

Abstract
New treatments are needed to address persistent unmet clinical needs for diffuse large B-cell lymphoma (DLBCL). Overexpression of transferrin receptor 1 (TFR1) is common across cancer and permits cell-surface targeting of specific therapies in preclinical and clinical studies of various solid tumors. Here we developed novel nanocarrier delivery of chemotherapy via TFR1-mediated endocytosis, assessing this target for the first time in DLBCL. Analysis of published datasets showed novel association of increased TFR1 expression with high-risk DLBCL cases. Carbon-nitride dots (CND) are emerging nanoparticles with excellent in vivo stability and distribution and are adaptable to covalent conjugation with multiple substrates. In vitro, linking doxorubicin (Dox) and transferrin (TF) to CND (CND-Dox-TF, CDT) was 10–100 times more potent than Dox against DLBCL cell lines. Gain- and loss-of-function studies and fluorescent confocal microscopy confirmed dependence of these effects on TFR1-mediated endocytosis. In contrast to previous therapeutics directly linking Dox and TF, cytotoxicity of CDT resulted from nuclear entry by Dox, promoting double-stranded DNA breaks and apoptosis. CDT proved safe to administer in vivo, and when incorporated into standard frontline chemoimmunotherapy in place of Dox, it improved overall survival (OS) by controlling patient-derived xenograft (PDX) tumors with greatly reduced host toxicities. Nanocarrier-mediated Dox delivery to cell-surface TFR1 therefore warrants optimization as a potential new therapeutic option in DLBCL.
Introduction
DLBCL comprises a third of non-Hodgkin lymphoma (NHL) in the United States, making it the most common hematologic malignancy (1). Frontline R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) is effective in ~60%, but patients with relapsed or refractory (rel/ref) disease following frontline therapy have poor prognosis, with only about 1 in 10 achieving long-term disease-free survival, typically requiring salvage chemoimmunotherapy followed by bone marrow transplantation (2). Overall, there is substantial unmet need in DLBCL, with at least one in three diagnosed patients ultimately dying. The anthracycline chemotherapeutic doxorubicin (Dox) remains the most active drug against DLBCL, serving as the backbone of R-CHOP and most other standard frontline combination treatment regimens, more than five decades after the compound’s introduction (3). Clinical use of Dox is limited by toxicities to bone marrow and cardiomyocytes, especially in patients with prior anthracycline exposure, resulting in lifetime cumulative and dose-dependent cardiotoxicity (4–6). Targeted delivery of Dox could alleviate unwanted effects by sparing non-malignant tissues while maintaining antitumor efficacy.
The transferrin receptor 1 (TFR1), also known as CD71, is a ubiquitous cell-surface receptor found at low levels in normal human tissue, serving as the point of entry for iron bound to its ligand transferrin (TF) (7). TF carrying two atoms of Fe3+ (holo-TF) undergoes clathrin-mediated endocytosis upon TFR1 binding, followed by Fe reduction and release to fuel metabolism and proliferative pathways. Tumors often meet high iron demands through TFR1 overexpression (8). TFR1 is expressed at higher levels in a variety of cancers, a well-established potential therapeutic window for targeted therapeutic delivery (9–18). Preclinical studies have exploited this in breast cancer (19–23), glioma, and melanoma (24–26). DLBCL has not previously been assessed for TFR1-targeted therapeutic delivery.
Carbon dots (CDs) are low-cost photoluminescent nanoparticles with a gaussian size distribution of 2–8 nm with varying mean diameters dependent on syntheses techniques (27). CDs have reduced toxicities and environmental hazards compared to first-generation quantum dots synthesized from semiconductor metals (28–30). Prior work demonstrates utility of CDs as imaging reagents through incorporation of photoluminescent moieties (31–34). Intravenous (i.v.) dosing results in homogeneous distribution of CDs to different organs including the bladder, kidney, liver, spleen, brain, and heart, followed by rapid excretion in urine (35–38). We have led recent efforts to develop third-generation nanoparticles called carbon nitride-dots (CNDs) which have a gaussian size distribution of 1–3.8 nm with a mean diameter of 2.4 nm, formed from C3N4 triazine polymers (39). CNDs have excellent properties as potential therapeutic scaffolds, including enhanced excitation-dependent photoluminescence, reduced size, and improved stability compared to CDs (40).
Here, we show association between TFR1 expression and reduced survival of DLBCL patients, pointing to TFR1 as a novel target to improve outcomes for high-risk cases. We describe CND-Dox-TF (CDT), a novel reagent for targeted CND-based delivery of Dox to tumors exploiting TFR1-mediated endocytosis. CDT has dramatically increased potency against DLBCL cell lines compared to Dox. In vivo, replacement of Dox with CDT in R-CHOP (R-nanoCHOP) significantly improves survival of NOD scid gamma (NSG) mice bearing DLBCL PDX tumors.
