

Abstract
Asthma is a common and ubiquitous chronic respiratory disease that is associated with airway inflammation and hyperreactivity resulting in airway obstruction. It is now accepted that asthma is controlled by a combination of host genetics and environment in a rather complex fashion; however, the link between sensing of the environment and development and exacerbation of allergic lung inflammation is unclear. Human populations expressing co-segregating D299G and T399I polymorphisms in the Toll-like receptor 4 (TLR4) gene are associated with a decreased risk for asthma in adults along with hyporesponsiveness to inhaled lipopolysaccharide (LPS), the TLR4 ligand. However, this data does not account for other human genetic or environmental factors. Using a novel mouse strain that expresses homologous human TLR4 polymorphisms (TLR4-SNP), we directly tested the effect of these TLR4 polymorphisms on in vivo responses to allergens using two models of induction. We report that intact TLR4 is required for allergic inflammation when using the ovalbumin and LPS model of induction as cellular and pathological benchmarks were diminished in both TLR4-SNP and TLR4-deficent mice. However, in the more clinically relevant model using House Dust Mite Extract (HDM) for induction, responses were enhanced in the TLR4-SNP mice, as evidenced by greater levels of eosinophilic inflammation, TH2 cytokine production, and HDM-specific IgG1 production compared to WT mice; however, mucus production and airway hyperreactivity were not affected. These results suggest that the TLR4 polymorphic variants (genes) interact differently with the allergic stimulation (environment).
Introduction
Asthma is a common chronic respiratory disease associated with airway inflammation and hyperreactivity that results in airway obstruction. In the U.S.A., asthma is prevalent among 7.7% of the total population with an increasing trend and has a mortality rate of 10.5 deaths per million (1). To date, there is no cure and 10% of asthmatics do not respond to current drug therapies (2). One sub-type of asthma is associated with allergic responses; asthma attacks can be triggered by environmental irritants or allergens such as tobacco smoke, house dust mites (HDM), and cockroaches (3, 4). It is now generally accepted that asthma is controlled by a combination of host genetics and environment in a complex fashion (5–7).
The “farm effect” hypothesis proposes that early-life exposure to farming environments, or environments with high microbial diversity and load, are causally related to a reduced incidence of asthma in children and persist into adulthood. These environments are rich in endotoxin activity(8–10), stimulated by bacterial lipopolysaccharide (LPS) as well as other lipid species. LPS is a Gram-negative bacterial outer membrane component that is found ubiquitously throughout the environment; however, its specific role is controversial as both the prevention and/or exacerbation of asthma has been reported depending on dose and timing of endotoxin exposure. Exposure to endotoxin during infancy and early childhood was correlated with decreased development of asthma due to, perhaps, endotoxin tolerance or increased TH1 immunity (9, 11, 12). Conversely, endotoxin exposure in adulthood is associated with an increased risk of symptoms associated with asthma, but the degree of correlation may also be modulated by a number of genetic and environmental factors including childhood endotoxin exposure (8, 13). While pure LPS is a useful experimental tool, in vivo animal models of allergic asthma have also employed bacterial inoculations or environmental dust as priming agents to treat groups of young mice leading to protection from asthma development (14–16). As such, it is apparent that having a rich and diverse microbiome is important for the prevention of asthma.
Toll-like receptor 4 (TLR4) is one of the most well studied pattern recognition receptors (PRR) and has as its primary stimulus LPS. TLR4 expression was shown to be required for allergic lung inflammation in mice using ovalbumin (OVA) (17) or house dust mite (HDM) (18, 19) as priming agents and the underlying mechanisms attributed to a lack of a predominate TH2 -priming environment. The Lambrecht group identified that TLR4 expression on epithelial cells was required for the maturation of downstream dendritic cells (DCs) to mediate allergic airway inflammation (18). However, a more recent study demonstrated that TLR4 was not always required for allergic inflammation stimulated by HDM (20). A significant proportion of Caucasians express co-segregating D299G and T399I single nucleotide polymorphisms (SNPs) in the TLR4 gene. Arbour et al. first demonstrated that inheritance of these relatively common SNPs were associated with hyporesponsiveness to inhaled LPS and decreased detection of LPS by airway epithelial cells (21). The impact of these SNPs on allergic asthma remains unclear. These SNPs have variably been associated with asthma risk in human populations (22). A German study determined that expression of both SNPs led to a decreased risk for asthma in adults depending on the abundance of endotoxin in the environment (22). However, the authors acknowledge that the studies were correlative and may be attributable to other genetic or environmental factors not directly related to TLR4 haplotype. Recently, Richard et al. generated a novel mouse strain (TLR4-SNP) that homozygously expresses co-segregating D298G and N397I SNPs in TLR4 (the murine homologs of the human SNPs) and confirmed that these mice exhibit LPS-hyporesponsiveness, increased susceptibility to Gram-negative infection, and altered susceptibility to influenza and respiratory syncytial virus (23).
Using this novel mouse strain, we directly tested the effect of these TLR-SNPs on in vivo responses to allergens. Interestingly, we found disparate allergic inflammatory responses depending on the allergen system. Responses to LPS/OVA were reduced in TLR4-SNP mice when compared to responses in wild-type (WT) mice. In contrast, allergic inflammation induced by the complex allergen HDM was enhanced. These results suggest that the TLR4 polymorphic variants (genes) interact differently with the allergic stimulation (environment).
Materials and Methods
Animals and Allergic Asthma Models:
C57BL/6J, C3H/HeOuJ, and C3H/HeJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and acclimated to specific pathogen-free conditions at University of Maryland-Baltimore (UMB). Both TLR4-SNP mice (23) and TLR4−/− mice (provided by S. Akira; Osaka University, Osaka, Japan and backcrossed >12 times onto a C57BL/6J background), were bred at the UMB Animal Facility. All animal experiments were performed after approval from the University of Maryland, Baltimore Institutional Animal Care and Use Committee (IACUC). All animal experiments were age (8–10 weeks) and sex-matched.
Endofit Ovalbumin (OVA, Invivogen Labs, low-endotoxin concentration) was resuspended in endotoxin-free PBS. The endotoxin level of reconstituted OVA was measured using a Limulus Amebocyte Lysate (LAL) assay (Associates of Cape Cod Incorporated) and was 4.0 EU/100 μg OVA dose. Protein-free Escherichia coli K235 LPS was prepared by the hot phenol-water method as previously reported (24). Following the Eisenbarth et al. protocol (17), mice were treated intranasally (i.n.) on each of three days with either 100 μg OVA or 100 μg OVA+ 0.1 μg LPS. On days +14, +15, +18, +19, all groups were treated with only 100 μg OVA. Mice were euthanized on day 21 as has been previously determined as the time point that correlates with the peak in eosinophil number (25, 26).
House Dust Mite (HDM) Extract (Greer Labs, Lot #: 360924, endotoxin concentration: 13.14 EU/50 μg HDM dose) was resuspended in endotoxin-free PBS at 1 mg/mL based on total protein concentration and endotoxin level was confirmed using a Limulus Amebocyte Lysate (LAL) assay (Associates of Cape Cod Incorporated). HDM-treated groups received 50 μg/treatment intranasally (i.n.) while control groups received endotoxin-free PBS (Millipore-Sigma). Mice were treated on day zero and every 5th day thereafter for a total of 4 treatments. Mice were euthanized on day 17.
Bronchoalveolar Lavage, Cell Counting, and Flow Cytometry:
On day +17, mice were anesthetized with 250 mg/kg of 2.5% Avertin (2, 2, 2-Tribromoethanol) by intraperitoneal (i.p.) injection. Tracheas were cannulated and lungs were lavaged with 1.0 mL of PBS. Total cells from the bronchoalveolar lavage (BAL) were counted using an automated cell counter. The cells were separated from the BAL fluid (BAL) by centrifugation and resuspended in PBS. The BAL was stored at −80° C, while the cells were used for cytospins in preparation for differential staining and counting or for flow cytometry.
Differential cell counting was performed under oil using a 100x objective by an investigator blinded to the experimental groups. Cellular content of BAL was examined on cytospin preparations stained with Diff-Quick (Dade Behring, Newark, DE) and differential counts were performed based on cell morphology and staining as described (27).
In preparation for flow cytometry, BAL cells were first stained with a LIVE/DEAD Fixable Stain (Invitrogen) for 30 min at 4°C in the dark. Fc receptors (FcR) on cells were blocked with TruStain FcX (anti-mouse CD16/32 Ab, Biolegend) for 15 min at room temperature and then immediately stained for cellular surface markers with fluorophore-conjugated antibodies against CD45, Siglec-F, CD11c, CD3, Ly6G (all Biolegend) at a concentration recommended by the manufacturer. Isotype-matched antibodies (Biolegend) and fluorophore minus one (FMO) controls were tested for all stains. Stained cells were analyzed using a LSRFortessa Cell Analyzer (Becton Dickinson) and FlowJo 10 software (Becton Dickinson).
Cytokine Monitoring in lung tissue and BAL:
BAL fluid was examined for cytokine production in The Cytokine Core Lab (Center for Innovative Biological Research (CIBR), UMSOM). Type 2-cytokine protein production was assessed by Luminex multiplex bead array assay (Millipore) as per manufacturers protocol and were designed to detect: IL-4, IL-5, IL-10, IL-13, IL-25, IL-33, CCL11, and RANTES. Plates were read using a Luminex MagPix reader and data was calculated using Luminex’s exponent software. Serum was used in an anti-HDM IgG1 ELISA (Chondrex Inc., Woodinville, WA). Blood was collected on day +12 by sub-mandibular bleed, allowed to clot, and separated serum was collected and stored at −80°C.
Lung Histology and Scoring:
After BAL lavage, mice were perfused with PBS and the whole heart-lung block was extracted. Lung tissue for histology was fixed in 10% formalin for 24 hours and then stored in 70% ethanol until processing by the Pathology Core in CIBR. Tissue was blocked in paraffin wax, sectioned, and stained with Hematoxylin & Eosin (H&E) to assess inflammation or stained with a Periodic Acid Schiff (PAS) Stain to assess production of mucus as described (27).
Sections used for immunohistochemistry were incubated first with 10% goat serum for 20 minutes, then stained with a 1:100 dilution of rabbit anti-mouse Ym1 antibody (StemCell Technologies) as previously described (25, 26).
Lung sections were evaluated by a pathologist blinded to the experimental conditions. H&E and PAS stains were evaluated as previously described (27): H&E; 0, no signs of inflammation; 1, light and dispersed infiltrate in only a few areas of the section; 2, modest infiltrate surrounding major vessels and airways, with little in distal airway unit; 3, moderate infiltrate surrounding <50% of distal airways and vessels; 4, heavy and focused eosinophilic infiltrate surrounding numerous distal vessels and airways; signs of increased alveolar macrophage numbers with macrophage fusions and ECM deposition. PAS; 0, no signs of mucus in airways or elevated numbers of PAS+ cells; 1, no mucus in airways, slight increase in numbers of PAS+ cells in a few major airways; 2, some mucus detectable in major airways, 3, mucus detected in major airways with <50% of distal airway epithelial cells PAS+ in distal airway units; 4, mucus plugging of airways observed in several airways, majority of distal airway epithelial cells are PAS+ in multiple distal airways per section.
Ym1 immunohistochemistry was evaluated by a pathologist blinded to treatment groups under a 40x objective, and scored according to the number of Ym1+ macrophages present and intensity of staining: 0, no staining; 1, single, isolated Ym1+ macrophages in alveolar spaces; 2, increased numbers of Ym1+ macrophages in alveolar spaces and Ym1+ airway epithelial cells; 3, foci of multiple Ym1+ macrophages in most alveolar spaces; and 4, multiple foci of intensely staining Ym1+ macrophages invading the airway epithelial layer and entering the airway space (26–28).
Measurement of Airway Hyperactivity:
Airway hyperactivity was measured using a SciReq Flexivent ventilator with FX2 Module. Prior to tracheostomy, mice were sedated with a mixture of Ketamine (100 mg/kg) and Xylazine (10 mg/kg). Mice were cannulated with 18G beveled cannulas and then paralyzed with a dose of pancuronium (1 mg/kg). Airway responsiveness was measured after nebulization of increasing concentrations of methacholine by Flexivent measurements (29, 30). Airway hyperreactivity was determined by calculating the fold change of resistance (Rrs) for each methacholine dosage over baseline (% of baseline).
Statistics
Statistical significance was determined using two-tailed Student’s t-tests, multiple unpaired t-test, Kruskal-Wallis test with Dunn’s multiple comparison’s test, or 2-way ANOVA with Tukey’s Multiple Comparison Test as indicated in the figure legends. All statistical tests, analysis, and graphs were calculated or generated using GraphPad Prism 9.0 software (La Jolla, CA).
