

An improved light-sheet microscope images live cells at sub 100nm axial resolution.
New light-microscopy techniques designed to improve spatial resolution are often less widely applicable than anticipated because of increased photo-toxicity in living samples. Writing in Nature Biotechnology, Bin Cao et al.1 report a technology that greatly increases the resolution of light-sheet microscopy without sacrificing its low photo-toxicity and high sensitivity. The method, called 3D interferometric lattice light-sheet (3D-iLLS) imaging, improves axial resolution through the coherent use of two detection objectives and structured illumination with optical lattices. The authors demonstrate live cell imaging with sub-100 nm axial resolution in a light-sheet format and single-molecule localization microscopy with sub-10 nm axial precision.
The idea of coherently using two opposing objectives for image formation dates to the early 1990s and was one of the first super-resolution concepts developed. By effectively doubling the numerical aperture and accessing light emitted from both sides of the sample, much higher resolution is possible than with a single objective system. For instance, if the two objectives covered all possible angles (i.e. a full sphere), then the resulting point-spread function of such a system would be a sphere as well (i.e., with the same resolution in any direction). Systems emerged both for laser scanning confocal microscopes2 and for widefield imaging3, and while different nomenclatures exist, they are most commonly referred to as 4Pi microscopes (referring to the 4Pi solid angle the two objectives should ideally cover). A renaissance of 4Pi microscopy has occurred in the field of localization microscopy, as the approach has enabled some of the highest axial localization precision to date4.
Now, Cao et al. elegantly apply the concept of 4Pi detection to light-sheet fluorescence microscopy (LSFM). In LSFM, the sample is illuminated with a sheet of light from the side, which avoids exciting out-of-focus fluorescence, while the emitted light is collected with a second objective perpendicular to the light sheet. LSFM is very gentle to the sample and has become a method of choice for sensitive live cell imaging. The desire to improve the resolving power in LSFM, in particular in the third, or ‘axial,’ dimension, has led microscope builders to adopt so-called propagation invariant beams that create lights sheets that are substantially thinner than the Gaussian light sheets that were initially employed. In particular, ‘lattice’ light sheets have been widely adopted5. However, even with advanced techniques, in practice axial resolution is no better than 350-1000 microns6. Further enhancing axial resolution using lattices or other engineered illumination beams would render these microscopes impractical as the light-sheets would become increasingly short, limiting the field of view, or they would generate excessive out-of-focus blur.
Rather than engineering the illumination part of the light-sheet microscope, Cao et al. focus on the detection optics. By using interferometric detection with two opposing objectives, they provide an orthogonal mechanism to improve axial resolution beyond the current state of the art. Figure 1A shows a simplified schematic of the setup. A thin light-sheet illuminates the sample along the joint focal plane of the two detection objectives. Fluorescence light from an emitter is captured by both objectives. Two mirrors guide the light to a beam splitter, which combines the two imaging paths and coherently interferes the fluorescence light on the camera. Importantly, even though fluorescence emission is incoherent, interference of a fluorescent photon with itself can occur if the two detection paths are of the same length (the path length difference must be smaller than the coherence length of fluorescence, which is on the order of microns). Minute phase differences between the two detection arms encode with interferometric precision the axial information of an emitter, which can be leveraged to increase the precision of particle localization and the resolution in 3D imaging.
Figure 1.
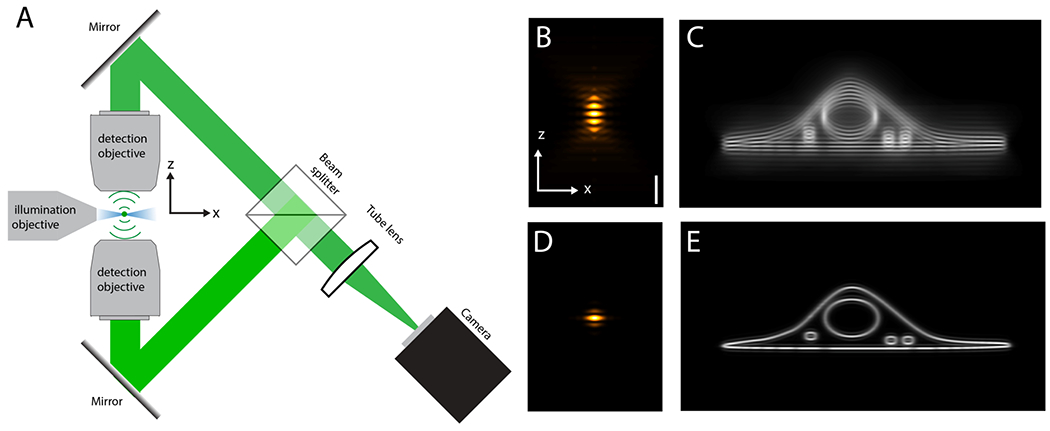
A Schematic and simplified setup for interferometric lattice light-sheet microscopy. Blue: Intensity of the light-sheet illumination. Green: fluorescence light from a single emitter. In a practical implementation, more mirrors than shown are needed for path length control and to prevent image rotation. B Detection point-spread function (PSF). C Cellular membranes imaged with the PSF shown in B. D overall PSF (detection and structured light-sheet illumination combined). E Same structure as in C imaged with PSF shown in D.