Materials and Methods
Carbon-Nitride Dot Synthesis
Anhydrous citric acid (BDH) was obtained from VWR (West Chester, PA). Urea was acquired from Eastman Kodak Company (NY, USA). Doxorubicin hydrochloride and holo-transferrin (human plasma) were from TCI America Inc. (OR, USA) and EMD Millipore Corp. (MA, USA), respectively. N-Hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were purchased from Millipore-Sigma (St. Louis, MO). 3500 Da molecular weight cut-off dialysis tubing was from Thermo-Scientific (Rockford, IL) while the 100–500 Da molecular weight cut-off tubing were bought from Spectrum Labs Inc., (CA, USA). The deionized (DI) water used was ultrapure (type I) water purified using a Millipore Direct-Q 3 water purification system acquired from EMD Millipore Corp. with a surface tension of 72.6 mN·m−1, a resistivity of 18 MΩ·cm and a pH of 6.6 ± 0.3 at 20.0 ± 0.5 °C. All chemicals were used as received.
The synthesis of carbon-nitride dots (CNDs) was performed using a simple hydrothermal microwave process using citric acid and urea as reported in our previous work (39). A summary of this synthesis involves a 0.5 g of each citric acid and urea dissolving in 25 mL of DI water for overnight vigorous stirring before a microwave thermal treatment for 7 min under 700 W. The resultant solid residue was sonicated in 20 mL water and centrifuged for 30 min twice to remove large particles out of the CNDs dispersion. Further, 0.2 μm filter membranes were used to filter the dispersion and the filtrate was dialyzed in a 100–500 Da dialysis tubing for 5 days against 4 L DI water with regular water change every 24 h. The water dispersion was evaporated to obtain the solid CNDs product. The characterization of the CNDs was performed to confirm the reproducibility and the same results were obtained as reported (39).
Synthesis and Characterization of Carbon-Nitride Dot-Doxorubicin-Transferrin (CND-Dox-TF)
The as-synthesized CNDs were used for the preparation of the conjugate. CNDs (8 mg) were first dissolved in 3 mL of phosphate buffered solution (PBS, pH 7.4 at 25 mM) and were mixed with EDC (17 mg in 1 ml PBS) before stirring at room temperature for 30 min. Then, NHS (10.2 mg in 1 mL PBS) was added to the above mixture and left for stirring for another 30 min. Then 6 mg of doxorubicin hydrochloride (Dox) was dissolved in a 0.5: 0.5 mL DMSO: PBS was added to the reaction mixture to be stirred for 30 min, before the addition of holo-transferrin (TF, 3 mg in 1 mL PBS). The reaction was stirred overnight and then the solution was transferred into a 3.5 kDa dialysis tubing to be dialyzed against 2 L DI water for 4 days with every 24 h water change, as previously established by us (40,41). Finally, the resultant dialyzed solution was freeze-dried to yield the lyophilized product.
The as-prepared CND-Dox-TF conjugate was subjected to different characterization techniques to confirm the existence of the said conjugate compound. UV-Vis absorption characterization was performed using a Cary 100 UV-Vis spectrophotometer (Agilent Technologies) in aqueous medium in a 1 cm quartz cuvette (Starna Cells). For the luminescent emission observations, a Horiba Jobin Yvon Fluorolog−3 spectrometer was used (in 1 cm path length quartz cuvette) using a slit width of 5 nm for both excitation and emission. OriginPro 9.1 was used to create the normalization of the emission spectra with the y-axis normalized to 1. A PerkinElmer Frontier with a universal ATR sampling accessary was used to record Fourier transform infrared (FTIR) spectra using air as the background. The samples were also analyzed through mass spectroscopy using matrix-assisted laser desorption ionization time of flight (MALDI-TOF) (Bruker).
Prognostic Correlation
Overall survival analysis based on TFRC expression for previously untreated DLBCL patients was performed using the SurvExpress online tool (42) for both the Lenz (GEO ID# GSE10846) (43) and Reddy (European Genome-phenome Archive at the European Bioinformatics Institute (EGAS00001002606) (44), data access kindly provided by the Sandeep Dave Lab, Duke University) datasets. Analysis for both datasets was conducted using the Maximize Risk Groups function in the SurvExpress online tool (42).
Cell Culture
All cells lines were verified by STR fingerprinting and assessed for mycoplasma contamination. Culture media for SU-DHL4, BJAB, and Riva (purchased from DSMZ); Farage and Toledo (purchased from ATCC); HBL1 and Karpas-422 (kind gift of H.-G. Wendel Lab, MSKCC); and A20 (kind gift of J. Rosenblatt Lab, UM) were RPMI 1640 supplemented with 10% fetal bovine serum (FBS), Penicillin/Streptomycin (P/S), and mycoplasma inhibitor plasmocin prophylactic (P/P) (ant-mpp). OCI-Ly19 (purchased from ATCC) was cultured in IMEM supplemented with 20% FBS, P/S and P/P. HEK293 and 3t3 (purchased from ATCC) was cultured in DMEM supplemented with 10% FBS, P/S, and P/P.