Results
Induction of allergic inflammation with LPS and OVA is reduced in TLR4-SNP mice
Mice expressing the co-segregating D298G and N397I SNPs in TLR4 (TLR4-SNP) exhibited LPS-hyporesponsiveness and increased susceptibility to Gram-negative infection (23). To determine the impact of these polymorphisms on allergic disease, we first evaluated responses in the TLR4-dependent allergenic model developed by Eisenbarth et al. (17). Wild-type (WT) C57BL/6J, TLR4-SNP, and TLR4−/− mice (males and females) were primed with OVA alone or OVA and LPS (OVA+LPS) (Figure 1A); all mice were challenged with OVA. On day +21, all mice were euthanized to assess the level of allergic inflammation. BAL fluid samples were obtained from all groups and used for differential cell counts (Figure 1B–E). No significant differences in total cell number were detected among groups after bronchoalveolar lavage (BAL) (Figure 1C). WT mice treated with both OVA+LPS responded with a significant increase in the numbers (Figure 1D) and percentage of eosinophils (12% ± 3.9% vs. 53% ± 3.7%, p< 0.0001) (Figure 1F) and a concomitant decrease in the numbers (Figure 1E) and percentage of macrophages (78.7% ± 5.5% vs. 27.5% vs 3.7%, p<0.0001) (Figure 1G) in the BAL when compared to mice treated with OVA only. Modest increases were apparent in the percentages of neutrophils and lymphocytes of OVA+LPS-treated WT groups. A statistically significant difference in the increase of lymphocyte percentage was detected in both WT (13.1± 1.1% vs. 5.9% ± 1.2%, p< 0.01, data not shown) and TLR4-SNP (15.1± 2.3% vs. 7.2% ± 1.5%, p< 0.01) mice receiving OVA+LPS when compared to the OVA-only group, while the increase in neutrophil percentage was not statistically significant (data not shown); however, after OVA+LPS treatment, neutrophil percentage was significantly greater in WT mice (7.2± 1.1%) than in TLR4-SNP (1.1± 0.3%, p< 0.003) and TLR4−/− (0.8± 0.3% p< 0.01) mice of the same treatment (not shown). In the TLR4-SNP mice, OVA+LPS stimulated a modest increase in the percentage of eosinophils (12% ± 4.1% vs. 31% ± 7.9%) (Figure 1D) and a modest decrease in the percentage of macrophages reduction (78% ± 5.9% vs. 53% ± 7.4%) (Figure 1E) compared to the OVA only-treated TLR4-SNP group; however, the increase in the percentage of lymphocytes between OVA+LPS and OVA-only groups among TLR4-SNP mice was statistically significant and mirrored the same 2-fold increase in lymphocytes as the OVA+LPS-treated WT group. These OVA+LPS-induced changes were less dramatic when compared to WT mice and did not achieve statistical significance (Figure 1D–E). Of note, the eosinophil response to OVA+LPS in TLR4−/− mice was substantially diminished (2.3% ± 0.8% vs. 6.3% ± 2.0%) when compared to its OVA only control group, however the lack of increase in the percent of eosinophils was significantly different when compared to responses in the WT (p<0.0001) mice. No sex differences between males (closed symbols) and females (open symbols) were apparent in any treatment groups.
Figure 1: Induction of allergic asthma with OVA and LPS treatment is dependent on TLR4 expression.
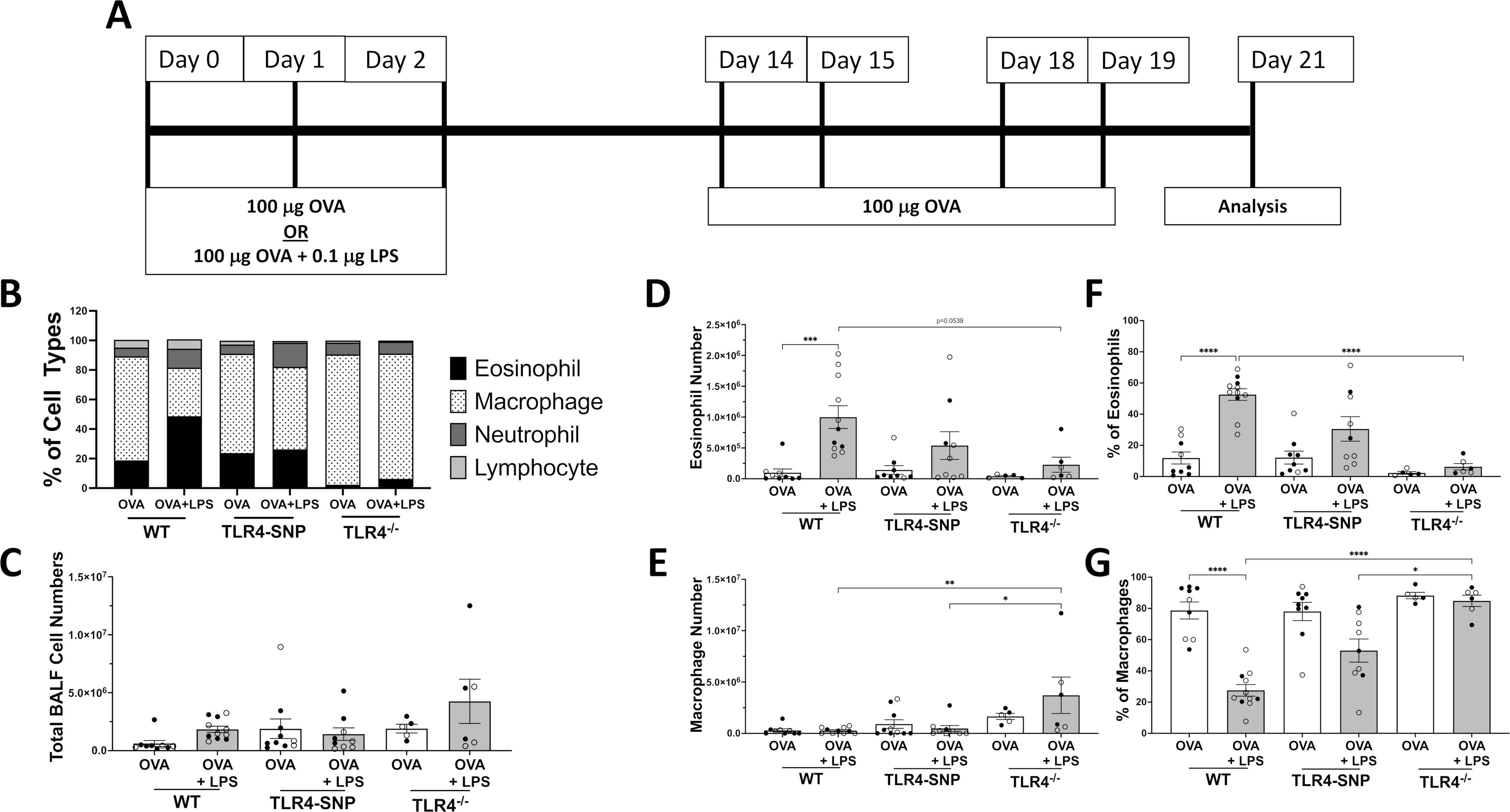
WT, TLR4-SNP, or TLR4−/− mice were treated with either Ovalbumin (OVA), Lipopolysaccharide (LPS), or a combination of both according to the timeline (A). Groups of mice were initially treated with 100 μg of OVA or OVA + 0.1 μg of LPS for 3 days, and then all groups were treated with 100 μg of OVA for 4 days prior to euthanasia on day +21. Lungs were lavaged with 1 mL of PBS to obtain Bronchoalveolar Lavage (BAL); this was used to prepare cytospins followed by differential cell counting to determine the percentages of eosinophils, macrophages, neutrophils, and lymphocytes in each treatment group. (B) is a representative summation of 1 individual trial to describe the cell type composition and percentages of BALF. (C) shows total BALF cell counts from 3 independent trials, (D-G) shows the individual total cell number (D, E) and individual cell percentage (F, G) of eosinophils and macrophages of individual mice from 3 independent trials (n=4–5/treatment group/trial for OVA-treated groups, n=5/treatment group/trial for OVA-LPS-treated groups) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM.
TLR4-SNP mice have reduced inflammatory pathology in response to OVA +LPS
Lung sections stained with H&E and PAS were examined and scored by a pathologist blinded to the identity of the experimental groups to determine the level of inflammation and mucus production, respectively (Figures 2 and 3). Similar to BAL cell counts, all groups primed with OVA only had minimal inflammation (Figure 2A) with average H&E scores of less than 1.0 (Figure 2B). WT mice treated with OVA+LPS had the greatest level of inflammation with a mean score of 3.2 ± 0.20 when compared to all other groups with 4 out of 11 mice scoring maximal inflammation, 4. In contrast, TLR4-SNP mice treated with OVA+LPS had a mean H&E score of 1.8 ± 0.36 with 0 of 9 mice exhibiting a maximal inflammation score. OVA+LPS-induced responses were dramatically curtailed in TLR4−/− mice. Similar to the inflammatory response, mice treated with only OVA demonstrated minimal mucus production with average PAS scores ranging ~0–0.5 (Figure 3B). The WT and TLR4-SNP mice showed similar increases in PAS positivity in response to OVA+LPS with average PAS scores of 1.18 ± 0.18 and 1.00 ± 0.24, respectively. However, in TLR4−/− mice OVA+LPS-induced mucus production was greatly reduced with an average PAS score of 0.33 ± 0.21.
Figure 2: Mice expressing TLR4-deficiencies have reduced inflammatory pathology after treatment with OVA and LPS.

Lung sections from WT, TLR4-SNP, and TLR4−/− mice were fixed, blocked, and then stained with Hematoxylin & Eosin to determine cellular infiltrate at 10X and 20X lens magnification (A). Lung sections were then evaluated by a pathologist blinded to the experimental conditions and scored on a scale of 0–4 (B). Scoring is aggregated from 3 independent trials (n=5 mice/treatment group/trial) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM.
Figure 3: Mice that lack TLR4 produce less mucus after treatment with OVA and LPS.
