

Abstract
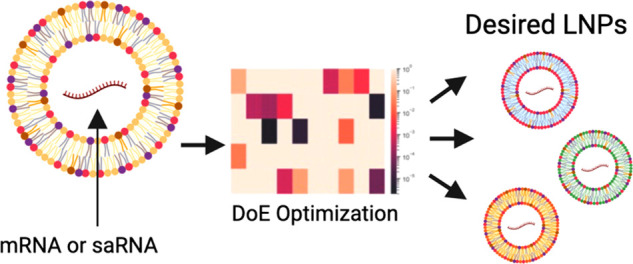
Lipid nanoparticles (LNPs) are the leading technology for RNA delivery, given the success of the Pfizer/BioNTech and Moderna COVID-19 mRNA (mRNA) vaccines, and small interfering RNA (siRNA) therapies (patisiran). However, optimization of LNP process parameters and compositions for larger RNA payloads such as self-amplifying RNA (saRNA), which can have complex secondary structures, have not been carried out. Furthermore, the interactions between process parameters, critical quality attributes (CQAs), and function, such as protein expression and cellular activation, are not well understood. Here, we used two iterations of design of experiments (DoE) (definitive screening design and Box–Behnken design) to optimize saRNA formulations using the leading, FDA-approved ionizable lipids (MC3, ALC-0315, and SM-102). We observed that PEG is required to preserve the CQAs and that saRNA is more challenging to encapsulate and preserve than mRNA. We identified three formulations to minimize cellular activation, maximize cellular activation, or meet a CQA profile while maximizing protein expression. The significant parameters and design of the response surface modeling and multiple response optimization may be useful for designing formulations for a range of applications, such as vaccines or protein replacement therapies, for larger RNA cargoes.
Keywords: lipid nanoparticle (LNP), self-amplifying mRNA (saRNA), mRNA (mRNA), protein expression, design-of-experiment (DoE), cytokine response, definitive screening design, Box−Behnken Design
1. Introduction
Lipid nanoparticles (LNPs) are the leading technology for nonviral nucleic acid delivery. In particular, LNPs have been clinically approved to facilitate efficient delivery of ribonucleic acid (RNA)-based medicines, including the mRNA (mRNA)–LNP vaccines from Pfizer-BioNTech and Moderna against coronavirus disease 2019 (COVID-19)1,2 and small interfering RNA (siRNA)–LNP therapeutics like patisiran, which treats polyneuropathies induced by hereditary transthyretin amyloidosis.3 Early research on RNA vaccines has demonstrated that naked RNA is quickly degraded after administration by cellular ribonucleases (RNases).4,5 LNPs slow down the degradation process to ensure RNA stability while also promoting cellular internalization via endocytosis and allowing intracellular release of RNA into the cytoplasm for translation by cellular machinery.6,7 The loading of mRNA cargo into nanoparticles is a spontaneous process wherein lipids undergo rapid mixing with RNA and collectively self-assemble into particles approximately 100 nm in diameter.8
The composition of LNPs typically includes an ionizable cationic lipid and three neutral helper lipids: phospholipid, cholesterol, and lipid-anchored polyethylene glycol (PEGylated lipid). The ionizable cationic lipids are electrostatically complexed with polyanionic RNA to enable its encapsulation by the neutral lipids and facilitate cellular uptake and endosomal escape.8,9 Phospholipids may play a role in membrane fusion and endosomal escape, but they mainly provide structural integrity to the particles.7,10 Similarly, cholesterol enhances stability and fluidity of the LNP and improves intracellular delivery.7,11 PEGylated lipids introduce a steric barrier at the surface of particles to increase colloidal stability by reducing aggregation and to “shield” particles from being bound by serum proteins in vivo including opsonins, which mark LNPs for removal by phagocytic systems.7,12 Consequently, the “PEG shield” may also interfere with cellular internalization and intracellular release by preventing lysosomal membrane destabilization after uptake by target cells.7,12
LNPs were initially optimized for formulating siRNA (∼23 nt) and have recently evolved to encapsulate larger RNA-agents, including mRNA (∼1000 nt).13−15 Recently, there has been increased activity for even larger RNA payloads, such as self-amplifying mRNA (saRNA).16 saRNA is a promising alternative to mRNA as it has been shown to induce immune responses with up to 100-fold lower doses and extended protein expression in vivo compared to mRNA.6,17,18 saRNA based on an alphavirus genome contains genes encoding the alphavirus RNA replication machinery where genes encoding viral structural proteins are replaced with a gene of interest. Upon entry into the cytoplasm, saRNA is translated by host cell machinery to assemble into the alphavirus replicase for producing multiple identical copies of the original saRNA strand, thus resulting in exponentially greater protein expression.6,16 However, saRNA (∼10 000 nt) is larger than mRNA (∼1000 nt) and has more secondary structure, making it more difficult to encapsulate and deliver.6 The inherent chemical and structural differences between mRNA and saRNA in terms of length, stability, and charge density suggests that LNP delivery formulations for saRNA may require conditions significantly different from those developed for mRNA delivery.19,20 Previous studies have optimized LNP compositions and production processes for siRNA and mRNA delivery using design of experiment (DoE) methodologies; however, there are limited reports available for optimizing LNP systems for larger payloads like saRNA. Furthermore, the composition and production parameters of LNPs can have profound effects on its physicochemical properties, including LNP size, charge, and morphology.21 However, there is limited understanding of how the LNP composition and formulation processes affects the final physicochemical properties that in turn influence protein expression and cellular activation. Therefore, understanding both the isolated and interactive effects of these parameters are important for developing a generalized approach to rapid and robust optimization of different RNA formulations.
To investigate the influence of LNP composition and formulation parameters for mRNA and saRNA delivery, we utilized a DoE approach to evaluate the parameters in a large multidimensional design space for efficient screening and examination of main effects, interactions, and second-order effects. We used the three leading, FDA-approved ionizable lipids in our study, including DLin-MC3-DMA (MC3), used in patisiran, ALC-0315, used in the Pfizer/BioNTech COVID-19 vaccine, and SM-102, used in the Moderna COVID-19 vaccine, as these lipids have not yet been explored for saRNA. Furthermore, in addition to standard analytical characterization techniques like dynamic light scattering and encapsulation efficiency, we also characterized the RNA integrity and proinflammatory cytokine secretion in response to the LNP formulations. Using formulation conditions of the original mRNA–LNPs as a starting point, we designed two libraries of LNPs using definitive screening design and Box–Behnken design (BBD), respectively, to improve the physicochemical properties, protein expression, and cytokine response in vitro. Through this approach, we optimized three different LNP-systems to maximize protein expression and trigger low or high levels of IL-6 response in vitro. These optimized formulations were used to encapsulate both mRNA and saRNA to validate the predicted response modeling and compare the two RNA types.
2. Materials and Methods
2.1. In Vitro Transcription
RNA transcripts were synthesized from constructs encoding firefly luciferase in either mRNA with tobacco mosaic virus (TMV) 5′ and 3′ UTRs22 or in saRNA with the nonstructural proteins of the Venezuelan equine encephalitis virus (VEEV), as previously described.20 Plasmid DNA (pDNA) was transformed into Escherichia coli, cultured in 100 mL of LB with 50 μg/mL ampicillin (Gibco, Thermo Fisher Scientific), and purified using a Plasmid Plus Maxiprep Kit (QIAGEN). pDNA concentrations and purity were measured on a NanoDrop One (Thermo Scientific). pDNA was linearized using SapI for 2 h at 37 °C and heat inactivated at 65 °C for 20 min. Uncapped RNA transcripts, used for iteration A, were synthesized using 1 μg of linearized DNA template in a MEGAScript T7 reaction (Invitrogen, ThermoFisher Scientific), and capped RNA transcripts, used for iteration B and validation of the optimized formulations, were synthesized using 750 ng of linearized DNA in a mMessage mMachine T7 reaction (Invitrogen, ThermoFisher Scientific), according to the manufacturer’s protocol. Transcripts were purified by overnight precipitation with LiCl at −20 °C, pelleted by centrifugation at 13 000 × g and 4 °C for 15 min, washed one time with 70% EtOH, centrifuged at 13 000 × g and 4 °C for 5 min, and resuspended in Tris-EDTA buffer (Sigma-Aldrich), then stored at −70 °C.
