an adrenal crisis (4). Acute initial presentation, frequency of respiratory symptoms, and a positive sweat-chloride test led to the diagnosis of systemic PHA1 in our patient.
In vitro screening of several mutant genes has led to advances in understanding the physiology of the mineralocorticoid receptors and ENaC. Therefore, the underlying molecular pathology of renal and systemic PHA1 has been associated with mutations in the mineralocorticoid receptor and ENaC subunit genes, SCNN1A, SCNN1B, and SCNN1G, respectively (11). Aldosterone plays a central role in electrolyte homeostasis and maintenance of fluid in the distal nephron. Loss-of-function mutations in two key components of the aldosterone response-the mineralocorticoid receptor and ENaC-lead to PHA1, a rare genetic disease of aldosterone resistance characterized by salt loss, dehydration, failure to thrive, hyperkalemia, and metabolic acidosis (14).
The prickly heat-like skin rash in our patient, which was typically aggravated by salt deprivation, is a characteristic feature of systemic PHA1. As a consequence of ENaC expression, this manifestation is consistent with recent studies that have reported that skin manifestations are common in systemic PHA1 patients, as are other phenotypic features, such as cholelithiasis, polyhydramnios, and hypercalciuria leading to hypocalcemia (18,19,20,21,22,23,24).
A sweat-chloride test is positive in both systemic PHA1 and cystic fibrosis because both disorders lead to salt loss from the sweat glands. The gold standard for a diagnosis of cystic fibrosis is based on a clinical diagnosis plus an abnormal sweat-chloride test and the determination of a mutation in the cystic fibrosis transmembrane conductance regulator gene CFTR. Therefore, based on our patient’s initial clinical presentation and her symptom improvement after salt supplementation and K exchange resins, cystic fibrosis was a less-likely diagnosis and a CFTR mutation analysis was not necessary in this case (25). CFTR encodes for a membrane protein and chloride channel, which play important roles in transepithelial salt absorption and secretion in some epithelial tissues, such as the intestine, airways and sweat glands, and reproductive system. Studies have shown that CFTR modulates the function of ENaC in salt absorption, but the mechanism is still controversial. CFTR mutations lead to an inadequacy in the CFTR-ENaC relationship, which may lead to increased Na+ absorption in the airways, resulting in hypersecretion of mucus, decreased mucociliary clearance, and bacterial colonization (26,27,28).
PHA1 was first reported by Cheek and Perry in 1958 as a sporadic occurrence with a severe salt-wasting syndrome. Since 1958, the literature has mainly consisted of case reports or case series but the number of patients with PHA1 has increased over the past decade.. Based on HGMD, more than 71 NR3C2, 42 SCNN1A, 47 SCNN1B, and 20 SCNN1G mutations have been reported. Most of the reported mutations were single-base deletions/insertions, or splice-site mutations with severe courses while missense mutations usually follow a mild course. Several new mutations have been reported with phenotypic variations (23,25). In 2017, Nur et al (24) reported one case and reviewed 27 others from clinical presentation to follow-up and outcome. In 2018, Guran (26) summarized the diagnosis and current management of PHA1.
To the best of our knowledge, this is the first recognized case of PHA1 with a new mutation that results in a frameshift and premature protein termination (p. Val245Glyfs*65), which leads to a sequence variant designated as c729_730delAG in SCNN1A exon four. This finding confirms the hypothesis that an autosomal-recessive or a sporadic PHA1 systemic type is a genetically heterogeneous disease involving other unidentified genes (15). However, null variant (frame-shift), in SCNN1A is a known mechanism of disease, single nucleotide deletion in the same coding region has been classified as pathogenic SCNN1A:c.729delA which leads to different frameshift (p.Val304TrpfsTer4). This reported mutation is associated with lung symptoms (recurrent lower respiratory tract infections) without development of chronic lung disease, which is a similar finding to our case (29).
Renal type 1 PHA1, an autosomal-dominant disease resulting from heterogeneous mutations in the NR3C2 gene that encodes for the mineralocorticoid receptor, results in a condition of aldosterone not being able to bind to its receptor. Severe phenotype has been reported due to mutations in both alleles in the NR3C2 gene. Approximately 50 distinct mutations have been reported, but there have been no reports on genotype/phenotype correlation (11). In contrast, systemic type 1 PHA1 is an autosomal-recessive disorder that typically presents during the neonatal period with severe symptoms, such as hyponatremia, hyperkalemia, and metabolic acidosis. This condition can be diagnosed by significantly elevated plasma aldosterone levels and renin activity with the symptoms persisting into adulthood (15,17,18).
Treatment of PHA1 should be considered a challenging emergency, and life-saving measures are needed to rectify dehydration, replace Na loss, and correct hyperkalemia and acidosis during the acute phase (4).
