

Abstract
Mas-related G protein-coupled receptor X1 (MRGPRX1) is a human sensory neuron-specific receptor and potential target for the treatment of pain. Positive allosteric modulators (PAMs) of MRGPRX1 have the potential to preferentially activate the receptors at the central terminals of primary sensory neurons and minimize itch side effects caused by peripheral activation. Using a high-throughput screening (HTS) hit, a series of thieno[2,3-d]pyrimidine-based molecules were synthesized and evaluated as human MRGPRX1 PAMs in HEK293 cells stably transfected with human MrgprX1 gene. An iterative process to improve potency and metabolic stability led to the discovery of orally available 6-(tert-butyl)-5-(3,4-dichlorophenyl)-4-(2-(trifluoromethoxy)phenoxy)thieno[2,3-d]pyrimidine (1t), which can be distributed to the spinal cord, the presumed site of action, following oral administration. In a neuropathic pain model induced by sciatic nerve chronic constriction injury (CCI), compound 1t (100 mg/kg, po) reduced behavioral heat hypersensitivity in humanized MRGPRX1 mice, demonstrating the therapeutic potential of MRGPRX1 PAMs in treating neuropathic pain.
Graphical Abstract
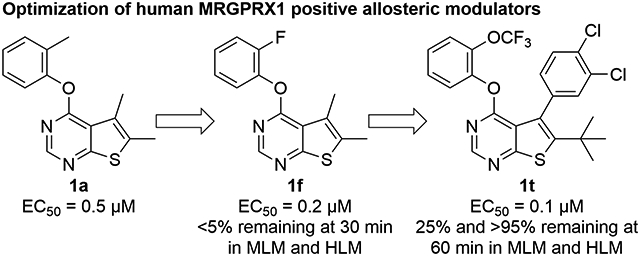
INTRODUCTION
Mas-related G protein-coupled receptor X1 (MRGPRX1) is expressed specifically in the small diameter dorsal root ganglion (DRG) sensory neurons and is implicated in the modulation of nociception.1 MRGPRX1 has gained increased interest as a promising therapeutic target partly based on the analgesic effects of BAM8–22 (Figure 1),2 a proteolytic product derived from proenkephalin A with full agonist activity at human MRGPRX1 and its closest rodent orthologues, mouse MRGPRC11 and rat MRGPRC.3 DRG sensory neurons have both centrally and peripherally projecting axons. The analgesic effects of MRGPRX1 agonists are thought to be partially mediated by downstream inhibition of N-type calcium channels resulting in reduced synaptic inputs to the spinal cord dorsal horn neurons.4 On the other hand, activation of peripheral MRGPRX1 localized in nerve terminals in the skin may generate an itch sensation.5 These dual effects produced by direct activation of MRGPRX1 at the central and peripheral terminals of DRG neurons may limit the therapeutic utility of small molecule orthosteric MRGPRX1 agonists6, 7 for systemic treatment. Interestingly, BAM22, another endogenous peptide-based MRGPRX1 agonist, was found to be upregulated at spinal dorsal horn during persistent pain after nerve injury or tissue inflammation, while it was undetectable in the skin.8 These findings have presented a unique therapeutic opportunity for positive allosteric modulators (PAMs) of MRGPRX1, which can potentiate the agonist effects of the endogenous agonist at the central terminals of sensory neurons without activating peripheral MRGPRX1, thus minimizing itch side effects.
Figure 1.
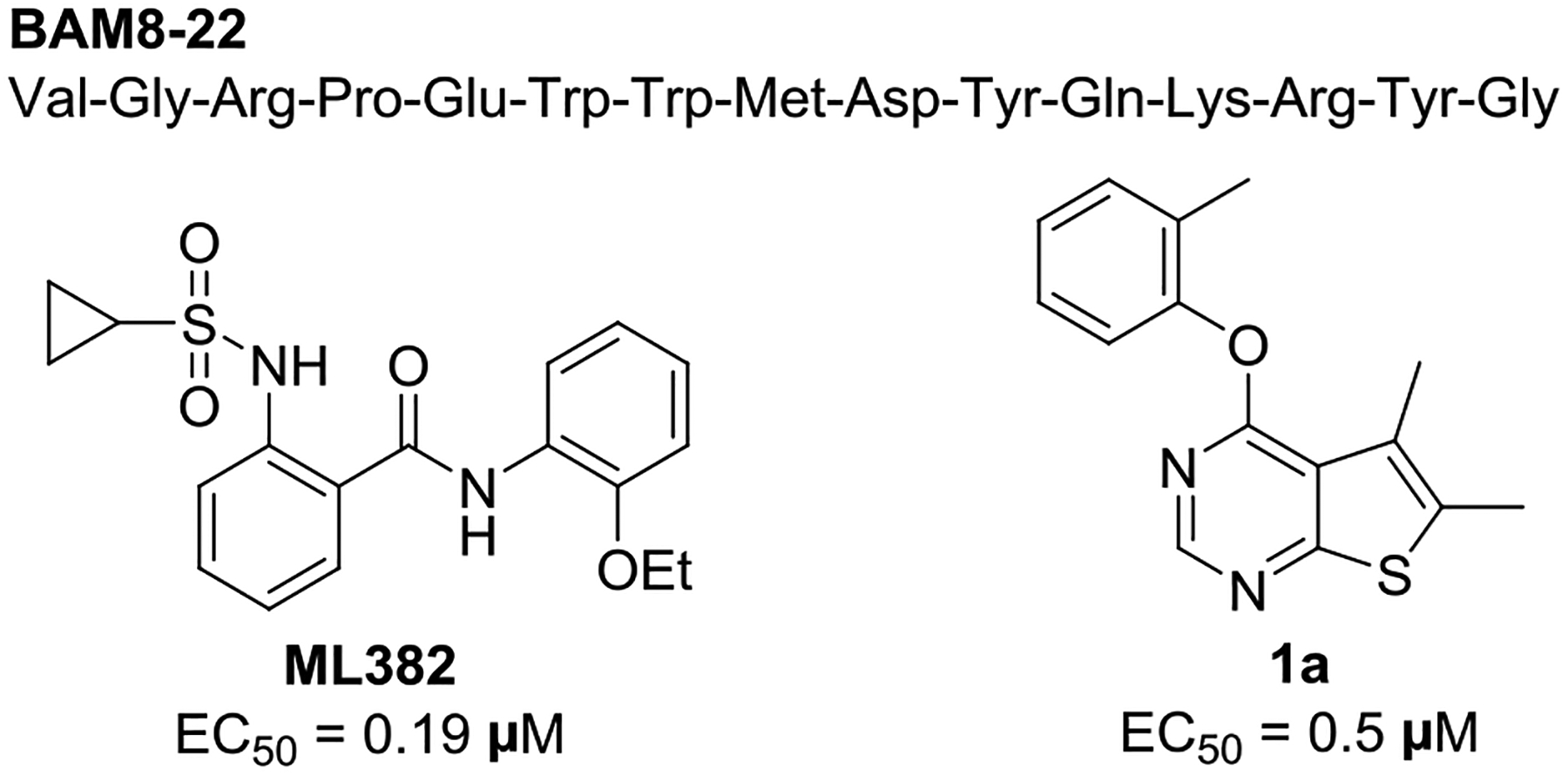
Chemical structures of BAM8-22, ML382, and compound 1a.
2-[(Cyclopropylsulfonyl)amino]-N-(2-ethoxyphenyl)benzamide, also known as ML382, is a prototype MRGPRX1 PAM identified by the Molecular Libraries Probe Production Centers Network (MLPCN).9 ML382 was found to inhibit pain hypersensitivity after injury following intrathecal injection, and systemic administration of ML382 did not elicit itch side effects.8 These findings confirm the presence of endogenous MRGPRX1 agonists in the spinal cord during persistent pain conditions and demonstrate the therapeutic potential of MRGPRX1 PAMs as novel analgesic agents devoid of the peripheral itch side effects. However, ML382 produces analgesic effects only when injected intrathecally presumably due to its poor distribution to the spinal cord, limiting its therapeutic utility in treating chronic neuropathic pain. This prompted us to explore structurally distinct MRGPRX1 PAMs with improved pharmacokinetics properties and in vivo analgesic efficacy following oral administration.
To this end, we conducted high throughput screening of Eisai’s compound library in HEK293 cells stably transfected with human MRGPRX1 by using FLIPR Calcium 4 Assay Kit (Molecular Devices) to measure intracellular calcium responses.10 We identified 5,6-dimethyl-4-(o-tolyloxy)thieno[2,3-d]pyrimidine 1a as a potent MRGPRX1 PAM with an EC50 value of 0.5 μM. Although compound 1a is less potent than ML382, its lack of a hydrogen bond donor prompted us to use this hit compound as a molecular template in an attempt to identity MRGPRX1 PAMs with improved pharmacokinetics properties. Here, we describe the design, synthesis, and structure−activity relationships (SAR) of a new series of human MRGPRX1 PAMs using compound 1a as a molecular template. Our optimization efforts aimed at improving not only MRGPRX1 PAM potency but also metabolic stability led to the discovery of an orally available MRGPRX1 PAM, potentially valuable as a new lead for treating neuropathic pain.
CHEMISTRY
Compounds 1a-p were synthesized by reacting 4-chloro-thieno[2,3-d]pyrimidines 2a-f with various phenols pretreated with NaH (Scheme 1).
Scheme 1.