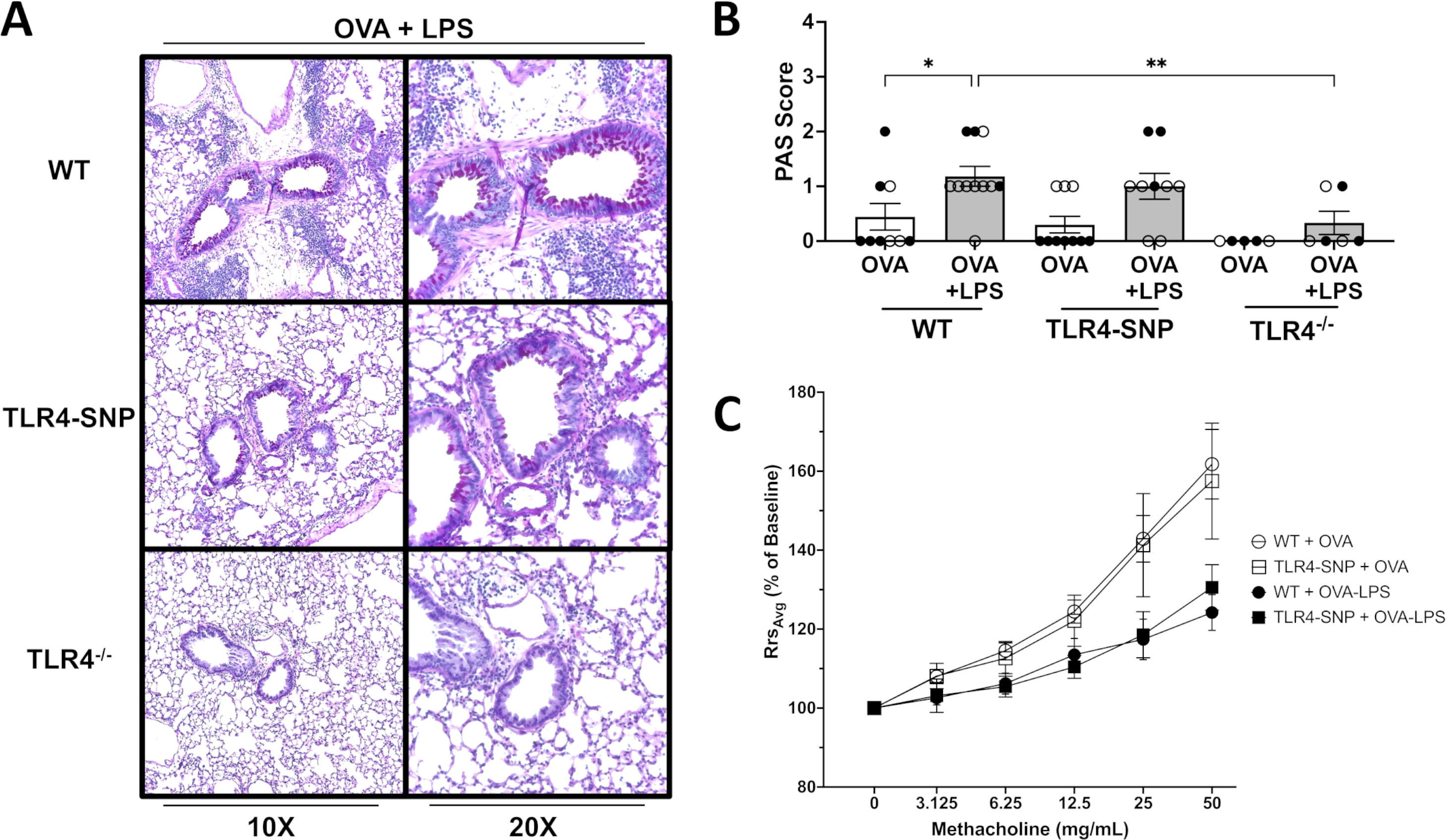
Lung sections from WT, TLR4-SNP, and TLR4−/− mice were fixed, blocked, and then stained with a Periodic Acid Schiff Stain to determine mucous production at 10X and 20X lens magnification (A). Lung sections were then evaluated by a pathologist blinded to the experimental conditions and scored on a scale of 0–4 (B). Scoring is aggregated from 3 independent trials (n=5 mice/treatment group/trial) with each point representing an individual mouse. Airway hyperreactivity was measured using a SciReq Flexivent Ventilator (C). Mice were sedated prior to tracheostomy and then paralyzed. Airway resistance was measured after nebulization of increasing concentrations of methacholine by Flexivent measurements. Airway hyperreactivity was determined by calculating the fold change of resistance (Rrs) for each methacholine dose over baseline (% of baseline). Data is aggregated from 2 independent trials (n=4–5/treatment group/trial for OVA-treated groups, n=5/treatment group/trial for OVA-LPS-treated groups) with each point representing the mean value of 2 independent trials. Open circles designate females and closed circles designate males. Scoring data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Airway hyperreactivity data analyzed by Kruskal-Wallis test with Dunn’s multiple comparison’s test with the following p-values (not shown). 50 mg: WT + OVA vs WT + OVA-LPS, p=0.0018 (**); 25 mg: WT + OVA vs WT + OVA-LPS, p=0.0313 (*). Error bars represent mean ± SEM.
To determine if airway mechanics were altered between WT and TLR4-SNP mice during allergic inflammation, we used a Mouse Flexivent Ventilator to monitor airway hyperreactivity (AHR). On day +21, all groups were sedated, paralyzed, cannulated, and nebulized with increasing concentration of methacholine. No significant difference in responsiveness were apparent between TLR4-SNP and WT mice that received either OVA-only or OVA-LPS Treatment (Figure 3C). We noted that both OVA-only-treated groups appeared to have greater reactivity when compared to OVA-LPS-treated groups; either OVA-only group reacted sooner at lower doses of methacholine and also had a trend of stronger bronchoconstriction of the small airways at higher doses (25 mg/mL and 50 mg/mL.) At 50 mg/mL of methacholine, WT and TLR4-SNP groups that received only OVA had a mean percent increase of only 161.8% ± 8.8% and 157.5% ± 14.7%, respectively, while WT and TLR4-SNP groups that received OVA-LPS had a mean percent increase of 124.2% ± 4.5% and 130.6% ± 5.8%, respectively. At 25 mg/mL of methacholine, WT and TLR4-SNP groups that received OVA only had a mean percent increase of 142.9% ± 5.9% and 141.3% ± 13.1%, respectively, while WT and TLR4-SNP groups that received OVA-LPS had a mean percent increase of 117.4% ± 5.1% and 118.6% ± 5.8%, respectively. A statistical significance was observed between WT groups receiving OVA and OVA-LPS at both 25 and 50 mg/mL of methacholine; however statistical significance was not observed between OVA and OVA-LPS groups of TLR4-SNP mice at any methacholine dosage.
We also analyzed the BAL for cytokines and chemokines associated with allergic inflammation (Figure 4) using a Luminex Multiplex Assay. Treatment of WT mice with OVA+LPS resulted in an increase in the levels of IL-5 and CCL11 and a significant increase in levels of IL-4 and RANTES protein when compared to OVA alone. This increase in all cytokines and chemokine levels was reduced in TLR4-SNP mice, with a significant decrease in IL-5 and RANTES and absent in the TLR4−/− mice. Other cytokines analyzed were all below detection of the assays including IL-13, IL-33, and IL-10. Taken together, these results demonstrate that the development of allergic inflammation by OVA+LPS is dependent on the expression of TLR4, and that allergic responses are reduced in the LPS-hyporesponsive TLR4-SNP mice.
Figure 4: Mice expressing TLR4-deficiencies have reduced type 2 cytokines after treatment with OVA and LPS.
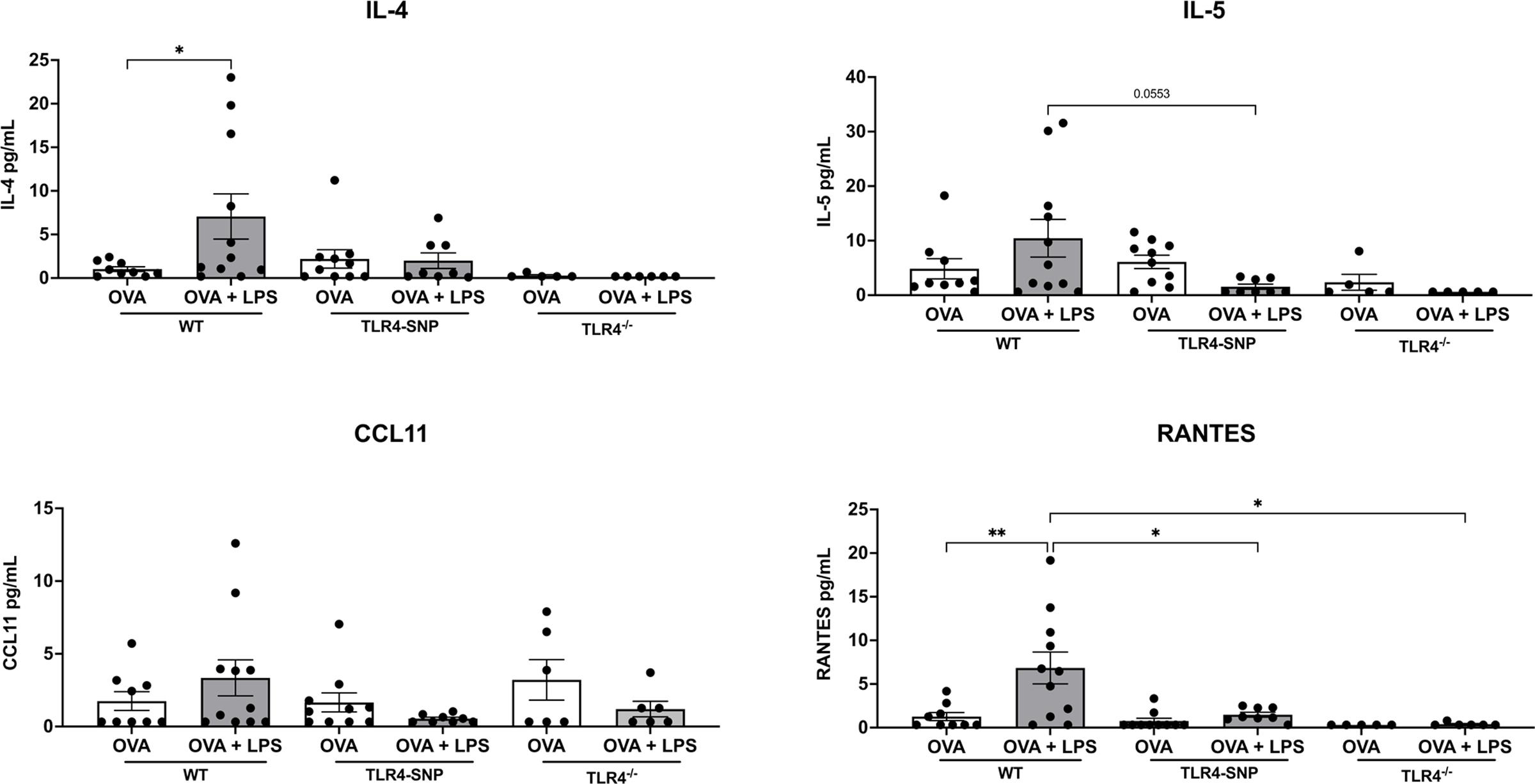
BAL fluid samples isolated from the indicated mice were analyzed for cytokines and chemokines including IL-4, IL-5, CCL11, and RANTES using Luminex Assays at the cytokine core lab at UMSOM’s Center for Innovative Biological Research. Data were aggregated from 2 independent trials (n=5 mice/treatment group/experiment) with each point representing an individual mouse. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005. Error bars represent mean ± SEM.
TLR4-SNP mice have increased numbers of eosinophils after HDM Treatment
We next sought to examine if TLR4 polymorphisms influence responses to the clinically relevant, complex human allergen, house dust mite (HDM). TLR4 has been reported to be required to induce allergic inflammation using a HDM model of induction (18, 19). However, another report suggested that responses to HDM are not absolutely dependent upon TLR4; instead, allergic inflammation was dependent on the precise nature, timing, and dose of HDM used (20). Therefore, we first wanted to address these differing reports and identify if TLR4 was required for the induction of allergic inflammation when using our HDM-model as we saw in the OVA-LPS model. To test the ability of our HDM to stimulate allergic inflammation in models of TLR4-deficiency, we used the C57BL/6 TLR4−/− and the spontaneous TLR4 mutant strain C3H/HeJ (31) in our HDM Model (Figure 5A). Groups of WT and TLR4-deficient mice were each administered either HDM or PBS on day 0 and then every 5 days thereafter for a total of 4 treatments (Figure 5A). All groups were euthanized 48 hours after the last HDM exposure (Day +17). We found that in two-independent experiments using either TLR4−/− or C3H/HeJ mice, HDM induced eosinophilic inflammation in the TLR4-deficent strains that was similar to the responses in matched HDM-treated C57BL/6J and C3H/HeOuJ, respectively (Figure S1). Differential counting of cells in the BAL from HDM-treated TLR4-deficent mouse strains (TLR4−/− and C3H/HeJ) revealed an increased trend in the percentages of eosinophils compared to PBS-treated controls, however HDM-treated C3H/HeJ mice had a low sample size (Figure S1D, E). This suggests that unlike the OVA-LPS model (Figure 1), the HDM model does not require expression of TLR4 for induction of allergic inflammation.
Figure 5: TLR4-SNP mice accumulate more eosinophils after exposure to HDM extract by differential cell counting.