Cell Viability
For 24–48 hr assessments, cells were seeded at 5000 cells per well in a 96-well plate under serial dilutions of drug. For delayed drug effect viability, cells were seeded at 500,000 cells per well in a 6-well plate on day 0 and treated with drug for 24 hrs, after which cells were washed x2 and plated in normal cell media without drug. Viability was assessed using Cell Titer Glo (Promega #G7573) according to the manufacturer’s protocol. Luminescence was measured using BioTek Synergy HT plate reader. EC50s were calculated using nonlinear fit regression analysis in GraphPad Prism 8. Apoptosis assessment was conducted with BD Biosciences reagent (#559763) by the manufacturer’s instructions using Attune NxT flow cytometer.
Antibodies
Cell Signaling Technology: CD71 (#13113S), Phospho-Histone H2A.X (#9718S), β-Actin (#4970S). Thermofisher: CD71 (#MA532500), Cyclophilin B (#PA1–027A).
Protein extraction, quantification, and immunoblotting
Cells were seeded at 500,000/mL and incubated as indicated. Proteins were extracted using RIPA (VWR, Radnor, PA, USA), Phosphatase Halt (Thermo #78428). Proteins were quantified using the BCA assay (Thermofisher Scientific, Waltham, MA, USA) with 20 ug loaded per lane for Western blotting. All blots were developed using autoradiography film (VWR, Radnor, PA, USA) or Li-Cor Odyssey Fc imaging system after incubation with antibodies indicated above. Densitometric analysis conducted using Li-Cor affiliated ImageStudio software, with all analyses normalized to loading controls. All antibodies were used per the manufacturer recommended dilutions.
Microscopy
Cells were seeded at 50,000 cells per well in a 12-well plate and treated with either vehicle or drug. BJAB cells were fixed with 4% paraformaldehyde and permeabilized with 1% NP40 followed by staining for DAPI (Thermofisher Scientific, Waltham, MA, USA). Cells were then imaged (60×) using a Leica DM4 B microscope. HEK293 cells had the nucleus stained for DAPI (Thermo #R37605) or GFP (Thermo #C10602) per the manufacturer protocol. Cells were then live-imaged (63× objective) using Leica Sp5 confocal microscope. Images were collected and analyzed using the ImageJ software.
TFR1 Overexpression
Human TFRC cDNA (HsCD00044911) was purchased from the DNASU Plasmid Repository (Tempe, AZ, USA) and was subsequently cloned into pLVX-IRES-ZsGreen1 vector (Clontech Laboratories, Mountain View, CA, USA). The recombinant plasmid, together with packaging/envelope plasmids psPAX2 and pMD2.G (Addgene, Cambridge, MA, USA), were co-transfected into HEK293 cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) following manufacturer’s instructions. Cell media was changed at 24 hours after transfection, and viral particles were collected at 48 hours and 72 hours post transfection.
For viral transduction, BJAB and Farage cells were infected with harvested virus by spinoculation. Briefly, the cells were spun at 1800 rpm for 45 mins at room temperature. Cells were infected twice per day for a total of 4 infections. Fresh media was replenished the day after infections and cells were expanded. BD FACSAria II cell sorter was used to sort GFP positive cells. Cells transduced with empty vector were used as negative controls.
Binding Assay
Briefly, 500,000 cells were taken up and spun down at 800g x 5 minutes, washed twice with cold PBS and hereon after moved to ice. Cells were incubated with holo-TFCF®568 (25 ug/mL) and CDT (500nM) for 30 minutes on ice, followed by two washes with cold PBS, and then measured using an Attune NxT flow cytometer using a YL1 laser.
Inhibitors
Holo-Transferrin (616397–500MG-M) was purchased from Millipore Sigma. Fluorescently conjugated Holo-Transferrin (CF®405S and CF®568) were purchased from Biotium. Dynasore (S8047) was purchased from Selleckchem. Rituximab, Cyclophosphamide, Doxorubicin, Vincristine and Prednisone were kindly provided by the Sylvester Comprehensive Cancer Center chemotherapy pharmacy.
In vivo studies
All animal studies were performed under the approval of the University of Miami Institutional Animal Care and Use committee. All mice in this study were NOD scid gamma (NSG) males > 8 weeks of age. For tumor-bearing experiments, we obtained DLBCL PDX DFBL-75549 tumor model and engrafted mice through surgical dorsal tumor implantation. We measured tumor volume (TV) by ultrasound (Vevo 3100, Visualsonics) with a predetermined survival endpoint of TV ≥ 1500 mm3. A continuous body weight loss of >20% was also a predetermined survival endpoint. For dose-finding experiments, mice were dosed intravenously on day 0, day 14, and day 24 and observed for changes in body weight. For R-nanoCHOP vs. R-CHOP tumor-bearing experiments, mice were dosed with all drugs intravenously once on day 1 of every 21 days, with exception to Prednisone administered orally. The following drug doses were used: Rituximab 20 mg/kg, Cyclophosphamide 40 mg/kg, Doxorubicin 3.3 mg/kg, CDT 33 mg/kg, Vincristine 0.5 mg/kg, Prednisone 0.2 mg/kg.
