

Abstract
Gene editing mediated by CRISPR/Cas9 systems is due to become a beneficial therapeutic option for treating genetic diseases and some cancers. However, there are challenges in delivering CRISPR components which necessitate sophisticated delivery systems for safe and effective genome editing. Lipid nanoparticles (LNPs) have become an attractive nonviral delivery platform for CRISPR-mediated genome editing due to their low immunogenicity and application flexibility. In this review, we provide a background of CRISPR-mediated gene therapy, as well as LNPs and their applicable characteristics for delivering CRISPR components. We then highlight the challenges of CRISPR delivery, which have driven the significant development of new, safe, and optimized LNP formulations in the past decade. Finally, we discuss considerations for using LNPs to deliver CRISPR and future perspectives on clinical translation of LNP-CRISPR gene editing.
Keywords: lipid nanoparticles, CRISPR/Cas9, genome editing, nanomedicine, gene therapy
Introduction
Gene therapy aims to deliver RNA- or DNA-based drugs to target the genetic cause of disease, rather than to alleviate downstream pathways or resulting symptoms. Gene therapies generally involve silencing pathological genes, expressing therapeutic proteins, or corrective editing of disease-causing genes.1−3 Silencing pathological genes is typically achieved by siRNAs, microRNAs, or oligonucleotides such as antisense oligonucleotides (ASOs).4 Therapeutic proteins can be expressed within cells by introducing therapeutic DNA or RNA molecules.4 Corrective gene editing is of significant interest, as there are over 6500 known monogenic diseases, less than 5% of which have viable treatments.5 While correcting the underlying genetic causes of such diseases is an exciting prospect, gene therapies are often limited by a lack of safe and effective delivery methods.
Corrective editing of pathologically mutated genes can be achieved using zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regularly interspaced short palindromic repeats (CRISPR). ZFNs are fusion proteins with multiple zinc-finger based domains attached to a bacterial enzyme that recognize up to 18 base pairs. Although the assembly of these domains produces ZFNs with high affinity to a specific sequence, their production is time-consuming and often expensive. TALENs rely on the same bacterial protein as ZFNs but pair with a TALE repeat that specifically binds to individual base pairs. The resulting TALEN system is easier to customize than ZFNs but still requires the design and synthesis of a new protein for each target site. In contrast, CRISPR-based systems use a designable guide RNA that directs a bacterial-derived gene-editing protein (such as a nuclease, a base editor, a transposase/recombinase, or a prime editor) to the desired genomic target.4,6 Compared to proteins, the relative ease and versatility of customizing guide RNAs allow higher throughput targeting of multiple genomic locations and expand the potential therapeutic applications of CRISPR-based systems.
CRISPR/Cas9-mediated genome editing has become a popular approach since it is a robust and versatile tool that targets DNA and can provide long-lasting therapeutic benefits after a single treatment. CRISPR systems typically include an endonuclease protein, such as Cas9, and a dual-guide RNA consisting of a trans-activating CRISPR RNA (tracrRNA) and a crRNA that guides the endonuclease to its target using sequence complementarity. While dual-guide RNAs are still extensively used for in vitro and ex vivo experiments, engineered single-guide RNAs (sgRNAs, or gRNAs for simplicity) are used for most in vivo studies. gRNAs can be designed to target a specific genomic sequence of interest to knockout a gene by inducing insertion or deletion mutations (indels) via nonhomologous end joining (NHEJ), or to knock-in a donor sequence via homology-directed repair (HDR).3 Recently, the CRISPR/Cas9 system has been engineered to expand its range of functions. For instance, introducing point mutations into either of the Cas9 nuclease domains (D10A or H840A) creates a Cas9 nickase (nCas) that induces single-stranded nicks instead of double-stranded breaks. nCas9 can also be complexed with other functional domains to generate base editors or prime editors, which introduce targeted and precise single-base or small corrections, respectively.7−9 Catalytically inactive Cas9 protein can also be tethered to transcriptional activators or suppressors to regulate gene transcription.7 Furthermore, CRISPR/Cas9 systems can be designed to target mRNAs of a pathologically mutated gene or the RNA genomes of viruses to block RNA replication and protein synthesis. Using dCas13, this approach has been used to block replication of the SARS-CoV-2 virus by targeting its viral RNA genome and its subsequence mRNA products.10 CRISPR-mediated genome editing can also be used in modifying gene expression by altering DNA methylation and chromatin configuration and has many other uses and applications.6,7
The therapeutic impact of CRISPR and other gene therapies cannot be achieved without safe and effective delivery methods. Despite first entering clinical trials in the early 1990s, only 20 nucleic acid based therapeutics have been approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA),11 as the safety of gene therapies is a consistent barrier to their clinical translation. In 1999, a 17 year old patient suffered a fatal immune response following the administration of an adenovirus vector containing therapeutic DNA.12 This tragedy emphasized the need for safer delivery methods for gene therapies that do not involve the use of viruses. In this review, we discuss recent advances in LNP-based delivery that have established the technology as a safe nonviral delivery platform for CRISPR gene-editing machinery.
Delivery Vectors for Nucleic Acids
Viral vectors have been extensively used by researchers as tools for nucleic acid delivery. These systems transfer genetic material using engineered viruses such as retroviruses, adenoviruses, and adeno-associated viruses.13 However, the suitability of viral vectors for therapeutic applications is limited by their potential to induce inflammatory and adverse immunogenic responses. The long-lasting expression of nucleic acid constructs delivered by viral transfection also makes gene-editing applications using this delivery system challenging, since sustained expression may increase the likelihood of adverse effects caused by recombination with the host genome or pathological insertional mutagenesis.13 Consequently, nonviral vectors are becoming a popular alternative. They are less immunogenic, easier to assemble, form stable complexes with nucleic acids, provide protection against serum nucleases, and can be more easily scaled-up for industrial commercialization.2,13 Nonviral vectors also have lower restrictions on payload size and packaging which is integral for the delivery of large nucleic acids.2,13
Nanoparticles can be structurally divided into two families: nanospheres and nanocapsules. Nanospheres have a homogeneous matrix throughout the particles that hold active compounds, whereas nanocapsules have a core–shell structure where the inner core contains the payload.14 Lipid-based nanoparticles contain lipid moieties in their structures and have tremendous biomedical potential in drug delivery and gene therapy.15 Lipid-based nanoparticles are typically nanospheres that encapsulate therapeutic compounds such as nucleic acids or proteins.16 Compared to viral and other nonviral nanoparticle systems, lipid-based nanoparticles offer advantages including ease of formulation, spontaneous self-assembly, high potency, high biocompatibility, larger payload capacity, and versatility in design for specific applications.16 LNPs are currently one of the most popular and sophisticated nonviral delivery platforms. They have enabled clinical translation of the siRNA drug ONPATTRO (patisiran)17 and the mRNA-based COVID-19 vaccines developed by Pfizer-BioNTech and Moderna.18
In 1987, Felgner et al. published the earliest report of a lipid-mediated DNA transfection procedure where cationic lipids were used to encapsulate DNA. The cationic lipids complexed with DNA through electrostatic interactions with the negatively charged phosphate backbone, and enabled cellular uptake by interacting with negative charges on cell membranes.19 Extensive research has improved LNPs as a delivery vector since this application of cationic lipids for cellular transfection. LNP formulations have been optimized to improve particle stability, increase circulation time, reduce toxicity, and lower immunogenicity.20−22 Tissue- and cell-specific targeting has also been achieved by modifying LNP compositions. These features mean LNPs are a robust method for drug delivery, as evidenced significantly by the 2018 approval of the LNP-siRNA drug ONPATTRO.
ONPATTRO contains siRNA targeting TTR mRNA to reduce hepatic transthyretin protein translation for treatment of hereditary transthyretin (ATTR) amyloidosis.23 ATTR amyloidosis is caused by the accumulation of misfolded TTR proteins in tissues.23 After being tested in 225 patients, ONPATTRO demonstrated promising phase 3 clinical trial results (NCT01960348) and received an FDA approval as the first nonviral systemic gene therapy.23−25 The drug includes a novel ionizable cationic lipid, DLin-MC3-DMA (MC3), which drives encapsulation, cellular uptake, and endosomal release of the siRNA.26 The ONPATTRO formulation is often used by other researchers as a reference during evaluation of novel ionizable cationic lipids and LNP formulations or for delivery of other cargoes.27 The successful clinical translation of LNPs for siRNA delivery has recently expanded to include mRNA with the approval of LNP-mRNA COVID-19 vaccines developed by Pfizer-BioNTech and Moderna. Current research is heavily focused on the development and optimization of LNPs for other applications including CRISPR/Cas9-based gene editing.
LNP Composition for Nucleic Acid Delivery
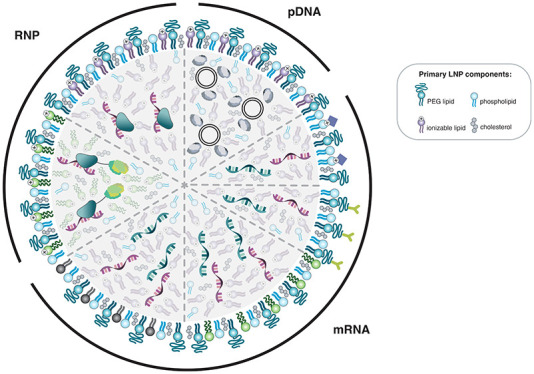
LNPs are a common and popular tool for therapeutic nucleic acid delivery. They typically include four primary lipid components: ionizable cationic lipids, polyethylene glycol (PEG) lipids, zwitterionic phospholipids, and cholesterol16,28,29 (Figure 1). LNPs primarily enter cells via the endocytosis pathway (Figure 2).3,30 These primary components confer unique functional elements to LNP systems that work synergistically to enable payload encapsulation, transport, and delivery.
Figure 1.

Visual representation of examples of different lipid nanoparticle (LNP) formulations for CRISPR/Cas9 delivery in plasmid, mRNA, and ribonucleoprotein (RNP) forms. The main components of LNP formulations are shown with specific examples of modifications used to enhance the delivery of different types of cargo. (A) Protamine is used to condense plasmid DNA (pDNA) to increase the encapsulation efficiency of pDNA within LNPs. (B) Ionizable cationic lipids conjugated with a ligand (top) and PEG lipids conjugated with antibodies (bottom) to mediate delivery to targeted cells expressing the corresponding receptors. (C) Biodegradable or bioreducible ionizable lipids are used to improve encapsulation and intracellular release with mRNA delivery. (D) Dendrimer ionizable lipids are used to enhance mRNA delivery. (E) Cationic bioreducible lipids are used in conjunction with a polyanionic macromolecule to increase encapsulation efficiency for RNP delivery. (F) Ionizable cationic lipids used in the absence of solvent enable efficient encapsulation and delivery of the active RNP complex.
Figure 2.

Intracellular delivery of CRISPR/Cas9 components in plasmid, mRNA, and ribonucleoprotein (RNP) forms using lipid nanoparticles (LNPs). LNPs are taken up by cells via endocytosis (A–C). As the pH of the endosomes decreases, ionizable cationic lipids gain a positive charge and mediate cargo release. (A) Plasmid DNA (pDNA) enters the nucleus where it is transcribed by RNA polymerases. The transcribed mRNA returns to the cytoplasm where it is translated and complexes with gRNAs to form RNPs. The resulting RNPs, which contain a nuclear localization signal enter the nucleus to induce gene editing. (B) Cas9 mRNA must be translated in the cytoplasm and ultimately complex with gRNAs to form RNPs before entering the nucleus to induce gene editing. (C) LNP-mediated delivery of preformed RNP complexes can directly enter the nucleus to induce gene editing without any in cell production of Cas9.