Renal PHA1 can be managed with salt supplementation, which is often decreased over the lifetime of the patient. Although systemic PHA1 requires aggressive management, including salt supplementation, intensive fluid administration, K exchange resins, and dietary manipulation to reduce hyperkalemia, the use of indomethacin may be beneficial in some patients and when treating systemic complications of the disease. The course of the disease is often interrupted by recurrent episodes of salt-loss crisis, with hyperkalemia associated with nausea, vomiting, and difficulty feeding, which might require gastrostomy/jejunostomy feeding, as well as a chronic pulmonary syndrome, with or without associated chest infections (18,19).
Na supplementation may be given in the form of NaHCO3, NaCl (1 g contains ~17 mEq Na), or Na citrate. Up to 110 mEq/kg/d can be given. Generally, it is difficult for patients to tolerate a high dose of salt orally so the dose intake can be divided and mixed with baby formula and food. It may need to be administered by nasogastric tube/jejunostomy in poor tolerance situations (18,26).
Severe hyperkalemia and acidosis can be managed by the intravenous slow infusion of NaHCO3 and then oral replacement when the condition stabilizes. A combination of glucose and insulin infused slowly at the usual ratio of 1 unit of insulin to every 5 g dextrose is an effective measure by which to reduce serum K. Salbutamol infusion or continuous nebulization has been added as adjunctive therapy. In severe persistent hyperkalemia, peritoneal dialysis is used, after which calcium chloride is infused to protect the heart from arrhythmias until the serum K levels are normalized.
K exchange resins, which act as K-binding agents, are widely used in hyperkalemia management. The most commonly used K-binding agents are Na polystyrene sulfonate and calcium polystyrene sulfonate. Usually the Na-containing resin is preferred to the calcium-containing resin because it simultaneously corrects both hyponatremia and hyperkalemia. It is used orally or rectally at approximately 1-2 g/kg/dose every six hours when high doses administered orally cannot be tolerated. In these cases, rectal administration of the drug is a good option (20,26).
Genomic testing for a carrier state in the asymptomatic siblings and the parents is highly recommended to detect homozygosity or compound heterozygosity (8). One limitation to the present study was that we did not measure testosterone levels during the patient’s hospitalization.
Conclusion
Adrenal crisis during the neonatal period highlights the importance of early screening for PHA1 to facilitate early recognition, proper treatment, close follow-up, and genetic counseling for the family. Parents should be advised to seek genetic counseling and testing when planning future pregnancies.
Footnotes
Ethics
Informed Consent: Informed consent for all tests was obtained from the patient’s parents.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices - Concept - Design - Data Collection or Processing - Analysis or Interpretation - Literature Search - Writing: All authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Zennaro MC, Hubert EL, Fernandes-Rosa FL. Aldosterone resistance: structural and functional considerations and new perspectives. Mol Cell Endocrinol. 2012;350:206–215. doi: 10.1016/j.mce.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 2.Genetics Home Reference. US National Library of Medicine. Pseudohypoaldosteronism type 1. Last Accessed Date: 12 June, 2019. Available from: [Internet] https://ghr.nlm.nih.gov/condition/pseudohypoaldosteronism-type-1.
- 3.Geller DS, Zhang J, Zennaro MC, Vallo-Boado A, Rodriguez-Soriano J, Furu L, Haws R, Metzger D, Botelho B, Karaviti L, Haqq AM, Corey H, Janssens S, Corvol P, Lifton RP. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol. 2006;17:1429–1436. doi: 10.1681/ASN.2005111188. [DOI] [PubMed] [Google Scholar]
- 4.Attia NA, Marzouk YI. Pseudohypoaldosteronism in a neonate presenting as life-threatening hyperkalemia. Case Rep Endocrinol. 2016;2016:6384697. doi: 10.1155/2016/6384697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13:248–250. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 6.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999;104:19–23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riepe FG, Finkeldei J, de Sanctis L, Einaudi S, Testa A, Karges B, Peter M, Viemann M, Grötzinger J, Sippell WG, Fejes-Toth G, Krone N. Elucidating the Underlying Molecular Pathogenesis of NR3C2 Mutants Causing Autosomal Dominant Pseudohypoaldosteronism Type 1. J Clin Endocrinol Metab. 2006;91:4552–4561. doi: 10.1210/jc.2006-1161. [DOI] [PubMed] [Google Scholar]
- 8.Al-Shaikh HAJ. Pseudohypoaldosteronism in Two Omani Siblings with a novel mutation in the SCNN1A gene. J Endocrinol Diab. 2017;4:1–4. [Google Scholar]