Formalin-fixed paraffin-embedded tissue sections, produced per standard protocols, were used to make H&E-stained and immunohistochemistry (IHC) pathology slides. Antibodies were used as per above, when applicable.
Statistical analysis
Two tailed Student t test was carried out for all data using the GraphPad t test calculator, with P < 0.05 considered statistically significant with a 95% confidence interval. Area under the curve (AUC) carried out for delayed onset toxicity assessments using the GraphPad AUC function with subsequent Student t test carried out based off total area, SEM and n values, with P < 0.05 considered statistically significant with a 95% confidence interval. All experiments reported are the mean triplicate or quadruplicate ± SEM of three independent replicates unless otherwise stated in the figure legend. OS analysis employed log-rank (Mantel-Cox) statistics in Prism 8 software, with p < 0.05 considered significant.
Results
High TFRC in DLBCL correlates with inferior outcome after frontline therapy
TFR1 expression correlates with worse clinical outcomes in solid tumor malignancies (14–18). Though work nearly 40 years ago suggested worse prognosis in aggressive lymphomas with high TFR1 expression, this has not been analyzed in DLBCL as currently defined and treated. We examined expression of TFRC, the gene encoding TFR1, in two independent published DLBCL gene-expression datasets. Analysis of chip-based gene-expression data from Lenz and colleagues on 414 previously untreated DLBCL tumors showed significantly worse overall survival (OS) after frontline therapy for patients with high TFRC (p=0.025, HR 1.44 (95%CI 1.05–1.97), Fig. 1A) (42,43). All these patients were treated with either R-CHOP (233) or CHOP (181). More recently, Reddy et al. performed RNA-seq on 756 newly diagnosed DLBCL cases from patients uniformly treated with rituximab-containing standard frontline combination therapy. High TFRC expression again carried significantly worse OS in these data (p=0.005, HR 1.48 (95%CI 1.12–1.95), Fig. 1B) (44). A known marker of highly proliferative cells, high TFRC identifies DLBCL cases, under current diagnostic criteria, at high risk to be failed by R-CHOP and other standard frontline treatments.
Figure 1. TFRC expression in DLBCL patients correlates with poor overall survival.

A) Lenz et al. Kaplan-Meier overall survival (OS) analysis of high (red) vs. low (green) TFRC expression in 414 newly diagnosed untreated DLBCL patients. B) Reddy et al. Kaplan-Meier OS analysis of high (red) vs. low (green) TFRC gene expression from 756 DLBCL patients. Dotted lines indicate median OS. Lenz et al. median OS high = 4.99 years, low = 10.62 years (log-rank high/low HR 1.462, 95% CI 1.041 to 2.053). Reddy et. al median OS high = 7.840 years, low = 10.40 years (log-rank high/low HR 1.476, 95% CI 1.087 to 2.006).
Generation of CND-Dox-TF
We synthesized CNDs from urea and citric acid using our previously published hydrothermal microwave technique (39). We confirmed resulting CNDs consist of a tris-s-triazine structure containing C and N, with high abundance of amine, amide, and carboxylic functional groups (Supplementary Fig. S1A–B). We used 1-ethyl-3-(3-dimethylaminopropyl (EDC)/N-Hydroxysuccinimidev (NHS) bioconjugation to form carbodiimide bonds between CND carboxylic (COOH) groups and amino (NH2) groups on Dox and holo-TF, forming stable covalent bonds, yielding the CDT chemotherapeutic (Fig. 2A–B) (40). We used ultraviolet (UV) absorption and photoluminescent spectra analyses of the individual CDT components to confirm the presence of each in CDT (Fig. 2C; Supplementary Fig. S2A–E). Fourier-transform infrared (FTIR) spectra analysis comparing unconjugated CNDs vs. CDT confirmed the structural bond presence of Dox and TF in CDT (Supplementary Fig. S2F). To assess long-term compound stability, we performed photoluminescent spectra analyses on stock unconjugated and conjugated CND compounds stored at −20 Celsius conditions for > 1 year, confirming the stable conjugation of each component (Supplementary Fig. S2G–H). We illustrate successful synthesis and stability of CND compounds and the novel CDT chemotherapeutic nanocarrier designed for targeted delivery of Dox to TFR1-expressing cells.
Figure 2. CND-Dox-TF synthesis and validation.

A) Schematic of CND-Dox-TF synthesis. B) Chemical structure of CND-Dox-TF conjugate showing carbodiimide bonds to Dox and holo-TF. C) Normalized photoluminescence emission spectra of CND-Dox-TF excited at specific excitation wavelengths related to each single component (TF-280, CNDs-330–370, Dox-480 nm) which confirms the presence of each individual component in the nanocarrier conjugate.