Ionizable Cationic Lipids
Ionizable cationic lipids have significantly improved nucleic acid encapsulation and delivery by LNPs. Initially, permanently cationic lipids were used in LNP formulations. Their positive charge interacts with negatively charged nucleic acids to drive nucleic acid loading into particles. The standard commercial reagent for cellular transfection Lipofectamine is made from a permanently cationic lipid which results in a high delivery efficiency in vitro; however, its utility in vivo is limited due to rapid plasma clearance, immune activation, and adverse toxicity.31 These effects are caused by the permanent charge of the cationic lipid, which interacts with negatively charged serum proteins and leads to rapid clearance of the nanoparticles from circulation. Permanently cationic lipids can also interact with endogenous anionic lipids and disrupt cell membranes resulting in toxic effects.32 The strong charge interaction between permanently cationic lipids and nucleic acids further leads to poor nucleic acids release following cellular uptake, which decreases their bioavailability.
Ionizable cationic lipids were developed to overcome the limitations associated with permanently cationic lipids. The incorporation of ionizable cationic lipids in LNP formulations enables efficient payload encapsulation, improves in vivo circulation time and cellular uptake, and permits endosomal cargo release.16,29,33 LNP formulation typically occurs at acidic pH, where ionizable cationic lipids are protonated and capable of binding negatively charged cargoes. A common method of LNP preparation involves microfluidic mixing of a lipid-containing organic solvent and an aqueous solvent containing nucleic acids. When the two phases are combined, the nucleic acids interact with positively charged ionizable lipid head groups and form the core LNP structure. Hydrophobic interactions between the lipid tails and hydrophilic interactions between lipid head groups and aqueous buffer drive formation of the outer LNP surface.
Formulation of LNPs containing proteins is more complex: Denaturation and resultant loss of activity can occur in acidic conditions or in the presence of organic solvents, and encapsulation efficiency is dependent on protein charges which are highly variable and dictated by amino acid sequence and secondary structure. While Cas9 protein is cationic, with a net charge of +22, complexation with gRNA produces a ribonucleoprotein (RNP) with an overall negative net charge.34 Ionizable cationic lipids are therefore crucial for encapsulation of all forms of CRISPR/Cas9 components (plasmid DNA, mRNA, or RNP).
In addition to driving particle encapsulation, ionizable cationic lipids can increase circulation half-life and improve the efficiency of endocytosis by target cells. At physiological pH (approximately 7.4), ionizable lipids are uncharged, which prevents sequestration by immune cells.33 Upon endocytosis, LNPs are taken up into endosomes35 that undergo progressive acidification and contain lysosomal enzymes that can degrade payloads.33 Ionizable lipids provide protection from degradation by enabling rapid cargo release. As endosomal acidification also causes ionizable lipid protonation, binding of protonated head groups with negatively charged lipids in the endosomal membrane causes membrane rupture28,33 and cargo release into the cytosol (Figure 2). The inclusion of ionizable lipids in LNP formulations for delivery of CRISPR/Cas9 is essential as they ensure that a higher proportion of CRISPR/Cas9 components escape endosomal degradation and are released into the cytosol.
Modifications to ionizable cationic lipid chemistry can significantly influence the potency and safety of LNP.36−39 Examples of modified ionizable lipids include those that are biodegradable or bioreducible, as well as dendrimer ionizable lipids.
Biodegradable or bioreducible ionizable lipids incorporate labile elements (including disulfide linkages or ester bonds) that are rapidly broken down into nontoxic products in intracellular environments.40,41 Efficient degradation of ionizable lipids used in LNP formulations means treatment carries a reduced risk of cytotoxicity, especially in multidosing regimens. Incorporation of biodegradable or bioreducible lipids into LNP systems is particularly useful for delivery of CRISPR/Cas9 components, because highly efficient ionizable-lipid-mediated transfection increases the likelihood of effective gene editing in target tissues and cells, while minimal toxicity means that LNP therapeutics are safe and repeated administration is plausible. Gene editing in the liver has been enabled by delivery of Cas9 mRNA and gRNA using LNPs containing bioreducible lipids, where on-target editing was detected in the absence of off-target effects and overt toxicity.42−44 Degradable ionizable lipids have also successfully delivered mRNA to B lymphocytes and lung tissues,41,45,46 indicating that this lipid class could be useful for Cas9 mRNA delivery to cells that are not easily transfected by conventional LNPs. Bioreducible lipids have also been used for Cas9 plasmid and RNP delivery.47,48
Dendrimers are three-dimensional macromolecules comprised of polymeric branches anchored to a central core.49,50 The branches and core molecules can be uniquely functionalized, so dendrimers can be designed for specific applications.51 Dendrimer lipids with ionizable amine cores have been incorporated into LNP formulations to enable nucleic acid encapsulation and delivery. These formulations have been used both independently from52 and in combination with other LNP excipients and have successfully delivered siRNA, mRNA, and Cas9 RNP to hepatocytes, liver endothelial cells, hepatocellular carcinomas, and extrahepatic tissues.28,53−56 Tissue- and cell-type-specific delivery can be tuned by altering the ratios at which dendrimer ionizable lipids are combined with other LNP components and by modifying the branches conjugated to the ionizable amine.28,53 Bioreducible chemistry can also be incorporated into the design of dendrimer ionizable lipids. These combined elements, which confer both safety and targeted delivery, mean that LNP systems prepared with dendrimer ionizable lipids may be well-suited for CRISPR/Cas9 applications. LNPs incorporating dendrimer ionizable lipids have been used to deliver both Cas9 mRNA and RNP and have achieved potent editing and homology-directed repair both in vitro and in vivo.28,56
PEG Lipids
PEG lipids improve LNP stability, regulate particle size, decrease immunogenicity, and increase circulation time.16,57 PEG lipids are comprised of PEG molecules attached to lipid head groups. When PEGylated lipids are used to prepare LNPs, the outer LNP surface is coated with PEG molecules. This coating preserves particle size by preventing particle aggregation during formulation, storage, and in circulation,11,16,57,58 and it also increases systemic circulation time by preventing opsonization and phagocytosis.57 Opsonization occurs when opsonins bind the LNP surfacem which triggers recognition and subsequent clearance by phagocytes. For example, complements C3, IgG, and the serum opsonin fibrinogen can coat the particles’ surface which are then recognized and sequestered by Kupffer cells.59 PEGylated lipids have a hydrophilic property, and their incorporation into LNP formulations create a barrier of water molecules around the outer surface that sterically hinders and protects against opsonization and particle aggregation. Therefore, PEG lipids are an especially important LNP component for in vivo applications by allowing to maintain the administered dose concentration in circulation. As a result, a higher circulation half-life can contribute to a higher percentage of CRISPR/Cas9 molecules delivered to target cells for efficient on-target genome editing. The concentrations and combinations of PEG lipids in LNP formulations can be altered to modify LNP delivery efficiency.
Zwitterionic Phospholipids
Zwitterionic phospholipids are mostly present in the outer lipid layer of LNPs and are used to improve structural stability and delivery efficiency. Combinations of phospholipids (also called helper lipids) can be added to LNP formulations to modify their biophysical characteristics including particle size and surface charge to promote optimal encapsulation, stability, and endosomal release.60
Cholesterol
Cholesterol is used in LNP formulations to increase particle stability by filling gaps between the phospholipids. Cholesterol also promotes membrane fusion during cellular uptake by augmenting the activity of positively charged lipids and promoting destabilization of the cellular bilayer during fusion.16,60
The ratios at which the four components of LNP formulations are combined also impacts the functionality of the particles. Optimization of the molar ratios of the components in ONPATTRO involved the testing of over three hundred ionizable lipids in thousands of combinations with different excipients. The final formulation contains a molar ratio of 50/10/38.5/1.5 MC3 (ionizable lipid)/DSPC (phospholipid)/cholesterol/PEG-lipid.18 The Pfizer-BioNTech and Moderna COVID-19 LNP-mRNA vaccines underwent a similarly rigorous testing process to identify optimal formulas.18
Inherent Targeting of LNPs toward the Liver
LNPs interactions with blood proteins upon intravenous administration determines where they will ultimately be localized. Serum proteins that adsorb on onto the surface of LNPs act as ligands for their cognate receptors on cell surfaces.61−63 Once they enter circulation, LNPs are opsonized by electrolytes, lipids, apolipoproteins, immunoglobulins, coagulation, and other factors creating a specific “biomolecular corona”64 that plays a role in the distribution of LNPs. One of the most important serum proteins that coat the LNPs surface is apolipoprotein E (ApoE). ApoE is recognized by low-density lipoprotein (LDL) receptors, which highly expressed on hepatocytes. When LNPs are systemically administered, serum ApoE adsorbed onto the surface of the particles creates an inherent receptor-mediated uptake into hepatoma cells and hepatocytes.61,64,65 Yan et al. demonstrated the hepatic clearance of liposomes via ApoE as a ligand–receptor-mediated uptake process using ApoE-deficient mice.61 LNPs also passively accumulate in the liver as a consequence of physiology since the liver is a highly perfused organ with fenestrated capillaries.65
This endogenous ligand–receptor-mediated targeting suggests that similar approaches using exogenous ligands may be useful for LNP targeting to different cell and tissue types.65,66 For example, the PEG-conjugated ligand anisamide interacts with sigma receptors expressed by murine melanoma cells (B16F10) and has been used to increase LNP-mediated siRNA delivery to lung tumors and metastases.67 However, the efficacy of anisamide for LNP targeting may be limited by the subcellular organization of sigma receptors and the impact of anisamide on LNP endocytosis.68
In other studies, incorporating small-molecule ligands into LNP formulations has enabled particle targeting and improved cellular uptake. The small-molecule ligand strophanthidin has been incorporated in LNP formulations to improve in vitro transfection of cells derived from different tissue types. PEG-conjugated strophanthidin improved LNP uptake in cell lines derived from cervix, ovary, lung, pancreas, liver, prostate, and breast.69 In a library screen of phage-displayed peptides that bind endothelia of tumor vasculature,70 the nanopeptide LyP-1 bound tumor cells, tumor lymphatics, and macrophages associated with tumors.71,72 LyP-1-conjugated liposomes showed enhanced uptake into SPC-A1 cells and mouse metastatic lymph nodes.73 Polymeric conjugation with hyaluronan, a natural polysaccharide ligand for CD44 receptors expressed by multiple types of cancer cells, has also been explored for LNP targeting.74 Hyaluronan-conjugated LNPs have efficiently delivered siRNA to glioblastoma multiforme (GBM) cell lines and primary glioma samples isolated from GBM patients.74
Antibodies are another common method used for LNP targeting. Murine CD4+ T lymphocytes targeted with siRNA-LNPs conjugated to the anti-CD4 monoclonal antibody efficiently bind and take up anti-CD4-conjugated particles and induce silencing in the blood, spleen, inguinal lymph nodes, and bone marrow.75
In vivo tissue targeting has also been achieved using a strategy termed selective organ targeting (SORT) that uses a fifth permanently cationic lipid for direct delivery of CRISPR components to the lung, spleen, and liver of mice76 (Table 1).