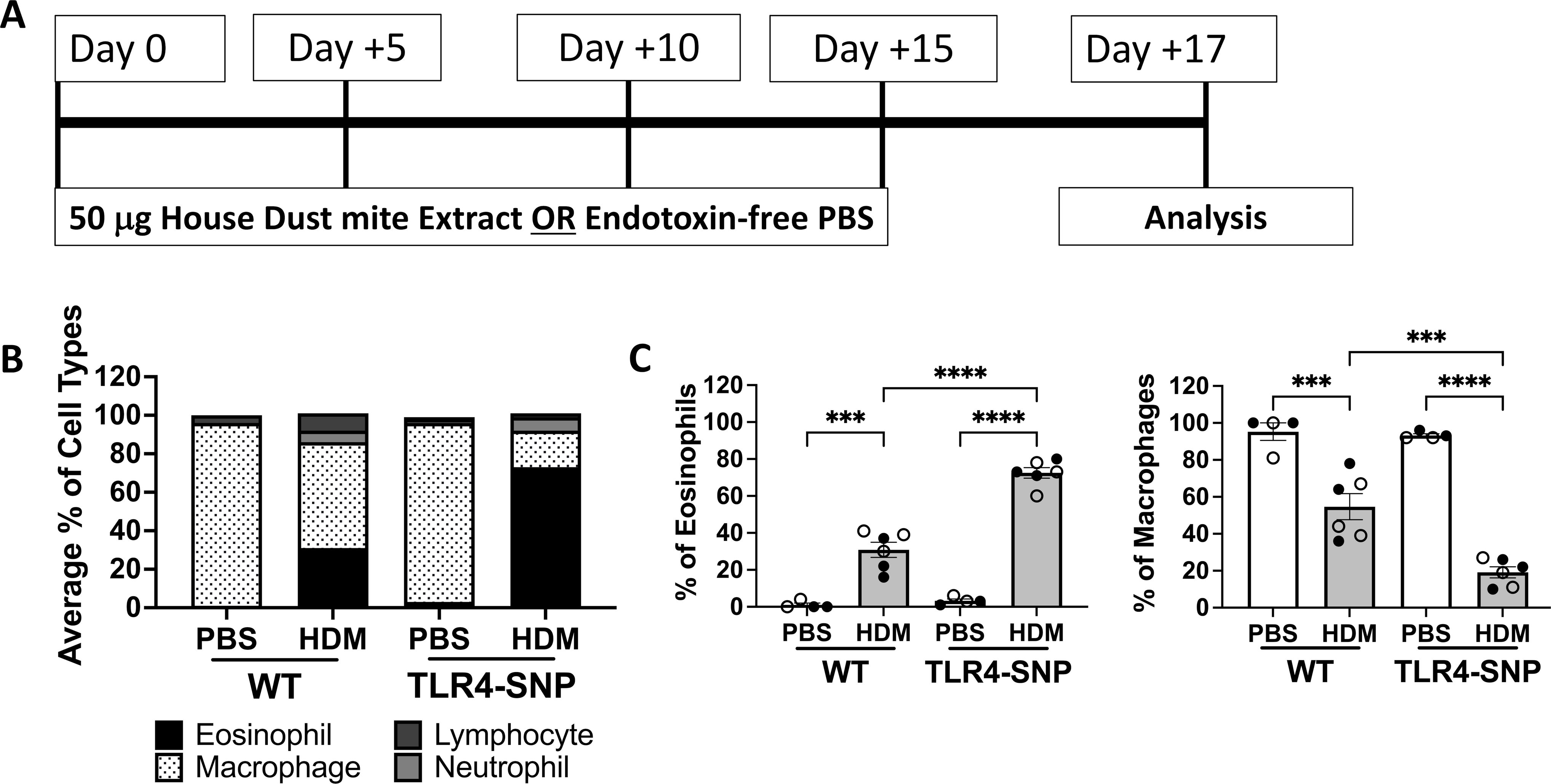
WT and TLR4-SNP mice were treated with either HDM extract or endotoxin-free PBS according to the timeline (A). Groups of mice were treated with PBS or 50 μg of HDM on days 0, +5, +10, +15, with euthanasia on day +17. Lungs were lavage with 1 mL of PBS to obtain BAL and was used for cell counts by Cytospin followed by differential counting (B, C). The mean percentages of eosinophils, macrophages, neutrophils, and lymphocytes in each treatment group (B) are representative of 3 individual trials. The percentages of eosinophils and macrophages (C) were aggregated from 3 independent trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM.
To determine if expression of the TLR4 SNPs influence the outcome of allergic inflammation in our HDM model (Figure 5A), we initially used differential cell counting (Figure 5B, C) and then flow cytometry (Figure 6) to assess eosinophilic inflammation. The TLR4-SNP mice treated with HDM demonstrated a significant increase (2-fold) in eosinophil percentages when compared to WT mice treated with HDM by differential counting (30% ± 4.1% vs. 72% ± 2.9%, p<0.0001, Figure 5C) with a concomitant decrease in macrophage representation (54% ± 7.0% vs. 19% ± 2.9%, p=0.0008, Figure 5C). Additionally, differential cell counting revealed a 2-fold increase in mean lymphocyte percentage between PBS and HDM-treated WT groups (3.0% ± 1.1% vs. 6.7% ± 0.9%, p<0.05, data not shown), whereas TLR4-SNP mice had a slight increase in mean lymphocyte percentage between PBS and HDM-treated groups (4.4% ± 1.3% vs. 7.3% ± 0.9%). Mean neutrophil percentages were not affected by HDM-treatment in WT mice (8.4% ± 4.2% vs. 7.2% ± 1.5%, data not shown), while TLR4-SNP mice showed a 5-fold increase between treatment groups that was not statically significant (0.8% ± 0.3% vs. 4.6% ± 1.7%). Using a multi-color staining flow cytometry approach (Figure S2), we also observed a significant increase in eosinophils (CD45+, Siglec-F+, CD11c−) in TLR4-SNP mice when compared to WT mice treated with HDM (65% ± 2.6% vs. 33% ± 4.6%, p<0.0001, Figure 6A, B). As in the OVA-LPS model, no differences due to sex were seen after HDM-treatment.
Figure 6: Mice expressing TLR4 SNPs accumulate more eosinophils after exposure to HDM extract by flow cytometry analysis.
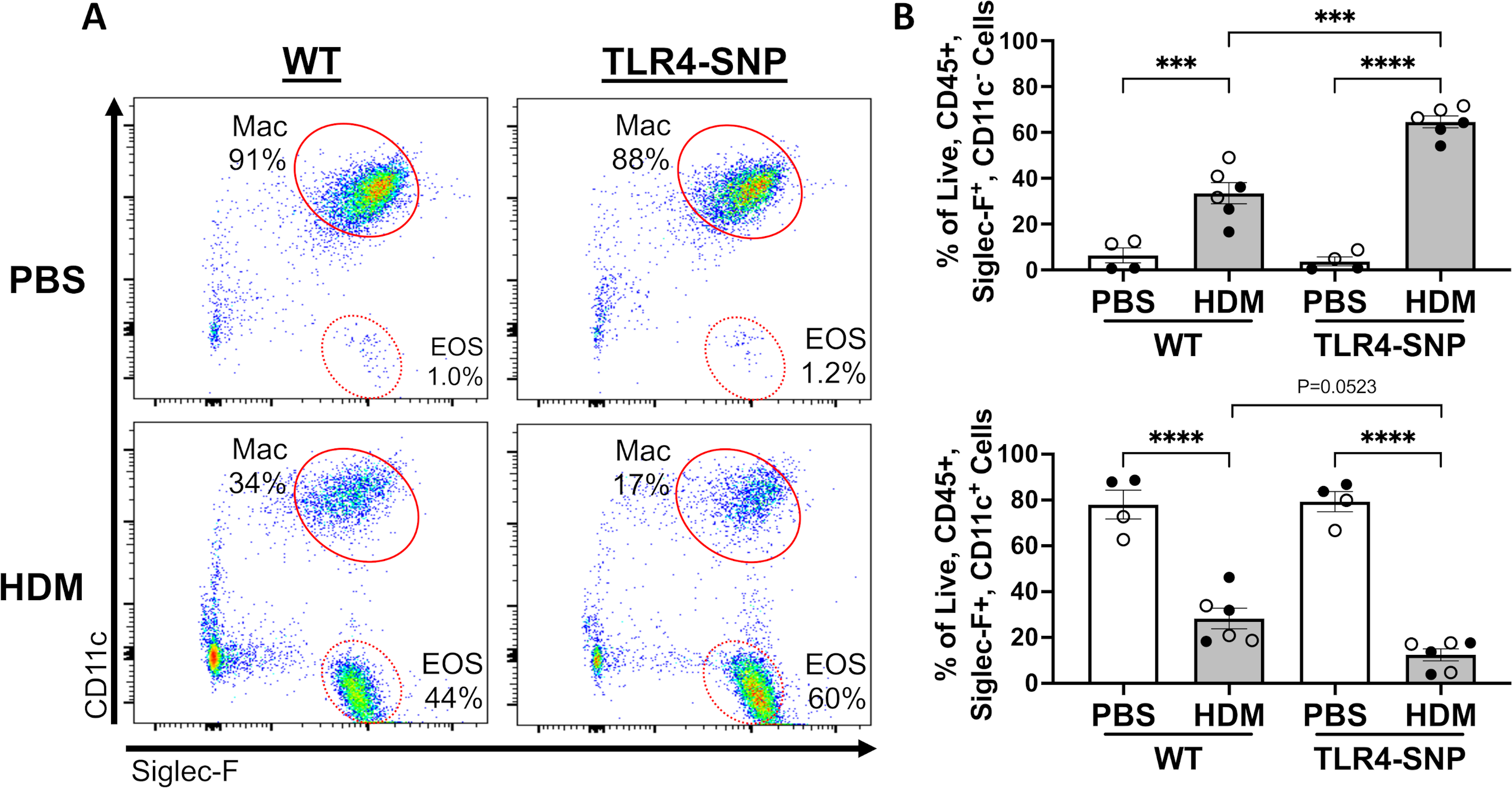
The lungs of WT and TLR4-SNP mice were lavaged with 1 mL of PBS to obtain BAL and was used for cell counts by Flow Cytometry (A, B) on day +17. Flow Cytometry data are gated on Live, CD45+ cells. A. Representative dot plots for Siglec F versus CD11c staining on gated cells. B. Flow cytometry data from 3 independent trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) were aggregated with each point representing an individual mouse. Open shapes, females. Closed shapes, males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001.
TLR4-SNP mice have increased inflammatory pathology in response to HDM
This increase in eosinophils in the BAL of TLR4-SNP mice was accompanied by increases in lung histopathology. On day +17, lung sections from all mice were prepared for staining with H&E and PAS to assess allergic inflammation and mucus production within the airways, respectively. Examination of H&E sections demonstrated an increase in cellular infiltrate surrounding the small airways of both HDM-treated mouse strains (Figure 7A–C) compared to PBS-treated mice. Moreover, an increase in eosinophilic infiltrate was observed in HDM-treated TLR4-SNP mice. Lung sections were evaluated by a pathologist blinded to the experimental conditions and both stains were evaluated on a 0–4 scale as previously described (27). PBS treated WT and TLR4-SNP group had little to no inflammation with a mean H&E score of 0.69 ± 0.18 and 0.08 ± 0.08, respectively. Both WT and TLR4-SNP showed significant increase in peribronchial and perivascular inflammation in response to HDM compared to the PBS-treated controls. The inflammatory infiltrate was dominated by eosinophils as evidenced upon high power examination (Figure 7C). Strikingly, the HDM-treated TLR4-SNP mice had significantly greater inflammation than the HDM-treated WT mice; the TLR-SNP group demonstrated a mean H&E score of 2.93 ± 0.27 with 6 of 16 mice scoring maximal pathology, in contrast to the WT mice that had a mean H&E score of 2.1 ± 0.19 with 0 of 17 mice scoring maximal pathology (Figure 7D). Analysis of the PAS-stained sections revealed an increase in mucus in both HDM-treated groups when compared to PBS-treated controls (Figure 8). However, unlike the inflammatory response, no difference in mean PAS scores was apparent between WT and TLR4-SNP groups that received HDM with mean PAS scores of 1.53 ± 0.19 and 1.87 ± 0.26, respectively (Figure 8B), although we noted that 5 of 15 TLR4-SNP mice scored 3 on the PAS scale while only 1 of 17 WT mice did so. These results suggest that TLR4-SNPs support greater eosinophilic inflammation of the lung after HDM treatment, with less of an effect on mucus production.
Figure 7: Mice expressing TLR4 SNPs have increased inflammatory pathology after treatment with HDM.

Lung sections from WT and TLR4-SNP mice on day +17 were fixed, blocked, and then stained with Hematoxylin & Eosin to determine cellular infiltrate at 10X (A), 40X (B), and 100X (C) lens magnifications. Lung sections were then evaluated by a pathologist blinded to the experimental conditions and scored on a scale of 0–4 (D). Scoring is aggregated from 3 independent trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM.
Figure 8: Airway hyperreactivity, but not mucus production, is altered in mice expressing TLR4 SNPs after treatment with HDM.