CDT is exponentially more potent than Dox against DLBCL cell lines
We next compared the activity of CDT to single-agent Dox in vitro. DLBCL lines SU-DHL-4, BJAB, Riva, Farage, OCI-Ly19, HBL1, Karpas-422, and Toledo were dramatically more sensitive to CDT (CND-Dox-TF) than molar-equivalent Dox (Fig. 3A, Supplementary Fig. S3A). Unconjugated (CND) and single-conjugate CND compounds (CND-Dox and CND-TF) had weak or no negative effects on cell viability (Supplementary Fig. S3A), suggesting CDT activity depends on dual conjugation of Dox and TF to CNDs. Dox may have several different cellular effects, with nuclear entry and DNA damage considered most important against tumors (45). Additionally, onset of apoptosis from Dox DNA damage may be delayed beyond its terminal half-life (46). We therefore treated BJAB, Farage, SU-DHL4, and Riva cells with CDT or Dox for 24 hours at a range of doses, followed by drug washout and continuous cell viability tracking over 6 days. Strikingly, 10 nM CDT induced rapid cytotoxicity, with complete loss of cell viability that never recovered over the time-course, an effect seen only with much higher doses of Dox (Fig. 3B; Supplementary Fig. S3B). Flow cytometry confirmed entry into apoptosis in CDT-treated BJAB and Farage cells at significantly lower doses compared to Dox (Fig. 3C; Supplementary Fig. S3C). Western blot analysis of γ-H2AX, the classic DNA-damage marker, further confirmed rapid onset of double-stranded DNA breaks by CDT at dramatically lower doses than Dox in BJAB cells (Fig. 3D). CDT therefore is exponentially more potent than Dox against DLBCL cells, inducing rapid DNA damage and onset of apoptosis.
Figure 3. CND-Dox-TF has enhanced in vitro cytotoxicity compared to Dox.

A) 48-hour viability assays for DLBCL cell lines plated in serial dilutions of Dox and CND-Dox-TF (left) and mean EC50 ±SEM calculated from three independent 48-hour viability experiments (right). (EC50 500 nM means no significant activity up to that concentration.) B) Time course viability response of BJAB cell line treated with Dox or CND-Dox-TF. Cells were plated in drug for 24 hours then washed out and re-plated in normal media with daily viability assessment. Data normalized to DMSO-treated controls. Statistical analysis reflective of AUC comparing the same dose across treatment groups. Shown are triplicate mean ±SEM. C) AnnexinV-PE 7-AAD 24-hour apoptosis assay for BJAB cell line at a range of CND-Dox-TF and Dox doses (left) with percentage of late-apoptotic cells representative of triplicate mean ±SEM (right). D) Immunoblot assessment of γ-H2AX (17 kD) for BJAB cells exposed as indicated to Dox or CND-Dox-TF. ****P < 0.0001; ***P < 0.001 **P < 0.01; *P < 0.05; ns, nonsignificant (t test). For densitometric analysis, all samples normalized to loading control first followed by normalization to DMSO. • = accurate densitometric evaluation not possible given overly strong signal.
CDT promotes rapid nuclear entry by Dox after TFR1-mediated endocytosis
To investigate whether CDT activity was due to TF binding to cell-surface TFR1, we overexpressed TFRC in high-TFR1 expressing BJAB and low-TFR1 expressing Farage cell lines (Fig. 4A, Supplementary Fig. S4A–B) and treated them with CDT (Fig. 4B). Baseline TFR1 in BJAB was too high for TFRC introduction to increase it significantly (1.13× increase) causing no significant difference in CDT sensitivity. Farage cells, however, with 2.64× increased expression after TFRC introduction, became dramatically more sensitive to CDT, consistent with a TFR1-dependent mode of action. Multiple attempts to create stable RNAi TFRC knockdown clones of DLBCL lines were unsuccessful. Although this prevented assessment of TFR1 reduced expression effect on drug activity, it further illustrates the essential nature of the protein in DLBCL cells. As an alternate loss-of-function approach, we employed flooding of TFR1 with its natural ligand holo-TF to reduce availability of binding by CDT. We co-incubated BJAB and Farage cells with CDT and the maximum non-toxic dose of Holo-TF (Supplementary Fig. S4C). This caused a significant negative shift in CDT sensitivity in both lines (Fig. 4C). As a second alternate loss-of-function assessment, we co-incubated BJAB and Farage cells with CDT and the clathrin-mediated endocytosis inhibitor dynasore (47). Single agent dynasore at concentrations known to inhibit endocytosis (15 μM, 50 μM) negatively affected viability of BJAB and Farage cells somewhat (Fig. 4D, top panel). Normalized for baseline dynasore effect on viability, 100 nM CDT had greatly reduced efficacy in both lines (Fig. 4D, bottom panel). We also found no noticeable change in TFR1 expression in response to CDT or non-toxic CND-TF treatment by 24-hours, consistent with rapid TFR1 recycling back to the cell surface, as previously described (48–50) even after exposure to toxic CDT (Supplementary Fig. 4D). To further investigate cellular events triggered upon CDT exposure, we took advantage of the inherent red fluorescence of CNDs and Dox with fluorescent confocal microscopy. While unconjugated CND and CND-Dox yielded little nuclear colocalization in BJAB, nuclear colocalization of CDT and Dox was similar, with CDT doing so at 10-fold lower concentration (Supplementary Fig. S5). Because the large nucleus-to-cytoplasm ratio of lymphoma cells limits microscopic evaluation of intracellular events, we employed embryonic kidney HEK293 cells for further assessments. After confirming HEK293 cells were an appropriate system (Supplementary Fig. S6A), we treated them with CDT and Dox and repeated fluorescent