Table 1. Summary of In Vitro, In Vivo, and Clinical Trial Studies Utilizing LNPs to Deliver CRISPR/Cas in the Forms of Plasmid DNA (pDNA), mRNA, and Ribonucleoprotein (RNP).a.
| dosing
regimen |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LNP system | gene target | model system | no. of doses | dose | route/target tissue | gene editing | off-target editing | therapeutic benefit | durability | toxicity |
| pDNA | ||||||||||
| reporter pDNA delivery using LNP containing DLin-KC2-DMA and unsaturated PCs80 | N/Ab | HepG2; Hep3B; PC12; MCF7; MDA-231; JHOC-9; primary embryonic mesenchymal cells | 1 | 0.75–6.0 μg/mL pDNA (cell lines); 4 μg/mL pDNA (primary cells) | N/Ab | N/Ab | N/Ab | N/Ab | N/Ab | >85% cell viability |
| white leghorn chicken embryos | 1 | 10 μg/mL pDNA (10–50 nL) | absence of developmental abnormalities | |||||||
| lipofectamine 2000 and RNAiMAX34 | EGFP | U2OS-EGFP | 1 | 750 ng Cas9 pDNA; 250 ng sgRNA pDNA | N/Ab | 38% | N/Ab | loss of EGFP in 30–40% of cells | N/Ab | moderate cellular toxicityd |
| EGFP | HEK293-(disrupted) EGFP | 1 | 700 ng Cas9 pDNA; 250 ng sgRNA pDNA | 7–10% HDR | N/Ab | restoration of EGFP expression | moderate cellular toxicityd | |||
| EGFP | U2OS-EGFP | 1 | 250 ng total pDNA (Cas9 D10A nickase/sgRNA pDNA) | N/Ab | N/Ab | loss of EGFP in 20–25% of cells | minimal cellular toxicitye | |||
| EMX1; CTLA2; VEGF | HEK293T | N/Ab | 700 ng Cas9 pDNA; 50 ng linear PCR product sgRNA | 18%; 36%; 46% | ∼0.1%; <0.1%; ∼0.1–1% | N/Ab | minimal cellular toxicitye | |||
| pDNA condensation within LNP using protamine and CS21 | Plk1 | A375 | 1 | 1 μg/mL pDNA | N/Ab | 16.1% | N/Ab | N/Ab | N/Ab | minimal cellular toxicityc |
| A375 tumor-bearing BALB/c nude mice | 8 | 10 μg pDNA/mouse | intratumoral injection/Implanted A375 tumor | 3% | N/Ab | reduced PLK1 expression; 67% suppression of tumor growth | N/Ab | N/Ab | ||
| novel ionizable lipid for CRISPR/Cas pDNA delivery81 | PLK1 | HepG2-Luc | 1 | 600 ng/μL pDNA | N/Ab | N/Ab | N/Ab | 32% reduction in PLK1 mRNA | N/Ab | >85% cell viability |
| HepG2-Luc-xenograft tumor-bearing BALB/c nude mice | 8 | 0.5 mg/kg pDNA | intratumoral injection; xenograft tumor | significant reduction in tumor growth rate; reduced PLK1 mRNA; Tumor apoptosis | no significant changes in body weight, liver-to-body weight ratio, AST/ALT or other serum biochemical markers | |||||
| mRNA | ||||||||||
| bioreducible LNP42 | GFP | HEK293-GFP | 1 | 20–500 ng/mL Cas9 mRNA | N/Ab | N/Ab | N/Ab | 90% GFP knockdown | N/Ab | >90% cell viability |
| Pcsk9 | C57BL/6 mice | 1 | 0.6 mg/kg Cas9 mRNA | tail vein injection/liver | 80% reduction of serum PCSK9 | no signs of overt inflammation or changes in AST/ALT or total bilirubin | ||||
| ABEmax (adenine base editor) mRNA delivery using LNP84 | Pcsk9 | C57BL/6J mice | 1 | 1.0, 1.5, or 3.0 mg/kg total RNA | tail vein injection/liver | 4, 12, or 51% base editingg | 20 A-to-G transitions in transcriptome; None detected in gDNA | significant reduction in plasma PCSK9 and LDL | N/Ab | transient increases in AST/ALT |
| 2 | 1.5 or 3.0 mg/kg total RNA | 40 or 67% base editingg | N/Ab | significant reduction in plasma PCSK9 and LDL | ||||||
| PCSK9 | cynomolgus monkeys | 1 | 0.75 or 1.5 mg/kg total RNA | intravenous injection/liver | 2% or 28% base editingg | up to 0.27% indel frequency; no off-target editing detected | 26% reduction in serum PCSK9; 9% reduction in serum LDL | N/Ab | transient increases in AST/ALT and cytokines (resolved by 7 days/24h) | |
| 2 | 3 or 24% base editingg | no off-target editing detected | 39% reduction in serum PCSK9; 19% reduction in serum LDL | transient increases in AST/ALT and cytokines (resolved by 7 days/24h); detectable SpCas9 IgG antibodies | ||||||
| SaKKH–CBE3 (cytidine base editor) mRNA delivery using LNP86 | Pah | Pahenu2 PKU model mice | 1 | 3.0 mg/kg total RNA | tail vein injection/liver | 10.7% base editing | no increase in C-to-U transitions in transcriptome; no above background C-to-T transitions in gDNA | 5.5% (1 dose) or 10.8% (2 doses) of RNA sequencing reads suggest PAH enzyme restoration; l-Phe reduced below therapeutic threshold | N/Ab | modest increase in serum cytokines |
| 2 | 18.8% base editing | N/Ab | N/Ab | |||||||
| NTLA-2001 biodegradable LNP delivery system44,93 | TTR | huTTR transgenic mice | 1 | 0.3–3 mg/kg total RNA | tail vein injection/liver | 70% | N/Ab | >97% reduction of serum TTR | 12 months | no significant changes in body weight or cytokines |
| 4 | 0.5 mg/kg total RNA | ∼55%f | N/Ab | N/Ab | ||||||
| 3 | 0.5 mg/kg total RNA | ∼45%d | ||||||||
| Ttr | Sprague–Dawley rats | 1 | 1, 2, or 5 mg/kg total RNA | tail vein injection/liver | ∼70% | N/Ab | >90% reduction of serum TTR | N/Ab | ||
| TTR | human primary hepatocytes | 1 | 0.007–15.517 nM total RNA | N/Ab | >93.7% | none detected | >95% reduction of TTR | N/Ab | N/Ab | |
| cynomolgus monkeys | 1 | 3 or 6 mg/kg total RNA | intravenous injection/liver | 73% | N/Ab | >95% reduction of serum TTR | 12 months | |||
| cell-selective mRNA delivery using PBA-derived LNP87 | HPV18E6 | HeLa cells | 1 | 83–660 ng/mL Cas9 mRNA | N/Ab | 18.7% | N/Ab | inhibition of HeLa cell growth | N/Ab | >80% viability with scramble sgRNA treatment |
| bioreducible lipidoid LNP43 | Angptl3 | C57BL/6 mice | 1 | 3.0 mg/kg total RNA | tail vein injection/liver | 38.5% | none detected | reductions in serum ANGPTL3 (65.2%), LDL-C (56.8%), and TG (29.4%) | 150 days | no changes in AST/ALT; modest increase in serum cytokines (resolved by 48h) |
| novel, multitailed pH-dependent ionizable lipid-containing LNP83 | DMD1 | myoblasts derived from DMD iPS cells | 1 | 2 μg total RNA | N/Ab | exon skipping detected | partial cleavage at two off target sites | restoration of DMD expression | N/Ab | N/Ab |
| DMD1 | hEx45KI-mdx44 (DMD model) mice | 1 | 1.0–10.0 mg/kg total RNA | dorsal saphenous vein injection (single limb perfusion)/ hindlimb muscle | dose-dependent exon skipping detected in multiple hindlimb muscles | N/Ab | restoration of DMD | N/Ab | increased plasma GLDH (resolved by 7 days) | |
| 1 | 10 μg Cas9 mRNA (with 10 μg sgRNA) | intramuscular injection/tibialis anterior muscle | ∼10% exon skipping | N/Ab | ∼1.1% DMD recovery | 12 months | mild immune cell infiltration and fibrosis at injection site; no GLDH elevation; transient cytokine elevation (resolved by 7 days) | |||
| 2 | ∼13% exon skipping | ∼2.6% DMD recovery | N/Ab | N/Ab | ||||||
| 3 | ∼15% exon skipping | ∼4.0% DMD recovery | ||||||||
| 6 | N/Ab | restoration of DMD in 38.5% of muscle fibres; increase in regenerating myofibers | ||||||||
| dendrimer ionizable lipid-containing LNP for selective organ targeting (SORT)76 | stop cassette (SV40 polyA) | tdTom (Ai9) reporter mice | 1 | 2.5 mg/kg total RNA | tail vein injection/multiple organs | red fluorescence in liver/lung/spleen | N/Ab | N/Ab | N/Ab | no changes in AST/ALT, CREA, or serum cytokines; no overt tissue injury detectedi |
| Pten | C57BL/6 mice | 1 | 2.5 mg/kg total RNA (4.0 mg/kg for spleen) | 11.6%-13.9% (liver); 15.1% (lung); observed in spleen | clear cytoplasm due to Pten loss | |||||
| Pcsk9 | C57BL/6 mice | 3 | 2.5 mg/kg total RNA | ∼60% (liver) | 100% reduction in liver and serum Pcsk9; increased liver-to-body weight ratio | |||||
| novel ionizable amino lipid-containing LNPs and extrahepatic ASSET targetingh38 | Plk1 | GBM 005 | N/Ab | 0.5 μg/mL total RNA | N/Ab | 84% | N/Ab | N/Ab | N/Ab | no reduction in cell viability with scramble sgRNA treatment |
| PLK1 | OV8 | 1.0 μg/mL total RNA | 91% | |||||||
| Plk1 | GBM-005-bearing C57BL/6JOlaHsd mice | 1 | 0.05 mg/kg total RNA | intratumoral injection to hippocampus/GBM 005 intracranial tumor | 68% | N/Ab | Plk1-dependent tumor apoptosis; 50% inhibition in tumor growth; 30% improved survival | N/Ab | no evidence of clinical toxicity or changes in AST/ALT, cytokines, or blood counts | |
| PLK1 | OV8-bearing athymic nude-Foxn1nu mice | 0.75 mg/kg total RNA | intraperitoneal injection | 82% | strong inhibition in tumor growth; 80% improved survival | |||||
| ABE8.8 (adenine base editor) mRNA delivery using LNP85 | PCSK9 | human primary hepatocytes | 1 | 125–2500 ng/mL total mRNA | N/Ab | >60% base editing | none detected | 55% reduction in PCSK9 expression | N/Ab | N/Ab |
| cynomolgus monkey primary hepatocytes | 250–5000 ng/mL total mRNA | ∼70% base editing | <1% off-target editing | N/Ab | ||||||
| Pcsk9 | C57BL/6 mice | 1 | 0.05–2.0 mg/kg total mRNA | tail vein injection/liver | 70% base editing | N/Ab | N/Ab | N/Ab | N/Ab | |
| PCSK9 | cynomolgus monkeys | 1 | 1.0 mg/kg total mRNA | intravenous injection/liver | 63% base editing | 0.5% indel frequency; <1% off-target editing | 81% reduction of blood PCSK9; 63% reduction of blood LDL | N/Ab | transient increases in AST/ALT (resolved by 2 weeks) | |
| 3.0 mg/kg total mRNA | 66% base editing | 0.2% indel frequency | 90% reduction of blood PCSK9; 60% reduction of blood LDL | 8 months | ||||||
| NTLA-2002 biodegradable LNP delivery system95 | KLK-B1 | huKLK-B1 transgenic mice | 1 | N/Ab | N/Ab | ∼70% | N/Ab | >90% reduction of plasma kallikrein and elimination of captopril-induced vascular leakage | N/Ab | N/Ab |
| cynomolgus monkeys | 1 | ∼70% | >95% reduction in kallikrein protein | 15 months | ||||||
| RNP | ||||||||||
| dendrimer ionizable lipid-containing LNP for selective organ targeting (SORT)76 | stop cassette (SV40 polyA) | tdTom (Ai9) reporter mice | 1 | 1.5 mg/kg sgRNA (Cas9/RNA = 2:1 n/n) | tail vein injection/multiple organs | red fluorescence in liver/lung | N/Ab | N/Ab | N/Ab | N/Ab |
| Pten | C57BL/6 mice | 1 | 2.7% (liver); 5.3% (lung) | None detected | ||||||
| bioreducible LNP89 | EGFP | HEK293-EGFP | N/Ab | 25 nM Cas9 (with 25 nM sgRNA) | N/Ab | 70% reduction in EGFP expression | N/Ab | N/Ab | N/Ab | >85% cell viability (up to 50 nM protein)h |
| lipofectamine 2000 and RNAiMAX34 | EGFP | U2OS-EGFP | N/Ab | 100 nM Cas9 (with 50 nM sgRNA) | N/Ab | 58% editing;i ∼8–11% HDRj | no loss of expression observed with off-target sgRNA | loss of EGFP expression in 65%i or 80%j of cells | N/Ab | minimal/emoderate toxicityd |
| EMX1; CTLA2; VEGF | HEK293T | 100 nM Cas9 (with 100 nM sgRNA) | 36–38% editingj | <1% indel frequency | N/Ab | minimal toxicitye | ||||
| GFP; EMX1 | Atoh1-GFP transgenic mice | 1 | 10 μmol Cas9 (with 5 μmol sgRNA) | scala media injection | indels detected in GFP; EMX1(7) | N/Ab | 13%7 or 20%8 loss of GFP expression | N/Ab | healthy expression of Myo7a and morphologically normal nucleid,e | |
| ionizable and permanently cationic lipid-containing LNP for Cas9 RNP encapsulation and selective organ targeting28 | GFP | HeLa-GFP | 1 | 24 nM sgRNA (Cas9/sgRNA = 1/3 n/n) | N/Ab | 74.2% | N/Ab | near-complete loss of GFP expression | 5 days | N/Ab |
| stop cassette (SV40 polyA) | tdTom (Ai9) reporter mice | 1 | 1 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | intramuscular injection/muscle | N/Ab | N/Ab | td-Tom fluorescence near injection site | N/Ab | N/Ab | |
| 0.15 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | intracranial injection/brain | |||||||||
| 1.5 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/liver | td-Tom fluorescence in liver | ||||||||
| tail vein injection/lung | td-Tom fluorescence in lung | |||||||||
| Pten | C57BL/6 mice | 1 | 1.5 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/liver | 2.8–3.6% indel frequency | none detected (indels measured in lung) | N/Ab | N/Ab | N/Ab | |