2.2. LNP Formulation
According to the conditions stipulated in Table 1, the ethanol phases were prepared at a concentration of 25 mM in ethanol by solubilizing (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino)butanoate (DLin-MC3-DMA, BroadPharm), ((4-hydroxybutyl)azanediyl)di(hexane-6,1-diyl) bis(2-hexyldecanoate) (ALC-0315, BroadPharm), heptadecan-9-yl 8-((2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino)octanoate (SM-102, BroadPharm), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC, Avanti Polar Lipids), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids), cholesterol (plant-derived, Avanti Polar Lipids), and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG-2000, Avanti Polar Lipids). The aqueous-phase RNA was prepared by diluting RNA transcripts in 25 mM sodium acetate according to the pH and RNA type specified in Table 1. The LNPs were formulated by microfluidic mixing as previously described.23,24 The T-tube mixing channel used an outlet tubing of 0.040 in. ID and an inlet tubing of 0.010 in. inner diameter (ID) for the ethanol phase and 0.020 in. ID for the aqueous phase. The ethanol and RNA phases were combined by microfluidic mixing using the Pump 33 DDS Syringe Pump (Harvard Apparatus) at a flow rate ratio of 1:3, respectively, using total flow rates (TFR) between 2 and 16 mL/min and temperatures between 4 and 37 °C, as specified in Table 1. The mixing effectively reduced the ethanol concentration to 25% upon leaving the micromixer. Immediately after mixing, the LNPs were diluted 10-fold with PBS and were purified using sterilized MWCO 10 kDa centrifugal filters (Amicon, Millipore Sigma) at 4 °C and 3214 × g for up to 4 h or until the concentration of RNA was ∼100 μg/mL. Fresh LNPs were used for dynamic light scattering and RiboGreen analysis, then stored at −70 °C until nanoparticle tracking analysis and bioanalyzer analysis. Formulations for iteration A were prepared in duplicate, while formulations for iteration B and the validation experiment were prepared in triplicate.
Table 1. Input and Outputs from Iteration A.
| experimental
inputs | |||
|---|---|---|---|
| factor | levels | ||
| N/P ratio | 5 | 10 | 15 |
| phospholipid type | DOPE | DSPC | |
| phospholipid content (mol %) | 10 | 15 | 20 |
| ionizable lipid type (corresponding pKa) | DLin-MC3-DMA (6.4) | ALC-0315 (6.09) | SM-102 (6.75) |
| ionizable lipid content (mol %) | 30 | 40 | 50 |
| DMG-PEG-2000 content (mol %) | 0 | 1.25 | 2.5 |
| total flow rate | 2 mL/min | 9 mL/min | 16 mL/min |
| ambient temperature during formulation (°C) | 4 | 20 | 37 |
| aqueous-phase pH | 3 | 5 | 7 |
| RNA type | mRNA | saRNA | |
| experimental
outputs | |
|---|---|
| critical quality attributes | analytical method |
| size | dynamic light scattering |
| PDI | dynamic light scattering |
| EE | RiboGreen assay |
| charge | dynamic light scattering |
| % filled particles | RiboGreen assay/nanoparticle tracking analysis |
| % full RNA transcripts | BioAnalyzer |
2.3. LNP Encapsulation Efficiency
The Quant-iT Ribogreen RNA assay kit (Invitrogen, Thermo Fisher Scientific) was used to determine the percentages of encapsulated RNA and RNA concentrations, as previously described.6 To quantify the unencapsulated amount of RNA in the LNPs, the samples were diluted to an expected total RNA concentration of 10 μg/mL in 1× TE buffer before addition of the RiboGreen dye. To quantify the total amount of RNA, the samples were diluted to an expected total RNA concentration of 10 μg/mL in 1× TE buffer containing 0.5% (v/v) Triton X-100 (Sigma-Aldrich) before addition of the dye. Standard solutions were prepared in 1× TE containing 0.5% (v/v) Triton X-100 at final RNA concentrations of 0–2.5 μg/mL. The assay was carried out according to the manufacturer’s protocol. Fluorescence intensities were measured at an excitation of 480 nm and emission of 520 nm. Dosing for transfection was based on the calculated encapsulated dose.
2.4. Physicochemical Characteristics of LNPs
2.4.1. Dynamic Light Scattering
LNP size, polydispersity index (PDI), and zeta potential were measured using the Zetasizer Nano (Malvern Instruments) and Zetasizer 7.12 software (Malvern) with the following settings: material refractive index of 1.4, absorbance of 0.01, dispersant viscosity of 0.882 cP, refractive index of 1.33, and dielectric constant of 79. LNPs were diluted 10-fold in PBS and equilibrated at room temperature (RT) prior to analysis in a plastic cuvette for particle size measurements or in a Dip cell for zeta potential measurements. Three measurements of up to 100 runs were collected for each sample until the value equilibrated.
2.4.2. Nanoparticle Tracking Analysis
Nanoparticle tracking analysis (NTA) was done using the Malvern Nanosight NS300 (Malvern Pananalytical) and NanoSight 3.4 software (Malvern Instruments) to measure the particle concentration. Samples were diluted 100- to 10 000-fold in 1× PBS to 10–40 particles per frame, and then three 30 s videos were taken for analysis. The number of transcripts per μg of RNA was calculated using the following equation:
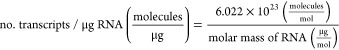 |
The number of full RNA transcripts per LNP was calculated using the following equation:
 |
2.5. Quality of Encapsulated RNA
RNA was isolated from the LNPs and analyzed for RNA quality. To lyse the LNPs, a 200 μL sample was added to an equal volume of nuclease-free water containing 2% (v/v) Triton X-100 and incubated at room temperature for 5 min. RNA was purified using the Monarch 50 μg RNA Cleanup Kit (New England Biolabs), according to the manufacturer’s protocol. RNA was eluted into 6 μL, and the concentration was adjusted to 25 ng/μL to 250 ng/μL using nuclease-free water. RNA quality was measured using the Bioanalyzer RNA 6000 Nano Kit (Agilent Technologies), 2100 Bioanalyzer Instrument (Agilent Technologies), and Bioanalyzer 2100 Expert software (Agilent Technologies), according to the manufacturer’s protocol. The peaks of full mRNA and saRNA transcripts occurred at elution times of 34–37 s and 51–67 s, respectively. All peaks appearing outside these ranges were considered to indicate truncated RNA. The area of the full transcript peaks and total peak area was used to calculate RNA integrity.
2.6. Quantification of In Vitro Protein Expression and Cellular Activation
Luciferase expression and cytokine response were measured in transfected HeLa cells (ATCC). It is important to reiterate that the objective of our study was focused on determining which process parameters should be manipulated to achieve a specific LNP profile, and the in vitro model was used to validate the ability for the optimized LNPs to enable sufficient protein expression and detectable immune response. HeLa cells were selected as our in vitro model system because they have been shown to be effectively transfected with LNPs and could exhibit a quantifiable inflammatory response.25 Previous studies demonstrated their relevance to capture LNP uptake and RNA endosomal escape, and their ability to accurately predict in vivo performance.26,27 Prior to transfection, HeLa cells (ATCC) were maintained in complete Dulbecco’s modified Eagle’s medium (cDMEM) (Gibco, Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS), 5 mg/mL l-glutamine, 10% GlutaMAXTM, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Thermo Fisher Scientific). Cells were confirmed to be mycoplasma-free and then plated at a density of 35 000 cells/well in a clear 96-well plate ay 18–24 h prior to transfection and cultured at 37 °C and 5% CO2. A 500 ng dose of fLuc-saRNA was used per well in a volume of 100 μL of PBS, which was added to a well already containing 50 μL of transfection medium (DMEM with 5 mg/mL l-glutamine). A high RNA dose was used to ensure a strong inflammatory response for detection by the immunoassay platforms used. Cells were allowed to transfect for 4 h, and then the media was replaced with 100 μL of cDMEM. At 24 h after the initial transfection, 70 μL of supernatant was removed from each well and stored at −70 °C for quantification of cellular activation. The remaining supernatant and cells were used for firefly luciferase assays. ONE-GloTM D-luciferin reagent (Promega) was prepared according to the manufacturer’s protocol. The remaining 30 μL of supernatant in each well was used, and the cells were lysed at 36 °C for 10 min. Next, 30 μL from each well was transferred to a white 96-well plate and analyzed on a plate reader, and background from the supernatant of a cell-only control was subtracted from the measurements. For iteration B, the U-PLEX multiplex immunoassay platform (Meso Scale Discovery) was used to screen the stored supernatants for proinflammatory cytokines (IFN-γ, IL-12p70, IL-13, IL-18, IL1β, IL-2, IL-4, IL-5, IL-6, and TNF-α) according to the manufacturer’s protocol. A 25 μL aliquot of each supernatant was used for the platform, and duplicates of an 8-point standard curve were prepared on each plate. For validation of the optimized LNP formulations, the IL-6 Human Elisa Kit (Invitrogen, ThermoFisher Scientific) was used to screen the stored supernatants according to the manufacturer’s protocol.