confocal assessment. We found that at a lower dose, CDT entered the cytoplasm more efficiently, with significantly improved nuclear colocalization compared to Dox (Fig. 4E). Rapid nuclear Dox accumulation seems unlikely if it remains attached to the bulky TF protein. We therefore hypothesized Dox, either alone or attached to the CND, separates from TF following TFR1-mediated endocytosis. To test this, we synthesized CDT using cyan-fluorophore tagged TF (CDTcy). After confirming similar potency of CDTcy compared to CDT (Supplementary Fig. S6B), we treated HEK293 cells with CDTcy, staining the nucleus green to prevent fluorescence overlap (Fig. 4F). We found that TFcy (blue) did not colocalize with red (Dox, CND) fluorescence in the nucleus, suggesting TF uncoupling in the cytoplasm prior to Dox reaching the nucleus. These data provide detailed insight to the mechanism of CDT therapy, demonstrating cellular entry through TFR1, subsequent loss of TF from the reagent and rapid entry of Dox into the nucleus.
Figure 4. CND-Dox-TF mechanism of action.

A) Immunoblot analysis of TFR1 (90 kD) expression in DLBCL cell lines with cyclophilin B (24 kD) loading control (top) and BJAB and Farage cells infected and FACS sorted for TFRC overexpression (bottom). B) 48-hour cell viability assays corresponding to cell lines depicted in A plated in serial dilutions of CND-Dox-TF. Data shown are mean quadruplicate ±SEM with P values corresponding to Uninfected vs. TFRC EC50 values. C) 48-hour cell viability assay of BJAB and Farage cell lines plated in serial dilutions of Dox, CND-Dox-TF, CND-TF, and CND-Dox-TF + a constant 250 uM concentration of competitive holo-TF in each well. Data shown are mean quadruplicate ±SEM with P values corresponding to CND-Dox-TF vs. CND-Dox-TF + a constant 250uM holo-TF EC50 values. D) 48-hour viability response of BJAB and Farage cells treated with dynasore for 48-hours (top panel) and treated with CND-Dox-TF (100nM) + 15 uM or 50 uM dynasore for 48-hours (bottom panel). Shown are mean triplicate ±SEM. E) Fluorescent confocal microscopy images (60× objective) of HEK293 cells incubated for 24 hours with CND-Dox-TF (50 nM), CND (500 nM), CND-Dox (500 nM) or Dox (500 nM). Quantitation of overlap (mean triplicate ±SEM), right panel, of blue nuclear (DAPI) with red fluorescence from both Dox and CNDs. F) Fluorescent confocal microscopy images (63×) of HEK293 cells incubated for 24 hours with a Cyan-TF labeled CND-Dox-TF (50 nM), Doxorubicin (500 nM), and DMSO control. Green fluorescence corresponds to nucleus, blue corresponds to TF and red fluorescence corresponds to inherent signal emitted from Dox +/− CND. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns, nonsignificant (t test). For densitometric analysis, all samples normalized to loading control first. Samples in A) top panel normalized to highest-expressing cell line BJAB. Samples in A) bottom panel normalized to uninfected basal condition cell line respectively. Scale bar 50 μm.
Safe and effective dosing of CDT to PDX DLBCL-bearing mice
We next tested the safety and efficacy of CNDs in NOD scid gamma (NSG) mice. While Dox at 2.47 mg/kg already had a significant effect on body weight, the maximum tolerated dose (MTD) of Dox was confirmed to be 3.3 mg/kg, leading to ~20% weight loss (survival endpoint) (51–53). A single dose of 82.5 mg/kg of CDT in non-tumor bearing NSG mice was too toxic, while 33.0 mg/kg was well-tolerated (Supplementary Fig. 7A). Importantly, we found dosing, equimolar to CDT 33.0 mg/kg, of CND-Dox (0.44 mg/kg) or CND (0.25 mg/kg) were non-toxic with no significant effects on body weight. Histology of heart, liver and kidney showed no necrosis from CDT treatment (Supplementary Fig. 7B). We next assessed CDT toxicity and efficacy in a DLBCL PDX model using the working dose (WD) of 33.0 mg/kg. The PDX was established from a DLBCL tumor, germinal-center B-cell (GCB) subtype, excised from the spleen of a 57-year-old male with no prior history of treatment. Immunohistochemistry (IHC) analysis of the DLBCL PDX tumor confirmed TFR1 expression (Supplementary Fig. 8A). We engrafted NSG mice with these DLBCL PDX tumors and randomized them at average tumor size 150 mm3 to 4 groups of 10: MTD Dox, WD CDT, molar equivalent CND-Dox (0.44 mg/kg) or molar equivalent CND (0.25 mg/kg). Mice were treated i.v. x1 with each drug on days 0, 14, and 24. Treatment with CDT resulted in similar anti-tumor efficacy compared to Dox, as shown through OS and tumor volume assessments, with CND-Dox and CND having no activity (Supplementary Fig. 8B–C). We have observed that treatment with Dox results in an expected continuous decline of body weight, with nadir by day 8, followed by a return to starting weight by day 10–14. We mimicked CND-conjugate treatment to Dox treatment as dictated by these body weight changes with eventual return to starting weight triggering re-treatment. Both CDT- and Dox-treated mice followed this expected trend following the first dose, but after two additional doses, Dox treated mice experienced irreversible weight loss, while CDT treated mice did not (Supplementary Fig. 8D). These initial in vivo studies showed CNDs can be safely administered in vivo. Importantly, we identify a working dose of our full CDT therapy that carries anti-lymphoma efficacy similar to single-agent Dox, while preliminarily appearing better tolerated. Since Dox is never used as a single agent clinically, relatively weak antitumor activity in this experiment was not unexpected but justified further evaluation as part of clinically relevant combination therapy.