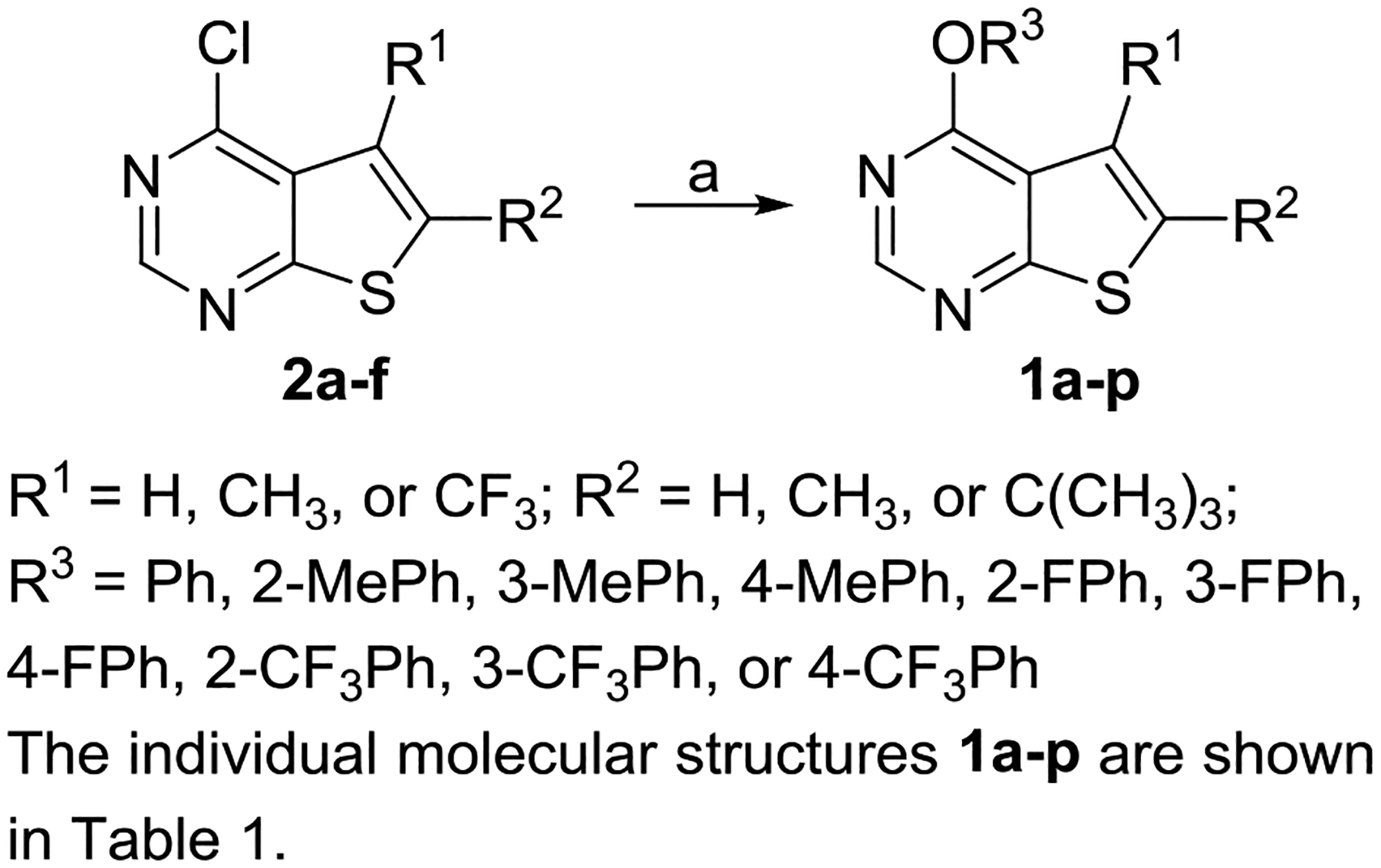
Synthesis of Compounds 1a-pa
aReagents and conditions: (a) R3OH, NaH, DMF, 0 °C to rt, 33–93%.
Compound 1q was synthesized in three steps starting with a one-pot synthesis of thieno[2,3-d]pyrimidin-4-ol 4 via Gewald reaction.11 Compound 4 was subsequently converted to 4-chloro derivative 5 by treating with phosphoryl chloride. Nucleophilic substitution with 2-fluorophenol afforded the desired product 1q.
Although the synthetic route developed for compound 1q (Scheme 2) described above takes only three steps to the desired product, it suffered from the poor yield in the first step involving the four-component reaction, presumably due to the steric hindrance by the tert-butyl group in starting material 3. This prompted us to explore alternative routes to compounds 1r-t containing a tert-butyl group at the 6-position. As shown in scheme 3, 6-(tert-butyl)-4-chlorothieno[2,3-d]pyrimidine 2f was first converted into the 6-methoxy derivative 6. Compound 6 was brominated at its 5-position by NBS to give compound 7. Suzuki coupling of compound 7 with aryl boronic acids gave compounds 8a-b. After converting 8a-b to 4-chloro derivative 10a-b, the desired product 1r-t was obtained by reacting them with either 2-fluorophenol or 2-trifluoromethoxyphenol.
Scheme 2.
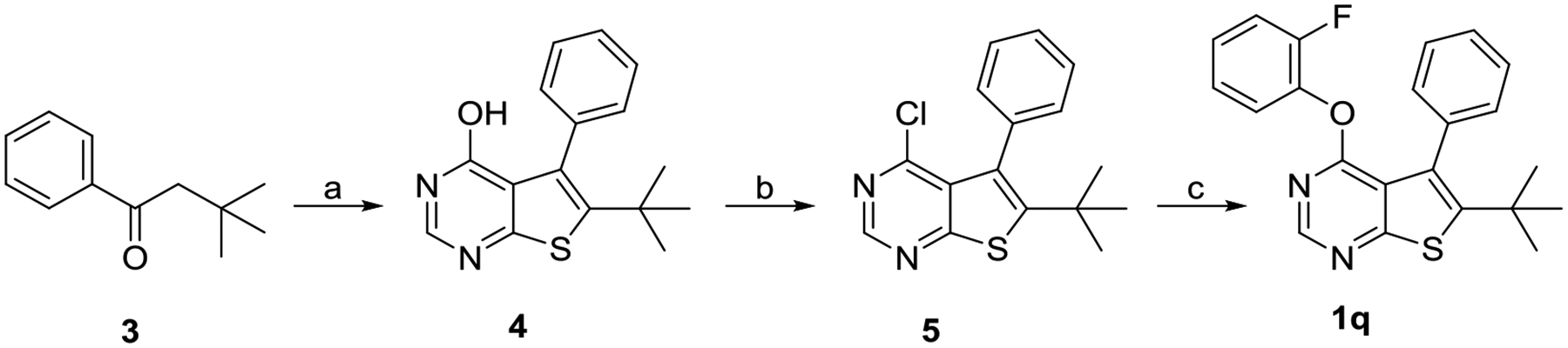
Synthesis of Compound 1qa
aReagents and conditions: (a) ethylcyanoacetate, S8, formamide, L-proline, Et2NH, 170 °C, 4% (b) POCl3, 100 °C, 66%; (c) 2-fluorophenol, NaH, DMF, 0 °C to rt, 40%.
Scheme 3.
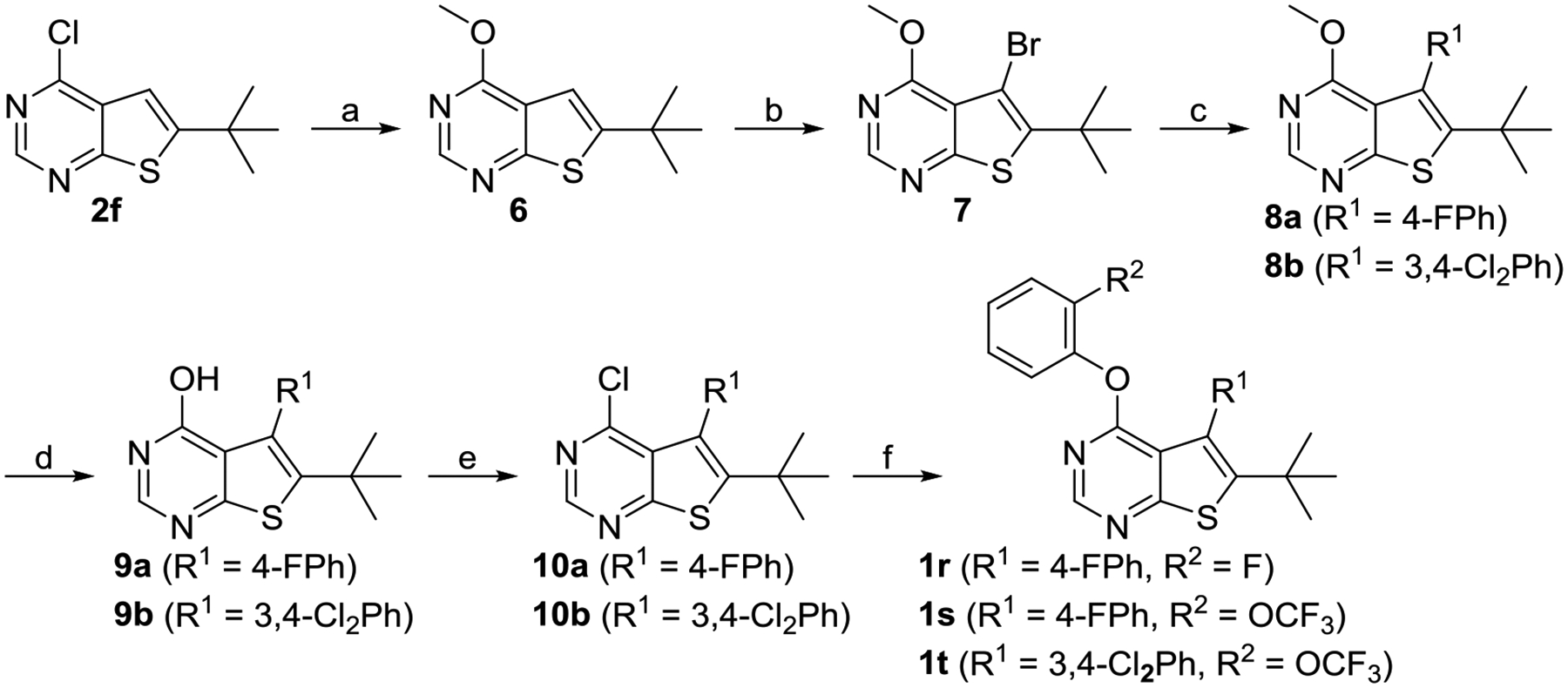
Synthesis of Compounds 1r-ta
aReagents and conditions: (a) MeONa, MeOH, 0 °C to rt, 98%; (b) NBS, AcOH, 55 °C, 85%; (c) R1B(OH)2, PdCl2(PPh3)2, K2CO3, DMF, 80 °C, 92% for 8a, 75% for 8b; (d) BBr3, DCM, rt, 41% for 9a, 93% for 9b; (e) POCl3, 110 °C, 61% for 10a, 95% for 10b; (f) 2-fluorophenol or 2-(trifluoromethoxy)phenol, NaH, DMF, 0 °C to rt, 77% for 1r, 65% for 1s, 69% for 1t.
RESULTS AND DISCUSSION
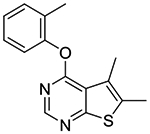
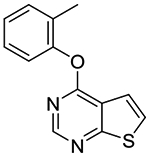
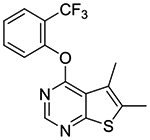
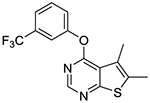
In vitro experiments measuring PAM activity at MRGPRX1 were performed with a FLIPR assay using HEK293 cells stably transfected with MRGPRX1 in the presence of EC20 (5–15 nM) of BAM8–22, a MRGPRX1 agonist devoid of opioid receptor affinity displayed by BAM22. The results are summarized in Table 1. All of the compounds with a submicromolar EC50 value displayed Emax ≥95% relative to the maximum response achieved by BAM8–22 alone. It should be noted that compound 1b was also identified as an active allosteric MRGPRX1 activator by the NIH Molecular Libraries Probe Centers Network (MLPCN).12 In our assay, compound 1b displayed 20-fold lower potency compared to compound 1a, indicating an important role played by the 5- and 6-methyl groups of 1a in the PAM potency. Thus, we tested 5,6-dimethylthieno[2,3-d]pyrimidine derivatives 1c-k possessing various phenoxy groups at the 4-position. Compound 1c containing an unsubstituted phenoxy group showed 4-fold loss in potency compared to 1b. m-Tolyloxy and p-tolyloxy derivatives 1d and 1e displayed substantially lower potency. We also explored fluoro and trifluoromethyl substitutions at the 4-phenoxy group in a systematic manner (compounds 1f-k). As observed with methyl substitutions, ortho-substituted derivatives 1f and 1i exhibited greater potency than meta- and para-substituted derivatives 1g-h and 1j-k. Among these, compound 1f containing a 2-fluorophenoxy group at the 4-position showed the most potent PAM activity.