2.7. Design of Experiment
DoE was carried out using JMP 13 software (SAS Institute). Duplicates were used in iteration A (DoE 1), and triplicates were used for iteration B (DoE 2). For iteration A, a definitive screening design (DSD) was created with 7 three-level quantitative factors (N/P ratio, phospholipid content, DMG-PEG-2000 content, ionizable lipid content, TFR, temperature, and buffer pH), 2 two-level qualitative factors (phospholipid type and RNA type), and 1 three-level qualitative factor (ionizable lipid type), resulting in a large 10D design space (Table 1). DoE was used to reduce the number of possible formulations (37 × 22 × 3 = 26 244) to a more practical number of 26 formulations. Of the 10 formulation parameters tested in iteration A, four parameters were selected and tested with narrower ranges of levels and more center point values to detect effects more reliably). For iteration B, a Box–Behnken design (BBD) was created with 3 three-level quantitative factors (phospholipid content, ionizable lipid content, and aqueous-phase pH) and 1 three-level qualitative factors (ionizable lipid type), resulting in a 4D design space in which the number of possible formulations (32 × 32 = 81) was reduced to 26 formulations (Table 2). Given the number of CQAs under study and our lack of mechanistic understanding of the exact RNA–LNP formation processes, the development of a holistic model-based DoE is currently unrealistic.28,29 Additional complex, nonlinear interactions between CQAs and formulation parameters are thus anticipated but could not be accurately captured at the present.30
Table 2. Inputs and Outputs from Iteration B.
| experimental
inputs | |||
|---|---|---|---|
| factor | levels | ||
| phospholipid content (mol %) | 12.5 | 15 | 17.5 |
| ionizable lipid type (corresponding pKa) | D-Lin-MC3-DMA (6.4) | ALC-0315 (6.09) | SM-102 (6.75) |
| ionizable lipid content (mol %) | 35 | 40 | 45 |
| aqueous-phase pH | 4 | 5 | 6 |
| Fixed Parameters | |||
| N/P ratio | 10 | ||
| phospholipid type | DOPE | ||
| DMG-PEG-2000 content (mol %) | 1.25 | ||
| total flow rate (mL/min) | 16 | ||
| ambient temperature during formulation (°C) | 20 | ||
| experimental
outputs | |
|---|---|
| critical quality attributes | analytical method |
| size | dynamic light scattering |
| PDI | dynamic light scattering |
| EE | RiboGreen Assay |
| charge | dynamic light scattering |
| % filled particles | RiboGreen assay/nanoparticle tracking analysis |
| % full RNA transcripts | BioAnalyzer |
| protein expression | luciferase assay |
| cellular activation | MSD cytokine assay |
2.8. Statistical Analysis
The primary analysis of both iterations A and B were conducted in Python, using analysis of variance (ANOVA), linear regression, and Spearman’s correlation methods. The scikit-learn module and statsmodel package were used to conduct this analysis, and the seaborn library was used for plotting.31,32 Model statistical significance was defined as p-values less than 0.05, and lack-of-fit was considered insignificant at a level of 0.1.
2.9. Response Surface Modeling and Multiple Response Optimization
For iteration B, response surface modeling was carried out using second-order ordinary least square (OLS) regression based on the scikit-learn module and statsmodel package. Ionizable lipid types were converted into a continuous variable based on lipid pKa values (Tables 1 and 2). Explanatory variables were coded from −1 to 1. Box-cox transformation of response variables did not strongly improve model accuracy.33 The optimization of simultaneous responses was realized by introducing an overall desirability function (see section 3.4.). Optimal operating conditions were then obtained using the scipy library and BFGS optimization algorithm.34
3. Results and Discussion
3.1. Overview of the DoE
The purpose of the first iteration, iteration A, was to screen for significant formulation parameters and select appropriate levels for the second DoE iteration. On the basis of previous studies, desirable LNPs tended to have the following physicochemical characteristics: particle size (Z-average diameter) between 80 and 100 nm, polydispersity index (PDI) less than 0.2, encapsulation efficiency (EE) at least 80%, and neutral zeta potential.8,21,35−38 Nanoparticle formulations that were optimized for mRNA delivery were compiled from previous studies and used to determine the appropriate parameters and range of levels for iteration A (Figure 1, Table 1, and Supplementary Table 7).8,21,35−37 Linear regression models and ANOVA were used to assess main and potential interaction effects in Iteration A. The second iteration, iteration B, was designed using significant parameters and ranges detected in iteration A with an objective to fine-tune the LNP systems for both high protein expression and varied levels of cellular activation in vitro (Table 2 and Supplementary Table 8). The effects of the parameters were captured using polynomial OLS regression models to detect any quadratic and interaction relationships. As expected, all LNPs in iteration B had improved physicochemical particle characteristics compared to iteration A, as all 26 formulations exhibited sizes in the range of 76–126 nm, PDI below 0.2, neutral charge (−5 to +5 mV), and EE of >50%. With regards to function, 17 of 26 formulations of fLuc saRNA–LNPs exhibited 2.5- to 16-fold higher levels of protein expression compared to Lipofectamine 3000 mediated saRNA delivery. Cellular activation was detected for all 26 formulations at total cytokine levels ranging from 350 to 1100 pg/mL. Finally, three optimized LNP formulations were determined using a desirability function to (1) minimize IL-6 response, (2) maximize IL-6 response, and (3) optimize critical quality attributes (CQAs). Model predictions of the CQAs, protein expression, and IL-6 response were validated by using the optimized LNP formulations to encapsulate both mRNA and saRNA.
Figure 1.

Composition of LNPs including ionizable lipid, phospholipid, cholesterol, and PEGylated lipid encapsulating RNA of varying size; firefly luciferase mRNA (fLuc mRNA, ∼1700 nt) or self-amplifying RNA (fLuc saRNA, ∼9300 nt).
3.2. Screening for Significant Parameters of LNP Physicochemical Properties
3.2.1. PEGylated Lipids are Crucial for Stabilization of LNP
The absence of DMG-PEG-2000 resulted in large and unstable particles (220–3564 nm, PDI 0.199–1.000) (Figure 2A,B). Unexpectedly, conclusions for other parameters could not be sufficiently drawn from these unstable formulations and were subsequently removed from analysis; thus, 15 of 26 formulations in iteration A were viable for analysis. The PEG moiety of DMG-PEG-2000 is a polyether consisting of ethoxy units that orient to the surface of LNPs due to its hydrophilic nature. PEG generates a hydrated environment that introduces a steric barrier against neighboring nanoparticles for effective stabilization and prevention of aggregation and fusion.39−41
Figure 2.

Statistical analysis from definite screening design experiments (Iteration A). (A, B) Box plot displaying LNP size and PDI as a function of PEG lipid content. (C) Heat map of correlations (p-value) from ANOVA carried out on the restricted data set. Explanatory variables with p-value < 0.1 are excluded from ANOVA model. (D) Heat map of regression coefficients for main and quadratic effects detected by ANOVA.
LNPs containing PEG did not significantly vary in size, PDI, encapsulation efficiency, charge, or RNA integrity regardless of the level of PEG content (Figure 2C). In other words, the presence of PEG was crucial for improved physicochemical properties of LNPs; however, the level has potential to be further minimized below 1.25%, which is the lowest level tested in this study. Why is it ideal to minimize the PEGylated lipid content in LNPs? There have been recorded cases of anaphylactic shock due to PEG-induced hypersensitivity reactions (HRs); in fact, PEG is considered one of the possible causes of anaphylaxis associated with COVID-19 vaccines like the Pfizer-BioNTech and Moderna mRNA vaccines.42−46 Thus, it is of interest to minimize the PEGylated lipid content in LNPs or to develop future RNA formulations that are stabilized by non-PEG lipids.