R-nanoCHOP prolongs overall survival compared to R-CHOP in DLBCL PDX-Bearing Animals
The 5-drug combination R-CHOP administered once on day 1 of every 21-day cycle is standard frontline therapy for DLBCL. In a clinically relevant assessment of our therapy, we replaced Dox in R-CHOP with CDT (R-nanoCHOP). We engrafted 22 NSG mice with DLBCL PDX tumors (Supplementary Fig. 8A) and randomized them to two groups of 11 at tumor engraftment for R-CHOP or R-nanoCHOP every 21 days. Of note, one mouse in the R-nanoCHOP group died prior to initiation of treatment and was excluded from further analysis. R-nanoCHOP (n=10) and R-CHOP (n=11) resulted in essentially identical tumor-volume control while both groups were alive (Fig. 5A, Supplementary Fig. 9A). R-nanoCHOP-treated mice, however, tolerated treatment with dramatically less weight loss than R-CHOP-treated animals (Fig. 5B, Supplementary Fig. 9B). This led to R-nanoCHOP-treated mice having significantly improved overall survival, tolerating an average of 2 additional treatment cycles compared to R-CHOP-treated mice (p <0.0001, Fig. 5C). Histology of R-nanoCHOP and R-CHOP treated mice showed minimal effects on vital organs of interest (Fig. 6A, Supplementary Fig. 9C). A decrease in cellularity in bone marrow and subtle evidence of hepatotoxicity, indicated by increased lobular inflammation, was seen in both treatment groups. Because animals were sacrificed by CO2 euthanasia as required by ethical considerations at ≥20% weight loss, necroscopy did not identify specific therapy-related causes of death for mice in either group. The greatly reduced toxicity of CDT compared to Dox with at least equal antitumor activity was encouraging, but a potential caveat is the species difference between host and tumor in these experiments. We wanted to confirm CDT, containing human holo-TF, interacted similarly with murine and human TFR1. Assessment of TFR1 expression using an antibody reactive to the protein from both species showed expression in the murine B-lymphoma line A20 similar to BJAB cells, while 3T3 non-transformed murine fibroblasts had lower expression (Fig. 6B). Like human DLBCL lines, A20 cells were dramatically more sensitive to CDT than to unconjugated Dox (Fig. 6C, CDT EC50 0.59 nM, Dox EC50 10.64 nM). This strongly suggests similar binding to murine and human TFR1 by CDT. For further confirmation, we exposed BJAB and A20 cells to 500 nM CDT or 25 ug/mL fluorophore-labeled human holo-TF (holo-TF CF®568) for 30 minutes and analyzed red fluorescence by flow cytometry (Fig. 6D). These results confirmed similar strong binding to both human BJAB and murine A20 cells by both reagents. In sum, human and murine TFR1 is bound similarly by human holo-TF, including as part of the CDT conjugate. The novel therapeutic regimen R-nanoCHOP has promising anti-lymphoma activity with a favorable toxicity profile that allowed administration of additional treatment cycles, prolonging overall survival.
Figure 5. R-nanoCHOP improves overall survival compared to R-CHOP in DLBCL PDX-bearing NSG mice.

22 NSG mice were implanted with DLBCL PDX tumor (S8B) and randomized to two groups. After death of one animal prior to engraftment, remaining animals were treated with R-CHOP (n=11) or R-nanoCHOP (n=10) once on day 1 of every 21 days. Predetermined survival endpoints were tumor volume ≥ 1500 mm3, weight loss > 20%, or other signs of morbidity. A) Average tumor volume ±SEM measured twice weekly via ultrasound. B) Average daily body weight ±SEM of surviving animals. C) Overall survival of all treatment groups. ****P < 0.0001 (Mantel-Cox). A-C are mean ±SEM of at least ten technical replicates representative of one independent experiment.