Table 1.
| Cmpd | Structure | EC50 (μM)a (Emax)b |
Cmpd | Structure | EC50 (μM)a (Emax)b |
|---|---|---|---|---|---|
| 1a |
|
0.48
± 0.04 (118%) |
1k |
|
>100 |
| 1b |
|
14.5
± 1.1 (88%) |
1l |
|
4.2 ± 1.1 (95%) |
| 1c |
|
1.6
± 0.4 (89%) |
1m |
|
1.7 ± 0.2 (94%) |
| 1d |
|
25 (40%) |
1n |
|
1.5 ± 0.2 (90%) |
| 1e |
|
2.7
± 1.5 (88%) |
1o |
|
2.1 ± 0.4 (94%) |
| 1f |
|
0.22
± 0.02 (112%) |
1p |
|
0.32 ± 0.06 (105%) |
| 1g |
|
30 (55%) |
1q |
|
0.34 ± 0.07 (95%) |
| 1h |
|
1.1
± 0.2 (83%) |
1r |
|
0.30 ± 0.13 (145%) |
| 1i |
|
0.52
± 0.03 (95%) |
1s |
|
0.23 ± 0.05 (143%) |
| 1j |
|
>100 | 1t |
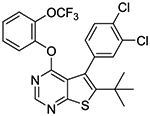
|
0.10 ± 0.01 (114%) |
Values are mean ± standard error of the mean (SEM) of at least three independent experiments performed in quadruplicate.
Emax values were defined as response elicited by maximum concentration of test compounds in the presence of EC20 concentration of BAM8–22 normalized to that elicited by 250 nM (>EC100) of BAM8–22 alone.
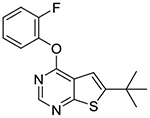
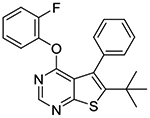
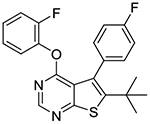
In the subsequent set of compounds, we retained the 2-fluorophenoxy group at the 4-position and modified the 5- and 6-positions of the thieno[2,3-d]pyrimidine ring. Compound 1l unsubstituted at the 5- and 6-positions showed much weaker potency. This is consistent with the substantial loss of potency observed in compound 1b as compared to 1c. The removal of one methyl group either from 5- or 6-position from 1a also resulted in loss of potency as seen in compounds 1m and 1n. Replacement of the 5-methyl group with a trifluoromethyl group (compound 1o) did not improve the potency either. Interestingly, introduction of a tert-butyl group into the 6-position (compounds 1p-r) led to submicromolar potency.
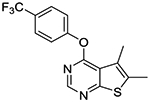
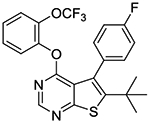
In the last stage, we incorporated a 2-(trifluoromethyl)phenoxy group into the 4-position of the 6-(tert-butyl)thieno[2,3-d]pyrimidine scaffold as seen in compounds 1s and 1t. Compound 1t represents one of the most potent MRGPRX1 PAMs within this series. We also confirmed that compound 1t has no intrinsic agonist activity at concentrations up to 100 μM in the absence of BAM8–22 (Supporting information Figure S1).
Structural optimization detailed above was guided by not only PAM potency but also metabolic stability. Table 2 summarizes the metabolic stability (%remaining after 30 and 60 min of incubation) of selected compounds in mouse and human liver microsomes. Compound 1f was extensively metabolized within 30 min in both human and mouse microsomes. Notably, the loss of the parent compound was only observed in the presence of NADPH, suggesting CYP450-mediated oxidation involved in the metabolism of 1f. Metabolite identification was attempted for compound 1f using HR-MS analysis. A sample from compound 1f exposed to human liver microsomes for 60 min exhibited a new peak representing [MH+ + 16] (291.0612), suggesting an insertion of an oxygen atom into the parent compound (Figure 2). Fragmentation of the Phase I metabolite using MS/MS spectroscopy showed a major peak for a fragment at 170.0256, corresponding to the oxidized thiophene moiety (Figure 2), indicating that the CYP450-mediated oxidation was primarily taking place on the thieno[2,3-d]pyrimidine moiety of compound 1f.
Table 2.
Metabolic Stability in Mouse and Human Liver Microsomes
| Cmpd | %remaining | |||
|---|---|---|---|---|
| 60 min | 60 min | |||
| 1f | <5 | <5 | ||
| 1o | <5 | <5 | ||
| 1p | <5 | 61 | ||
| 1q | <5 | 78 | ||
| 1r | <5 | 88 | ||
| 1s | 5 | >95 | ||
| 1t | 25 | >95 | ||
Figure 2.
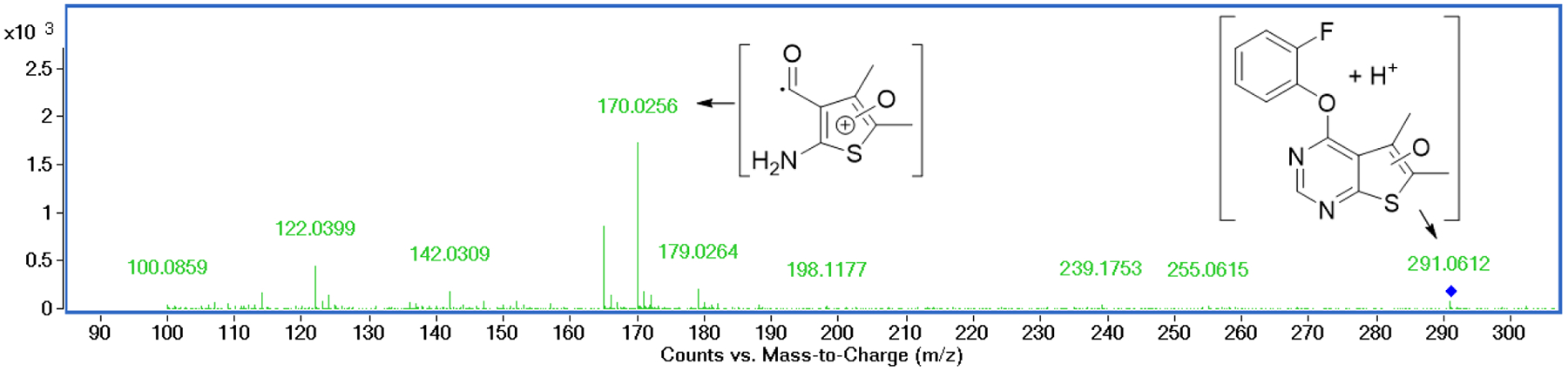
Identification of the major metabolite of compound 1f incubated in human liver microsomes. MS/MS spectroscopy of the major metabolite [MH+ + 16] shows a fragment corresponding to the oxidized thiophenium moiety.
Compound 1o was designed to make the thieno[2,3-d]pyrimidine ring electron-deficient by the electron-withdrawing effects of the 5-trifluoromethyl group. Although it showed slightly better stability in human liver microsomes, it failed to show any improvement over compound 1f in mouse liver microsomes. Interestingly, compounds 1p-r possessing a tert-butyl group at the 6-position showed substantially improved stability in human liver microsomes, presumably by protecting the thieno portion from oxidation. As seen with compound 1f, however, these three compounds were extensively metabolized within a period of 30 min in mouse liver microsomes. Given these species-dependent metabolic stability data, it is conceivable that another site is being metabolized in mouse liver microsomes. Indeed, upon incorporation of a 2-(trifluoromethoxy)phenoxy group into the 4-position (compound 1s), a slight improvement in stability was achieved against mouse liver microsomes. It should be also noted that compound 1s was completely stable in human liver microsomes. Finally, compound 1t, in which a 3,4-dichlorophenyl is introduced into the 5-position, showed substantial improvement in stability against mouse liver microsomes with 25% remaining at 60 min.
Given its potent PAM activity and good metabolic stability, we selected compound 1t for in vivo pharmacokinetics studies in mice. In addition to plasma and brain levels, we measured drug levels in spinal cord (Figure 3), which is the putative site of action for MRGPRX1 PAMs to attenuate spinal nociceptive transmission. As shown in Table 3, substantial levels of 1t were detected in plasma (Cmax = 38.98 ± 5.06 nmol/mL; AUC0-last = 108.53 ± 1.18 nmol*h/mL) following oral administration (100 mg/kg). In contrast, its distribution to the brain was substantially restricted. Compound 1t, however, was distributed to the spinal cord (Cmax = 4.60 ± 0.60 nmol/mL; AUC0-last = 13.79 ± 0.14 nmol*h/mL) with a spinal cord-to-plasma ratio of 0.13. While Cmax in the spinal cord is more than 40-fold greater than the EC50 value of compound 1t, additional studies will be required to determine the precise PK/PD relationship, including evaluation of dose-response and quantification of unbound drug concentrations in the spinal cord tissues.
Figure 3.