- 9.Sartorato P, Lapeyraque AL, Armanini D, Kuhnle U, Khaldi Y, Salomon R, Abadie V, Di Battista E, Naselli A, Racine A, Bosio M, Caprio M, Poulet-Young V, Chabrolle JP, Niaudet P, De Gennes C, Lecornec MH, Poisson E, Fusco AM, Loli P, Lombès M, Zennaro MC. Different inactivating mutations of the mineralocorticoid receptor in fourteen families affected by type I pseudohypoaldosteronism. J Clin Endocrinol Metab. 2003;88:2508–2517. doi: 10.1210/jc.2002-021932. [DOI] [PubMed] [Google Scholar]
- 10.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Välimäki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riepe FG. Clinical and Molecular Features of Type 1 Pseudohypoaldosteronism. Horm Res. 2009;72:1–9. doi: 10.1159/000224334. [DOI] [PubMed] [Google Scholar]
- 12.Hubert EL, Teissier R, Fernandes-Rosa FL, Fay M, Rafestin-Oblin ME, Jeunemaitre X, Metz C, Escoubet B, Zennaro MC. Mineralocorticoid Receptor Mutations and a Severe Recessive Pseudohypoaldosteronism Type 1. J Am Soc Nephrol. 2011;22:1997–2003. doi: 10.1681/ASN.2011030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Autosomal Recessive Pseudohypoaldosteronism Type 1; Genetic and Rare Diseases Information Center (GARD) of National Center for Advancing Translational Sciences (NCATS), USA. Available at: [Internet] https://www.dovemed.com/diseases-conditions/autosomal-recessive-pseudohypoaldosteronism-type-1/
- 14.Pathare G, Hoenderop JG, Bindels RJ, San-Cristobal P. A molecular update on pseudohypoaldosteronism type II. Am J Physiol Renal Physiol. 2013;305:1513–1520. doi: 10.1152/ajprenal.00440.2013. [DOI] [PubMed] [Google Scholar]
- 15.Viemann M, Peter M, López-Siguero JP, Simic-Schleicher G, Sippell WG. Evidence for genetic heterogeneity of pseudohypoaldosteronism type 1: identification of a novel mutation in the human mineralocorticoid receptor in one sporadic case and no mutations in two autosomal dominant kindreds. J Clin Endocrinol Metab. 2001;86:2056–2059. doi: 10.1210/jcem.86.5.7449. [DOI] [PubMed] [Google Scholar]
- 16.Silva N, Costa M, Silva A, Sá C, Martins S, Antunes A, Marques O, Castedo S, Pereira A. A case of systemic pseudohypoaldosteronism with a novel mutation in the SCNN1A gene. Endocrinol Nutr. 2013;60:33–36. doi: 10.1016/j.endonu.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Manipriya R, Umamaheswari B, Prakash A, Binu N. Rare cause of hyperkalemia in the newborn period: report of two cases of pseudohypoaldosteronism Type 1. Indian J Nephrol. 2018;28:69–72. doi: 10.4103/ijn.IJN_258_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, Frazer S, Savill G, Wright NP, Makaya T, Mushtaq T. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010. doi: 10.1530/EDM-13-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and in disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 20.Porter J, Kershaw M, Kirk J, Trevelyan N, Shaw NJ. The use of sodium resonium in pseudohypoaldosteronism. Arch Dis Child. 2003;88:1138–1139. doi: 10.1136/adc.88.12.1138-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanukoglu A, Hanukoglu I. In systemic pseudohypoaldosteronism type 1 skin manifestations are not rare and the disease is not transient. Clin Endocrinol (Oxf) 2018;89:240–241. doi: 10.1111/cen.13730. [DOI] [PubMed] [Google Scholar]
- 22.Akkurt I, Kuhnle U, Ringenberg C. Pseudohypo-aldosteronism and cholelithiasis: coincidence or pathogenetic correlation? Eur J Pediatr. 1997;156:363–366. doi: 10.1007/s004310050615. [DOI] [PubMed] [Google Scholar]
- 23.Kuhnle U, Nielsen MD, Tietze HU, Schroeter CH, Schlamp D, Bosson D, Knorr D, Armanini D. Pseudohypoaldosteronism in eight families: Different forms of inheritance are evidence for various genetic defects. J Clin Endocrinol Metab. 1990;70:638–641. doi: 10.1210/jcem-70-3-638. [DOI] [PubMed] [Google Scholar]
- 24.Nur N, Lang C, Hodax JK, Quintos JB. Systemic Pseudohypoaldosteronism Type I: a case report and review of the literature. Case Rep Pediatr. 2017;2017:7939854. doi: 10.1155/2017/7939854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahluwalia GK, Dasouki M, Lennon A. Phenotypic variation of autosomal recessive pseudohypoaldosteronism type I: a case in point. Clin Case Rep. 2014;2:326–330. doi: 10.1002/ccr3.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guran T, Sahin I, Cayir A. Diagnosis and management of Pseudohypoaldosteronism type 1 in children. 2018 Internal Medicine Review. 2018;4:1–27. [Google Scholar]
- 27.Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5:123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy MM, Quinton PM. Functional interaction of CFTR and ENaC in sweat glands. Pflugers Arch. 2003;445:499–503. doi: 10.1007/s00424-002-0959-x. [DOI] [PubMed] [Google Scholar]
- 29.Schaedel C, Marthinsen L, Kristoffersson AC, Kornfält R, Nilsson KO, Orlenius B, Holmberg L. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the alpha-subunit of the epithelial sodium channel. J Pediatr. 1999;135:739–745. doi: 10.1016/s0022-3476(99)70094-6. [DOI] [PubMed] [Google Scholar]