3.2.2. Physicochemical Properties of LNPs
The TFR and ambient temperature may affect the level of aggregation of neutral lipids during formulation, resulting in larger LNPs. We observed that increasing TFR significantly reduced the hydrodynamic diameter of particles (Figure 2C,D). Previous studies have suggested that at a higher TFR the increased turbulence in the microfluidic device may create smaller fluid eddies, allowing the lipids and RNA molecules to be distributed more uniformly in the mixing tube.35,47 Consequently, fewer molecules are available in each eddy to assemble into particles, resulting in smaller LNPs.47 In other words, the greater turbulence produced by a higher TFR may prevent neutral lipids from aggregating before encapsulating the RNA molecules. Temperature also had a significant role in affecting particle size (Figure 2C). LNPs were smaller as temperature increased during formulation (Figure 2D). Low temperatures likely induced phospholipids to adopt more rigid structures and form larger aggregates prior to encapsulating RNA, resulting in larger particles.48 As described in a previous report on lipid vesicle aggregation induced by cooling, newly formed LNPs may continue aggregating under low temperatures following formulation.48
Furthermore, the aqueous-phase pH may be tuned to significantly alter particle size (Figure 2C). Increasingly acidic (lower) pH resulted in smaller particles (Figure 2D). MC3, ALC-0315, and SM-102 have pKa values of 6.4, 6.09, and 6.75, so there were likely more opportunities for electrostatic interactions at pH 3 and 5. The ionizable lipids are more highly charged at lower pH levels, resulting in strong interactions between the anionic RNA molecules and ionizable cationic lipids. We hypothesize that more frequent electrostatic interactions may result in rapid particle formation and smaller particles. Finally, size was negatively correlated with a higher N/P ratio (Figure 2D), similar to trends reported elsewhere.49,50
Interestingly, the significant parameters for size did not overlap with those for PDI, which was affected by phospholipid type and content, ionizable lipid type, and RNA type (Figure 2C). PDI was highest in DSPC-containing LNPs that were formulated with low phospholipid content (Figure 2D and Supplementary Figure 1). Conical lipids, like DOPE, tend to adopt an inverted hexagonal H(II) phase, while cylindrical lipids, like DSPC, tend to adopt a lamellar phase.51 As previously noted, DOPE is preferential over DSPC for providing improved packing properties in the inverted micellar environment surrounding RNA and more consistently sized particles.51 In a previous study, DOPE-containing LNPs were reported to form uniformly spherical particles compared to the DSPC-containing counterpart.52 It has been established that phospholipids are crucial for providing structural integrity to LNPs, so by extension increasing the level of phospholipid content likely resulted in more compacted and stable particles.
EE was significantly affected by temperature, phospholipid content, and ionizable content (Figure 2C,D). EE was generally highest at a midrange ambient temperature (20 °C) with midrange phospholipid content (15%) and midrange ionizable lipid content (40%) (Supplementary Figure 1).
3.2.3. Comparison of mRNA and saRNA
The integrity of encapsulated RNA and PDI was significantly different between the mRNA and saRNA (Figure 2C,D and Supplementary Figure 3). fLuc saRNA (∼9300 nt) is over 5-fold larger than fLuc mRNA (∼1700 nt), so we hypothesize the two RNAs may form different secondary and tertiary structures with varying stabilities. Thus, saRNA may have been more susceptible to degradation during the formulation process. Furthermore, during the in vitro transcription reaction, there was likely more mRNA that was completely transcribed than saRNA due to the large difference in RNA size, length of reaction, and limited amount of nucleotides in the reaction. By extension, saRNA molecules may have been present in diverse lengths during formulation, consequently producing more polydisperse LNPs than the mRNA counterpart. However, structural integrity for both RNA types were affected by the ionizable lipid type and content, and temperature (Figure 2C,D).
Our results show that the same LNP technology can be used interchangeably between saRNA and mRNA, although larger payloads are more fragile and increasingly likely to degrade during the encapsulation process and to influence the PDI. This implies that saRNA can replace current mRNA-optimized LNP systems with relative ease and minimal optimization required in terms of process conditions. We also hypothesize that optimal conditions for saRNA or large RNA payloads can also be applied to smaller payloads such as mRNA. These results motivate development of a gentle process that yield LNPs with high encapsulation efficiency of saRNA and maintain integrity.
3.2.4. Selection of Significant Parameters for Iteration B
Moving forward, only saRNA and DOPE lipid were used for iteration B, as saRNA was seemingly more challenging to encapsulate and preserve during processing, and DOPE was shown to be advantageous compared to DSPC according to iteration A. In addition, multiple pairwise comparisons from iteration A enabled us to fix the processing temperature, N/P ratio, and TFR at optimal values (Table 2, Supplementary Figure 1). Thus, the ionizable lipid content and type, pH, and phospholipid content were kept as variables for in vitro optimization as these parameters are crucial for LNP formation, and many interactions effects are anticipated. The continuous variable ranges were also reduced by half and centered around the preferential level for fine-tuning purposes using BBD of experiments (Table 2). This design was selected as it allows accurate estimations of interactions and quadratic effects with a limited number of experimental runs.53 As compared to other conventional response surface methods, such as central composite design, BBD is rotatable and more suited when three factors are available.
The temperature was also fixed as there is limited practicality for scale-up of formulation processes at either 4 or 37 °C, and the TFR used in our system is unique to the T-junction setup and may not apply to other lab-scale microfluidic or scale-up technologies, such as jet impingement mixers.
3.3. Optimization of saRNA–LNP Function In Vitro
3.3.1. RNA–LNP Quality Attributes
The results of BBD allowed development of accurate response surface models based on polynomial regression (see the “Material and Methods” section) and was employed for iteration B as the objective was to fine-tune process parameters for protein expression and cellular activation, as opposed to screening formulations in a wide design space.
Improving model fitting would certainly require better control of process variability or measurement errors. The lack of fit for PDI and zeta potential is well-explained by the narrow range of values observed in iteration B. However, these quality attributes remain in acceptable ranges throughout the entire design space, with an averaged PDI of 0.13 and zeta potential of −2.5 mV.
We observed that the effect of pH was pervasive in all measured physicochemical properties of LNPs. Increasing pH resulted in significantly smaller particles (Figure 3A). Ionizable lipid content and phospholipid content had a quadratic effect on size, such that particles were largest at 40% ionizable lipid content and 15% phospholipid content (Figure 3B,C). Particles were smallest when SM-102 was used (Supplementary Figure 2A). This phenomenon was not observed in iteration A, likely because effects from TFR were more dominant. Notably, EE was significantly affected by interactive effects between pH and ionizable lipid content. EE significantly decreased as ionizable lipid content increased; however, when interactions with pH is considered, EE is the highest at either low pH and high ionizable lipid content or high pH and low ionizable lipid content (Figure 3 and Supplementary Table 2). Presumably, low pH may have enabled better complexation between the ionizable lipid and RNA, thereby, resulting in high EE. Conversely, at high pH levels, that is, above the pKa of the ionizable lipids, there are weak interactions with the RNA such that ionizable lipids do not contribute to the encapsulation process; therefore, a lower level of ionizable lipids may allow other lipid components to better encapsulate the RNA. Nevertheless, future studies should carry out a structural analysis of the RNA complexation with each ionizable lipid to elucidate these results. Moreover, at low pH, EE was highest when SM-102 was used due to interactive effects (Supplementary Table 2). Although all the ionizable lipids have pKa above 5, SM-102 has the highest pKa of 6.75. We hypothesize relatively more SM-102 molecules were in the protonated form, allowing for better complexation with RNA molecules during formulation, thus allowing better encapsulation.54 RNA integrity was best preserved at pH 5 (Figure 3C). As previously observed, this phenomenon occurs because RNA phosphodiester bond hydrolysis is lowest at pH 4–5.55 Alkaline hydrolysis occurs at pH > 6, while acid hydrolysis occurs at pH < 2. The approximated number of full-length RNA transcripts per particle was mainly affected by pH and ionizable lipid type (Supplementary Table 4). This number increased as pH decreased and was highest when MC3 was used. However, the approximated values (39–67 molecules per particle) were higher than anticipated (1–10 molecules per particle). It is likely that the threshold for particle detection in the NTA analyses was set too high, thereby underestimating the particle number. Setting the threshold in NTA is arbitrary, so these counts should not be considered absolute but rather a relative measure for comparison between formulations.