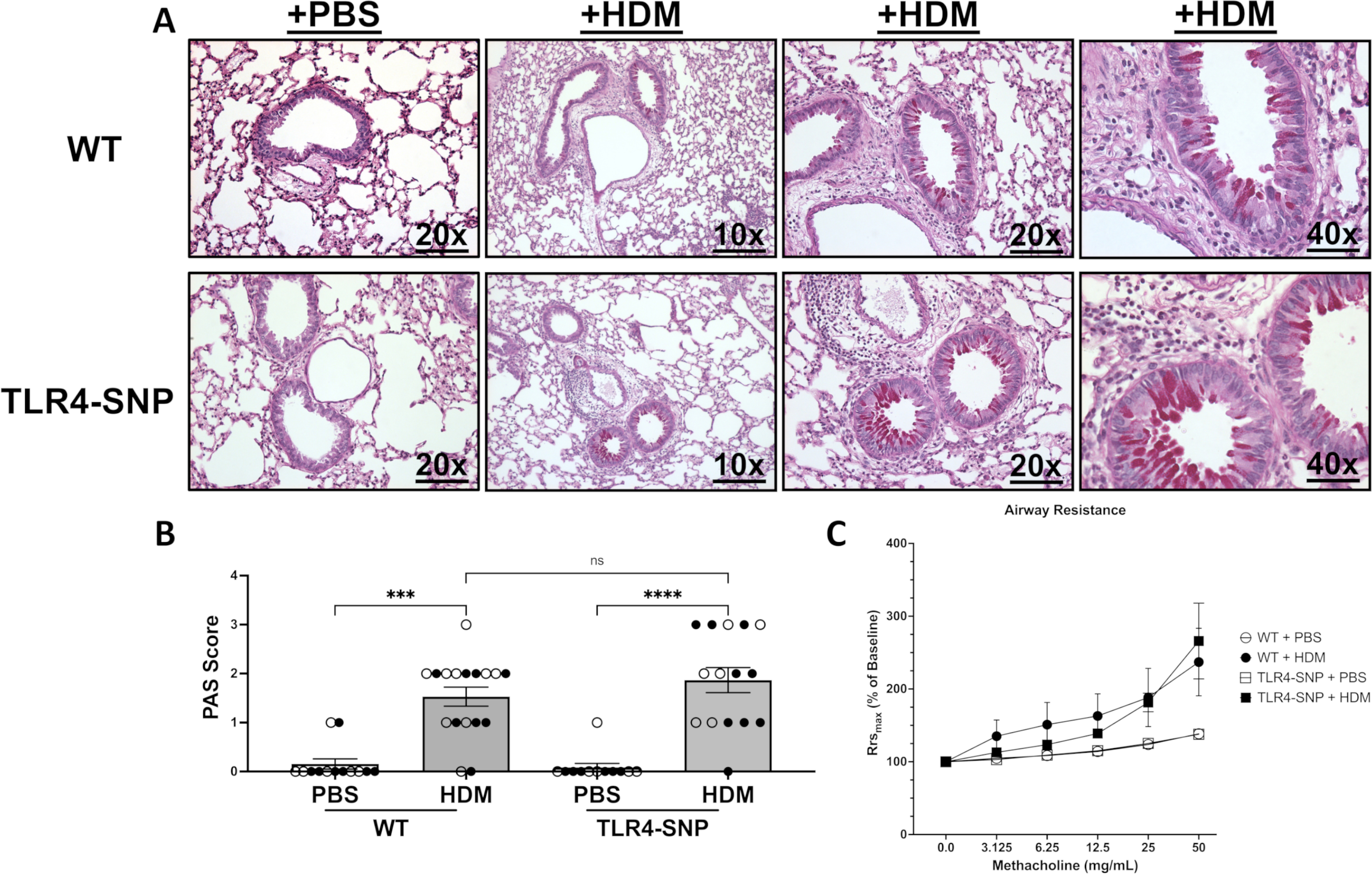
Lung sections from WT and TLR4-SNP mice on day +17 were fixed, blocked, and then stained with a Periodic Acid Schiff Stain to determine mucous production at 10X, 20X, and 40X lens magnification (A). Lung sections were then evaluated by a pathologist blinded to the experimental conditions and scored on a scale of 0–4 (B). Scoring is aggregated from 3 independent trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM. Airway hyperreactivity was measured using a SciReq Flexivent Ventilator (C). Mice were sedated prior to tracheostomy and then paralyzed. Airway resistance was measured after nebulization of increasing concentrations of methacholine by Flexivent measurements. Airway hyperreactivity was determined by calculating the fold change of resistance (Rrs) for each methacholine dose over baseline (% of baseline). Data is aggregated from 2 independent trials (n=4/trial for PBS-treated groups, n=6/trial for HDM-treated groups) with each point representing the mean value of 2 independent trials. Open circles designate PBS-treated mice and closed circles designate HDM-treated mice. All data was analyzed by Kruskal-Wallis test with Dunn’s multiple comparison’s test with the following p-values (not shown). 50 mg: WT + PBS vs WT + HDM, p=0.0432 (*), TLR4-SNP + PBS vs. TLR4-SNP + HDM, p = 0.0033(**); 25 mg: WT + PBS vs WT + HDM, p=0.0531, TLR4-SNP + PBS vs. TLR4-SNP + HDM, p = 0.0045(**); 12.5 mg: WT + PBS vs WT + HDM, p=0.0453 (*), TLR4-SNP + PBS vs. TLR4-SNP + HDM, p = 0.0204(*); unnoted interactions are non-significant. Error bars represent mean ± SEM.
Type 2 cytokines and IL-4-mediated responses are increased HDM-treated TLR4-SNP mice
To determine whether the TLR4 SNPs modified hallmarks of Type 2 inflammation, we first compared cytokine production between WT and TLR4-SNP mice by measuring the concentrations of IL-4, IL-5, IL-13, and IL-25, IL-33 in the BAL on day +17. The BAL of both HDM-treated groups demonstrated an increase in IL-4 and IL-5 concentrations when compared to PBS controls (Figure 9). IL-13, IL-25, IL-33, IL-12, and IL-10 were all below the limit of detection (not shown). Interestingly, HDM-treated TLR4-SNP mice showed a significant increase in IL-4 concentration and an increase in IL-5 that was non-significant when both cytokines were compared to HDM-treated WT mice. Furthermore, we observed a 2.4-fold increase in HDM-specific IgG1 in the serum of TLR4-SNP mice compared to WT mice (0.07 ± 0.01 vs. 0.17 ± 0.04), respectively; Interestingly, this increased IgG1 response is consistent with the observed increases in IL-4 concentration in BAL (Figure 9B). We did not detect any differences in total IgE between treatment groups or any HDM-specific IgE in the serum of any HDM-treated mice (data not shown). Increased production of IgE has been reported to be directly correlated with higher concentrations of proteases in HDM preparations, however, our HDM extract (Greer) had relatively lower levels of proteases compared to other manufacturers (32).
Figure 9: Type 2 cytokines and HDM-specific IgG1 production are increased in TLR4-SNP mice after treatment with HDM.
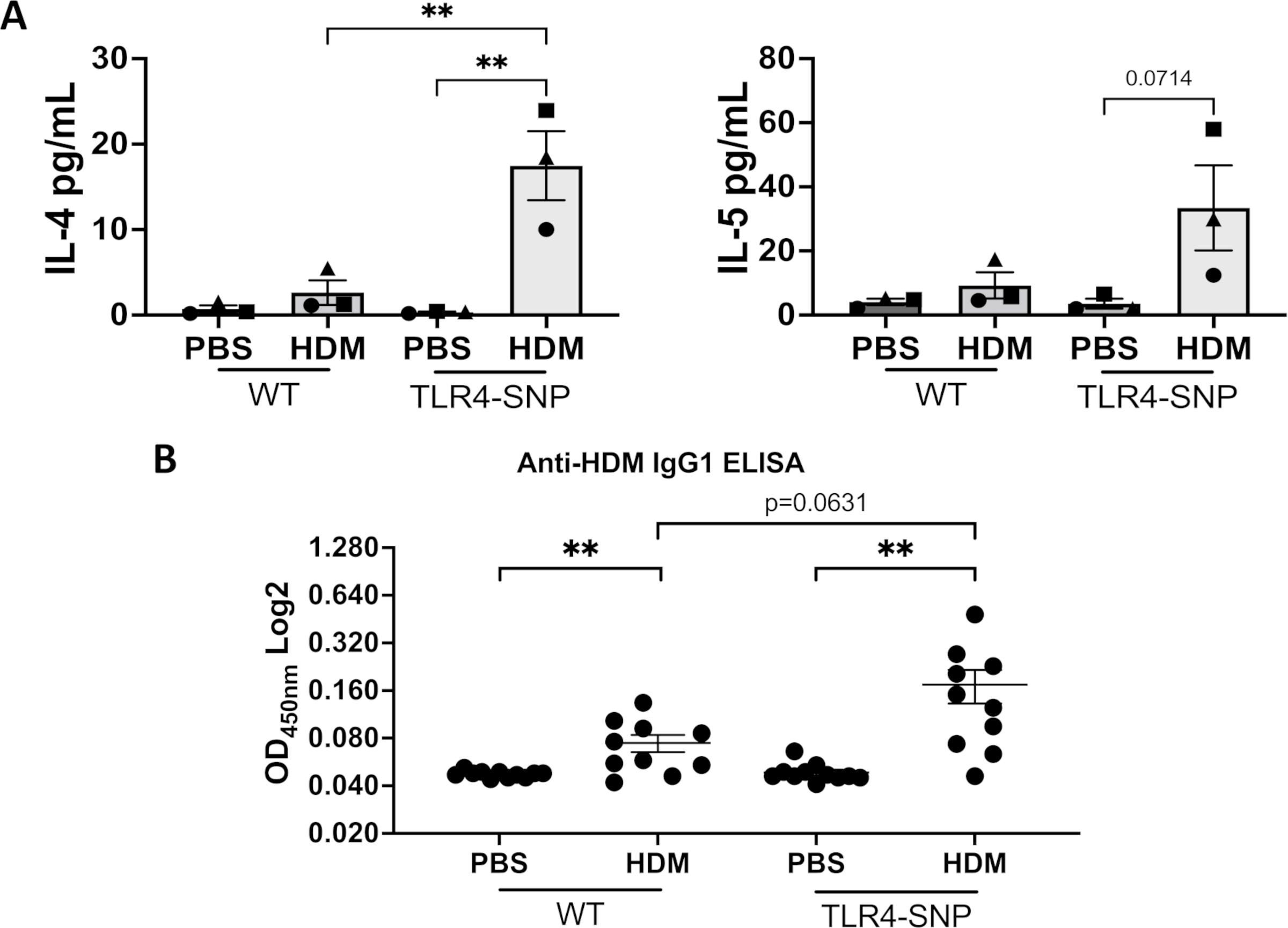
BAL samples were used to determine induction of allergic inflammation and was collected on day +17 from WT and TLR4-SNP mice. BAL fluid was tested for IL-4 and IL-5 using specific Luminex assays at the cytokine core lab at UMSOM’s Center for Innovative Biological Research (A). Serum was collected on day +12 and used in an anti-HDM IgG1 ELISA (B). The IL-4 and IL-5 BAL fluid charts (A) shows the mean cytokine concentration for 3 independent trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) with each shape representing the mean cytokine concentration of 1 independent trial. The anti-HDM IgG1 ELISA (B) is the aggregate of 2 independent trials (n=5/trial for PBS-treated groups, n=5/trial for HDM-treated groups) on log scale with each point representing an individual mouse. BALF data was analyzed by 2-way ANOVA with Tukey’s Multiple Comparisons Test: *, P<0.05; **, P<0.005. IgG1 ELISA data was analyzed by Multiple unpaired t-test: *, P<0.05; **, P<0.005. Error bars represent mean ± SEM.
We previously reported that M2a (Ym1+) macrophages were induced during allergic inflammation in response to allergens in an IL-4Rα-dependent manner, and that such macrophages were sufficient to enhance eosinophilic inflammation (26, 27). Therefore, we examined the presence of Ym1+ macrophages in the lungs by IHC (Figure 10). Using immunohistochemistry, lung sections from all mice on day +17 were stained with anti-Ym1 antibody to quantify Ym1 within the small airways. In agreement with our earlier report, both WT and TLR4-SNP groups that were treated with HDM had increased Ym1 positivity lining the airways compared to PBS-treated groups (Figure 10A, B). However, TLR4-SNP mice that received HDM exhibited much greater Ym1 positivity than WT mice of the same treatment (4.0 ± 0.00 vs 2.67 ± 0.42, p=0.010), with 6 of 6 mice scoring the maximal score of Ym1 positivity compared to 2 of 6 mice attaining the maximal score, respectively (Figure 10C). Taken together, these results suggest that TLR4-SNPs contribute to elevated IL-4 and IL-5 production in response to HDM leading to enhanced eosinophilic inflammation, M2a macrophage differentiation, and HDM-specific IgG1.
Figure 10: Immunohistochemistry reveals increased Ym1 positivity within the small airways of TLR4-SNP Mice.