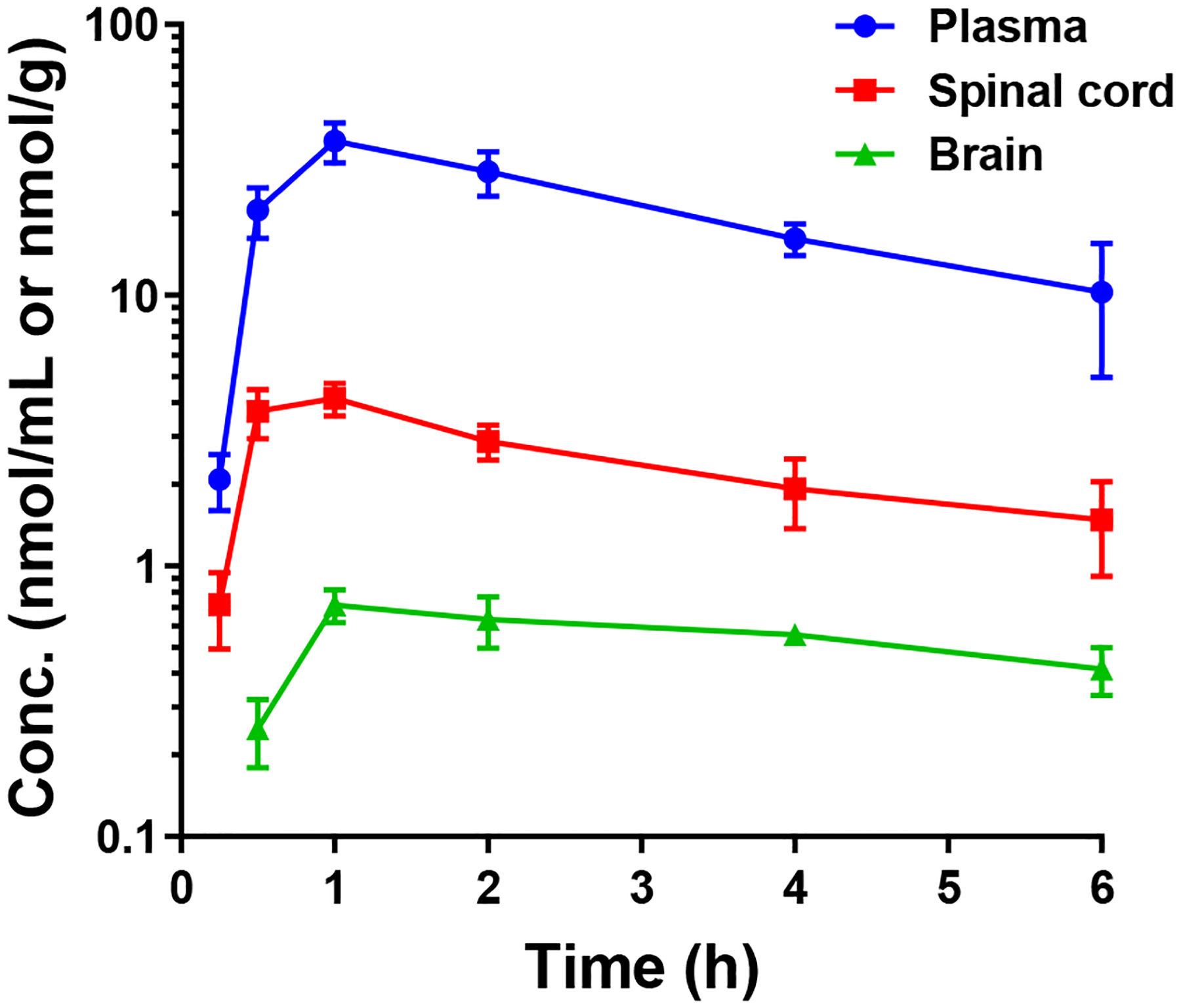
Plasma, spinal cord, and brain levels of compound 1t in mice following oral administration (100 mg/kg).
Table 3.
In Vivo Pharmacokinetic Profiles of Compound 1t
| Tmax (h) | Cmax (nmol/mL or nmol*h/g) |
AUC0-last (nmol*h/mL or nmol*h/g) |
Tissue-to-plasma ratio (by AUC) | |
|---|---|---|---|---|
| Plasma | 1 | 38.98 ± 5.06 | 108.53 ± 1.18 | |
| Brain | 1 | 0.82 ± 0.05 | 2.37 ± 0.03 | 2.2% |
| Spinal cord | 1 | 4.60 ± 0.60 | 13.79 ± 0.14 | 12.7% |
To assess its selectivity as a PAM, compound 1t was tested in HEK293 cells stably expressing MRGPRX2 in the presence of EC20 concentration of C48/80,13 a known MRGPRX2 orthosteric agonist, using Fluo-4 AM-based calcium detection assay (Supporting Information Figure S2). Compound 1t showed no MRGPRX2 PAM activity at 5 μM, indicating at least 50-fold selectivity for MRGPRX1. Subsequently, a broader off-target activity profile of compound 1t was evaluated by screening against a panel of 44 selected targets (Eurofins Cerep SafetyScreen 44) recommended by major pharmaceutical companies.14 At 10 μM, compound 1t was found to inhibit 5 targets (Supporting Information Table S1) by more than 50% (50−64%) including hERG channel. Although these off-target activities were detected at a concentration 100-fold greater than its EC50 value for MRGPRX1, future structural optimization efforts should aim to improve not only MRGPRX1 PAM potency but also selectivity over these targets.
To examine if compound 1t elicits any adverse effect, spontaneous behaviors were video-recorded during 0–1 hour post-drug in awake, free-moving humanized MRGPRX1 mice, in which human MRGPRX1 is specifically expressed in nociceptive DRG neurons and replacing the murine homolog, MrgprC11.8 Compared to vehicle, compound 1t at this oral dose (100 mg/kg) caused no overt adverse effects including itch side effects as evident from the lack of spontaneous scratch bouts (Supporting Information Table S3). In the open field test, neither compound 1t (100 mg/kg, po) nor vehicle significantly impaired exploratory performance in naïve MRGPRX1 mice as compared to pre-drug (Supporting Information Figure S3A–E). Furthermore, naïve MRGPRX1 mice treated with compound 1t (100 mg/kg, po) showed no difference in performance on the rotarod test when compared to vehicle-treated group and pre-treatment values (Supporting Information Figure S3F). Collectively, no signs of acute toxicity were observed after oral administration of compound 1t (100 mg/kg) in naïve MRGPRX1 mice, presumably due to its limited penetration to the brain and/or sensory neuron specific expression of MRGPRX1.
Compound 1t was subsequently tested for its pain inhibitory effects in vivo following oral administration (100 mg/kg, po) in the sciatic nerve chronic constriction injury (CCI) model of neuropathic pain15 using humanized MRGPRX1 mice. As shown in Figure 4, sciatic nerve CCI decreased paw withdrawal latency (PWL) to radiant heat stimuli in the hind paw (left) ipsilateral to the injury on days 12 and 26 post-injury, as compared to pre-injury level, suggesting heat hypersensitivity. The contralateral (right) PWL was not significantly changed after injury. Importantly, compound 1t (100 mg/kg, po), but not vehicle, significantly increased ipsilateral PWLs from pre-drug level at 1 and 2 hours post-drug on day 12 (Figure 4A) and at 2 hours post-drug on day 26 post-injury (Figure 4B). There was no significant change of PWLs on the contralateral hind paw after drug treatment.
Figure 4.
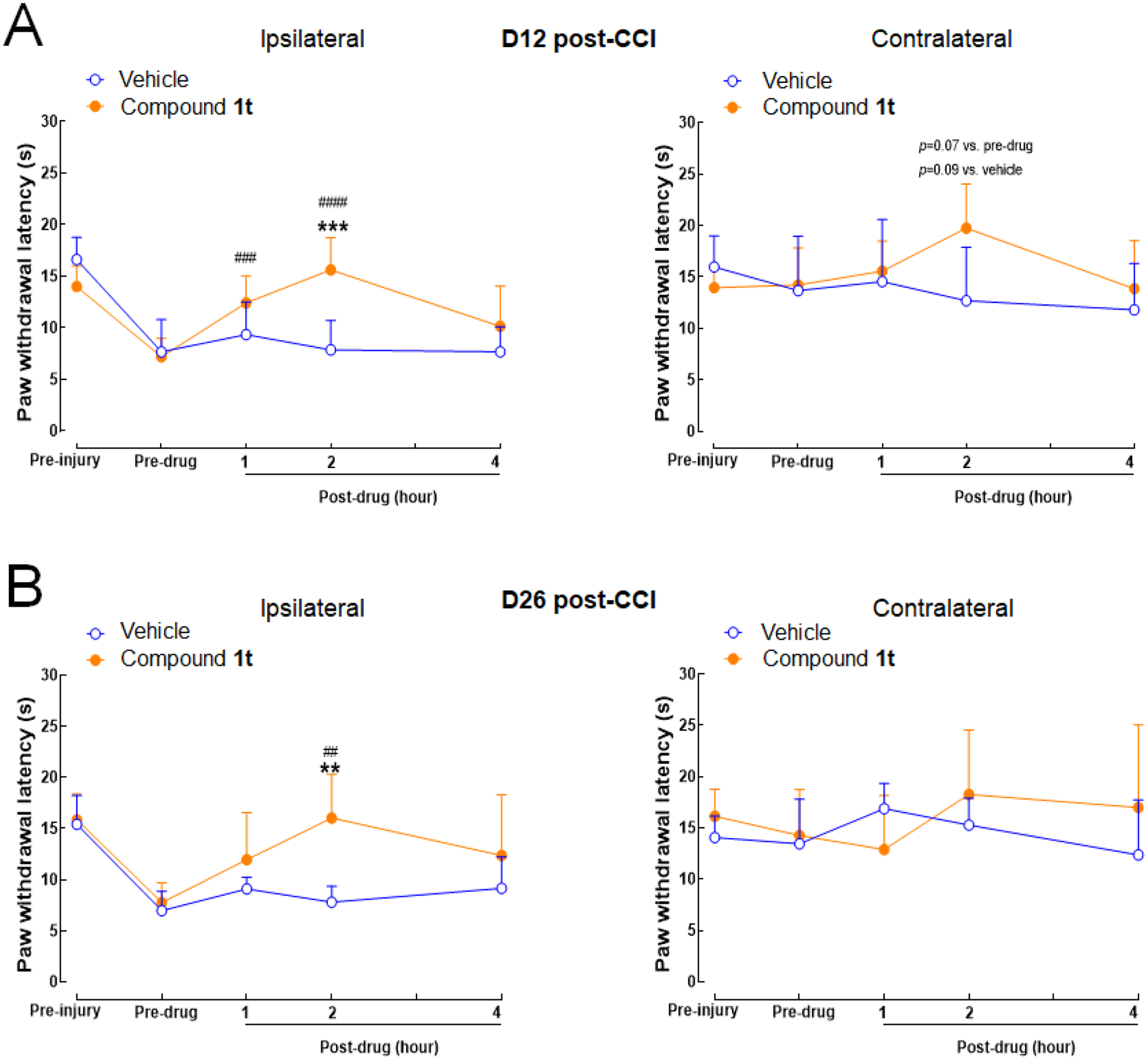
Oral administration of compound 1t attenuated heat hypersensitivity in the ipsilateral hind paw of MRGPRX1 mice subjected to sciatic nerve CCI. (A) Paw withdrawal latency (PWL) to noxious heat stimuli was measured with the Hargreaves test. PWLs were increased from pre-drug level after treated with compound 1t (100 mg/kg, po, n=5), but not those treated with vehicle (n=4) on day 12 post-CCI, and (B) on day 26 post-CCI (n=5/group). ** P<0.01, *** P<0.001 vs. vehicle; ## P<0.01, ### P<0.001, #### P<0.0001 vs. pre-drug. Two-way mixed-model ANOVA with Bonferroni post hoc test. Data are expressed as mean + SD.