Figure 3.

Response surface plot from Box–Benhken design experiments (Iteration B). IL-6 cytokine release (heat map) as a function of LNP size (A), encapsulation efficiency (B), and RNA integrity (C), predicted using second-order ordinary least square regression. Nondisplayed explanatory variables are fixed at center point, and ALC-0315 is herein selected (see Table 2). Black circles indicate the local maxima for protein expression, and black squares indicate the local minima for protein expression.
3.3.2. Protein Expression and Cellular Activation of LNPs
Protein expression and cellular activation levels were not strongly correlated with individual LNP CQAs, suggesting more complex interactions between formulation parameters, LNP structure, and in vitro activity (Figure 4D,E). We observed that the ionizable lipid type played a critical role in protein expression, as MC3-containing LNPs did not enable potent protein expression relative to the other ionizable lipids (Figure 4A). In addition, the highest levels were observed in LNPs containing ALC-0315, followed by those containing SM-102 (Figure 4A). Once MC3-containing LNPs were removed from the analysis, protein expression could be modeled well by the different process parameters (R2 = 0.95, see Supplementary Table 6). It is important to note that further testing, such as cryo-electron microscopy or SAXS measurements, could provide crucial structural insight and would allow a better mechanistic understanding of RNA–LNP function and formation. For instance, the effects of these parameters on LNP morphological shape, inner structure, and RNA microenvironment might better explain the variability observed in potency and cellular activation. Furthermore, looking at the structure–activity relationship would be insightful for elucidating the significant interactions between ionizable lipid content and protein expression.
Figure 4.

LNP protein expression profile and statistical analysis from BBD experiments (Iteration B). (A) Box plot displaying protein expression (RLU) as a function of ionizable lipid type. (B) Response surface plot for protein expression for LNP containing ALC-0315 ionizable lipid. (C) Response surface plot for protein expression for LNP containing SM-102. Heat map plot displaying Spearman correlation’s coefficient (D) and p-values (E) between CQAs. LNPs containing MC3 ionizable lipid are excluded from the protein expression analysis in B–E due to low expression levels.
Despite the lack of model prediction with inclusion of MC3-containing LNPs, significant statistical correlations can still be detected (Figure 4). Protein expression increased with higher ionizable lipid content and pH, and this effect was strongest when SM-102 was used (Figure 4B,C). This was expected as the number of full-length transcripts per particle followed the same trend, and mRNA loading per particle was previously shown to be a critical factor for enhanced mRNA functional delivery of LNPs (Supplementary Table 4).56 Of particular interest, protein expression was positively correlated with IL-6 cytokine release but negatively correlated with IL-18 level (Figure 4D,E). For instance, toll-like receptor 4 (TLR4) activation, potentially related with IL-18 release, can explain the reduced potency as it is known to block RNA translation and LNP uptake in cells.57,58 In addition, the activation of these proinflammatory cytokines was not correlated with RNA integrity level, showcasing cellular activation likely originated from LNP properties or lipid-RNA interactions.
The immunogenicity of LNPs can be affected by numerous factors, including the particles’ physicochemical properties, therapeutic payload, surface-decorating moieties, and cargo identity. Many components of LNPs may be recognized as foreign and trigger intricate immune responses. For iteration B, total cytokine levels in vitro ranged from 350 to 1100 pg/mL, which was over 2-fold less than the response to Lipofectamine 3000 complexed saRNA (2300 pg/mL). Interestingly, IL-6 cytokines made up nearly 100% of the total cytokines measured. IL-18 was detected in all 26 formulations; however, the levels were too low for robust statistical predictions in the final LNP optimization. Several parameters were statistically significant in affecting release of IL-6 cytokine in vitro (Figure 4D,E and Supplementary Table 5). IL-6 secretion increased with aqueous-phase pH and was highest when SM-102 was used (Figure 4B,C). High immunogenicity is negatively correlated with low EE (Figure 4D,E), suggesting the presence of unencapsulated RNA may drive IL-6 response. Levels were also highest at phospholipid content of 15% (Supplemental Table 5). IL-6 activates inflammatory cells during acute fever reactions and increases vascular permeability to cause swelling and redness.59 Vaccine immunogenicity can result in some observed adverse events,60 and interestingly, IL-6 has previously been shown to positively correlate with enhanced RNA vaccine immunogenicity.61 Thus, being able to tune LNP composition and process parameters to induce IL-6 or not is highly advantageous. Unlocking the RNA technology for gene therapies applications, or chronic administrations, will certainly require efforts to reduce this innate immune activation.62
3.4. Optimized LNP Formulations
The optimized LNP formulations were obtained using a desirability function (objective function), such that the maximum desirability is achieved when the conditions are as follows: RNA integrity exceeding 50%, size below 100 nm, PDI below 0.2, EE exceeding 80%, and protein expression maximized. Immunogenicity is either minimized (in desirability function 1) or maximized (desirability function 2). Regardless of in vitro measurement, a third function (desirability function 3) is defined to maximize RNA integrity. The multiresponse problem is solved using the approach introduced by Derringer and Suich.63 This approach, combined with response surface methodology, is a powerful tool to find optimal balance between multiple antagonist responses.64
More precisely, each relevant response surface model is transformed into a response function d, such as
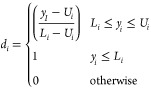 |
when the response is minimized, or
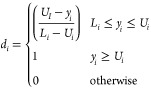 |
when the response is maximized, where Ui is the upper limit (or a target value) and Li is the lower limit (or a target value).
These individual functions are then aggregated into an overall desirability measure D, ranging from 0 to 1. D is herein defined as the weighted geometric mean:
with wi defined as the relative weight of each response function di to optimize.
In this study, the overall desirability D is then optimized for the complete design space with identical weights (Figure 5). Optimal process conditions are displayed in Table 3.
Figure 5.

Multiple response surface plot from BBD (Iteration B) based on desirability function optimization. IL-6 cytokine release is minimized in panels A–C (Desirability (1)) and maximized in panels D–F (Desirability (2)). In panels G–I, only physical CQAs are optimized (Desirability (3)). Nondisplayed explanatory variables are fixed at center point (see Table 2).
Table 3. Optimal Conditions for saRNA–LNP Formulation with Regards to Cellular Activation or LNP CQAs.
| input | optimal value 1: minimize cellular activation | optimal value 2: maximize cellular activation | optimal value 3: optimize CQAs |
|---|---|---|---|
| phospholipid content (mol %) | 15.9 | 17.5 | 17.5 |
| aqueous-phase pH | 4.53 | 6 | 5.25 |
| ionizable lipid type | ALC-0315 | SM-102 | ALC-0315 |
| ionizable lipid content (mol %) | 45 | 45 | 35 |
| Fixed Parameters | |
| N/P ratio | 10 |
| phospholipid type | DOPE |
| DMG-PEG-2000 content (mol %) | 1.25 |
| total flow rate (mL/min) | 16 |
| ambient temperature during formulation (°C) | 20 |
Depending on the objective of the desirability function, distinct preferential conditions appear (Table 3). In all cases, the overall desirability is high (greater than 0.8), suggesting satisfactory optimization. The predicted LNP- and RNA-related attributes further confirm appropriate levels of quality assurance at optimal conditions (Table 4). Even though the target of 50% for RNA integrity is herein not achieved, as currently defined for BNT162b mRNA vaccine specification, RNA integrity remains acceptable given the higher fragility of self-amplifying RNA molecules.65,66
Table 4. Model Predictions and Experimental Values of CQAs, Cellular Activation, and Protein Expression for the Optimized LNP Formulations.
| optimal
1 |
optimal
2 |
optimal
3 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| output | predicted | mRNA | saRNA | predicted | mRNA | saRNA | predicted | mRNA | saRNA |
| EE (%) | 81.42 | 76.73 | 77.97 | 80.27 | 82.00 | 87.91 | 92.03 | 86.10 | 92.29 |
| size (nm) | 104.83 | 82.62 | 87.72 | 92.83 | 85.56 | 85.51 | 100.00 | 81.70 | 81.45 |
| RNA integrity (% full transcript) | 48.74 | 73.87 | 54.80 | 42.27 | 73.10 | 47.65a | 49.43 | 84.23 | 52.30 |
| IL-6 release (pg/mL) | 780 | 872 | 771 | 1424 | 1324 | 1478 | 881 | 823 | 782 |
| protein expression (RLU) | 2.01 × 107 | 5.65 × 104 | 2.04 × 107 | 2.42 × 107 | 3.61 × 105 | 1.17 × 107 | 1.80 × 107 | 3.55 × 104 | 1.32 × 107 |
| overall desirability | 0.8228 | 0.9165 | 0.9203 | ||||||
One outlier replicate was removed from analysis due to highly apparent degradation by RNase.