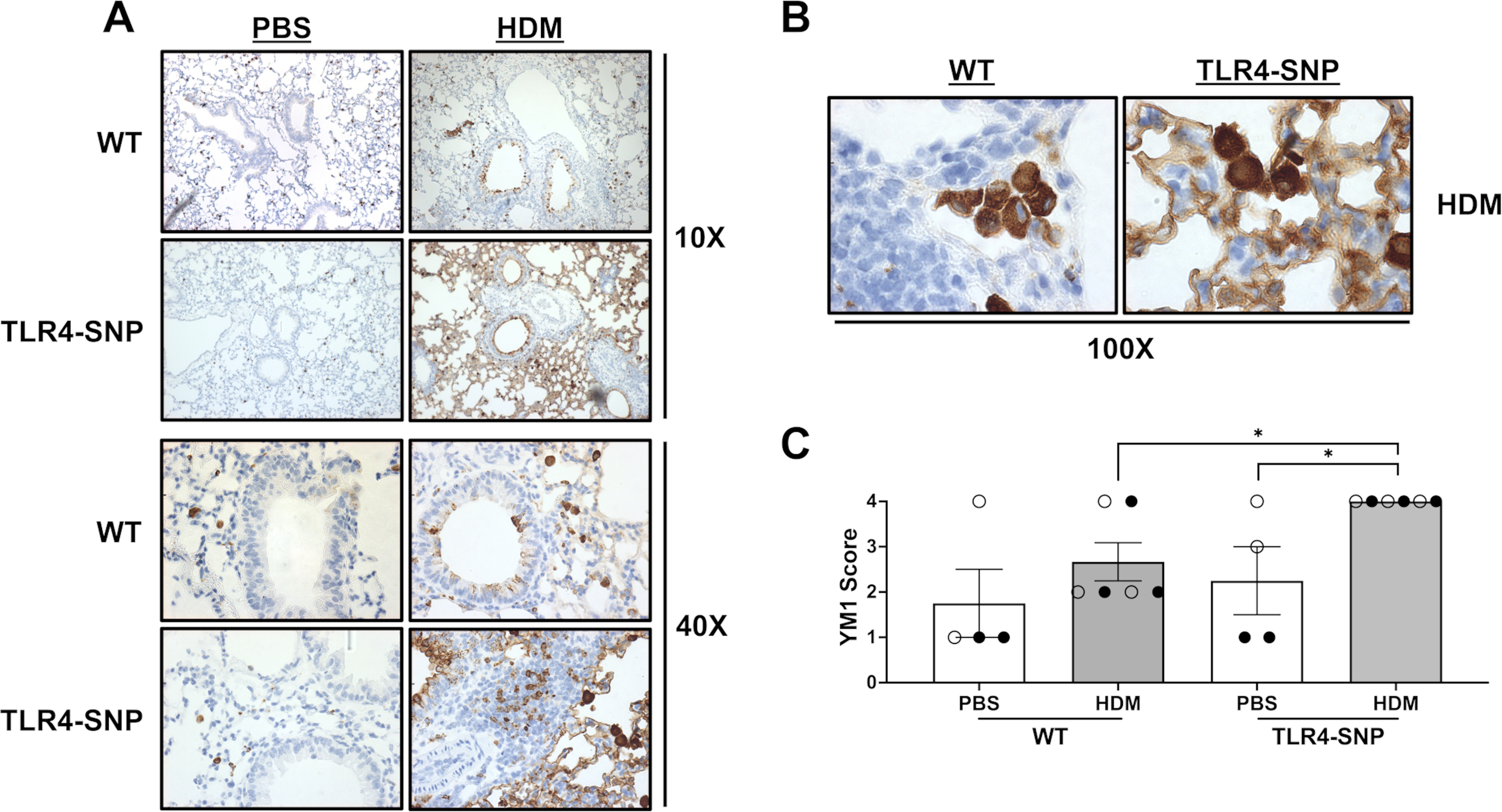
Lung sections from WT and TLR4-SNP mice on day +17 were fixed, blocked, and then stained with anti-YM1 to determine presence of YM1+ cells at 10X (A), 40X (A), and 100X (B) magnifications. Lung sections were then evaluated by a pathologist blinded to the experimental conditions and scored on a scale of 0–4 (C). Scoring is representative of 3 individual trials (n=4/treatment group/trial for PBS-treated groups, n=6/treatment group/trial for HDM-treated groups) with each point representing an individual mouse. Open circles designate females and closed circles designate males. All data was analyzed by a two-tailed t-test: *, P<0.05; **, P<0.005; ***, P<0.0005; ****, P<0.0001. Error bars represent mean ± SEM.
Airway hyperreactivity is not altered in TLR4-SNP mice
To determine if airway mechanics were altered between TLR4-SNP and WT mice during HDM-induced allergic inflammation, we used a Mouse Flexivent Ventilator to monitor AHR. On day +17, all groups were sedated, cannulated, paralyzed, and nebulized with increasing concentration of methacholine. Both HDM-treated groups responded to lower doses of methacholine when compared to PBS-treated groups and suggests increased AHR; however, no significant difference in responsiveness between TLR4-SNP and WT mice was detected (Figure 8). Taken together, these data indicate that polymorphisms in the TLR4 receptor contribute to the enhancement of some allergic manifestations (eosinophilic inflammation, TH2 cytokines, IgG1), but not others (mucus production, airway hyperreactivity) induced by the allergen HDM.
Discussion
In this report, we sought to determine the impact of co-segregating D298G and N397I polymorphisms (SNPs) in TLR4 on the development of allergic inflammation under uniform conditions. Through the use of a novel mouse that expresses homologous human TLR4 polymorphisms (TLR4-SNP), we show that the impact of these SNPs on the development of allergic inflammation in mice is dependent on the model used to mediate the inflammatory response. We report that when using the OVA and LPS model of allergic inflammation, TLR4 expression is required for induction to occur as cellular and pathological benchmarks were diminished in both TLR4-SNP and TLR4-deficent mice. However, in the more clinically relevant model using HDM for induction, allergic responses were enhanced as TLR4-SNP mice displayed greater levels of eosinophilic inflammation, TH2 cytokine production, and HDM-specific IgG1 production when compared to WT mice. TLR4-SNP mice did not exhibit any increase in mucus production and airway hyperreactivity after allergic induction. These results suggest that the TLR4 polymorphic variants (genes) interact differently with the allergic stimulation (environment).
The study by Michel et al. showed a significant relationship between natural endotoxin (also interchangeably used as lipopolysaccharide (LPS) in this discussion) exposure and the clinical manifestation of allergic disease in humans (33); however, the relationship between endotoxin and induction of allergic disease has always been controversial and dual-sided. Publications over the years have had somewhat disparate conclusions leading to the more complex idea that the variance in the outcome of allergic disease due to endotoxin exposure may be due to a variety of factors (8–13). Several reports have suggested that the timing of LPS exposure in the environment is capable of modulating the development of asthma leading to differential clinical outcomes. Moreover, the climate of study sites and bacterial species of LPS used to generate experimental data may explain the variance in endotoxin studies.
The data we report here is in agreement that endotoxin is capable of inducing the development of asthma as WT mice treated with OVA and LPS had greater allergic inflammation and mucus production than OVA-only treated mice. Yet, it is quite possible that LPS tolerance and priming is more important in protection against the development of asthma. It has been reported that tolerance to LPS leads to a decrease in TLR4 expression on the surface (34), which could lead to reduced allergic inflammation (35). Richard et al. generated a novel mouse strain (TLR4-SNP) that homozygously expresses co-segregating D298G and N397I SNPs in TLR4 (the murine homologs of the human SNPs) and confirmed that these mice exhibit LPS-hyporesponsiveness, increased susceptibility to Gram-negative infection, and altered susceptibility to influenza and respiratory syncytial virus (23). Additionally, the authors reported that TLR4-SNP mice had reduced TLR4 surface expression on in vitro cultured macrophages, as previously reported on human epithelial cells expressing these SNPs (21, 36). Our data shows that TLR4-SNP mice in the OVA+LPS model had reduced allergic inflammation when compared to WT mice. It is possible that in an LPS tolerance model of inflammation where LPS is provided to young mice prior to allergen exposure, an enhanced development of asthma may be observed in the TLR4-SNP mice as their TLR4 surface levels are already in a reduced state.
Several studies have also reported that children 12 months or younger, exposed to higher endotoxin level concentrations (>10,000 EU/g of allergen), develop increased risk of wheezing or display exacerbated wheezing episodes when compared to children exposed to lower levels (37, 38); children exposed to higher endotoxin levels also had a higher prevalence of pediatric respiratory infections (38). On the other hand, exposure to endotoxin during infancy and early childhood were also correlated with decreased development of asthma due to, perhaps, endotoxin tolerance or increased TH1 immunity (9, 11, 12). Farmers exposed to higher levels of endotoxin also had increased risk of asthma, yet the relationship between endotoxin levels and adult asthma was also influenced by the level of exposure to endotoxin at early-life (8, 13). In an animal model using Piebald–Virol–Glaxo rats, bacterial LPS provided immediately before or shortly after primary allergen sensitization inhibited the increase of ovalbumin-specific IgE levels, yet exposure to LPS several days after allergen challenge led to the exacerbation of the allergic inflammation (39). Taken together, these observations suggest that the timing of the exposure to endotoxin is critical in modulating the allergic response.
When directly measuring airway responses, we observed no differences in airway reactivity to methacholine between WT and TLR4-SNP mice. Unexpectedly, the OVA-only-treated groups showed greater airway hyperreactivity when compared to OVA-LPS-treated groups as either OVA-only group reacted sooner at lower doses of methacholine and showed a trend of stronger bronchoconstriction of the small airways at higher doses (25 mg/mL and 50 mg/mL). These results suggest that endotoxin is having an impact on AHR in this model. Mice given OVA-only are still capable of generating TH2 responses which may be due to the small amounts of endogenous endotoxin contamination (17, 40). The fact that the OVA-treated WT mice had ~15% eosinophils in the BAL while the TLR4−/− mice had little to none supports this contention. One plausible explanation is that our introduction of exogenous LPS is mediating an increase in the TReg population, as high doses of LPS are capable of inducing these cells while low doses of exogenous LPS are not (40); however, this report used the OVA-Alum model in conjunction with LPS as opposed to intranasal OVA instillation without alum. Reports using the same installation method as used in the current study suggest that our OVA-LPS model is “pre-programing” our model towards a more regulatory T cell state. Pre-exposure to low doses of LPS prior to sensitization leads to increased TReg populations, yet these models used young or neonatal mice as opposed to adult mice. Consequently, the timeline for the assessment of these characterizations are much longer than the model we report here (41, 42). While our OVA/LPS-model in this report does not directly address the question of LPS tolerance or “pre-programming,” our model can be adapted in future experiments to understand if LPS tolerance provides protection towards the induction of asthma. Moreover, only adult mice were used for all the data reported; as such, it is unclear if these results would be similar if younger mice were used instead since many studies have revealed that the very early childhood window is critical in determine the long-term outcome of asthma. In a more recent human study, endotoxin predictors and associations of health outcomes significantly differed across climate regions across the entire United States. Endotoxin was associated with a higher prevalence of wheeze outcomes in “cooler” areas, while in “hotter” regions, it is associated with a higher prevalence of wheeze and asthma, but lower prevalence of sensitization to inhalant allergens (43). Beyond climate, the authors could not elucidate the environmental determinate that led to the differences in clinical outcomes.
Our data shows that induction of the allergic response is also dependent on the type of allergic model used as our OVA-LPS model required a fully proficient TLR4 receptor, while our HDM-model did not show TLR4-dependence for allergic inflammation to occur and the lot of HDM used contained relatively low levels of endotoxin. Furthermore, a comparison of HDM from different manufactures showed that endotoxin levels did not correlate with severity of inflammation and Derp1 was more closely correlated instead with inflammation (44) along with IgE production (32). Additionally, it is also becoming quite clear that there is considerable variation in HDM sensitization timelines, HDM lot preparations (Derp1, protein, endotoxin concentration) used by investigators in the field, and in the reporting of endotoxin levels with HDM dosages that all contribute to the difficulty in comparing the phenotypic response of an animal model without knowing those possible limitations. TLR4-dependency is also another controversial topic as TLR4 expression has been reported to be required for allergic lung inflammation in mice using either ovalbumin (OVA) (17) or house dust mite (HDM) (18, 19) as priming agents. However, the results of our HDM-model is in agreement with Ishii et al. suggesting that responses to HDM are not absolutely dependent upon TLR4; instead, allergic inflammation was dependent on the precise nature, timing, and dose of HDM used (20), though, our HDM-model does not address this area.