| tail vein injection/lung | 7.6–13% indel frequency | none detected (indels measured in liver) | ||||||||
| Stop cassette (SV40 polyA); p53; Pten; Eml4; Alk; RB1 | tdTom (Ai9) reporter mice | 1 | 0.33 mg/kg each sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/lung | 1.1% (p53) 3.4% (Pten); 7.7% (Eml4); 1.1% (Alk); 7.5% (Rb1) | N/Ab | td-Tom fluorescence in lung | N/Ab | N/Ab | |
| p53; Pten; RB1 | C57BL/6 mice | 3 | 2.5 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/liver | 8.6%; 7.9%; 13.3% | N/Ab | Ki67-positive tumors in liver and abdominal cavity | 20 weeks | N/Ab | |
| Eml4/Alk rearrangement | C57BL/6 mice | 1 | 2 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/lung | 16.3% (Eml4); 4.5% (Alk) | N/Ab | successful chromosomal rearrangement; Ki67-positive tumors in lung | 16 weeks | N/Ab | |
| 2 | 1.5 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | 13.1% (Eml4); 3.5% (Alk) | 24 weeks | |||||||
| DMD | ΔEx44 DMD mice | 3 | 1.0 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | intramuscular injection/tibialis anterior muscle | N/Ab | N/Ab | 4.2% restoration of DMD | N/Ab | N/Ab | |
| Pcsk9 | C57BL/6 mice | 3 | 2.5 mg/kg sgRNA (Cas9/sgRNA = 1/3 n/n) | tail vein injection/liver | 5.7% | N/Ab | reduction of PCSK9 in serum and liver tissue | N/Ab | N/Ab | |
A375 = human amelanotic melanoma cell line; ALT = alanine transaminase; Angptl3 = angiopoietin like 3; AST = aspartate aminotransferase; Atoh1 = atonal BHLH transcription factor 1; CREA = creatinine; CS = chondroitin sulfate; CTLA2 = cytotoxic T lymphocyte antigen 2; DLin-KC2-DMA = 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane; DMD = dystrophin; DMD1 = dystrophin gene; EGFP = enhanced green fluorescent protein; EMX1; empty spiracles homeobox 1; GBM 005 = human glioblastoma cell line; GLDH = Glutamate dehydrogenase; HDR = Homology-directed repair; HEK293 = human embryonic kidney cell line; HeLa = human papillomavirus-related endocervical adenocarcinoma cell line; Hep3B = human hepatocellular carcinoma cell line; HepG2 = human hepatoblastoma cell line; HPV18E6 = human papillomavirus 18 E6; Icam-2 = Intercellular adhesion molecule 2; JHOC-9 = human ovarian adenocarcinoma cell line; KLK-B1 = prekallikrein; l-Phe = l-phenylalanine; LDL = low-density lipoprotein; MCF7 = human breast carcinoma cell line; MDA-231 = human epithelial breast adenocarcinoma cell line; Myo7a = myosin VIIA; n/n = mol/mol; OV8 = human ovarian adenocarcinoma cell line; Pah = phenylalanine hydroxylase; PC12 = rat pheochromocytoma cell line; PC = phosphatidylcholine; Pcsk9 = proprotein convertase subtilisin/kexin type 9; PKU = phenylketonuria; Plk1 = serine/threonine-protein kinase; TTR = transthyretin; U2OS = human osteosarcoma cell line; VEGF = vascular endothelial growth factor.
Not reported
Qualitative live/dead cell assay.
Lipofectamine 2000
Lipofectamine RNAiMAX.
Cumulative editing.
Respectively (relative to dose).
Anchored secondary scFv enabling targeting (ASSET); OV8 peritoneal xenograft targeting only.
Measured following two injections of mCherry mRNA (1.0 mg/kg).
Measured following 0–50 nM treatment of HeLa cells with Cre recombinase containing LNP.
Particle surface modifications used for LNP targeting must be undertaken carefully since the orientation of conjugated ligands and changes to particle size and surface charge caused by ligand conjugation may hinder cellular uptake and reduce formulation efficacy.
Using LNPs to Deliver CRISPR Components for Gene Editing
Challenges of Nucleic Acid Delivery
The benefit of gene editing lies in the ability of the approach to permanently target the root cause of genetic diseases, which could result in the prevention or treatment of hereditary or acquired diseases and disorders.4,13 However, the direct injection of nucleic acids (e.g., plasmid DNA or mRNA encoding Cas9) for in vivo gene editing faces multiple challenges. DNA and RNA molecules are large polynucleotides that are hydrophobic, negatively charged, and unstable on their own.29 These physiochemical characteristics prevent their spontaneous entry into cells by creating a repulsive effect with the cell membranes. Nucleic acids also have a short half-life in circulation because of serum nuclease activity making it difficult for unprotected nucleic acids to reach specific target tissues. These nucleic acids go through rapid renal clearance and may also induce immunostimulatory effects via interactions with pattern recognition receptors.1,2,4,77
Overcoming these challenges warrants sophisticated packaging and delivery systems. An important advantage of using LNPs as drug carriers is their ability to escape recognition by the innate immune system and have higher circulation time.78 These features are especially useful for delivering hydrophobic drugs with short circulation half-lives such as nucleic acids and proteins. Sufficient circulation time enables LNPs (including those containing CRISPR components in either nucleic acid or protein forms) to reach target tissues and induce efficient on-target therapeutic genome editing.
CRISPR components in a variety of formats can be delivered to cells using LNPs. The most common approaches are encapsulating (1) plasmid DNA (pDNA) encoding both Cas9 protein and gRNA or pDNA encoding Cas9 protein in combination with gRNA oligos, (2) Cas9 mRNA and gRNA, and (3) Cas9/sgRNA (protein/RNA) RNP complex (Figure 2). There are advantages and limitations to each of these techniques, so each approach must use a unique set of LNP-specific formulation criteria to ensure optimal compatibility without compromising function.
Commercially available transfections reagents originally designed for delivery of plasmids and siRNAs can be used to deliver plasmid-based Cas9/gRNA, RNA mixtures of Cas9 and sgRNA, and even RNPs to cell lines such as HEK293FT, U2OS, mouse ESCs, N2A, and A549.79 However, in vivo applications of these transfection reagents, including lipofectamine, are limited by their cytotoxic and inflammatory effects.79 The development of novel synthetic ionizable cationic lipids and LNP formulations has overcome many of these barriers and has made LNP-mediated therapeutic gene editing a realistic prospect.79
LNP Delivery of pDNA Encoding Cas9 and gRNA
CRISPR/Cas9 gene-editing machinery can be delivered using pDNA forms of both Cas9 and gRNA. There are several key considerations for LNP–pDNA delivery for gene editing. Plasmids must be transcribed within the nucleus, so LNP formulations of pDNA must facilitate nuclear entry (Figure 2A). This can be achieved in actively dividing cells when the nuclear membrane is compromised during mitosis, but the relatively large size of CRISPR plasmids (>10 000 bp) can hinder encapsulation efficiency. Thus, the negative charge of poorly encapsulated pDNA can interfere with negatively charged cell membranes.21 Therefore, LNP formulations must overcome these barriers to encapsulation and cellular uptake.
To optimize LNP–pDNA delivery, Kulkarni et al. used an LNP formulation containing DLin-MC3-DMA and replaced the saturated helper lipid distearoylphosphatidylcholine (DSPC) with unsaturated phosphatidylcholine (PC) helper lipids (Table 1). The performance of formulations containing different ionizable cationic lipids in combination with unsaturated PC helper lipids was also evaluated. In primary embryonic mesenchymal cells isolated from chicken embryos, the most efficient formulation contained DLin-KC2-DMA and 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC). This formulation achieved ∼90% transfection efficiency and >85% cell viability compared to just 50% transfection and 33% cell viability using lipofectamine.80
Despite these limitations, commercial transfection reagents Lipofectamine 2000 and RNAiMAX have been used as proof-of-concept delivery systems for CRISPR/Cas9 pDNA. Using these reagents, targeting an EGFP reporter gene in U2OS-EGFP cells achieved gene editing and resulted in the loss of EGFP fluorescence34 (Table 1). CRISPR/Cas9 pDNA transfection using RNAiMAX was also tested for three endogenous genes (EMX1, CTLA2, and VEGF) in HEK293T cells and resulted in editing efficiencies between 18 and 46%34 (Table 1).
Zhang et al. overcame several previously identified barriers to pDNA delivery: plasmid volume for encapsulation, cell membrane penetration, cytotoxicity, and nonspecific interactions with serum or extracellular proteins.21 Encapsulation efficiency was increased by condensing the plasmids’ volume using chondroitin sulfate and protamine to form a compact LNP core (Figure 1A and Table 1). Their LNP formulation consisted of cholesterol, a permanently cationic lipid (DOTAP), and a helper lipid (DOPE). The latter two lipids improved transfection by creating electrostatic interactions with cell membranes. Further modification with DSPE-PEG improved stability and solubility, reduced toxicity, increased half-life, and lowered immunogenicity.21 The DOTAP/DOPE ratio (optimal at 0.8:1) significantly impacted particle size, polydispersity index (a measurement of particle size distribution), and zeta potential (a fundamental parameter of particle stability representing the magnitude of LNP surface charge that affects electrostatic interactions between particles).21 This formulation was used to treat cancer cells (A375, PC3, and MCF-7) that overexpress PLK-1, a master gene regulator in mitosis. In A375 cells, flow cytometry results showed ∼47% transfection efficiency followed by high-throughput sequencing that detected between 1 and 20 indels in PLK-1. When compared to Lipofectamine 2000, this formulation achieved a higher genome editing efficiency both in vitro and in vivo.21 A375-tumor bearing BALB/c nude mice received intratumoral injection of the formulation which reduced expression of PLK-1 and suppressed tumor growth by 67%.21 Together, these strategies improved encapsulation efficiency, resultant cellular uptake, CRISPR/Cas9 expression, and ultimately genome editing efficiency.
LNP-mediated delivery of CRISPR/Cas9 pDNA targeting PLK1 has also achieved gene editing in vitro and in vivo in the absence of plasmid condensation reagents. In a proof-of-concept study, Li et al. used a reporter gene system to transfect HepG2-Luc cells and HepG2-Luc-xenograft tumor-bearing mice with a novel ionizable LNP formulation81 (Table 1). This formulation effectively encapsulated CRISPR/Cas9 pDNA targeting PLK1 using an additional incubation step to reassemble and stabilize the LNPs.81 Resultant PLK1 editing led to 32% mRNA knockdown in vitro and significantly reduced tumor growth rate, reduced PLK1 mRNA, and increased tumor cell apoptosis in vivo.81
LNP Delivery of Cas9 mRNA and gRNA Simultaneously or Separately
As an alternative to pDNA delivery, research is becoming increasingly focused on Cas9 mRNA delivery. LNPs can be used to coencapsulate Cas9 mRNA and gRNA into single particles, or to encapsulate them separately.42,82 Co-encapsulation of Cas9 mRNA and gRNA may achieve more effective genome editing than separate encapsulation, as it leads to a higher probability that the optimal gRNA and Cas9 mRNA ratio will be delivered to each cell for sufficient complexation42,82 (Figure 2B). Many LNP formulations for targeted genome editing have been developed to coencapsulate Cas9 mRNA and gRNA simultaneously, but separate encapsulation methods have also been successful38, 42 83.