As expected, pH and ionizable lipid type play a pivotal role in overall process optimization. The combination of an acidic buffer (pH 4.53) with ALC-135 prevents IL-6 cytokine release, while the combination of a higher pH value (pH 6.00) along with SM-102 lead to highly immunogenic systems. Interestingly, operating at intermediate pH values (5.24) with low level of ionizable lipid (35 mol %) seems to be the preferred approach to maximize RNA integrity and encapsulation (Table 4). We also observed that the predicted number of RNA molecules per particle was ∼2-fold higher for formulations with minimized immunogenicity (optimal condition 1, Table 4) than for formulations with maximized immunogenicity optimal condition 2, Table 4).
3.5. Validating the Optimized LNP Formulations for mRNA and saRNA Delivery
The optimized LNP formulations were used to encapsulate both fLuc mRNA and saRNA to validate the CQA prediction model (Tables 3 and 4, Supplemental Table 9). This external validation procedure confirms the robustness and accuracy of the developed prediction models, and showcases the utility of our approach for simultaneous product and process development. In addition, results show that LNPs encapsulating either mRNA or saRNA were comparable in EE and size, as expected based on conclusions from Iteration A, as well as the level of IL-6 released (Table 4). PDI for LNPs encapsulating both RNA types were also less than 0.2 and had a neutral charge. Again, these results indicate that the same LNP technology can be used interchangeably between the two RNA types to achieve the desired LNP physicochemical profile. Furthermore, although saRNA is more fragile than mRNA, it can still result in 30- to 370-fold higher levels of protein expression. When compared to the prediction model, saRNA–LNPs showed the same magnitude of protein expression as the predicted values and all physicochemical characteristics were within range of the prediction interval (Table 4), except for RNA integrity of mRNA–LNPs, as expected.
Overall, these results establish significant parameters for a more rational design of RNA–LNP formulation with regard to achieving a specific profile of CQAs, protein expression and IL-6 response. There may be applications of RNA technology that require LNP formulations to have minimal cellular activation, such as protein replacement therapy or gene transfer, whereas maximizing cellular activation may be more relevant in the context of vaccine design and cancer treatment.67 In the future, it may even be possible to design and select for the most relevant inflammatory pathways for certain formulations by LNP composition and process alone. For example, it has been shown that IL-6 is integral to the immunogenicity of RNA vaccines; thus, an LNP formulation that induces IL-6 would be highly desirable for this application.61 Furthermore, the physical attributes, such as size and PDI, and in vitro cellular activation can be decoupled, which makes it possible to tune the activation profile by selecting for LNP composition while maintaining an equivalent CQA profile. While previous studies have shown that in vitro observations do not necessarily predict in vivo efficacy,27 we believe that this study is the first step to understanding the role of these design parameters on protein expression and cellular activation, and the DoE approach and analytical methods used here can be extended to in vivo evaluation in the future.
4. Conclusion
Here, we optimized LNP compositions with the three leading, FDA-approved ionizable lipids (MC3, ALC-0315, and SM-102) for encapsulation of saRNA, protein expression, and cellular activation. We observed that PEG is necessary to preserve CQAs such as size and PDI and that while similar manufacturing processes can be used for RNA cargoes of varying size, it is more challenging to preserve the integrity of larger RNA payloads, like saRNA, compared to mRNA. We defined the design space for both protein expression and cytokine activation and identified three optimal formulations for minimizing cellular activation, maximizing cellular activation, or preserving only physical CQAs. These results are useful to inform LNP formulations that may require disparate inflammatory profiles, such as vaccines and protein replacement therapies and to provide insight for efficient encapsulation of larger RNA cargoes, like saRNA, that may also be applicable to other large RNA systems such as Cas9 or multigene expression systems.
Acknowledgments
A.K.B., H.H.L., and S.K.V.S. were funded by the Natural Sciences and Engineering Research Council (2021-02931), Canadian Foundation for Innovation, and BC Knowledge Development Fund (41164), Michael Smith Foundation for Health Research (2021-1544), and startup funds provided by the Michael Smith Laboratories and School of Biomedical Engineering at UBC. H.H.L. and S.K.V.S. were supported by a BioTalent Canada award. Z.K. also gratefully acknowledges additional financial supportfrom the Department of Health and Social Care using UK Aid funding as managed by EPSRC (EP/R013764/1). The views expressed in this publication are those of the author(s) and not necessarily those of the Department of Health and Social Care. Funding via the Wellcome Leap R3 program to Z.K. is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.2c00032.
Responses for DoE iterations A and B, selected electropherograms, response surface methodology results, and a list of all formulations tested (PDF)
Author Contributions
∥ H.H.L. and S.D. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): A.K.B. is a co-founder of VaxEquity.
Supplementary Material
References
- Jackson L. A.; Anderson E. J.; Rouphael N. G.; Roberts P. C.; Makhene M.; Coler R. N.; McCullough M. P.; Chappell J. D.; Denison M. R.; Stevens L. J.; Pruijssers A. J.; McDermott A.; Flach B.; Doria-Rose N. A.; Corbett K. S.; Morabito K. M.; O’Dell S.; Schmidt S. D.; Swanson P. A. 2nd; Padilla M.; Mascola J. R.; Neuzil K. M.; Bennett H.; Sun W.; Peters E.; Makowski M.; Albert J.; Cross K.; Buchanan W.; Pikaart-Tautges R.; Ledgerwood J. E.; Graham B. S.; Beigel J. H. An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. N Engl J. Med. 2020, 383 (20), 1920–1931. 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack F. P.; Thomas S. J.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; Perez J. L.; Pérez Marc G.; Moreira E. D.; Zerbini C. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; Maier M. A.; Manoharan M.; Fitzgerald K.; Jayaraman M.; Barros S.; Ansell S.; Du X.; Hope M. J.; Madden T. D.; Mui B. L.; Semple S. C.; Tam Y. K.; Ciufolini M.; Witzigmann D.; Kulkarni J. A.; van der Meel R.; Cullis P. R. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14 (12), 1084–1087. 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- Wayment-Steele H. K.; Kim D. S.; Choe C. A.; Nicol J. J.; Wellington-Oguri R.; Watkins A. M.; Parra Sperberg R. A.; Huang P. S.; Participants E.; Das R. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucleic Acids Res. 2021, 49 (18), 10604–10617. 10.1093/nar/gkab764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi N.; Tuyishime S.; Muramatsu H.; Kariko K.; Mui B. L.; Tam Y. K.; Madden T. D.; Hope M. J.; Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Release 2015, 217, 345–51. 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; McKay P. F.; Yus B. I.; Aldon Y.; Shattock R. J. Inside out: optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26 (9), 363–372. 10.1038/s41434-019-0095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Lee R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. 