A fundamental problem of 4Pi microscopy is excessive sidelobes, which are residual local intensity maxima outside of the central lobe of the point-spread function (Figure 1B). These sidelobes occur because the two opposing objectives cannot cover the full solid angle. The strength and number of sidelobes increases when using objectives with lower numerical aperture.The sidelobe problem has been solved for 4Pi laser scanning microscopes with objectives of very high numerical aperture and two photon excitation7, or in widefield implementations with structured illumination8. For the application of the 4Pi concept to light-sheet microscopy, only moderate numerical aperture (here 1.1) detection objectives can be used, as higher numerical aperture objectives would not allow the insertion of the additional illumination objective due to steric hindrance. Using objectives with an NA of 1.1, the detection point-spread function has many sidelobes (Figure 1B), which would result in data riddled with ghost images (see also Figure 1C). Cao et al. solved this issue by using structured lattice light-sheets. This suppresses sidelobes both physically (by virtue of the thin light-sheet) and computationally (structured illumination image post processing)8, yielding a much clearer image (see also Figure 1D–E).
The authors present dual color time-lapse imaging in U2OS cells of microtubules and mitochondrial dynamics with axial resolution better than 100 nm at moderate temporal resolution (~1minute/volume). The imaging reveals the hollow structure of the mitochondria (labeled with a mitochondria membrane marker), and microtubules that are tightly packed in the z-direction are resolved — neither of which is visible with traditional lattice light-sheet imaging.
For single-molecule localization, it is necessary to quantify the small phase differences between the two detection arms. The authors achieve this by rapidly modulating the path length difference between the two arms with a piezo-electric actuator and collecting fluorescence with two cameras (not shown in Figure 1A). They demonstrate tracking of single mRNAs in live Hela cells with sub-10 nm precision and 50-ms temporal resolution.
Overall, this is an exciting development, as it enables an unprecedented combination of 3D resolution, imaging speed, sensitivity, and low photo-toxicity. Indeed, this work marks a rare application of 4Pi microscopy to live cell imaging. Nevertheless, formidable challenges remain. Refractive index differences may introduce point-spread functions that differ between different parts of the sample, making it difficult to computationally remove sidelobes. Indeed, in the presented work, small residual sidelobes are still visible, hinting at the difficulties posed by the heterogeneity of the sample. Additionally, the 4Pi detection arm must maintain alignment with interferometric precision over extended time periods, which is a serious engineering challenge. Thus, further work will be needed to make interferometric lattice light-sheet microscopy a widely applicable tool. Such work may include adding adaptive optics to the detection path, using reagents to balance the refractive index of the sample, and the development of novel high numerical aperture lenses and automatic alignment routines for the microscope system. Given the potential of LSFM with drastically improved axial resolution, meeting these challenges would offer rich rewards.
Acknowledgments:
The author would like to thank Dr. James Manton for help with simulating the 4Pi PSF used in Figure 1B&D.
References:
- 1.Cao B, Wang G, Li J & Pertsinidis A 3D Interferometric Lattice Light-Sheet Imaging. 10.1038/s41587-021-01042-y (2021). [DOI] [PMC free article] [PubMed]
- 2.Hell SW, Stelzer EHK, Lindek S & Cremer C Confocal microscopy with an increased detection aperture: type-B 4Pi confocal microscopy. Opt. Lett 19, 222–224 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Gustafsson MG, Agard DA & Sedat JW I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J Microsc 195, 10–16 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Shtengel G et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proceedings of the National Academy of Sciences 106, 3125–3130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B-C et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang B-J, Dean KM & Fiolka R Systematic and quantitative comparison of lattice and Gaussian light-sheets. Optics express 28, 27052–27077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hell SW, Lindek S & Stelzer EHK Enhancing the Axial Resolution in Far-field Light Microscopy: Two-photon 4Pi Confocal Fluorescence Microscopy. Journal of Modern Optics 41, 675–681 (1994). [Google Scholar]
- 8.Shao L et al. I5S: Wide-Field Light Microscopy with 100-nm-Scale Resolution in Three Dimensions. Biophysical journal 94, 4971–4983 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]