This report is the first study to use novel TLR4-SNP mice (23) in a model of allergic inflammation and is the first to directly examine the impact of polymorphisms without other possible biases. A significant proportion of Caucasians express co-segregating D299G and T399I single nucleotide polymorphisms (SNPs) in the TLR4 gene, yet it is still unclear if expression of these SNPs have an effect on the manifestation of allergic asthma. In a German population, these SNPs have variably been associated with asthma risk as it was determined that expression of both SNPs led to a decreased risk for asthma in adults depending on the abundance of endotoxin in the environment (22). However, the authors acknowledge that the studies were correlative and may be attributable to other genetic or environmental factors not directly related to TLR4 haplotype. Our most interesting finding was that HDM-induced allergic inflammation was markedly exacerbated in TLR4-SNP mice. This data suggests that TLR4-SNP mice have greater IL-4 production and thus more robust IL-4-mediated responses during allergic inflammation as evidenced by greater levels of eosinophilic inflammation, TH2 cytokine production, and HDM-specific IgG1 production compared to WT mice (45). However, we were unable to detect any IL-13 in the BAL of TLR4-SNP mice which also supports the idea that mucus production and airway hyperreactivity were not affected as these mechanisms are markedly impacted by IL-13 (46, 47). These results suggest that the TLR4-SNPs influence IL-4 production induced by HDM, however, we do not yet know the precise cellular and molecular mechanisms by which an LPS-hyporesponsive TLR4 would stimulate greater IL-4 production.
TLR4 on epithelial cells is critical for mediating downstream DC recruitment and eventual TH2 cell activation (18, 48). TLR4-SNP mice have reduced surface expression of TLR4 on macrophages and reduced signaling to LPS in vitro (23) and humans with these SNPs show reduced expression on epithelial cells (21, 36). These findings would predict a reduction in allergic inflammation in the TLR4-SNP mice. However, in the present study, TLR4-SNP mice exhibited a slightly diminished capacity to mediate allergic inflammation in the OVA-LPS model (Figure 1), but an increased response in the HDM model (Figure 5, 6). Activated epithelial cells produce a number of chemokines and cytokines that participate in the recruitment and polarization of TH2 mediated responses, such as IL-13, IL-33, and IL-25 to mediate mast cell and basophil recruitment; however, these cytokines were all below the limit of detection in BALF through our Luminex panel. The absence of detectable IL-13 may possibly be due to IL-13 complexing with soluble IL-13Rα2 to avoid detection (49), and IL-33 may not be produced at detectable levels as higher levels of IL-33 are associated with necrotic cells which we did not detect (50, 51). Bronchial epithelial cells transfected with a Asp299Gly and Thr399Ile TLR4 alleles and exposed to exogenous LPS produced smaller amounts of IL-10 in cell culture relative to WT, with a more robust production of IL-13 and IL-5 (36). Although TLR4 is a very important innate receptor in allergic inflammation, PAR2 has also been shown to be required for the development of allergic inflammation as PAR2−/− mice had decreased eosinophil counts after HDM treatment (52). Interestingly, airway epithelial cells are also able to secrete thymic stromal lymphopoietin (TSLP) and this is dependent on the activation of PAR2 (53). Human DCs primed with TLSP and co-cultured with naive allogeneic T cells acquired an inflammatory TH2-like phenotype with production of IL-4, IL-5, IL-13, and TNF-α but not IL-10 (54). As such, it may be possible that our LPS-hyposensitive TLR4-SNP mice may not produce a robust IL-10 response in response to allergen, and IL-10 mediated regulatory mechanisms are not fully able to control the polarization of T cells towards type 2 allergic inflammation. Additionally, TLR4-SNP mice do not appear to be deficient in the ability to produce chemokines as our gene expression data using total lung homogenate from the mice of our HDM model also showed an increase in the fold-expression of CCL11 (eotaxin-1), an eosinophil attractant, in TLR4-SNP mice treated with HDM compared to PBS-controls and also to the same fold-difference between PBS and HDM-treated WT groups (data not shown). Taken together, while the hyporesponsive TLR4-SNP mice may have reduced expression and signaling on epithelial cells, it is possible that other compensatory PRR receptor and ligand combinations, such as PAR2 (52) or Dectin-1(55, 56), may be in play to overcome the hyporesponsive TLR4 on epithelial cells.
One possible mechanism to explore in future studies is the impact of TLR4-SNPs on TH2 priming by TLR4+ Dendritic Cells (DCs) in response to HDM. TLR4+ DCs are required for the development on TH2 response as TLR4-deficent mice had defective DC signaling leading to reduced Type 2 cytokine production when challenged with OVA (57). As such, the DC compartment within TLR4-SNP mice may have developmental defects leading to a reduction in T cell activation; however, this is unlikely as we report that Type 2 cytokines were detectable and not completely abolished as one would expect. Rather, we speculate that in our HDM model, TLR4-SNP DCs are unable to direct T cell development towards a TH1 or TH17 response or that induction of an allergen specific regulatory (TReg) response is impaired which may lead to enhanced production of TH2 cytokines, namely IL-4. In the absence of CD11c+ DCs, endogenous or adoptively transferred CD4+ TH2 cells did not produce interleukin (IL)-4, IL-5, and IL-13 in response to OVA aerosol (58); however, CD11b+ DCs and, to a lesser extent, monocyte-derived DCs were reported to be fundamental for TH2 priming after HDM-exposure (59). TLR4-SNP mice mirror the human phenotype and are hyposensitive in nature to LPS (23); thus, it may be possible that our current detection methods will not reveal a reduction in the TH1 cytokines, IFN-γ and IL-12p40, that would be suggestive of an imbalance in the TH1/TH2 axis.
It has been reported that asthma development is attenuated in IL-17R-deficeint mice, but if the disease is allowed to progress, IL-17 is able to reduce eosinophil recruitment and also TH2 cytokine production (60). McAlees et al. reported that TLR4 expression on various cell types could give rise to a divergence in airway inflammation. TLR4 on hematopoietic cells was critical for neutrophil-mediated inflammation following both LPS and allergen priming with HDM and OVA, while TLR4 on airway epithelial cells was necessary for eosinophilic inflammation (61). Although our study did not examine the various cell types that express TLR4, we did not find a significant expansion of neutrophils, drivers of Type 17 asthma (62, 63), in HDM-treated groups nor was a difference in neutrophilia detected between WT and TLR4-SNP mice. Additionally, TLR4-SNP mice primed with HDM had distinctly increased in eosinophils, contrary to the findings of McAlees et al. Eosinophils are now considered to have a greater role in mediating and resolving Type 2 allergic inflammation beyond just being downstream effector cells. As our data reports a significant increase in the population of these cells, it would be interesting to understand if the expansion of eosinophils are aiding in increased recruitment of TH2 cells to the lung or even polarizing the immune system towards greater TH2 immunity (64). However, the reason for the differences between our studies is unclear, but one possible explanation is the differing use of HDM and OVA/LPS animal models for inducing allergic inflammation.
In conclusion, our study is the first to directly examine the impact of TLR4 polymorphisms on allergic inflammation in a controlled experimental setting using novel TLR4-SNP mice exposed to two well-defined allergens (23). We show that the impact of these SNPs on the development of allergic inflammation in mice is dependent on the model used to mediate the inflammatory response. In the OVA+LPS model of induction, TLR4 expression was required for the development of asthma. However, in the more clinically relevant model using HDM for induction, allergic responses were enhanced as TLR4-SNP mice displayed greater levels of eosinophilic inflammation, TH2 cytokine production, and HDM-specific IgG1 production when compared to WT mice. However, TLR4-SNP mice did not exhibit any increase in mucus production and airway hyperreactivity after allergic induction. The data we present here is a small snapshot in the understanding of how human D299G and T399I polymorphisms in TLR4 affect human health and well-being. Given the differing reports of the requirement for TLR4 expression in order for development to occur, more studies are warranted to understand if the nature of the allergen, timing of administration, or perhaps endotoxin are capable of providing protection against or exacerbation of the development of asthma. Moreover, since endotoxin and induction of allergic disease have a seemingly complex relationship between each other, more studies are warranted to understand duality of this interaction and also the effect of LPS tolerance on the induction of asthma.
Supplementary Material
Key Points:
Severity of asthma in TLR4-SNP mice is dependent on the allergen system.
TLR4-SNP mice have reduced allergic inflammation when using the OVA-LPS model.
TLR4-SNP mice have enhanced allergic responses when using the HDM model.
Acknowledgements
The authors would like to acknowledge Dr. Hongjuan Gao and Dr. Nicolas Dorsey for critical discussions, Ms. Wendy Lai for technical contributions, and Dr. Chixiang Chen for help with biostatistics analysis. Cytokine Analysis, Statistical Consulting, and Histology were performed at the Center for Innovative Biomedical Resource Core Facilities at the University of Maryland, School of Medicine.
This work was supported by National Institutes of Health grants AI143845 (to ADK, SNV, and RMV), AI122631(ADK), AI095190 (SNV), and AI123371 (SNV) and Veteran’s Affairs Medical Center Merit Award BX001850 (ADK). MYF was supported in part by T32AI095190.
Footnotes
Disclosures
The authors declare that they have no conflicts of interest affecting the conduct of this research or the publication of these results.
References
- 1.Prevention C. f. D. C. a. National Data. In Most Recent National Asthma Data, https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm.