10.1016/j.addr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Kauffman K. J.; Dorkin J. R.; Yang J. H.; Heartlein M. W.; DeRosa F.; Mir F. F.; Fenton O. S.; Anderson D. G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15 (11), 7300–6. 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- Jayaraman M.; Ansell S. M.; Mui B. L.; Tam Y. K.; Chen J.; Du X.; Butler D.; Eltepu L.; Matsuda S.; Narayanannair J. K.; Rajeev K. G.; Hafez I. M.; Akinc A.; Maier M. A.; Tracy M. A.; Cullis P. R.; Madden T. D.; Manoharan M.; Hope M. J. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem., Int. Ed. Engl. 2012, 51 (34), 8529–33. 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhorn I. S.; Bakowsky U.; Polushkin E.; Visser W. H.; Stuart M. C.; Engberts J. B.; Hoekstra D. Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol. Ther 2005, 11 (5), 801–10. 10.1016/j.ymthe.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Lu J. J.; Langer R.; Chen J. A Novel Mechanism Is Involved in Cationic Lipid-Mediated Functional siRNA Delivery. Mol. Pharmaceutics 2009, 6 (3), 763–771. 10.1021/mp900023v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui B. L.; Tam Y. K.; Jayaraman M.; Ansell S. M.; Du X.; Tam Y. Y.; Lin P. J.; Chen S.; Narayanannair J. K.; Rajeev K. G.; Manoharan M.; Akinc A.; Maier M. A.; Cullis P.; Madden T. D.; Hope M. J. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther Nucleic Acids 2013, 2, e139 10.1038/mtna.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri M.; Nawaz M.; Papadimitriou A.; Angerfors A.; Camponeschi A.; Na M.; Holtta M.; Skantze P.; Johansson S.; Sundqvist M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10 (1), 4333. 10.1038/s41467-019-12275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup A.; Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet 2015, 16 (9), 543–52. 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damase T. R.; Sukhovershin R.; Boada C.; Taraballi F.; Pettigrew R. I.; Cooke J. P. The Limitless Future of RNA Therapeutics. Front Bioeng Biotechnol 2021, 9, 628137. 10.3389/fbioe.2021.628137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; McKay P. F.; Bouton C. R.; Hu K.; Samnuan K.; Shattock R. J. Innate Inhibiting Proteins Enhance Expression and Immunogenicity of Self-Amplifying RNA. Mol. Ther 2021, 29 (3), 1174–1185. 10.1016/j.ymthe.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A. B.; Lambert L.; Kinnear E.; Busse D.; Erbar S.; Reuter K. C.; Wicke L.; Perkovic M.; Beissert T.; Haas H.; Reece S. T.; Sahin U.; Tregoning J. S. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26 (2), 446–455. 10.1016/j.ymthe.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geall A. J.; Verma A.; Otten G. R.; Shaw C. A.; Hekele A.; Banerjee K.; Cu Y.; Beard C. W.; Brito L. A.; Krucker T.; O’Hagan D. T.; Singh M.; Mason P. W.; Valiante N. M.; Dormitzer P. R.; Barnett S. W.; Rappuoli R.; Ulmer J. B.; Mandl C. W. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (36), 14604–9. 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; Yilmaz G.; McKay P. F.; Becer C. R.; Shattock R. J. One Size Does Not Fit All: The Effect of Chain Length and Charge Density of Poly(ethylene imine) Based Copolymers on Delivery of pDNA, mRNA, and RepRNA Polyplexes. Biomacromolecules 2018, 19 (7), 2870–2879. 10.1021/acs.biomac.8b00429. [DOI] [PubMed] [Google Scholar]
- Blakney A. K.; Zhu Y.; McKay P. F.; Bouton C. R.; Yeow J.; Tang J.; Hu K.; Samnuan K.; Grigsby C. L.; Shattock R. J.; Stevens M. M. Big is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711. 10.1021/acsnano.0c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada T.; Kulkarni J. A.; Huynh A.; Chen S.; van der Meel R.; Tam Y. Y. C.; Cullis P. R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37 (3), 1120–1128. 10.1021/acs.langmuir.0c03039. [DOI] [PubMed] [Google Scholar]
- Blakney A. K.; Deletic P.; McKay P. F.; Bouton C. R.; Ashford M.; Shattock R. J.; Sabirsh A. Effect of complexing lipids on cellular uptake and expression of messenger RNA in human skin explants. J. Controlled Release 2021, 330, 1250–1261. 10.1016/j.jconrel.2020.11.033. [DOI] [PubMed] [Google Scholar]
- Kulkarni J. A.; Chen S.; Tam Y. Y. C. Scalable Production of Lipid Nanoparticles Containing Amphotericin B. Langmuir 2021, 37 (24), 7312–7319. 10.1021/acs.langmuir.1c00530. [DOI] [PubMed] [Google Scholar]
- Jeffs L. B.; Palmer L. R.; Ambegia E. G.; Giesbrecht C.; Ewanick S.; MacLachlan I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res. 2005, 22 (3), 362–72. 10.1007/s11095-004-1873-z. [DOI] [PubMed] [Google Scholar]
- Kulkarni J. A.; Myhre J. L.; Chen S.; Tam Y. Y. C.; Danescu A.; Richman J. M.; Cullis P. R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine 2017, 13 (4), 1377–1387. 10.1016/j.nano.2016.12.014. [DOI] [PubMed] [Google Scholar]
- Ding F.; Zhang H.; Cui J.; Li Q.; Yang C. Boosting ionizable lipid nanoparticle-mediated in vivo mRNA delivery through optimization of lipid amine-head groups. Biomater Sci. 2021, 9 (22), 7534–7546. 10.1039/D1BM00866H. [DOI] [PubMed] [Google Scholar]
- Whitehead K. A.; Matthews J.; Chang P. H.; Niroui F.; Dorkin J. R.; Severgnini M.; Anderson D. G. In Vitro-In Vivo Translation of Lipid Nanoparticles for Hepatocellular siRNA Delivery. ACS Nano 2012, 6 (8), 6922–6929. 10.1021/nn301922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeki M.; Fujishima Y.; Sato Y.; Yasui T.; Kaji N.; Ishida A.; Tani H.; Baba Y.; Harashima H.; Tokeshi M. Understanding the formation mechanism of lipid nanoparticles in microfluidic devices with chaotic micromixers. PLoS One 2017, 12 (11), e0187962 10.1371/journal.pone.0187962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C.; Pan S. Vesicle formation mechanisms: an overview. J. Liposome Res. 2021, 31 (1), 90–111. 10.1080/08982104.2020.1730401. [DOI] [PubMed] [Google Scholar]
- Paloncyova M.; Cechova P.; Srejber M.; Kuhrova P.; Otyepka M. Role of Ionizable Lipids in SARS-CoV-2 Vaccines As Revealed by Molecular Dynamics Simulations: From Membrane Structure to Interaction with mRNA Fragments. J. Phys. Chem. Lett. 2021, 12 (45), 11199–11205. 10.1021/acs.jpclett.1c03109. [DOI] [PubMed] [Google Scholar]
- Waskom M. L. seaborn: statistical data visualization. Journal of Open Source Software 2021, 6 (60), 3021. 10.21105/joss.03021. [DOI] [Google Scholar]
- Seabold S.; Perktold J. Statsmodels: Econometric and Statistical Modeling with Python. Proc. SciPy2010 2010, 92–96. 10.25080/Majora-92bf1922-011. [DOI] [Google Scholar]
- Hattab M. W. On the use of data transformation in response surface methodology. Quality and Reliability Engineering International 2018, 34 (6), 1185–1194. 10.1002/qre.2317. [DOI] [Google Scholar]
- Virtanen P.; Gommers R.; Oliphant T. E.; Haberland M.; Reddy T.; Cournapeau D.; Burovski E.; Peterson P.; Weckesser W.; Bright J.; et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17 (3), 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett K. J.; Higgins J.; Woods A.; Levy B.; Xia Y.; Hsiao C. J.; Acosta E.; Almarsson O.; Moore M. J.; Brito L. A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Controlled Release 2021, 335, 237–246. 10.1016/j.jconrel.2021.05.021. [DOI] [PubMed] [Google Scholar]