CONCLUSIONS
Chronic pain treatment remains a major medical challenge. There is a critical unmet medical need for new chronic pain therapies devoid of abuse potential and adverse effects. Restricted expression of human MRGPRX1 in primary nociceptive neurons provides a unique opportunity to develop analgesic agents targeting this receptor. The allosteric potentiation of MRGPRX1 appears to be a particularly attractive approach given the preferential upregulation of endogenous MRGPRX1 agonists at the central terminals of primary sensory neurons, eliminating the possibility of itch side effects mediated by peripheral receptors. Using a thieno[2,3-d]pyrimidine-based hit compound 1a identified from our HTS efforts as a molecular template, systematic structural optimization was carried out in an attempt to improve potency and pharmacokinetics properties. As described in this paper, our iterative efforts led to the discovery of more potent MRGPRX1 PAM 1t with improved metabolic stability and good oral bioavailability, making it suitable for chronic treatment. As a proof of principle, our results with compound 1t in a well-established neuropathic pain model represents the first report of in vivo efficacy achieved by an orally administered MRGPRX1 modulator in humanized MRGPRX1 mice. Studies are currently underway to not only further characterize compound 1t as an analgesic agent but also identify more potent MRGPRX1 PAMs using compound 1t as a new molecular template.
EXPERIMENTAL SECTION
General.
All solvents were reagent grade or HPLC grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Melting points were obtained on a Mel-Temp apparatus and are uncorrected. 1H NMR spectra were recorded at 400 or 500 MHz. 13C NMR spectra were recorded at 101 or 125 MHz. The HPLC solvent system consisted of deionized water and acetonitrile, both containing 0.1% formic acid. Preparative HPLC purification was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector using a Phenomenex Luna 5 μm C18 column (21.2 mm × 250 mm, 5 μm). Analytical HPLC was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector (detection at 220 nm) and an Agilent 6120 quadrupole MS detector. Unless otherwise specified, the analytical HPLC conditions involve 20% acetonitrile/80% water for 0.25 min followed by gradient to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min with a Luna C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min. Elemental analysis was performed by Atlantic Microlabs (Norcross, GA). All final compounds biologically tested were confirmed to be of >95% purity by the HPLC methods described above. All animal studies were conducted according to protocols approved by the Johns Hopkins University Animal Care and Use Committee.
5,6-Dimethyl-4-(o-tolyloxy)thieno[2,3-d]pyrimidine (1a).
To a solution of o-cresol (0.41 g, 3.78 mmol) in DMF (8 mL) at 0 °C was added NaH (60% w/w, 0.15 g, 3.78 mmol). After the mixture was stirred at 0 °C for 10 min, 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a (0.50 g, 2.52 mmol) was added in one portion as a solid. The reaction was stirred at 0 °C and was gradually warmed up to rt and stirred overnight. After ice water was added to quench the reaction, the mixture was concentrated and taken up in Et2O (40 mL). The mixture was washed with H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated. The resulting solid residue was triturated with EtOAc/hexanes (1:4) to afford 0.4 g of the desired product as a white solid (59% yield); mp 106–108 °C. 1H NMR (CDCl3) δ 2.20 (s, 3H), 2.53 (s, 3H). 2.59 (s, 3H), 7.15 (dd, J = 1.0 Hz, J = 7.6 Hz, 1H), 7.23 (m, 1H), 7.32 (m, 2H), 8.48 (s, 1H); 13C NMR (CDCl3) δ 13.6, 13.8, 16.5, 119.9, 122.2, 125.1, 126.0, 127.1, 130.6, 131.4, 132.6, 150.8, 152.2, 163.0, 167.7. LCMS: tR 2.23 min, m/z 271 [M + H]+.
4-(o-Tolyloxy)thieno[2,3-d]pyrimidine (1b).
Compound 1b was prepared as described for the preparation of 1a except 4-chlorothieno[2,3-d]pyrimidine 2b was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. White solid (82% yield); mp 63–64 °C. 1H NMR (CDCl3) δ 2.18 (s, 3H), 7.12–7.39 (m, 4H), 7.49–7.60 (m, 2H), 8.61 (s, 1H); 13C NMR (CDCl3) δ 16.3, 118.5, 118.9, 122.0, 125.5, 126.3, 127.2, 130.6, 131.5, 150.7, 153.4, 163.4, 169.5. LCMS: tR 1.98 min, m/z 243 [M + H]+.
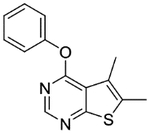
5,6-Dimethyl-4-phenoxythieno[2,3-d]pyrimidine (1c).
Compound 1c was prepared as described for the preparation of 1a except phenol was used in place of o-cresol. White solid (67% yield); mp 107–109 °C. 1H NMR (CDCl3) δ 2.51 (s, 3H), 2.56 (s, 3H), 7.20–7.33 (m, 3H), 7.44–7.51 (m, 2H), 8.49 (s, 1H); 13C NMR (CDCl3) δ 13.5, 13.6, 120.1, 121.8, 125.0, 125.6, 129.6, 132.6, 151.9, 152.2, 163.1, 167.7. LCMS: tR 2.16 min, m/z 257 [M + H]+.
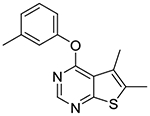
5,6-Dimethyl-4-(m-tolyloxy)thieno[2,3-d]pyrimidine (1d).
Compound 1d was prepared as described for the preparation of 1a except m-cresol was used in place of o-cresol. White solid (44% yield); mp 154–155 °C. 1H NMR (CDCl3) δ 2.41 (s, 3H), 2.51 (s, 3H), 2.55 (s, 3H), 6.99–7.07 (m, 2H), 7.11 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.7 Hz, 1H), 8.50 (s, 1H); 13C NMR (CDCl3) δ 13.6, 13.7, 21.4, 118.8, 120.2, 122.4, 125.1, 126.6, 129.4, 132.6, 140.0, 152.1, 152.3, 163.3, 167.8. LCMS: tR 2.29 min, m/z 271 [M + H]+.
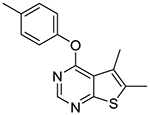
5,6-Dimethyl-4-(p-tolyloxy)thieno[2,3-d]pyrimidine (1e).
Compound 1e was prepared as described for the preparation of 1a except p-cresol was used in place of o-cresol. White powder (50% yield); mp 90–94 °C. 1H NMR (CDCl3) δ 2.40 (s, 3H), 2.50 (s, 3H), 2.56 (s, 3H), 7.11 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 8.49 (s, 1H); 13C NMR (CDCl3) δ 13.5, 13.6, 20.9, 120.1, 121.50, 121.52, 125.1, 130.19, 130.21, 132.5, 135.3, 150.0, 152.01, 152.03, 163.4, 167.6. LCMS: tR 2.26 min, m/z 271 [M + H]+.
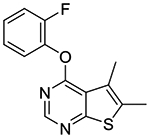
4-(2-Fluorophenoxy)-5,6-dimethylthieno[2,3-d]pyrimidine (1f).
Compound 1f was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol. White powder (75% yield); mp 143–145 °C. 1H NMR (CDCl3) δ 2.53 (s, 3H), 2.59 (s, 3H), 7.22–7.39 (m, 4H), 8.50 (s, 1H); 13C NMR (CDCl3) δ 13.57, 13.62, 116.8 (d, JCF = 18.3 Hz), 119.8, 124.0, 124.6 (d, JCF = 4 Hz), 125.0, 127.0 (d, JCF = 7 Hz), 133.0, 139.6 (d, JCF = 12.7 Hz), 151.77, 151.82, 154.5 (d, JCF = 249 Hz), 162.3, 167.9. LCMS: tR 2.22 min, m/z 275 [M + H]+.
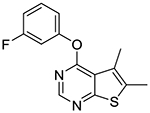
4-(3-Fluorophenoxy)-5,6-dimethylthieno[2,3-d]pyrimidine (1g).
Compound 1g was prepared as described for the preparation of 1a except 3-fluorophenol was used in place of o-cresol. White solid (62% yield); mp 158–161 °C. 1H NMR (CDCl3) δ 2.51 (s, 3H), 2.53–2.56 (m, 5H), 6.96–7.07 (m, 5H), 7.38–7.47 (m, 1H), 8.50 (s, 1H); 13C NMR (CDCl3) δ 13.6, 109.9 (d, JCF = 24.2 Hz), 112.7 (d, JCF = 20.9 Hz), 117.6 (d, JCF = 3.4 Hz), 120.1, 124.9, 130.4 (d, JCF = 9.3 Hz), 133.2, 151.8, 153.2 (d, JCF = 10.8 Hz), 162.6, 163.1 (d, JCF = 248 Hz), 168.0. LCMS: tR 2.16 min, m/z 275 [M + H]+.
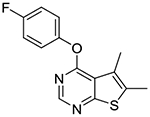
4-(4-Fluorophenoxy)-5,6-dimethylthieno[2,3-d]pyrimidine (1h).
Compound 1h was prepared as described for the preparation of 1a except 4-fluorophenol was used in place of o-cresol. White fluffy needles (54% yield); mp 137–140 °C. 1H NMR (CDCl3) δ 2.51 (s, 3H), 2.55 (s, 3H), 7.12–7.22 (m, 5H), 8.48 (s, 1H); 13C NMR (CDCl3) δ 13.6, 13.7, 116.3 (d, JCF = 23.6 Hz), 120.0, 123.3 (d, JCF = 8.3 Hz), 125.0, 132.9, 148.0 (d, JCF = 3 Hz), 151.8, 160.1 (d, JCF = 244 Hz), 163.1, 167.8. LCMS: tR 2.16 min, m/z 275 [M + H]+.
5,6-Dimethyl-4-(2-(trifluoromethyl)phenoxy)thieno[2,3-d]pyrimidine (1i).
Compound 1i was prepared as described for the preparation of 1a except 2-(trifluoromethyl)phenol was used in place of o-cresol. White powder (53% yield). mp 155–157 °C. 1H NMR (CDCl3) δ 2.52 (s, 3H), 2.55 (s, 3H), 7.34–7.45 (m, 2H), 7.64–7.70 (m, 1H), 7.77 (d, J = 7.9 Hz, 1H), 8.47 (s, 1H). 13C NMR (CDCl3) δ 13.3, 13.7, 120.0, 123.0 (q, JCF = 273 Hz), 123.4, 124.7, 125.1, 125.7, 127.2 (q, JCF = 4.8 Hz), 133.0, 133.2, 149.65, 151.6, 162.5, 168.1. LCMS: tR 2.29 min, m/z 325 [M + H]+.
5,6-Dimethyl-4-(3-(trifluoromethyl)phenoxy)thieno[2,3-d]pyrimidine (1j).
Compound 1j was prepared as described for the preparation of 1a except 3-(trifluoromethyl)phenol was used in place of o-cresol. White solid (71% yield); mp 120–126 °C. 1H NMR (CDCl3) δ 2.52 (s, 3H), 2.56 (s, 3H), 7.40–7.65 (m, 4H), 8.49 (s, 1H); 13C NMR (CDCl3) δ 13.6, 119.1 (q, JCF = 3.8 Hz), 120.1, 122.4 (q, JCF = 3.9 Hz), 123.5 (q, JCF = 272 Hz) 124.8, 125.5, 130.1, 132.1 (q, JCF = 32.9 Hz), 133.3, 151.6, 152.4, 162.5, 168.1. LCMS: tR 2.38 min, m/z 325 [M + H]+.