Liu et al. delivered Cas9 mRNA and gRNA simultaneously using an LNP formulation containing the bioreducible ionizable lipid (Figure 1C), BAMEA-O16B, to achieve fast and efficient genome editing both in vitro and in vivo(42) (Table 1). This strategy reduced cellular GFP expression by 90% in vitro in HEK293-GFP cells.42In vivo, tail vein injection of C56BL/6 mice with a formulation containing Cas9 mRNA and sgPCSK9 resulted in liver accumulation and reduction of serum PCSK9 by 80%.42 No signs of inflammation or obvious hepatocellular injury were observed, suggesting that LNPs prepared with BAMEA-O16B are a potential biocompatible in vivo delivery system for gene editing in the liver.42
LNPs have also been used for delivering adenine base editor mRNA and gRNA for in vivo targeting of PCSK9. An ONPATTRO-like LNP formulation encapsulated with base editor mRNA and a gRNA targeting PCSK9 in the liver has efficiently induced up to 67% editing in mice and up to 34% editing in macaques84 (Table 1). Importantly, the editing rates were low in nonhepatic tissues in both mice and macaques84 demonstrating the tissue specificity of the formulation. Although there was a transient increase in serum transaminase (which indicates hepatocellular injury) following LNP administration, animals were asymptomatic and elevated levels were quickly resolved.84 A similar study developed an ionizable cationic lipid formulation to deliver base editor mRNA and sgPCSK9 both in vitro (primary human and cynomolgus monkey hepatocytes) and in vivo (mouse and cynomolgus monkey livers)85 (Table 1). Intravenous infusion of a lower LNP dose (1.0 mg kg–1) resulted in a mean 63% base editing in the cynomolgus monkey liver accompanied by a mean 81% reduction in serum PCSK9 and a mean 65% reduction of serum LDL cholesterol.85 Subsequently, a higher dose (3.0 mg kg–1) was tested for durability which demonstrated a mean 66% base editing where serum PCSK9 reached its lowest level in 1 week and remained stable at 90% reduction for 8 months.85 In another study, a single administration of LNPs encapsulating a cytidine base editor mRNA and gRNA targeting the Pah gene in mice achieved 10.7% editing, with 5.5% of RNA sequencing reads (Table 1) suggesting Pah enzyme restoration in whole liver lysate.86 This rate was further increased to 18.8% editing and 10.8% functional enzyme restoration after re-dosing 1 week later.86 The dosing regimens were sufficient to reduce l-Phe levels below the therapeutic threshold and resulted in reversion of a PAH-associated fur color phenotype.86 These studies demonstrate the versatility of LNPs for delivery of various types of CRISPR gene editors.
Finn et al. developed the LNP system LNP-INT01 that achieved efficient encapsulation and delivery of Cas9 mRNA and gRNA (Table 1).44 LNP-INT01 contains a biodegradable ionizable lipid (LP01), a helper lipid, and PEG-DMG (Figure 1C).44 Liver bioaccumulation of LP01 was significantly reduced compared to levels observed with nonbiodegradable ionizable lipids such as MC3, and was cleared from the liver with a half-life of ∼6 h.44 The authors also found that highly modified gRNA increased durability in vivo by conferring protection from degradation until sufficient Cas9 protein is translated for RNP complexation within cells.44 By leveraging the LDL receptor-mediated uptake of LNPs in the liver, Finn et al. designed gRNAs targeting the mouse Ttr gene to evaluate the therapeutic potential of gene correction in treating hepatic amyloidosis.44 The transiency of CRISPR components was assessed by quantifying Cas9 mRNA and end-modified sgRNA in plasma and the liver, both of which were rapidly cleared and undetectable after 72 h.44 Rapid clearance is desirable for gene therapies that permanently alter the genomic DNA to avoid inducing long-term off-target effects. After just a single systemic administration at the highest dose, LNP-INT01 resulted in >97% serum Ttr knockdown in CD-1 mice and ∼70% editing efficiency for 12 months following treatment.44 Greater cumulative editing was observed when LNP-INT01 was administered weekly or monthly.44 Multidosing allows for titratable genome editing which is an important therapeutic feature for clinical translation of LNP-based gene therapy drugs. One of the significant attributes of ONPATTRO that contributed to its successful clinical translation is the ability to administer multiple doses of siRNA.
Lipid conjugation can be used to target LNPs to extrahepatic tissues. For example, conjugation of the cationic lipid BADP with phenylboronic acid (PBA) promotes LNP interaction with the cellular surface receptor sialic acid (SA) that is overexpressed in cancer cells (Figure 1B and Table 1). Encapsulating CRISPR/Cas9 mRNA with this formulation allows selective targeting of cancer cells for gene editing87 since PBA ligands on the surface of LNPs interact with SA expressed by cancer cells to facilitate their cellular uptake. PBA–BADP LNPs were used to target the HPV18E6 gene that is overexpressed in HeLa cervical cancer cells.87 This treatment induced indels in 18.7% of cells (a higher rate than the level of editing achieved using BADP LNPs without PBA) and effectively inhibited HeLa cell growth.87 The therapeutic benefit and relative safety of this intervention was also demonstrated by HeLa cell viability: PBA–BADP LNP-treated cells were 50% viable (indicating successful growth arrest induced by HPV18E6 editing), compared to the >80% viability measured in cells treated with negative control scramble mRNA (indicating the LNP system itself is relatively nontoxic).87 The highest dose of Cas9 mRNA most significantly reduced HeLa cell viability87 suggesting that LNP-mediated HPV18E6 knockout is effective for reducing cancer cell proliferation in this model system. These findings propose that cell selective gene targeting can be achieved through modifications of LNP formulations for targeted CRISPR/Cas9 mRNA delivery.
Qiu et al. used an LNP formulation containing the lipid 306–012B to deliver Cas9 mRNA and gRNA targeting ANGPTL3 (Table 1).43 306–012B was chosen after screening a library of novel bioreducible ionizable lipids for effective luciferase mRNA delivery in vivo using wild-type BALB/c mice.43 This lipid formulation produced the most robust liver bioluminescence in the library reporter screen. In the liver, it achieved a median editing rate of 38.5% and induced 65.2, 56.8, and 29.4% reductions in serum ANGPTL3, LDL cholesterol, and triglyceride levels, respectively.43 These results corresponded to over a 2-fold improvement compared to those achieved using MC3-based LNPs.43 Serum markers of liver function including aspartate aminotransferase (AST), alanine aminotransferase (ALT), and proinflammatory cytokine tumor necrosis factor alpha (TNF-α) were examined and no significant changes were observed.43 Serum cytokine levels were elevated at 6 h post-LNP administration but were transient and resolved by 48 h.43 Like the studies conducted by Liu et al. and Finn et al., this study demonstrates the utility of bioreducible LNP formulations for efficient genome editing in the liver.
Also using an in vivo lipid screening approach, Kenjo et al. identified a novel ionizable lipid, TCL053, for intramuscular mRNA delivery (Table 1).83 LNP formulations containing TCL053 were used to separately encapsulate Cas9 mRNA and gRNA and were tested in a humanized mouse model of Duchene muscular dystrophy (DMD) with a mouse Dmd1 exon 44 deletion and knock-in of human DMD1 exon 45.83 Treatment with this formulation resulted in ∼10% exon-skipping efficiency in the tibialis anterior muscle which was approximately 5-fold higher than exon skipping achieved using MC3-based LNPs.83 Serum cytokine levels were elevated 6 and 24 h postadministration, which resolved by 7 days postinjection.83 Due to the low immunogenicity of the formulation, safe readministration of the LNPs once, twice, or three times at 1 month intervals was evaluated and achieved cumulative DMD1 editing.83
In addition to targeting by ligand conjugation, LNP formulations can be modified to confer nonhepatic tissue specificity. For instance, selective organ targeting (SORT) involves the addition of a permanently cationic lipid to a traditional four component LNP system containing dendrimer ionizable lipids for extrahepatic targeting following intravenous injection in mice (Table 1).76 Increasing the molar percentage of the permanently cationic lipid DOTAP in the formulation shifted particle accumulation to the lung, whereas addition of 1,2-dioleoyl-sn-glycero-3-phsphate (18PA) led to spleen targeting.76 To demonstrate the therapeutic potential of SORT lipids, the tumor suppressor gene PTEN was targeted by encapsulating Cas9 mRNA and gRNA into multiple SORT LNP formulations. Formulations containing 50% DOTAP led to 15.1% editing in the lungs, whereas those containing 20% DODAP resulted in 13.9% editing in the liver.76 SORT lipids are also compatible with RNP delivery of CRISPR/Cas9. LNP–RNP formulations containing 7% DOTAP resulted in 2.7% editing in the liver whereas those with 55% DOTAP resulted in 5.3% editing in the lungs.76
Gene editing for cancer therapy has also been enabled by delivering Cas9 mRNA and gRNA to tumors using LNP systems. Incorporating safe ionizable lipids optimized for efficient in vivo mRNA delivery into LNPs36 (Table 1) induced targeted disruption of the mitotic regulator PLK1 at 68% of loci following intratumoral LNP injection, which reduced tumor growth and improved survival in glioblastoma-bearing mice.38 In addition, preliminary evaluation of liver toxicity and immunogenicity revealed no significant differences in liver enzymes and cytokine levels.38 Intraperitoneal injection of the same LNP formulation conjugated to EGFR targeting antibodies (Figure 1B) achieved 82% PLK1 editing in a metastatic ovarian adenocarcinoma model in which similar reduced tumor growth and improved survival rates were observed.38,88 These results demonstrate the efficacy of Cas9-mediated gene editing as a feasible strategy for cancer treatment and leverage the versatility of LNP systems for targeted delivery to diverse tumor types.
LNP Delivery of Cas9/gRNA RNP Complex
Compared to plasmid-based CRISPR/Cas9 DNA or mRNA, delivering CRISPR RNPs is predicted to result in fewer off-target effects, significantly higher editing efficiency, and up to 10-fold higher target specificity.34,79 Therefore, exhaustive efforts have been undertaken to optimize LNP formulations to effectively encapsulate and deliver CRISPR RNPs to desired targets. One common strategy is the addition of a polyanionic macromolecule to enhance RNP encapsulation.