- Hassett K. J.; Benenato K. E.; Jacquinet E.; Lee A.; Woods A.; Yuzhakov O.; Himansu S.; Deterling J.; Geilich B. M.; Ketova T.; Mihai C.; Lynn A.; McFadyen I.; Moore M. J.; Senn J. J.; Stanton M. G.; Almarsson O.; Ciaramella G.; Brito L. A. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther Nucleic Acids 2019, 15, 1–11. 10.1016/j.omtn.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roces C. B.; Lou G.; Jain N.; Abraham S.; Thomas A.; Halbert G. W.; Perrie Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12 (11), 1095. 10.3390/pharmaceutics12111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedmi R.; Ben-Arie N.; Peer D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31 (26), 6867–75. 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Ryals R. C.; Patel S.; Acosta C.; McKinney M.; Pennesi M. E.; Sahay G. The effects of PEGylation on LNP based mRNA delivery to the eye. PLoS One 2020, 15 (10), e0241006 10.1371/journal.pone.0241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbuzenko O.; Barenholz Y.; Priev A. Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer. Chem. Phys. Lipids 2005, 135 (2), 117–29. 10.1016/j.chemphyslip.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Shi L.; Zhang J.; Zhao M.; Tang S.; Cheng X.; Zhang W.; Li W.; Liu X.; Peng H.; Wang Q. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale 2021, 13 (24), 10748–10764. 10.1039/D1NR02065J. [DOI] [PubMed] [Google Scholar]
- Erdeljic Turk V. Anaphylaxis associated with the mRNA COVID-19 vaccines: Approach to allergy investigation. Clin Immunol 2021, 227, 108748. 10.1016/j.clim.2021.108748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox F.; Khalib K.; Conlon N. PEG That Reaction: A Case Series of Allergy to Polyethylene Glycol. J. Clin Pharmacol 2021, 61 (6), 832–835. 10.1002/jcph.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylon K.; Dolle S.; Worm M. Polyethylene glycol as a cause of anaphylaxis. Allergy Asthma Clin. Immunol. 2016, 12, 67. 10.1186/s13223-016-0172-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenande E.; Garvey L. H. Immediate-type hypersensitivity to polyethylene glycols: a review. Clin. Exp. Allergy 2016, 46 (7), 907–22. 10.1111/cea.12760. [DOI] [PubMed] [Google Scholar]
- Banerji A.; Wickner P. G.; Saff R.; Stone C. A. Jr.; Robinson L. B.; Long A. A.; Wolfson A. R.; Williams P.; Khan D. A.; Phillips E.; Blumenthal K. G. mRNA Vaccines to Prevent COVID-19 Disease and Reported Allergic Reactions: Current Evidence and Suggested Approach. J. Allergy Clin Immunol Pract 2021, 9 (4), 1423–1437. 10.1016/j.jaip.2020.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z.; Hu Y.; Nie T.; Tang H.; Zhu J.; Chen K.; Liu L.; Leong K. W.; Chen Y.; Mao H. Q. Size-controlled lipid nanoparticle production using turbulent mixing to enhance oral DNA delivery. Acta Biomater 2018, 81, 195–207. 10.1016/j.actbio.2018.09.047. [DOI] [PubMed] [Google Scholar]
- Howard F. B.; Levin I. W. Lipid vesicle aggregation induced by cooling. Int. J. Mol. Sci. 2010, 11 (2), 754–61. 10.3390/ijms11020754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Li H.; Sun J.; Gao J.; Liu W.; Li B.; Guo Y.; Chen J. DC-Chol/DOPE cationic liposomes: a comparative study of the influence factors on plasmid pDNA and siRNA gene delivery. Int. J. Pharm. 2010, 390 (2), 198–207. 10.1016/j.ijpharm.2010.01.035. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Hui S.-W.; Frederik P.; Szoka F. C. Physicochemical characterization and purification of cationic lipoplexes. Biophys. J. 1999, 77 (1), 341–53. 10.1016/S0006-3495(99)76894-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltover I.; Salditt T.; Radler J. O.; Safinya C. R. An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery. Science 1998, 281 (5373), 78–81. 10.1126/science.281.5373.78. [DOI] [PubMed] [Google Scholar]
- Leung A. K.; Tam Y. Y.; Chen S.; Hafez I. M.; Cullis P. R. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B 2015, 119 (28), 8698–706. 10.1021/acs.jpcb.5b02891. [DOI] [PubMed] [Google Scholar]
- Beg S.; Akhter S.. Box–Behnken Designs and Their Applications in Pharmaceutical Product Development. In Design of Experiments for Pharmaceutical Product Development: Vol. I: Basics and Fundamental Principles; Beg S., Ed.; Springer Singapore: Singapore, 2021; pp 77–85. [Google Scholar]
- Carrasco M. J.; Alishetty S.; Alameh M. G.; Said H.; Wright L.; Paige M.; Soliman O.; Weissman D.; Cleveland T. E. t.; Grishaev A.; Buschmann M. D. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 2021, 4 (1), 956. 10.1038/s42003-021-02441-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt H. S.; Tate W. P. Primordial soup or vinaigrette: did the RNA world evolve at acidic pH?. Biol. Direct 2012, 7, 4. 10.1186/1745-6150-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Hunter M. R.; Sonzini S.; Pereira S.; Romanelli S. M.; Liu K.; Li W.; Liang L.; Yang B.; Mahmoudi N.; Desai A. S. Mechanistic Studies of an Automated Lipid Nanoparticle Reveal Critical Pharmaceutical Properties Associated with Enhanced mRNA Functional Delivery In Vitro and In Vivo. Small 2021, e2105832 10.1002/smll.202105832. [DOI] [PubMed] [Google Scholar]
- Lokugamage M. P.; Gan Z.; Zurla C.; Levin J.; Islam F. Z.; Kalathoor S.; Sato M.; Sago C. D.; Santangelo P. J.; Dahlman J. E. Mild Innate Immune Activation Overrides Efficient Nanoparticle-Mediated RNA Delivery. Adv. Mater. 2020, 32 (1), 1904905. 10.1002/adma.201904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Melicio L. A.; Fernandes R. K.; Rodrigues D. R.; Golim M. A.; Soares A. M. V. C. Interleukin-18 Increases TLR4 and Mannose Receptor Expression and Modulates Cytokine Production in Human Monocytes. Mediators of Inflammation 2015, 2015, 236839. 10.1155/2015/236839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Salinas L.; Verdugo-Rodriguez A.; Rodriguez L. L.; Borca M. V. The Role of Interleukin 6 During Viral Infections. Front. Microbiol. 2019, 10, 1057. 10.3389/fmicb.2019.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Chen Y.; Zhao Y.; Lung D. C.; Ye Z.; Song W.; Liu F.-F.; Cai J.-P.; Wong W.-M.; Yip C. C.-Y.; et al. Intravenous Injection of Coronavirus Disease 2019 (COVID-19) mRNA Vaccine Can Induce Acute Myopericarditis in Mouse Model. Clinical Infectious Diseases 2021, ciab707 10.1093/cid/ciab707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakney A. K.; McKay P. F.; Hu K.; Samnuan K.; Jain N.; Brown A.; Thomas A.; Rogers P.; Polra K.; Sallah H.; Yeow J.; Zhu Y.; Stevens M. M.; Geall A.; Shattock R. J. Polymeric and lipid nanoparticles for delivery of self-amplifying RNA vaccines. J. Controlled Release 2021, 338, 201–210. 10.1016/j.jconrel.2021.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.; Sorensen E. W.; Mintri S.; Rabideau A. E.; Zheng W.; Besin G.; Khatwani N.; Su S. V.; Miracco E. J.; Issa W. J. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6 (26), eaaz6893 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derringer G.; Suich R. Simultaneous Optimization of Several Response Variables. Journal of Quality Technology 1980, 12 (4), 214–219. 10.1080/00224065.1980.11980968. [DOI] [Google Scholar]
- Amdoun R.; Khelifi L.; Khelifi-Slaoui M.; Amroune S.; Asch M.; Assaf-ducrocq C.; Gontier E. The Desirability Optimization Methodology; a Tool to Predict Two Antagonist Responses in Biotechnological Systems: Case of Biomass Growth and Hyoscyamine Content in Elicited Datura starmonium Hairy Roots. Iranian Journal of Biotechnology 2018, 16 (1), 11–19. 10.21859/ijb.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi Z.A Piece of the COVID Vaccine Recipe. Public Citizen, 2021. https://www.citizen.org/article/a-piece-of-the-covid-vaccine-recipe.
- Tinari S. The EMA covid-19 data leak, and what it tells us about mRNA instability. BMJ. 2021, 372, n627. 10.1136/bmj.n627. [DOI] [PubMed] [Google Scholar]
- Zhang H.; You X.; Wang X.; Cui L.; Wang Z.; Xu F.; Li M.; Yang Z.; Liu J.; Huang P.; et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (6), e2005191118 10.1073/pnas.2005191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.