5,6-Dimethyl-4-(4-(trifluoromethyl)phenoxy)thieno[2,3-d]pyrimidine (1k).
Compound 1k was prepared as described for the preparation of 1a except 4-(trifluoromethyl)phenol was used in place of o-cresol. Light yellow needles (33% yield). mp 115–120 °C. 1H NMR (CDCl3) δ 2.52 (s, 3H), 2.55 (s, 3H), 7.36 (d, 2H), 7.73 (d, 2H), 8.49 (s, 1H). 13C NMR (CDCl3) δ 13.62, 13.65, 120.2, 122.2, 123.9 (q, JCF = 271 Hz), 124.8, 127.0 (q, JCF = 3.7 Hz), 127.8 (q, JCF = 32.9 Hz), 133.4, 151.66, 151.70, 154.9, 162.4, 168.2. LCMS: tR 2.35 min, m/z 325 [M + H]+.
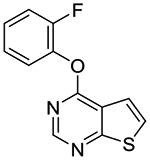
4-(2-Fluorophenoxy)thieno[2,3-d]pyrimidine (1l).
Compound 1l was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol, and 4-chlorothieno[2,3-d]pyrimidine 2b was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. White solid (36% yield). mp 104–106 °C. 1H NMR (CDCl3) δ 7.20–7.37 (m, 4H), 7.53 (d, J = 5.9 Hz, 1H), 7.57 (dd, J = 6.0, 0.6 Hz, 1H), 8.62 (s, 1H). 13C NMR (CDCl3) δ 116.9 (d, JC-F = 18.3 Hz), 117.0, 118.4, 118.7, 123.9, 124.8 (d, JCF = 3.7 Hz), 125.9, 127.2 (d, JCF = 7.1 Hz), 139.4 (d, JCF = 12.6 Hz), 153.1, 154.4 (d, JCF = 250 Hz), 162.8, 169.7. LCMS: tR 1.92 min, m/z 247 [M + H]+.
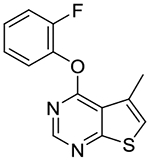
4-(2-Fluorophenoxy)-5-methylthieno[2,3-d]pyrimidine (1m).
Compound 1m was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol, and 4-chloro-5-methylthieno[2,3-d]pyrimidine 2c was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. Beige fluffy solid (93% yield). mp 98–102 °C. 1H NMR (CDCl3) δ 2.69 (s, 3H), 7.09 (s, 1H), 7.19–7.38 (m, 4H), 8.55 (s, 1H). 13C NMR (CDCl3) δ 17.0, 116.9 (d, JCF = 18.3 Hz), 118.6, 120.6, 124.0, 124.7 (d, JCF = 3.9 Hz), 127.1 (d, JCF = 6.9 Hz), 130.5, 139.5 (d, JCF = 12.6 Hz), 152.9, 154.5 (d, JCF = 249 Hz), 163.6, 170.4. LCMS: tR 2.07 min, m/z 261 [M + H]+.
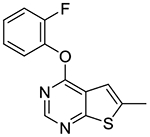
4-(2-Fluorophenoxy)-6-methylthieno[2,3-d]pyrimidine (1n).
Compound 1n was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol, and 4-chloro-6-methylthieno[2,3-d]pyrimidine 2d was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. White solid (84% yield). mp 135–138 °C. 1H NMR (CDCl3) δ 2.67 (s, 3H), 7.19–7.37 (m, 5H), 8.55 (s, 1H). 13C NMR (CDCl3) δ 16.4, 115.5, 116.9 (d, JCF = 18.5 Hz), 119.5, 123.9, 124.7 (d, JCF = 3.9 Hz), 127.1 (d, JCF = 7 Hz), 139.5 (d, JCF = 12.5 Hz), 140.8, 152.2, 154.4 (d, JCF = 250 Hz), 161.5, 169.5. LCMS: tR 2.07 min, m/z 261 [M + H]+.
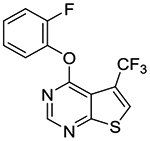
4-(2-Fluorophenoxy)-5-(trifluoromethyl)thieno[2,3-d]pyrimidine (1o).
Compound 1o was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol, and 4-chloro-5-(trifluoromethyl)thieno[2,3-d]pyrimidine 2e16 was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. White cake solid (90% yield). mp 65–66 °C. 1H NMR (CDCl3) δ 7.22 – 7.27 (m, 2H), 7.28 – 7.34 (m, 2H), 8.03 (s, 1H), 8.67 (s, 1H). 13C NMR (CDCl3) δ 114.4, 117.0 (d, JCF = 18.2 Hz), 121.1 (q, JCF = 270 Hz), 123.2 (q, JCF = 38 Hz), 123.7, 124.8 (d, JCF = 4.1 Hz), 127.5 (d, JCF = 7 Hz), 128.4 (q, JCF = 5.9 Hz), 139.2 (d, JCF = 12.7 Hz), 154.2, 154.3 (d, JCF = 250 Hz), 162.3, 170.3. LCMS: tR 2.13 min, m/z 315 [M + H]+.
6-(tert-butyl)-4-(2-fluorophenoxy)thieno[2,3-d]pyrimidine (1p).
Compound 1p was prepared as described for the preparation of 1a except 2-fluorophenol was used in place of o-cresol, and 6-(tert-butyl)-4-chlorothieno[2,3-d]pyrimidine 2f was used in place of 4-chloro-5,6-dimethylthieno[2,3-d]pyrimidine 2a. Clear oil (60% yield). 1H NMR (CDCl3) δ 1.51 (s, 9H), 7.21–7.35 (m, 5H), 8.54 (s, 1H). 13C NMR (CDCl3) δ 32.0, 35.4, 111.7, 116.9 (d, JCF = 18.3 Hz), 119.1, 123.9, 124.8 (d, JCF = 3.8 Hz), 127.1 (d, JCF = 7.2 Hz), 139.6 (d, JCF = 12.6 Hz), 152.3, 154.5 (d, JCF = 250 Hz), 158.1, 161.8, 169.0. LCMS: tR 2.41 min, m/z 303 [M + H]+.
6-tert-butyl-5-phenylthieno[2,3-d]pyrimidin-4-ol (4).
In a vial, 3,3-dimethyl-1-phenylbutan-1-one 3 (500 mg, 2.84 mmol), ethylcyanoacetate (483 mg, 4.26 mmol), sulfur (726 mg, 2.84 mmol), formamide (0.9 mL, 22.7 mmol), L-proline (65.3 mg, 0.57 mmol), and diethylamine (41.5 mg, 0.57 mmol) was heated to 170 °C and stirred for 6 h. After the vial cooled to rt, the residue was triturated with DCM. The resulting black solid was removed by filtration and the filtrate was concentrated and purified by Biotage Isolera One, using 30–50% EtOAc in DCM as eluent to give 35 mg of the desired compound as yellow solid (4% yield). 1H NMR (CDCl3) δ 1.24 (s, 9H), 7.25–7.31 (m, 2H), 7.34–7.46 (m, 3H), 10.91 (s, 1H). LCMS: tR 1.98 min, m/z 285 [M + H]+.
6-tert-butyl-4-chloro-5-phenylthieno[2,3-d]pyrimidine (5).
A solution of 4 (35 mg, 0.12 mmol) in POCl3 (0.5 mL) was stirred at 110 °C for 1 h. Excess POCl3 was removed in vacuo followed by co-evaporation with DCM, three times. The crude was purified by Biotage Isolera One using 0–10% EtOAc in DCM as eluent to give 25 mg of the desired compound as yellow oil (66% yield). 1H NMR (CDCl3) δ 1.30 (s, 9H), 7.28–7.32 (m, 2H), 7.38–7.47 (m, 3H), 8.76 (s, 1H). LCMS: tR 2.44 min, m/z 303 [M + H]+.
6-tert-butyl-4-(2-fluorophenoxy)-5-phenylthieno[2,3-d]pyrimidine (1q).
Sodium hydride (60% dispersion in mineral oil, 5.3 mg, 0.13 mmol) was added to a solution of 2-fluorophenol (14.8 mg, 0.13 mmol) in DMF (0.2 mL) at 0 °C and the mixture was stirred at 0 °C for 5–10 min. A solution of 5 (20 mg, 0.07 mmol) in DMF (0.3 mL) was then added slowly to the mixture via syringe at 0 °C. The reaction mixture was stirred and allowed to warm to rt overnight. A few drops of 10% KHSO4 were added to the mixture to quench the excess NaH and the mixture was then concentrated in vacuo. The crude residue was triturated in methanol and water to give 10 mg of desired compound as a light yellow solid (40% yield); Mp 81–82 °C. 1H NMR (CDCl3) δ 1.33 (s, 9H), 6.92 (dt, J = 8.4, 4.1 Hz, 1H), 7.03–7.17 (m, 3H), 7.27–7.44 (m, 5H), 8.50 (s, 1H); 13C NMR (CDCl3) δ 32.6, 36.4, 116.6 (d, JCF =18 Hz), 123.5, 124.4, 126.6 (d, JCF = 7 Hz), 127.4, 127.5, 129.1, 130.4, 137.1, 143.0 (d, JCF = 19 Hz), 151.0, 152.2, 154.1 (d, JCF = 250 Hz), 162.2, 165.9. LCMS: tR 2.64 min, m/z 379 [M + H]+.
6-tert-Butyl-4-methoxythieno[2,3-d]pyrimidine (6).
To a solution of 6-tert-butyl-4-chlorothieno[2,3-d]pyrimidine 2f (1.62 g, 7.15 mmol) in methanol (20 mL) at 0 °C was added 25 wt. % sodium methoxide in methanol (17.2 mmol, 3.92 mL). The mixture was stirred at 0 °C for 30 min, then at rt for 1 h. The solvent was removed in vacuo and water was carefully added to quench excess of sodium methoxide. The compound was extracted with EtOAc and the organic layer was dried over Na2SO4 and concentrated to give 1.56 g of the desired compound as a yellow oil which was used without further purification (98% crude yield). 1H NMR (CDCl3) δ 1.45 (s, 9H), 4.12 (s, 3H), 7.06 (s, 1H), 8.59 (s, 1H).
5-Bromo-6-tert-butyl-4-methoxythieno[2,3-d]pyrimidine (7).
To a solution of 6 (1.56 g, 7.02 mmol) in AcOH (30 mL) was added N-bromosuccinimide (3.75 g, 21.1 mmol) and the mixture was stirred at 55 °C overnight. The reaction was concentrated and the crude material was purified by Biotage Isolera One using 0–10% EtOAc in hexanes as eluent to give 1.8 g of the desired compound as a light-yellow solid (85% yield). 1H NMR (CDCl3) δ 1.59 (s, 9H), 4.14 (s, 3H), 8.59 (s, 1H).