Wang et al. demonstrated that their formulation containing bioreducible lipids can deliver polyanionic RNPs to target GFP-expressing HEK cells (Table 1).89 During endosomal release, bioreducible lipids augment the endosomal escape of LNP cargo, which induced the loss of eGFP signals with >50% efficiency.89 The nuclear localization signal (NLSs) on Cas9 allowed efficient nuclear translocation and subsequent genome editing. The authors demonstrate that using a combinatorial strategy to screen unique LNP formulations, including negatively supercharged proteins, and complexation of Cas9 protein with inherently polyanionic gRNAs may enable the development of a highly efficient delivery platform for gene-editing machinery.89
Similarly, Zuris et al. fused a polyanionic protein, (−30) GFP, to Cas9 (Figure 1E) and Cre tyrosine recombinase protein (a site-specific genome editing recombinase).34 They used this strategy to drive complexation with several common commercial cationic lipid reagents (including Lipofectamine RNAiMAX), which facilitated efficient protein delivery to cells (Table 1).34 The authors also showed that the inherent negative charge of gRNAs was sufficient to facilitate complexation of Cas9 protein with cationic lipids and resulted in up to 80% genome editing of human cells after one treatment in vitro.34 Delivering RNPs with this strategy achieved 20% genome editing within the inner ear hair cell population of live mice.34 The success of this in vivo application suggests therapeutic potential for hearing recovery using LNPs, especially since many genetic deafness diseases are caused by hair cell loss or dysfunction.34
RNP delivery is further complicated by the large size of the Cas9 protein (∼160 kDa),38 and the possibility of RNP denaturation during LNP formulation.28 These limitations mean that most nanoparticles used to deliver RNPs are less effective than their nucleic acid-containing counterparts, and are as a result limited to applications for which local administration is suitable.28 However, Wei et al. developed an approach to systemically deliver RNPs to muscle, liver, and lung tissues of mice using LNPs (Table 1).28 DOTAP was added to an established LNP formulation containing the dendrimer ionizable lipid 5A2-SC8, which resulted extrahepatic organ delivery.28 Upon intravenous injection, relative tissue-specific gene editing was achieved in different organs depending on the DOTAP percentage used in the formulations. Tissue-specific RNP editing was evaluated in a transgenic mouse model harboring the tdTomato gene. In this model, reporter expression is prevented by SV40 polyA stop cassettes. Fluorescence after targeted stop cassette editing was used to evaluate organ-specific editing. Intravenous injection of DOTAP-modified 5A2-SC8-containing LNPs produced observable Td-Tomato fluorescence for 7 days in the liver, where increasing the DOTAP percentage in the formulations from 5 to 60% caused a gradual shift in reporter fluorescence from liver to lung.28
It is important to emphasize that the methods (ethanol and acidic buffers) used to formulate the majority of LNPs denature RNPs and significantly increase LNP–RNP particle size (from 10 to 150 nm), which makes preparation challenging and the resulting formulations less effective. To resolve this issue, the authors used a neutral buffer (PBS) plus DOTAP to facilitate encapsulation and preserve the structure and stability the LNP–RNP complexes.28 While LNP formulations containing permanently cationic lipids may achieve potent transfection as well as efficient genome editing and can be suitable for in vitro applications, their safety for in vivo applications must be carefully evaluated to avoid in vivo toxicity.
An alternative approach has been developed using a novel method of LNP complexation that eliminates the use of solvents (such as ethanol) that may denature RNPs (Figure 1F). The approach was developed after observing that rapid mixing of ethanolic lipids with acidic buffer in the absence of nucleic acids produces liposomal vesicles, indicating that vesicle formation does not depend on the presence of nucleic acids.90,91 Dialysis against the same acidic buffer to remove ethanol does not disrupt vesicle structure. Subsequent mixing of the vesicles with nucleic acid and pH neutralization on the benchtop is sufficient to entrap siRNA, complete the process of particle formation, and generate particles with in vitro activity comparable to conventionally prepared LNP systems.91 Because interactions between polyanionic molecules and positively charged liposomes destabilize vesicle membranes and drive encapsulation in this system and because the exposure of LNP cargoes to acidic pH during particle loading is very brief, loading of Cas9 RNP (an overall negatively charged complex) into empty preformed vesicles using this approach circumvents the harsh conditions required by conventional LNP preparation and can efficiently encapsulate the active Cas9 RNP complex.
Clinical Trials of CRISPR/Cas9 LNP Platforms
Novel LNP formulations and methods of manufacturing are being developed to enable efficient delivery of CRISPR components for therapeutic gene editing. In addition to enabling the clinical translation of ONPATTRO, LNPs have a history of successful in vivo applications for delivering CRISPR/Cas9. Their inherent target specificity and accumulation in the liver makes them a promising delivery platform for treating genetic liver diseases. LNP delivery of CRISPR systems is relatively transient, allows the flexibility of repeated dosing, and is efficient and highly scalable, which makes this platform an attractive option for clinical translation. The first clinical trial using LNPs as delivery vector for CRISPR/Cas9 was initiated by Intellia Therapeutics in November 2020 (NCT04601051).92 NTLA-2001, delivered through intravenous infusion, is a CRISPR/Cas9 gene therapy targeting the TTR gene in hepatocytes for the treatment of hereditary transthyretin amyloidosis with polyneuropathy (ATTRv-PN) (Table 1). In 2021, interim study results were published which included six patients with ATTRv-PN.93 Three patients received a low dose of NTLA-2001 (0.1 mg/kg) whereas the remaining half received a high dose (0.3 mg/kg).93 At 28 days after the initial infusion, the mean reductions of serum TTR protein from baseline were 52 and 87% for the low- and high-dose groups, respectively.93 Post-treatment of NTLA-2001 adverse events were observed in three patients which were all mild (grade 1) in severity. Increased d-dimer levels (indicative of blood clotting) were observed 4–24 h postinfusion in five out of six patients which resolved in all cases by day 7.93 Currently, a larger phase I clinical trial that aims to recruit 73 patients is underway.92
In November 2021, Intellia Therapeutics announced a second clinical trial which aims to prevent angioedema attacks in patients with hereditary angioedema (HAE).94 The company’s CRISPR/Cas9 LNP platform, NTLA-2002, targets the KLKB1 gene (Table 1). Preclinical studies included both humanized KLKB1 mice and cynomolgus monkeys.95 A single administration of NTLA-2002 in mice resulted in ∼70% KLKB1 gene editing and a subsequent reduction of total plasma kallikrein protein of >90%.95 In the monkey, a single administration also resulted in ∼70% gene editing and >95% decrease in total kallikrein protein and activity.95
Considerations and Future Directions for Using LNP to Deliver CRISPR/Cas9
When designing LNP formulations for CRISPR/Cas9 gene editing, the specific and unique experimental application must be considered. In vitro applications primarily require lipids that enhance encapsulation and cellular uptake. In addition to requiring these characteristics, in vivo applications also depend on lipids that increase circulation time, can escape the innate immune system, minimize toxicity, and in some cases, interact with specific receptors on target cells. For hard-to-reach tissues, PEG lipids increase circulation time and prevent phagocytosis by macrophages in the blood to allow LNP formulations to persist in circulation. Studies that focus on in vivo intravenous delivery must consider formulations containing optimal ratios of these lipids.
LNP formulations primarily target hepatic tissues when systemically administered. Because of this inherent advantage, the first clinical trials for both LNP–siRNA and LNP–Cas9 mRNA delivery target a liver-related genetic disease. However, additional lipids have been incorporated into LNPs to achieve extrahepatic delivery in mice. The overall charge and size of the LNPs determine their biodistribution in vivo. For example, LNPs containing novel degradable ionizable lipids can facilitate splenic delivery and transfect B lymphocytes.41 The addition of a negatively charged lipid (18PA) can also result in spleen targeting.76 Lung specificity can be achieved in formulations with PEG-lipids at 7 mol % that encapsulate mRNA.45,46 Ligand conjugation of lipids can further achieve target specificity in in vitro in multiple cell lines69 including those representing cancer cells,87 as well as in lung tumors in vivo.67 Addition of permanently cationic lipids in SORT can also achieve delivery to the lung, spleen and liver.76 Nanoparticle size is another important factor in determining the biodistribution of LNPs after intravenous injections. Nanoparticles that are less than 10 nm in diameter may be removed by the kidneys via the glomerular capillaries whereas those larger than 200 nm will activate the complement system and are removed from the blood, accumulating in the liver and spleen.96,97 However, LNPs of ≤150 nm in size are capable of escaping fenestrated capillaries in the liver.97 LNP formulations of an appropriate size are necessary to achieve optimum CRISPR/Cas9 delivery and subsequent genome editing efficiency. Particle size further plays a role in colloidal formulation stability. Ultimately, the overall charge, particle size, lipid ratio, and receptor-mediated ligand or antibody conjugations emphasize the versatility of LNPs as a delivery platform suitable for gene therapy that can achieve desired target specificity in vivo.
The format of LNP-encapsulated cargo also plays a role in determining the most suitable approach to CRISPR/Cas9 delivery. Different lipid formulations have been developed to enhance delivery and subsequently increase gene-editing efficiency based on the format of the CRISPR components. Delivering pDNA, mRNA, or RNP offers advantages and disadvantages that have varying degrees of relevance for different applications of gene editing.
Designing and modifying pDNA constructs is straightforward and relatively inexpensive. pDNA delivery can be useful for screening a high number of gRNAs or nucleases to identify optimal sequences for targeting, editing, and evaluating off-target effects or in applications using dividing cells in vitro. However, the utility of pDNA is severely limited in postmitotic cells where translocation of pDNA across the nuclear membrane is exceedingly rare. The larger size and the less stringently regulated plasmid expression pathway (Figure 2A) is also a critical safety concern, because expression of DNA-disrupting CRISPR components is difficult to control postadministration. One of the major concerns surrounding CRISPR gene editing is the potential for off-target editing. This is an important factor to consider when selecting a delivery format, because the frequency of off-target effects may by determined in part by the format in which Cas9 editors are encoded. The poor regulation of Cas9 expression from pDNA vectors means there is a higher likelihood of off-target editing using these systems. Alternatively, delivering CRISPR components using RNP reduces the frequency of off-target editing by minimizing exposure of the genome to unbound Cas9 nuclease.38
Delivery of the mRNA form of CRISPR/Cas9 is supported by research to develop safe ionizable lipids that optimize mRNA encapsulation and delivery. Critical innovations to the design of RNA molecules have made therapeutic mRNA delivery possible and enabled the LNP-mRNA COVID-19 vaccines developed by Pfizer-BioNTech and Moderna. RNA nucleoside modifications, including 5-methylcytosine and pseudouridine, shield exogenous RNAs from recognition by the innate immune system while improving their translation and in vivo stability.98,99 Similarly, modifications to gRNAs used for CRISPR/Cas9-mediated gene editing improve stability without compromising editing efficiency. These modifications are particularly important for applications using Cas9 mRNA because they allow gRNAs to resist nuclease degradation during Cas9 protein translation, resulting in efficient downstream RNP complexation.44,100 The efficacy of LNP therapeutics that incorporate modified mRNA also varies depending on LNP composition and the cells and tissues targeted by the particles, indicating that interactions between components of these systems also partially determine their efficacy.101
An important consideration for delivering RNA forms of CRISPR components is delivery of Cas9 mRNAs and gRNAs at optimal ratios for efficient intracellular RNP assembly and targeting (Figure 2B). gRNAs are frequently encapsulated with and delivered in excess of Cas9 mRNA. Conversely, Cas9 RNPs are often assembled prior to delivery, which ensures that enough of each component is delivered in a single complex simultaneously to cells (Figure 2C). Previous work has found that a Cas9/gRNA molar ratio of 1:3 achieves robust gene editing and does not impact LNP size or zeta potential.28 When compared to more persistently expressed pDNA, RNPs are degraded rapidly after cellular delivery. This minimizes the potential for off-target effects and makes Cas9 RNP delivery a more desirable therapeutic option. LNP delivery results in transient payload expression that, when combined with short-lived mRNA and RNP cargoes, allows for flexible dose titration, multiple dosing, and, if required, immediate suspension of treatment.
It is also important to address that many therapeutic applications of the CRISPR/Cas9 system involves using the HDR pathway for gene correction, instead of NHEJ (which is useful primarily for gene knockout). This means an additional donor DNA template containing the desired edits must also be present. The success rate of HDR is largely dependent upon the availability of the donor template at close proximity to the Cas9 cut site. This complicates the delivery scheme by requiring encapsulation of an additional DNA component. Current research on LNP formulations usually focuses on creating indels with the NHEJ pathway or base editing, where the delivery of only Cas9 and gRNA are sufficient. LNP delivery systems that can efficiently deliver DNA repair templates with RNP to maximize the frequency of gene correction using HDR are the future of this approach.
The current pace of LNP research and the relevance of LNPs for delivering mRNA in the BioNTech/Pfizer and Moderna vaccines developed for SARS-CoV-292,102,103 will significantly contribute to the clinical translation of LNP systems. In addition to lipids developed for the current COVID-19 vaccines, extensive efforts have been made to develop safe and efficient ionizable lipids to optimize in vivo delivery. Intellia Therapeutics is currently leading the clinical translation of LNP–CRISPR therapeutics using mRNA, and it is likely that the clinical translation of new LNP–RNP systems will expand into multiple therapeutic areas in the future.