- 2.Barnes PJ, and Adcock IM. 2009. Glucocorticoid resistance in inflammatory diseases. Lancet 373: 1905–1917. [DOI] [PubMed] [Google Scholar]
- 3.Murrison LB, Brandt EB, Myers JB, and Hershey GKK. 2019. Environmental exposures and mechanisms in allergy and asthma development. J Clin Invest 129: 1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick S, Friend A, Dynes K, AlKandari F, Doust E, Cowie H, Ayres JG, and Turner SW. 2014. A systematic review of associations between environmental exposures and development of asthma in children aged up to 9 years. BMJ Open 4: e006554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burbank AJ, Sood AK, Kesic MJ, Peden DB, and Hernandez ML. 2017. Environmental determinants of allergy and asthma in early life. J Allergy Clin Immunol 140: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang HHF, Teo SM, Sly PD, Holt PG, and Inouye M. 2021. The intersect of genetics, environment, and microbiota in asthma-perspectives and challenges. J Allergy Clin Immunol 147: 781–793. [DOI] [PubMed] [Google Scholar]
- 7.Kauffmann F, and Demenais F. 2012. Gene-environment interactions in asthma and allergic diseases: challenges and perspectives. J Allergy Clin Immunol 130: 1229–1240; quiz 1241–1222. [DOI] [PubMed] [Google Scholar]
- 8.Carnes MU, Hoppin JA, Metwali N, Wyss AB, Hankinson JL, O’Connell EL, Richards M, Long S, Freeman LE, Sandler DP, Henneberger PK, Barker-Cummings C, Umbach DM, Thorne PS, and London SJ. 2017. House Dust Endotoxin Levels Are Associated with Adult Asthma in a U.S. Farming Population. Ann Am Thorac Soc 14: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ege MJ, Bieli C, Frei R, van Strien RT, Riedler J, Ublagger E, Schram-Bijkerk D, Brunekreef B, van Hage M, Scheynius A, Pershagen G, Benz MR, Lauener R, von Mutius E, Braun-Fahrländer C, and team PS. 2006. Prenatal farm exposure is related to the expression of receptors of the innate immunity and to atopic sensitization in school-age children. J Allergy Clin Immunol 117: 817–823. [DOI] [PubMed] [Google Scholar]
- 10.Stern DA, Riedler J, Nowak D, Braun-Fahrlander C, Swoboda I, Balic N, Chen KW, Vrtala S, Gronlund H, van Hage M, Valenta R, Spitzauer S, Von Mutius E, and Vercelli D. 2007. Exposure to a farming environment has allergen-specific protective effects on TH2-dependent isotype switching in response to common inhalants. J Allergy Clin Immunol 119: 351–358. [DOI] [PubMed] [Google Scholar]
- 11.Braun-Fahrländer C, Riedler J, Herz U, Eder W, Waser M, Grize L, Maisch S, Carr D, Gerlach F, Bufe A, Lauener RP, Schierl R, Renz H, Nowak D, von Mutius E, and Team A. a. E. S.. 2002. Environmental exposure to endotoxin and its relation to asthma in school-age children. The New England journal of medicine 347: 869–877. [DOI] [PubMed] [Google Scholar]
- 12.Gereda JE, Leung DY, Thatayatikom A, Streib JE, Price MR, Klinnert MD, and Liu AH. 2000. Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet 355: 1680–1683. [DOI] [PubMed] [Google Scholar]
- 13.Smit LA, Heederik D, Doekes G, Blom C, van Zweden I, and Wouters IM. 2008. Exposure-response analysis of allergy and respiratory symptoms in endotoxin-exposed adults. Eur Respir J 31: 1241–1248. [DOI] [PubMed] [Google Scholar]
- 14.Fujimura KE, Demoor T, Rauch M, Faruqi AA, Jang S, Johnson CC, Boushey HA, Zoratti E, Ownby D, Lukacs NW, and Lynch SV. 2014. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc Natl Acad Sci U S A 111: 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debarry J, Garn H, Hanuszkiewicz A, Dickgreber N, Blümer N, von Mutius E, Bufe A, Gatermann S, Renz H, Holst O, and Heine H. 2007. Acinetobacter lwoffii and Lactococcus lactis strains isolated from farm cowsheds possess strong allergy-protective properties. J Allergy Clin Immunol 119: 1514–1521. [DOI] [PubMed] [Google Scholar]
- 16.Fujimura KE, Johnson CC, Ownby DR, Cox MJ, Brodie EL, Havstad SL, Zoratti EM, Woodcroft KJ, Bobbitt KR, Wegienka G, Boushey HA, and Lynch SV. 2010. Man’s best friend? The effect of pet ownership on house dust microbial communities. J Allergy Clin Immunol 126: 410–412, 412.e411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, and Bottomly K. 2002. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med 196: 1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, and Lambrecht BN. 2009. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med 15: 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phipps S, Lam CE, Kaiko GE, Foo SY, Collison A, Mattes J, Barry J, Davidson S, Oreo K, Smith L, Mansell A, Matthaei KI, and Foster PS. 2009. Toll/IL-1 signaling is critical for house dust mite-specific helper T cell type 2 and type 17 [corrected] responses. Am J Respir Crit Care Med 179: 883–893. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Niikura Y, Kurata K, Muroi M, Tanamoto K, Nagase T, Sakaguchi M, and Yamashita N. 2018. Time-dependent distinct roles of Toll-like receptor 4 in a house dust mite-induced asthma mouse model. Scandinavian journal of immunology 87. [DOI] [PubMed] [Google Scholar]
- 21.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, and Schwartz DA. 2000. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25: 187–191. [DOI] [PubMed] [Google Scholar]
- 22.Werner M, Topp R, Wimmer K, Richter K, Bischof W, Wjst M, and Heinrich J. 2003. TLR4 gene variants modify endotoxin effects on asthma. J Allergy Clin Immunol 112: 323–330. [DOI] [PubMed] [Google Scholar]
- 23.Richard K, Piepenbrink KH, Shirey KA, Gopalakrishnan A, Nallar S, Prantner DJ, Perkins DJ, Lai W, Vlk A, Toshchakov VY, Feng C, Fanaroff R, Medvedev AE, Blanco JCG, and Vogel SN. 2021. A mouse model of human TLR4 D299G/T399I SNPs reveals mechanisms of altered LPS and pathogen responses. J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McIntire FC, Barlow GH, Sievert HW, Finley RA, and Yoo AL. 1969. Studies on a lipopolysaccharide from Escherichia coli. Heterogeneity and mechanism of reversible inactivation by sodium deoxycholate. Biochemistry 8: 4063–4067. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta P, Chapoval SP, Smith EP, and Keegan AD. 2011. Transfer of in vivo primed transgenic T cells supports allergic lung inflammation and FIZZ1 and Ym1 production in an IL-4Rα and STAT6 dependent manner. BMC Immunol 12: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ford AQ, Dasgupta P, Mikhailenko I, Smith EM, Noben-Trauth N, and Keegan AD. 2012. Adoptive transfer of IL-4Rα+ macrophages is sufficient to enhance eosinophilic inflammation in a mouse model of allergic lung inflammation. BMC Immunol 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly-Welch AE, Melo ME, Smith E, Ford AQ, Haudenschild C, Noben-Trauth N, and Keegan AD. 2004. Complex role of the IL-4 receptor alpha in a murine model of airway inflammation: expression of the IL-4 receptor alpha on nonlymphoid cells of bone marrow origin contributes to severity of inflammation. J Immunol 172: 4545–4555. [DOI] [PubMed] [Google Scholar]
- 28.Keegan AD, Shirey KA, Bagdure D, Blanco J, Viscardi RM, and Vogel SN. 2016. Enhanced allergic responsiveness after early childhood infection with respiratory viruses: Are long-lived alternatively activated macrophages the missing link? Pathog Dis 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gueders MM, Paulissen G, Crahay C, Quesada-Calvo F, Hacha J, Van Hove C, Tournoy K, Louis R, Foidart JM, Noel A, and Cataldo DD. 2009. Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflamm Res 58: 845–854. [DOI] [PubMed] [Google Scholar]
- 30.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, and Gelfand EW. 1997. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 156: 766–775. [DOI] [PubMed] [Google Scholar]
- 31.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162: 3749–3752. [PubMed] [Google Scholar]
- 32.Post S, Heijink IH, Petersen AH, de Bruin HG, van Oosterhout AJ, and Nawijn MC. 2014. Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS One 9: e91206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michel O, Ginanni R, Duchateau J, Vertongen F, Le Bon B, and Sergysels R. 1991. Domestic endotoxin exposure and clinical severity of asthma. Clin Exp Allergy 21: 441–448. [DOI] [PubMed] [Google Scholar]
- 34.Nomura F, Akashi S, Sakao Y, Sato S, Kawai T, Matsumoto M, Nakanishi K, Kimoto M, Miyake K, Takeda K, and Akira S. 2000. Cutting edge: endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J Immunol 164: 3476–3479. [DOI] [PubMed] [Google Scholar]
- 35.Natarajan S, Kim J, Bouchard J, Cruikshank W, and Remick DG. 2012. Pulmonary endotoxin tolerance protects against cockroach allergen-induced asthma-like inflammation in a mouse model. Int Arch Allergy Immunol 158: 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tulic MK, Hurrelbrink RJ, Prele CM, Laing IA, Upham JW, Le Souef P, Sly PD, and Holt PG. 2007. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J Immunol 179: 132–140. [DOI] [PubMed] [Google Scholar]
- 37.Park JH, Gold DR, Spiegelman DL, Burge HA, and Milton DK. 2001. House dust endotoxin and wheeze in the first year of life. Am J Respir Crit Care Med 163: 322–328. [DOI] [PubMed] [Google Scholar]
- 38.Gehring U, Bolte G, Borte M, Bischof W, Fahlbusch B, Wichmann HE, Heinrich J, System L. s. g. L.-R. F. o. t. I., and the Development of Allergies in C. 2001. Exposure to endotoxin decreases the risk of atopic eczema in infancy: a cohort study. J Allergy Clin Immunol 108: 847–854. [DOI] [PubMed] [Google Scholar]
- 39.Tulic MK, Wale JL, Holt PG, and Sly PD. 2000. Modification of the inflammatory response to allergen challenge after exposure to bacterial lipopolysaccharide. Am J Respir Cell Mol Biol 22: 604–612. [DOI] [PubMed] [Google Scholar]
- 40.Kumar S, and Adhikari A. 2017. Dose-dependent immunomodulating effects of endotoxin in allergic airway inflammation. Innate Immun 23: 249–257. [DOI] [PubMed] [Google Scholar]
- 41.Ding F, Liu B, Niu C, Wang T, Wang Y, Geng G, Tian D, Dai J, and Fu Z. 2020. Low-Dose LPS Induces Tolerogenic Treg Skewing in Asthma. Front Immunol 11: 2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding F, Fu Z, and Liu B. 2018. Lipopolysaccharide Exposure Alleviates Asthma in Mice by Regulating Th1/Th2 and Treg/Th17 Balance. Med Sci Monit 24: 3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mendy A, Wilkerson J, Salo PM, Cohn RD, Zeldin DC, and Thorne PS. 2018. Endotoxin predictors and associated respiratory outcomes differ with climate regions in the U.S. Environ Int 112: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Post S, Nawijn MC, Hackett TL, Baranowska M, Gras R, van Oosterhout AJ, and Heijink IH. 2012. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax 67: 488–495. [DOI] [PubMed] [Google Scholar]
- 45.Brusselle GG, Kips JC, Tavernier JH, van der Heyden JG, Cuvelier CA, Pauwels RA, and Bluethmann H. 1994. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy 24: 73–80. [DOI] [PubMed] [Google Scholar]
- 46.Munitz A, Brandt EB, Mingler M, Finkelman FD, and Rothenberg ME. 2008. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A 105: 7240–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wynn TA 2003. IL-13 effector functions. Annual review of immunology 21: 425–456. [DOI] [PubMed] [Google Scholar]
- 48.Tan AM, Chen HC, Pochard P, Eisenbarth SC, Herrick CA, and Bottomly HK. 2010. TLR4 signaling in stromal cells is critical for the initiation of allergic Th2 responses to inhaled antigen. J Immunol 184: 3535–3544. [DOI] [PubMed] [Google Scholar]
- 49.Zheng T, Liu W, Oh SY, Zhu Z, Hu B, Homer RJ, Cohn L, Grusby MJ, and Elias JA. 2008. IL-13 receptor alpha2 selectively inhibits IL-13-induced responses in the murine lung. J Immunol 180: 522–529. [DOI] [PubMed] [Google Scholar]
- 50.Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG, and Hamid Q. 2010. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol 125: 752–754. [DOI] [PubMed] [Google Scholar]
- 51.Cayrol C, and Girard JP. 2009. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A 106: 9021–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Boer JD, Van’t Veer C, Stroo I, van der Meer AJ, de Vos AF, van der Zee JS, Roelofs JJ, and van der Poll T. 2014. Protease-activated receptor-2 deficient mice have reduced house dust mite-evoked allergic lung inflammation. Innate Immun 20: 618–625. [DOI] [PubMed] [Google Scholar]
- 53.Kouzaki H, O’Grady SM, Lawrence CB, and Kita H. 2009. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol 183: 1427–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe N, Hanabuchi S, Soumelis V, Yuan W, Ho S, de Waal Malefyt R, and Liu YJ. 2004. Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4+ T cell homeostatic expansion. Nat Immunol 5: 426–434. [DOI] [PubMed] [Google Scholar]
- 55.Ito T, Hirose K, Norimoto A, Tamachi T, Yokota M, Saku A, Takatori H, Saijo S, Iwakura Y, and Nakajima H. 2017. Dectin-1 Plays an Important Role in House Dust Mite-Induced Allergic Airway Inflammation through the Activation of CD11b+ Dendritic Cells. J Immunol 198: 61–70. [DOI] [PubMed] [Google Scholar]
- 56.Ryu JH, Yoo JY, Kim MJ, Hwang SG, Ahn KC, Ryu JC, Choi MK, Joo JH, Kim CH, Lee SN, Lee WJ, Kim J, Shin DM, Kweon MN, Bae YS, and Yoon JH. 2013. Distinct TLR-mediated pathways regulate house dust mite-induced allergic disease in the upper and lower airways. J Allergy Clin Immunol 131: 549–561. [DOI] [PubMed] [Google Scholar]
- 57.Dabbagh K, Dahl ME, Stepick-Biek P, and Lewis DB. 2002. Toll-like receptor 4 is required for optimal development of Th2 immune responses: role of dendritic cells. J Immunol 168: 4524–4530. [DOI] [PubMed] [Google Scholar]
- 58.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, and Lambrecht BN. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med 201: 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, Hammad H, and Lambrecht BN. 2013. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38: 322–335. [DOI] [PubMed] [Google Scholar]
- 60.Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, and Schnyder B. 2006. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med 203: 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McAlees JW, Whitehead GS, Harley IT, Cappelletti M, Rewerts CL, Holdcroft AM, Divanovic S, Wills-Karp M, Finkelman FD, Karp CL, and Cook DN. 2015. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol 8: 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, Jia G, Ohri CM, Doran E, Vannella KM, Butler CA, Hargadon B, Sciurba JC, Gieseck RL, Thompson RW, White S, Abbas AR, Jackman J, Wu LC, Egen JG, Heaney LG, Ramalingam TR, Arron JR, Wynn TA, and Bradding P. 2015. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 7: 301ra129. [DOI] [PubMed] [Google Scholar]
- 63.Cosmi L, Liotta F, Maggi E, Romagnani S, and Annunziato F. 2011. Th17 cells: new players in asthma pathogenesis. Allergy 66: 989–998. [DOI] [PubMed] [Google Scholar]
- 64.Jacobsen EA, Lee NA, and Lee JJ. 2014. Re-defining the unique roles for eosinophils in allergic respiratory inflammation. Clin Exp Allergy 44: 1119–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.