6-(tert-butyl)-5-(4-fluorophenyl)-4-methoxythieno[2,3-d]pyrimidine (8a).
A suspension of 7 (200 mg, 0.66 mmol), 4-fluorophenylboronic acid (186 mg, 1.33 mmol), potassium carbonate (4.13 g, 29.9 mmol), and bis(triphenylphosphine)palladium(II) dichloride (46.6 mg, 0.07 mmol) in DMF (8 mL) was purged with N2 gas via an inserted long needle for 30 min. The mixture was then stirred at 80 °C overnight. After removing solvent in vacuo, water was added to the mixture. The mixture was extracted with dichloromethane twice and the combined extracts were washed with brine, dried over Na2SO4, and concentrated. The resulting white precipitate was removed by filtration and the filtrate was concentrated and purified by Biotage Isolera One using 5% EtOAc/DCM as eluent to give 190 mg of the desired compound as a thick brown oil (92% yield). 1H NMR (CDCl3) δ 1.27 (s, 9H), 3.69 (s, 3H), 7.02 – 7.12 (m, 2H), 7.19 – 7.26 (m, 2H), 8.56 (s, 1H).
6-tert-Butyl-5-(3,4-dichlorophenyl)-4-methoxythieno[2,3-d]pyrimidine (8b).
A suspension of 7 (3.00 g, 9.96 mmol), 3,4-dichlorophenylboronic acid (3.80 g, 19.9 mmol), potassium carbonate (4.13 g, 29.9 mmol), and bis(triphenylphosphine)palladium(II) dichloride (0.699 g, 1.00 mmol) in DMF (45 mL) was purged with N2 gas via an inserted long needle for 30 min. The mixture was then stirred at 80 °C overnight. After removing solvent in vacuo, water was added to the mixture. The mixture was extracted with dichloromethane twice and the combined extracts were washed with brine, dried over Na2SO4, and concentrated. The resulting white precipitate was removed by filtration and the filtrate was concentrated and purified by Biotage Isolera One using 5% EtOAc/DCM as eluent to give 2.73 g of the desired compound as a thick brown oil (75% yield). 1H NMR (CDCl3) δ 1.30 (s, 9H), 3.74 (s, 3H), 7.13 (dd, J = 2.0, 8.1 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 8.58 (s, 1H).
6-tert-Butyl-5-(4-fluorophenyl)thieno[2,3-d]pyrimidin-4-ol (9a).
To a solution of 8a (189 mg, 0.60 mmol) in DCM (6 mL) at 0 °C was added 1 M BBr3 solution in DCM (1.80 mL, 1.80 mmol). The mixture was stirred at 0 °C, then gradually warmed to rt and stirred until completion (overnight). The mixture was then cooled to 0 °C and methanol was added to quench excess of BBr3. After removal of solvents, the crude material was partitioned between water and dichloromethane. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The residual solid was purified by Biotage Isolera One using 20–35% EtOAc in hexanes with 2% AcOH as eluent to give 75 mg of the desired compound as a white solid (41% yield). 1H NMR (CDCl3) δ 1.25 (s, 9H), 7.05 – 7.12 (m, 2H), 7.22 – 7.27 (m, 2H), 7.77 (s, 1H), 10.59 (s, 1H).
6-tert-Butyl-5-(3,4-dichlorophenyl)thieno[2,3-d]pyrimidin-4-ol (9b).
To a solution of 8b (1.50 g, 4.08 mmol) in DCM (50 mL) at 0 °C was added 1 M BBr3 solution in DCM (20.4 mL, 20.4 mmol). The mixture was stirred at 0 °C, then gradually warmed to rt and stirred until completion (7 days). The mixture was then cooled to 0 °C and methanol was added to quench excess of BBr3. After removal of solvents, the crude material was partitioned between water and dichloromethane. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The residual solid was triturated with dichloromethane to give 1.27 g of the desired compound as a white solid (93% yield). 1H NMR (CDCl3) δ 1.28 (s, 9H), 7.14 (dd, J = 2.0, 8.1 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 7.76 (s, 1H), 11.25 (s, 1H).
6-tert-Butyl-4-chloro-5-(4-fluorophenyl)thieno[2,3-d]pyrimidine (10a).
A solution of 9a (70 mg, 0.23 mmol) in POCl3 (3 mL) was stirred at 110 °C for 1 h. Excess POCl3 was removed in vacuo followed by co-evaporation with DCM (three times). The residual material was passed through a short silica column using 5% EtOAc in hexanes as eluent to give 45 mg of the desired compound as a yellow solid (61% yield). 1H NMR (CDCl3) δ 1.30 (s, 9H), 7.08 – 7.18 (m, 2H), 7.24 – 7.34 (m, 2H), 8.77 (s, 1H).
6-tert-Butyl-4-chloro-5-(3,4-dichlorophenyl)thieno[2,3-d]pyrimidine (10b).
A solution of 9b (2.00 g, 5.66 mmol) in POCl3 (15 mL) was stirred at 110 °C for 1 h. Excess POCl3 was removed in vacuo followed by co-evaporation with DCM (three times). The residual material was passed through a short silica column using 0–10% EtOAc in DCM as eluent to give 2.00 g of the desired compound as a white solid (95% yield). 1H NMR (CDCl3) δ 1.33 (s, 9H), 7.17 (dd, J = 2.0, 8.3 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.79 (s, 1H).
6-(tert-butyl)-4-(2-fluorophenoxy)-5-(4-fluorophenyl)thieno[2,3-d]pyrimidine (1r).
To a solution of 2-fluorophenol (41.9 mg, 0.37 mmol) in DMF (1 mL) was added sodium hydride (60 % dispersion in mineral oil, 11.2 mg, 0.28 mmol) at 0 °C and the mixture was stirred at 0 °C for 5–10 min. A solution of 10a (60 mg, 0.19 mmol) in DMF (2 mL) was then slowly added to the mixture via syringe at 0 °C. The reaction mixture was gradually warmed up to rt and stirred overnight. A few drops of 10% KHSO4 was added to the mixture to quench excess of NaH and the mixture was concentrated in vacuo. The crude material was partitioned between water and EtOAc. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residual material was purified by Biotage Isolera One using 10% EtOAc in hexanes as eluent to give 60 mg of desired compound as a light tan solid (77% yield); mp 172–173 °C. 1H NMR (CDCl3) δ 1.33 (s, 9H), 6.94 (t, J = 8.0 Hz, 1H), 7.01 – 7.22 (m, 5H), 7.32 – 7.43 (m, 2H), 8.51 (s, 1H); 13C NMR (CDCl3) δ 32.6, 36.4, 114.5 (d, JCF = 21 Hz), 116.7 (d, JCF = 18 Hz), 120.4, 123.6, 124.4 (d, JCF = 4 Hz), 126.7 (d, JCF = 6.8 Hz), 127.9, 132.0 (d, JCF = 8.1 Hz), 133.0 (d, JCF = 3.5 Hz), 139.3 (d, JCF = 12.7 Hz), 151.6, 152.4, 154.1 (d, JCF = 249 Hz), 162.2, 162.3 (d, JCF = 247 Hz), 165.9. LCMS: tR 2.63 min, m/z 397 [M + H]+.
6-(tert-butyl)-5-(4-fluorophenyl)-4-(2-(trifluoromethoxy)phenoxy)thieno[2,3-d]pyrimidine (1s).
To a solution of 2-trifluoromethoxyphenol (35 mg, 0.31 mmol) in DMF (1 mL) was added sodium hydride (60 % dispersion in mineral oil, 12.5 mg, 0.31 mmol) at 0 °C and the mixture was stirred at 0 °C for 5–10 min. A solution of 10b (50 mg, 0.16 mmol) in DMF (1 mL) was then slowly added to the mixture via syringe at 0 °C. The reaction mixture was gradually warmed up to rt and stirred overnight. A few drops of 10% KHSO4 was added to the mixture to quench excess of NaH and the mixture was concentrated in vacuo. The crude material was partitioned between water and EtOAc. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residual material was purified by Biotage Isolera One using 0–10% EtOAc in hexanes as eluent to give 46 mg of desired compound as a yellow solid (65% yield); mp 127–129 °C. 1H NMR (CDCl3) δ 1.33 (s, 9H), 6.98 – 7.07 (m, 3H), 7.22 – 7.38 (m, 5H), 8.49 (s, 1H); 13C NMR (CDCl3) δ 29.6, 32.5, 36.4, 114.5 (d, JCF = 21.5 Hz), 120.2 (q, JCF = 259 Hz) 120.3, 122.3, 122.7, 124.0, 124.4, 126.5, 126.8, 127.6, 127.8, 127.9, 132.0 (d, JCF = 8.1 Hz), 132.8 (d, JCF = 3.7 Hz), 140.87, 140.90, 143.8, 151.7, 152.2, 152.4, 162.2, 162.3 (d, JCF = 246 Hz) 165.9. LCMS: tR 2.63 min, m/z 463 [M + H]+.
6-tert-butyl-5-(3,4-dichlorophenyl)-4-(2-(trifluoromethoxy)phenoxy)thieno[2,3-d]pyrimidine (1t).
To a solution of 2-trifluoromethoxyphenol (0.529 g, 2.97 mmol) in DMF (6 mL) was added sodium hydride (60 % dispersion in mineral oil, 0.119 g, 2.97 mmol) at 0 °C and the mixture was stirred at 0 °C for 5–10 min. A solution of 10b (0.920 g, 2.48 mmol) in DMF (15 mL) was then slowly added to the mixture via syringe at 0 °C. The reaction mixture was gradually warmed up to rt and stirred overnight. A few drops of 10% KHSO4 was added to the mixture to quench excess of NaH and the mixture was concentrated in vacuo. The crude material was partitioned between water and EtOAc. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residual material was purified by Biotage Isolera One using CHCl3 as eluent to give 0.880 g of desired compound as a white solid (69% yield); mp 85–89 °C. 1H NMR (CDCl3) δ 1.35 (s, 9H), 7.02 (m, 1H), 7.23–7.33 (m, 4H), 7.43 (d, J = 8.3 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 8.51 (s, 1H); 13C NMR (CDCl3) δ 32.6, 36.5, 119.9, 120.2 (q, JCF = 259 Hz), 122.4, 124.1, 126.2, 126.7, 127.7, 129.5, 129.9, 131.6, 131.8, 132.2, 137.0, 140.9, 143.7, 152.3, 152.4, 162.1, 166.0. LCMS: tR 2.98 min, m/z 513.1 [M + H]+. Anal. Calcd. for C23H17Cl2F3N2O2S: C, 53.81; H, 3.34; N, 5.46; S, 6.25; F, 11.10; Cl, 13.81. Found: C, 53.82; H, 3.41; N, 5.38, S, 6.23; F, 10.93; Cl, 13.87.
In vitro MRGPRX1 receptor assay.