Acknowledgments
This work acknowledges the support of the Canadian Institutes of Health Research (CIHR), and the Huntington Society of Canada (HSC) to S.B.T., as well as the funding support from a Nanomedicines Innovation Network (NMIN) grant (2019-T2-05), and a Michael Smith Foundation for Health Research (MSFHR) Scholar Award (number 16458) to C.J.D.R.
The authors declare the following competing financial interest(s): B.R.L. is a co-founder, shareholder, and CEO of Incisive Genetics Inc. The authors declare no other competing financial interests.
References
- Cullis P. R.; Hope M. J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Molecular Therapy 2017, 25 (7), 1467–1475. 10.1016/j.ymthe.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Aguado I.; et al. Nanomedicines to Deliver Mrna: State of the Art and Future Perspectives. Nanomaterials 2020, 10 (2), 364. 10.3390/nano10020364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski P. S.; et al. Delivering the Messenger: Advances in Technologies for Therapeutic Mrna Delivery. Molecular Therapy 2019, 27 (4), 710–728. 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Cullis P. R.; Van Der Meel R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Therapeutics 2018, 28 (3), 146–157. 10.1089/nat.2018.0721. [DOI] [PubMed] [Google Scholar]
- Brooks P. J.; et al. The Platform Vector Gene Therapies Project: Increasing the Efficiency of Adeno-Associated Virus Gene Therapy Clinical Trial Startup. Hum. Gene Ther. 2020, 31 (19–20), 1034–1042. 10.1089/hum.2020.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A. V.; Koblan L. W.; Liu D. R. Genome Editing with Crispr–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38 (7), 824–844. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- Brezgin S.; et al. Dead Cas Systems: Types, Principles, and Applications. International Journal of Molecular Sciences 2019, 20 (23), 6041. 10.3390/ijms20236041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. C.; et al. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533 (7603), 420–424. 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N. M.; et al. Programmable Base Editing of a•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551 (7681), 464–471. 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott T. R.; Dhamdhere G.; Liu Y.; Lin X.; Goudy L.; Zeng L.; Chemparathy A.; Chmura S.; Heaton N. S.; Debs R.; et al. Development of Crispr as an Antiviral Strategy to Combat Sars-Cov-2 and Influenza. Cell 2020, 181 (4), 865–876.e12. 10.1016/j.cell.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; et al. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16 (6), 630–643. 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Somia N.; Verma I. M. Gene Therapy: Trials and Tribulations. Nat. Rev. Genet. 2000, 1 (2), 91–99. 10.1038/35038533. [DOI] [PubMed] [Google Scholar]
- Bondì M. L.; Craparo E. F. Solid Lipid Nanoparticles for Applications in Gene Therapy: A Review of the State of the Art. Expert Opinion on Drug Delivery 2010, 7 (1), 7–18. 10.1517/17425240903362410. [DOI] [PubMed] [Google Scholar]
- Battaglia L.; Ugazio E. Lipid Nano- and Microparticles: An Overview of Patent-Related Research. J. Nanomater. 2019, 2834941. 10.1155/2019/2834941. [DOI] [Google Scholar]
- Khan I.; Saeed K.; Khan I. Nanoparticles: Properties, Applications and Toxicities. Arabian Journal of Chemistry 2019, 12 (7), 908–931. 10.1016/j.arabjc.2017.05.011. [DOI] [Google Scholar]
- Mitchell M. J.; et al. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discovery 2021, 20 (2), 101–124. 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14 (12), 1084–1087. 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- Schoenmaker L.; et al. Mrna-Lipid Nanoparticle Covid-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. 10.1016/j.ijpharm.2021.120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgner P. L.; et al. Lipofection: A Highly Efficient, Lipid-Mediated DNA-Transfection Procedure. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (21), 7413–7417. 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; et al. Nanoparticle Depots for Controlled and Sustained Gene Delivery. J. Controlled Release 2020, 322, 622–631. 10.1016/j.jconrel.2020.03.021. [DOI] [PubMed] [Google Scholar]
- Zhang L.; et al. Lipid Nanoparticle-Mediated Efficient Delivery of Crispr/Cas9 for Tumor Therapy. NPG Asia Materials 2017, 9 (10), e441–e441a. 10.1038/am.2017.185. [DOI] [Google Scholar]
- Kauffman K. J.; et al. Optimization of Lipid Nanoparticle Formulations for Mrna Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15 (11), 7300–7306. 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- Adams D.; et al. Patisiran, an Rnai Therapeutic, for Hereditary Transthyretin Amyloidosis. New England Journal of Medicine 2018, 379 (1), 11–21. 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- Wood H. FDA Approves Patisiran to Treat Hereditary Transthyretin Amyloidosis. Nat. Rev. Neurol. 2018, 14 (10), 570–570a. 10.1038/s41582-018-0065-0. [DOI] [PubMed] [Google Scholar]
- Apollo: The Study of an Investigational Drug, Patisiran (Aln-Ttr02), for the Treatment of Transthyretin (Ttr)-Mediated Amyloidosis. https://clinicaltrials.gov/ct2/show/NCT01960348 [accessed on 08/24/2021].
- Zhang X.; Goel V.; Robbie G. J. Pharmacokinetics of Patisiran, the First Approved Rna Interference Therapy in Patients with Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019, 60 (5), 573–585. 10.1002/jcph.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedic M.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified Mrna in the Sprague-Dawley Rat and Cynomolgus Monkey. Veterinary Pathology 2018, 55 (2), 341–354. 10.1177/0300985817738095. [DOI] [PubMed] [Google Scholar]
- Wei T.; Cheng Q.; Min Y.-L.; Olson E. N.; Siegwart D. J. Systemic Nanoparticle Delivery of Crispr-Cas9 Ribonucleoproteins for Effective Tissue Specific Genome Editing. Nat. Commun. 2020, 11 (1), 3232. 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton O. S.; et al. Bioinspired Alkenyl Amino Alcohol Ionizable Lipid Materials for Highly Potent in Vivo Mrna Delivery. Adv. Mater. 2016, 28 (15), 2939–2943. 10.1002/adma.201505822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens B. E.; Naguib Y. W.; Geary S. M.; Devor E. J.; Salem A. K. Nanoparticle-Based Delivery of Crispr/Cas9 Genome-Editing Therapeutics. AAPS J. 2018, 20 (6), 108. 10.1208/s12248-018-0267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion M. C.; Phillips N. C. Toxicity and Immunomodulatory Activity of Liposomal Vectors Formulated with Cationic Lipids toward Immune Effector Cells. Biochimica et Biophysica Acta (BBA) - Biomembranes 1997, 1329 (2), 345–356. 10.1016/S0005-2736(97)00126-0. [DOI] [PubMed] [Google Scholar]
- Hafez I.; Maurer N.; Cullis P. On the Mechanism Whereby Cationic Lipids Promote Intracellular Delivery of Polynucleic Acids. Gene Ther. 2001, 8 (15), 1188–1196. 10.1038/sj.gt.3301506. [DOI] [PubMed] [Google Scholar]
- Schlich M.; Palomba R.; Costabile G.; Mizrahy S.; Pannuzzo M.; Peer D.; Decuzzi P. Cytosolic Delivery of Nucleic Acids: The Case of Ionizable Lipid Nanoparticles. Bioeng. Transl. Med. 2021, 6 (2), e10213. 10.1002/btm2.10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris J. A.; et al. Cationic Lipid-Mediated Delivery of Proteins Enables Efficient Protein-Based Genome Editing in Vitro and in Vivo. Nat. Biotechnol. 2015, 33 (1), 73–80. 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.-B.; Dammer E. B; Ren R.-J.; Wang G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4 (1), 18. 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramishetti S.; et al. A Combinatorial Library of Lipid Nanoparticles for Rna Delivery to Leukocytes. Adv. Mater. 2020, 32 (12), 1906128. 10.1002/adma.201906128. [DOI] [PubMed] [Google Scholar]
- Jayaraman M.; et al. Maximizing the Potency of Sirna Lipid Nanoparticles for Hepatic Gene Silencing in Vivo. Angew. Chem., Int. Ed. 2012, 51 (34), 8529–8533. 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum D.; Gutkin A.; Kedmi R.; Ramishetti S.; Veiga N.; Jacobi A. M.; Schubert M. S.; Friedmann-Morvinski D.; Cohen Z. R.; Behlke M. A.; et al. Crispr-Cas9 Genome Editing Using Targeted Lipid Nanoparticles for Cancer Therapy. Science Advances 2020, 6 (47), eabc9450 10.1126/sciadv.abc9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj K. A.; et al. Branched-Tail Lipid Nanoparticles Potently Deliver Mrna in Vivo Due to Enhanced Ionization at Endosomal Ph. Small 2019, 15 (6), 1805097. 10.1002/smll.201805097. [DOI] [PubMed] [Google Scholar]
- Kim T.-I.; Kim S. W. Bioreducible Polymers for Gene Delivery. React. Funct. Polym. 2011, 71 (3), 344–349. 10.1016/j.reactfunctpolym.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton O. S.; et al. Synthesis and Biological Evaluation of Ionizable Lipid Materials for the in Vivo Delivery of Messenger Rna to B Lymphocytes. Adv. Mater. 2017, 29 (33), 1606944. 10.1002/adma.201606944. [DOI] [PubMed] [Google Scholar]
- Liu J.; et al. Fast and Efficient Crispr/Cas9 Genome Editing in Vivo Enabled by Bioreducible Lipid and Messenger Rna Nanoparticles. Adv. Mater. 2019, 31 (33), 1902575. 10.1002/adma.201902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu M.; Glass Z.; Chen J.; Haas M.; Jin X.; Zhao X.; Rui X.; Ye Z.; Li Y.; Zhang F.; Xu Q. Lipid Nanoparticle-Mediated Codelivery of Cas9Mrna and Single-Guide Rna Achieves Liver-Specific in Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (10), e2020401118 10.1073/pnas.2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn J. D.; et al. A Single Administration of Crispr/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In vivo Genome Editing. Cell Reports 2018, 22 (9), 2227–2235. 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- Kaczmarek J. C.; et al. Polymer-Lipid Nanoparticles for Systemic Delivery of Mrna to the Lungs. Angew. Chem., Int. Ed. 2016, 55 (44), 13808–13812. 10.1002/anie.201608450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek J. C.; et al. Systemic Delivery of Mrna and DNA to the Lung Using Polymer-Lipid Nanoparticles. Biomaterials 2021, 275, 120966. 10.1016/j.biomaterials.2021.120966. [DOI] [PubMed] [Google Scholar]
- Li Y.; et al. Intracellular Delivery and Biodistribution Study of Crispr/Cas9 Ribonucleoprotein Loaded Bioreducible Lipidoid Nanoparticles. Biomaterials Science 2019, 7 (2), 596–606. 10.1039/C8BM00637G. [DOI] [PubMed] [Google Scholar]
- Rui Y.; et al. Poly(Beta-Amino Ester) Nanoparticles Enable Nonviral Delivery of Crispr-Cas9 Plasmids for Gene Knockout and Gene Deletion. Molecular Therapy - Nucleic Acids 2020, 20, 661–672. 10.1016/j.omtn.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawker C. J.; Frechet J. M. J. Preparation of Polymers with Controlled Molecular Architecture. A New Convergent Approach to Dendritic Macromolecules. J. Am. Chem. Soc. 1990, 112 (21), 7638–7647. 10.1021/ja00177a027. [DOI] [Google Scholar]
- Bosman A. W.; Janssen H. M.; Meijer E. W. About Dendrimers: Structure, Physical Properties, and Applications. Chem. Rev. 1999, 99 (7), 1665–1688. 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- Zhou K.; et al. Modular Degradable Dendrimers Enable Small Rnas to Extend Survival in an Aggressive Liver Cancer Model. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (3), 520–525. 10.1073/pnas.1520756113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; et al. One-Component Multifunctional Sequence-Defined Ionizable Amphiphilic Janus Dendrimer Delivery Systems for Mrna. J. Am. Chem. Soc. 2021, 143 (31), 12315–12327. 10.1021/jacs.1c05813. [DOI] [PubMed] [Google Scholar]