HEK293 cells stably transfected with human MrgprX1 were plated in 96 well plates at 25,000 cell /well and incubated 2 days before imaging. On the day of imaging cells were incubated in 100 μL HBSS with 2 μM Fluo 4AM and 1% Trypan Red for 50 minutes at 37°C. The cells were then equilibrated for 10 minutes at room temperature before imaging. Test compounds were dissolved in HBSS and diluted in a serial dilution. Test compounds, BAM 8–22 (positive control) or HBSS (negative control) were added (50 μL into 100 μL) and cells were imaged on the FLIPR for 2 minutes. Data was exported as maximum – minimum fluorescent signal.
In vitro Metabolic Stability Studies.
The metabolic stability was evaluated using mouse and human liver microsomes as previously reported.17 The assay was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, 50 μM sodium citrate). Reactions, in triplicate, were initiated by addition of the liver microsomes to the incubation mixture (compound final concentration was 5 μM; 0.5 mg/mL microsomes). Controls in the absence of cofactors were carried out to determine the specific cofactor-free degradation. After 30 and 60 min of incubation, aliquots of the mixture were removed in triplicate and the reaction quenched by addition of three times the volume of ice-cold acetonitrile spiked with the internal standard. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method.
Metabolite identification studies.
Metabolite identification was performed on samples obtained from human liver microsomes. Samples (2 μL) were injected on an Agilent 1290 UPLC coupled to an Agilent 6520 quadrupole time of flight mass spectrometer. Metabolites were separated on an Agilent Eclipse Plus C18 1.8 μm, 2.1 × 100 mm column with a 5-minute gradient from 97.5/2.5 to 5/95 water + 0.1% formic acid/acetonitrile + 0.1% formic acid mobile phase. Analysis was performed in positive ion mode, with fragmentation by collision induced dissociation with a collision energy set at 25. Product ions were identified using MassHunter Qualitative analysis software.
In vivo pharmacokinetic studies in mice.
All pharmacokinetic studies in mice were conducted according to protocols approved by the Animal Care and Use Committee at Johns Hopkins University. Male CD 1 mice between 25 and 30 g were obtained from Harlan, Inc., and maintained on a 12-hour light-dark cycle with ad libitum access to food and water. Compound 1t was given to mice (n=3) by oral gavage at 100 mg/kg in a vehicle (5% dimethyl acetamide, 10% Cremophor EL, 10% Tween80, 25% PEG400, and 50% water). The mice were sacrificed at predetermined time points (0.25–6 h) post drug administration. For collection of plasma and spinal cord and brain, animals were euthanized with CO2, and blood samples were collected in heparinized microtubes by cardiac puncture. Spinal cords and brains were dissected and immediately flash frozen (−80 °C). Plasma was obtained by centrifugation of blood at 3,000 g for 15 minutes, and stored at −80 °C until LC/MS analysis.
Prior to extraction, frozen samples were thawed on ice. The calibration curves were developed using plasma and spinal cord from naïve animals as a matrix. Plasma samples (50 μL), were processed using a single liquid extraction method by addition of 300 μL of acetonitrile as with internal standard (losartan [5 μM]), followed by vortexing for 30 s and then centrifugation at 10000 rcf for 10 min. For spinal cord tissue, homogenized samples were vortexed and centrifuged as above. A 10 μL aliquot of supernatant was diluted with 40 μL water containing 0.5 μM losartan as internal standard. Extracts were centrifuged at 10000 rcf at 4 °C for 10 minutes. Supernatants were transferred to 250 μL polypropylene autosampler vials sealed with a Teflon cap. A volume of 3 μL was injected onto the ultra-performance liquid chromatography (UPLC) instrument for quantitative analysis by LC/MS/MS. Calibration curves over the 0.01–10 nmol/mL plasma and spinal cord were constructed from the peak area ratio of the analyte to the internal standard using linear regression with a weighting factor of 1/(nominal concentration). Correlation coefficient of greater than 0.99 was obtained in all analytical runs for all analytes.
Sciatic nerve chronic constrictive injury (CCI) model of neuropathic pain.
MRGPRX1 mice were generated as described in our previous study.8 MRGPRX1 mice (both sexes) were bred at the Johns Hopkins University, and had access ad libitum to food and water. Neuropathic pain was induced by CCI surgery to the sciatic nerve in adult mice (2- to 3-month-old, 20–30 g). Mice were anesthetized by inhalation of 2% isoflurane delivered through a nose cone. Skin was shaved, and small incision was made at the level of the mid-thigh. The left sciatic nerve was exposed by blunt dissection through the biceps femoris, and separated from the surrounding tissue. The nerve trunk proximal to the distal branching point was loosely tied with three nylon sutures (6–0 nonabsorbable braided silk suture, 18020–60, Fine Science Tools, CA, USA) until the epineurium was slightly compressed and minor twitching of the relevant muscles was observed. The ligatures were approximately 0.5 mm apart. The muscle layer was closed with 4–0 silk suture and the wound closed with suture.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by NIH grants UG3NS115718 (T.T.), R21NS101954 (TT), R01NS054791 (XD), R01NS070814 (Y.G.), R01NS117761 (Y.G.), R01NS110598 (Y.G.), 5T32NS070201 (I.B.), and the Johns Hopkins Institute for Clinical and Translational Research (ICTR), which is funded in part by UL1TR001079 from the National Center for Advancing Translational Sciences (NCATS). XD is an investigator of the Howard Hughes Medical Institute. Animal behavioral studies were facilitated by the Pain Research Core funded by the Blaustein Fund and the Neurosurgery Pain Research Institute at the Johns Hopkins University.
ABBREVIATIONS
- MRGPRX1
mas-related G-protein coupled receptor member X1
- DRG
dorsal root ganglion
- HTS
high throughput screening
- CCI
chronic constriction injury
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01709.
1H and 13C spectra of the final compounds, in vitro selectivity profile of compound 1t, and behavioral effects of compound 1t in naive MRGPRX1 mice (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
REFERENCES
- 1.Dong X; Han S; Zylka MJ; Simon MI; Anderson DJ A diverse family of gpcrs expressed in specific subsets of nociceptive sensory neurons. Cell 2001, 106, 619–632. [DOI] [PubMed] [Google Scholar]
- 2.Lembo PM; Grazzini E; Groblewski T; O’Donnell D; Roy MO; Zhang J; Hoffert C; Cao J; Schmidt R; Pelletier M; Labarre M; Gosselin M; Fortin Y; Banville D; Shen SH; Strom P; Payza K; Dray A; Walker P; Ahmad S Proenkephalin a gene products activate a new family of sensory neuron--specific gpcrs. Nat Neurosci 2002, 5, 201–209. [DOI] [PubMed] [Google Scholar]
- 3.Chen H; Ikeda SR Modulation of ion channels and synaptic transmission by a human sensory neuron-specific g-protein-coupled receptor, snsr4/mrgx1, heterologously expressed in cultured rat neurons. J Neurosci 2004, 24, 5044–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z; He SQ; Xu Q; Yang F; Tiwari V; Liu Q; Tang Z; Han L; Chu YX; Wang Y; Hin N; Tsukamoto T; Slusher B; Guan X; Wei F; Raja SN; Dong X; Guan Y Activation of mrgc receptor inhibits n-type calcium channels in small-diameter primary sensory neurons in mice. Pain 2014, 155, 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson SR; Gerhold KA; Bifolck-Fisher A; Liu Q; Patel KN; Dong X; Bautista DM Trpa1 is required for histamine-independent, mas-related g protein-coupled receptor-mediated itch. Nat Neurosci 2011, 14, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wroblowski B; Wigglesworth MJ; Szekeres PG; Smith GD; Rahman SS; Nicholson NH; Muir AI; Hall A; Heer JP; Garland SL; Coates WJ The discovery of a selective, small molecule agonist for the mas-related gene x1 receptor. J. Med. Chem 2009, 52, 818–825. [DOI] [PubMed] [Google Scholar]
- 7.Prchalova E; Hin N; Thomas AG; Veeravalli V; Ng J; Alt J; Rais R; Rojas C; Li Z; Hihara H; Aoki M; Yoshizawa K; Nishioka T; Suzuki S; Kopajtic T; Chatrath S; Liu Q; Dong X; Slusher BS; Tsukamoto T Discovery of benzamidine- and 1-aminoisoquinoline-based human mas-related g-protein-coupled receptor x1 (mrgprx1) agonists. J Med Chem 2019, 62, 8631–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z; Tseng PY; Tiwari V; Xu Q; He SQ; Wang Y; Zheng Q; Han L; Wu Z; Blobaum AL; Cui Y; Tiwari V; Sun S; Cheng Y; Huang-Lionnet JH; Geng Y; Xiao B; Peng J; Hopkins C; Raja SN; Guan Y; Dong X Targeting human mas-related g protein-coupled receptor x1 to inhibit persistent pain. Proc Natl Acad Sci U S A 2017, 114, E1996–E2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen W; Wang Y; Li Z; Tseng PY; McManus OB; Wu M; Li M; Lindsley CW; Dong X; Hopkins CR Discovery and characterization of 2-(cyclopropanesulfonamido)-n-(2-ethoxyphenyl)benzamide, ml382: A potent and selective positive allosteric modulator of mrgx1. ChemMedChem 2015, 10, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woszczek G; Fuerst E Ca2+ mobilization assays in gpcr drug discovery. Methods Mol Biol 2015, 1272, 79–89. [DOI] [PubMed] [Google Scholar]
- 11.Shi T; Kaneko L; Sandino M; Busse R; Zhang M; Mason D; Machulis J; Ambrose AJ; Zhang DD; Chapman E One-step synthesis of thieno[2,3-d]pyrimidin-4(3h)-ones via a catalytic four-component reaction of ketones, ethyl cyanoacetate, s8 and formamide. ACS Sustain Chem Eng 2019, 7, 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pubchem identifier: Aid 602413. https://pubchem.ncbi.nlm.nih.gov/bioassay/602413
- 13.McNeil BD; Pundir P; Meeker S; Han L; Undem BJ; Kulka M; Dong X Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowes J; Brown AJ; Hamon J; Jarolimek W; Sridhar A; Waldron G; Whitebread S Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat Rev Drug Discov 2012, 11, 909–922. [DOI] [PubMed] [Google Scholar]
- 15.Bennett GJ; Xie YK A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [DOI] [PubMed] [Google Scholar]
- 16.Taylor S; Murfin S; Coulter TS; Jaekel S; Aicher B; Kelter A-R; Krämer J; Kirchhoff C; Scheel A; Wӧlcke J Thienopyrimidines for pharmaceutical compositions. PCT Int. Appl. WO2006/136402, 2006.
- 17.Giancola JB; Bonifazi A; Cao J; Ku T; Haraczy AJ; Lam J; Rais R; Coggiano MA; Tanda G; Newman AH Structure-activity relationships for a series of (bis(4-fluorophenyl)methyl)sulfinylethyl-aminopiperidines and -piperidine amines at the dopamine transporter: Bioisosteric replacement of the piperazine improves metabolic stability. Eur J Med Chem 2020, 208, 112674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.