- Khan O. F.; et al. Ionizable Amphiphilic Dendrimer-Based Nanomaterials with Alkyl-Chain-Substituted Amines for Tunable Sirna Delivery to the Liver Endothelium in Vivo. Angew. Chem., Int. Ed. 2014, 53 (52), 14397–14401. 10.1002/anie.201408221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O. F.; et al. Dendrimer-Inspired Nanomaterials for the in Vivo Delivery of Sirna to Lung Vasculature. Nano Lett. 2015, 15 (5), 3008–3016. 10.1021/nl5048972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; et al. Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic Fah Mrna to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater. 2018, 30 (52), 1805308. 10.1002/adma.201805308. [DOI] [PubMed] [Google Scholar]
- Farbiak L.; et al. All-in-One Dendrimer-Based Lipid Nanoparticles Enable Precise Hdr-Mediated Gene Editing in Vivo. Adv. Mater. 2021, 33 (30), 2006619. 10.1002/adma.202006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk J. S.; et al. Pegylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Delivery Rev. 2016, 99, 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodle M. C.; Lasic D. D. Sterically Stabilized Liposomes. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes 1992, 1113 (2), 171–199. 10.1016/0304-4157(92)90038-C. [DOI] [PubMed] [Google Scholar]
- Aggarwal P.; et al. Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Advanced drug delivery reviews 2009, 61 (6), 428–437. 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Lee R. J. The Role of Helper Lipids in Lipid Nanoparticles (Lnps) Designed for Oligonucleotide Delivery. Adv. Drug Delivery Rev. 2016, 99, 129–137. 10.1016/j.addr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Yan X.; et al. The Role of Apolipoprotein E in the Elimination of Liposomes from Blood by Hepatocytes in the Mouse. Biochem. Biophys. Res. Commun. 2005, 328 (1), 57–62. 10.1016/j.bbrc.2004.12.137. [DOI] [PubMed] [Google Scholar]
- Cullis P. R.; Chonn A.; Semple S. C. Interactions of Liposomes and Lipid-Based Carrier Systems with Blood Proteins: Relation to Clearance Behaviour in Vivo. Adv. Drug Deliv Rev. 1998, 32 (1–2), 3–17. 10.1016/S0169-409X(97)00128-2. [DOI] [PubMed] [Google Scholar]
- Chonn A.; Semple S. C.; Cullis P. R. Association of Blood Proteins with Large Unilamellar Liposomes in Vivo. Relation to Circulation Lifetimes. J. Biol. Chem. 1992, 267 (26), 18759–18765. 10.1016/S0021-9258(19)37026-7. [DOI] [PubMed] [Google Scholar]
- Francia V.; et al. The Biomolecular Corona of Lipid Nanoparticles for Gene Therapy. Bioconjugate Chem. 2020, 31 (9), 2046–2059. 10.1021/acs.bioconjchem.0c00366. [DOI] [PubMed] [Google Scholar]
- Akinc A.; et al. Targeted Delivery of Rnai Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Molecular Therapy 2010, 18 (7), 1357–1364. 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara M. L.; Persano F.; Persano S. Advances in Lipid Nanoparticles for Mrna-Based Cancer Immunotherapy. Front. Chem. 2020, 8 (963), 589959. 10.3389/fchem.2020.589959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.-D.; Chono S.; Huang L. Efficient Oncogene Silencing and Metastasis Inhibition Via Systemic Delivery of Sirna. Molecular Therapy 2008, 16 (5), 942–946. 10.1038/mt.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasargyri A.; Kümin C. D.; Leroux J.-C. Targeting Nanocarriers with Anisamide: Fact or Artifact?. Adv. Mater. 2017, 29 (7), 1603451. 10.1002/adma.201603451. [DOI] [PubMed] [Google Scholar]
- Tam Y. Y. C.; et al. Small Molecule Ligands for Enhanced Intracellular Delivery of Lipid Nanoparticle Formulations of Sirna. Nanomedicine: Nanotechnology, Biology and Medicine 2013, 9 (5), 665–674. 10.1016/j.nano.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Porkka K.; et al. A Fragment of the Hmgn2 Protein Homes to the Nuclei of Tumor Cells and Tumor Endothelial Cells in Vivo. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (11), 7444–7449. 10.1073/pnas.062189599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P.; et al. A Tumor-Homing Peptide with a Targeting Specificity Related to Lymphatic Vessels. Nature Medicine 2002, 8 (7), 751–755. 10.1038/nm720. [DOI] [PubMed] [Google Scholar]
- Laakkonen P.; et al. Antitumor Activity of a Homing Peptide That Targets Tumor Lymphatics and Tumor Cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (25), 9381–9386. 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z.; et al. Lyp-1-Conjugated Pegylated Liposomes: A Carrier System for Targeted Therapy of Lymphatic Metastatic Tumor. J. Controlled Release 2012, 157 (1), 118–125. 10.1016/j.jconrel.2011.07.034. [DOI] [PubMed] [Google Scholar]
- Cohen Z. R.; et al. Localized Rnai Therapeutics of Chemoresistant Grade Iv Glioma Using Hyaluronan-Grafted Lipid-Based Nanoparticles. ACS Nano 2015, 9 (2), 1581–1591. 10.1021/nn506248s. [DOI] [PubMed] [Google Scholar]
- Ramishetti S.; et al. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano 2015, 9 (7), 6706–6716. 10.1021/acsnano.5b02796. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; et al. Selective Organ Targeting (Sort) Nanoparticles for Tissue-Specific Mrna Delivery and Crispr–Cas Gene Editing. Nat. Nanotechnol. 2020, 15 (4), 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzigmann D.; et al. Lipid Nanoparticle Technology for Therapeutic Gene Regulation in the Liver. Adv. Drug Delivery Rev. 2020, 159, 344–363. 10.1016/j.addr.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri A.; et al. Lipid-Based Nanoparticles as Pharmaceutical Drug Carriers: From Concepts to Clinic. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26 (6), 523–580. 10.1615/CritRevTherDrugCarrierSyst.v26.i6.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; et al. Delivery Strategies of the Crispr-Cas9 Gene-Editing System for Therapeutic Applications. J. Controlled Release 2017, 266, 17–26. 10.1016/j.jconrel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; et al. Design of Lipid Nanoparticles for in Vitro and in Vivo Delivery of Plasmid DNA. Nanomedicine 2017, 13 (4), 1377–1387. 10.1016/j.nano.2016.12.014. [DOI] [PubMed] [Google Scholar]
- Li C.; et al. Ionizable Lipid-Assisted Efficient Hepatic Delivery of Gene Editing Elements for Oncotherapy. Bioact Mater. 2022, 9, 590–601. 10.1016/j.bioactmat.2021.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. B.; et al. Non-Viral Crispr/Cas Gene Editing in Vitro and in Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9Mrna and Sgrna. Angew. Chem., Int. Ed. 2017, 56 (4), 1059–1063. 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenjo E.; Hozumi H.; Makita Y.; Iwabuchi K. A.; Fujimoto N.; Matsumoto S.; Kimura M.; Amano Y.; Ifuku M.; Naoe Y. Low Immunogenicity of Lnp Allows Repeated Administrations of Crispr-Cas9Mrna into Skeletal Muscle in Mice. Nat. Commun. 2021, 12 (1), 7101. 10.1038/s41467-021-26714-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothgangl T.; et al. In Vivo Adenine Base Editing of Pcsk9 in Macaques Reduces Ldl Cholesterol Levels. Nat. Biotechnol. 2021, 39 (8), 949–957. 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K.; et al. In Vivo Crispr Base Editing of Pcsk9 Durably Lowers Cholesterol in Primates. Nature 2021, 593 (7859), 429–434. 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- Villiger L.; et al. In Vivo Cytidine Base Editing of Hepatocytes without Detectable Off-Target Mutations in Rna and DNA. Nature Biomedical Engineering 2021, 5 (2), 179–189. 10.1038/s41551-020-00671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q.; et al. Cell-Selective Messenger Rna Delivery and Crispr/Cas9 Genome Editing by Modulating the Interface of Phenylboronic Acid-Derived Lipid Nanoparticles and Cellular Surface Sialic Acid. ACS Appl. Mater. Interfaces 2019, 11 (50), 46585–46590. 10.1021/acsami.9b17749. [DOI] [PubMed] [Google Scholar]
- Veiga N.; Goldsmith M.; Granot Y.; Rosenblum D.; Dammes N.; Kedmi R.; Ramishetti S.; Peer D. Cell Specific Delivery of Modified Mrna Expressing Therapeutic Proteins to Leukocytes. Nat. Commun. 2018, 9 (1), 4493. 10.1038/s41467-018-06936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; et al. Efficient Delivery of Genome-Editing Proteins Using Bioreducible Lipid Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (11), 2868–2873. 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; et al. Fusion-Dependent Formation of Lipid Nanoparticles Containing Macromolecular Payloads. Nanoscale 2019, 11 (18), 9023–9031. 10.1039/C9NR02004G. [DOI] [PubMed] [Google Scholar]
- Kulkarni J. A.; Thomson S. B.; Zaifman J.; Leung J.; Wagner P. K.; Hill A.; Tam Y. Y. C.; Cullis P. R.; Petkau T. L.; Leavitt B. R. Spontaneous, Solvent-Free Entrapment of Sirna within Lipid Nanoparticles. Nanoscale 2020, 12 (47), 23959–23966. 10.1039/D0NR06816K. [DOI] [PubMed] [Google Scholar]
- A Study to Evaluate Efficacy, Safety, and Immunogenicity of Mrna-1273 Vaccine in Adults Aged 18 Years and Older to Prevent Covid-19. https://clinicaltrials.gov/ct2/show/NCT04470427 [accessed on 08/24/2021].
- Gillmore J. D.; et al. Crispr-Cas9 in Vivo Gene Editing for Transthyretin Amyloidosis. New England Journal of Medicine 2021, 385 (6), 493–502. 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- Ntla-2002 in Adults with Hereditary Angioedema (Hae) (Ntla-2002). https://clinicaltrials.gov/ct2/show/NCT05120830 [accessed on 08/24/2021].
- Seitzer J. Ntla-2002: Crispr/Cas9-Mediated Gene Knockout of Klkb1 to Treat Hereditary Angioedema. Journal of Allergy and Clinical Immunology 2021, 147 (2), AB147. 10.1016/j.jaci.2020.12.531. [DOI] [Google Scholar]
- Hoshyar N.; et al. The Effect of Nanoparticle Size on in Vivo Pharmacokinetics and Cellular Interaction. Nanomedicine (Lond) 2016, 11 (6), 673–92. 10.2217/nnm.16.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaei M.; et al. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10 (2), 57. 10.3390/pharmaceutics10020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó K.; et al. Suppression of Rna Recognition by Toll-Like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of Rna. Immunity 2005, 23 (2), 165–175. 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Karikó K.; et al. Incorporation of Pseudouridine into Mrna Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Molecular therapy: the journal of the American Society of Gene Therapy 2008, 16 (11), 1833–1840. 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; et al. Structure-Guided Chemical Modification of Guide Rna Enables Potent Non-Viral in Vivo Genome Editing. Nat. Biotechnol. 2017, 35 (12), 1179–1187. 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed J. R.; et al. Lipid Nanoparticle Chemistry Determines How Nucleoside Base Modifications Alter Mrna Delivery. J. Controlled Release 2022, 341, 206–214. 10.1016/j.jconrel.2021.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann M. D.; et al. Nanomaterial Delivery Systems for Mrna Vaccines. Vaccines 2021, 9 (1), 65. 10.3390/vaccines9010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration . Comirnaty and Pfizer-Biontech Covid-19 Vaccine. https://www.fda.gov/coronavirus-disease-2019-covid-19/comirnaty-and-pfizer-biontech-covid-19-vaccine [accessed on 01/01/2022].