Figure 6. R-nanoCHOP treatment has favorable toxicity profile.
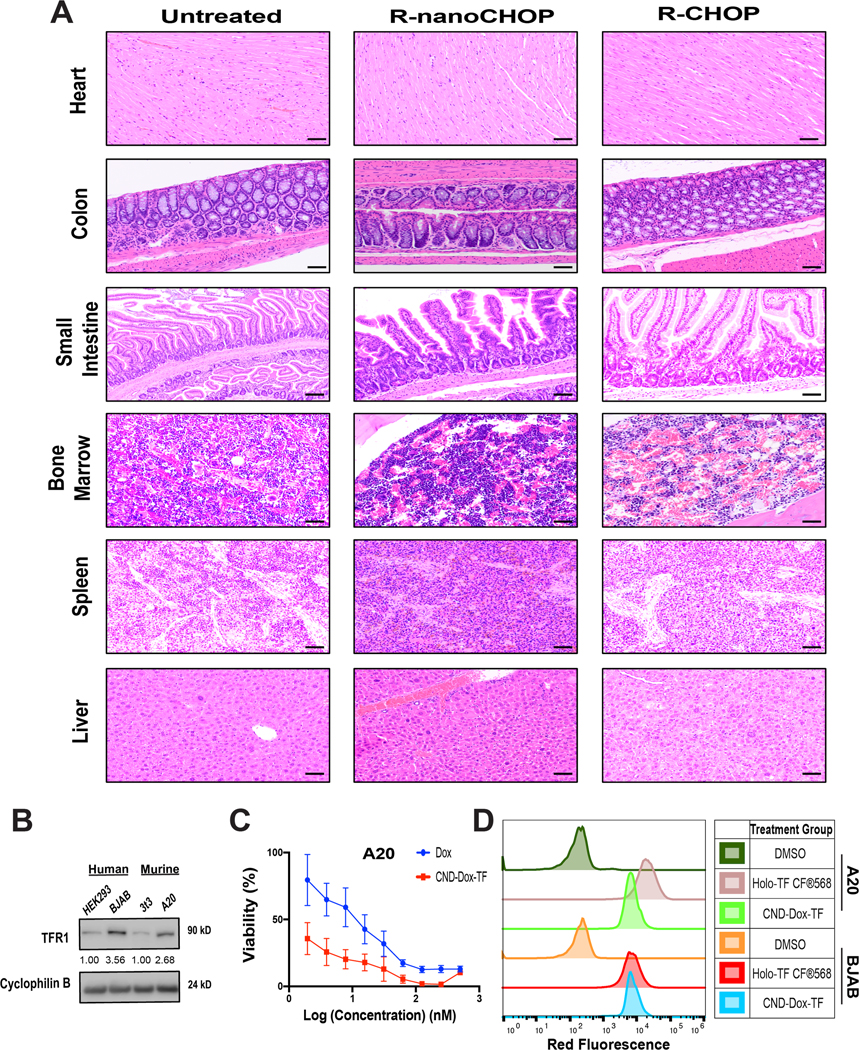
A) Heart, Colon, Small Intestine, Bone Marrow, Spleen, Liver H&E pathology collected from R-CHOP and R-nanoCHOP treated mice at predetermined survival endpoints. B) Immunoblot analysis of TFR1 (90 kD) expression in HEK293, BJAB, 3T3 and A20 cell lines. C) 48-hour viability assays for A20 cells plated in serial dilution of Dox and CND-Dox-TF. D) Flow cytometry binding assay of A20 and BJAB cells incubated for 30 minutes with DMSO, holo-TFCF®568, or CND-Dox-TF. Scale bar 50 μm (20× objective for all). For densitometric analysis, all samples normalized to loading control first followed by normalization to respective lower TFR1-expressing cell lines.
Discussion
Frontline R-CHOP results in long-term disease-free survival in up to 60% of DLBCL, but salvage of rel/ref patients has limited success (1,2). Recent advances in immunotherapy provide new options for subsets of patients, but costly and laborious ex vivo methodology, unfavorable clinical toxicities, and strict patient-eligibility requirements have hindered broad clinical implementation so far (54,55). Overexpression of cell-surface receptor TFR1 is well described across cancer and has been therapeutically investigated in various solid-tumor malignancies (9–18). Association between TFR1 and NHL aggressiveness was reported in 1983 (56), but the limited sample size, outdated methodologies, and heterogenous NHL diagnoses limit current application of these findings. Here, we link TFR1 overexpression to poor prognosis in DLBCL in two well-known large datasets from pretreatment biopsies of patients treated with standard therapies with curative intent (Fig. 1). TFR1-targeted therapy is therefore an opportunity to treat high-risk DLBCL tumors in a novel fashion.
Targeting TFR1 has been the focus of previously developed anti-cancer therapeutic compounds, either utilizing the receptor as an entry point to deliver toxic cargo, or simply blocking TFR1’s growth-promoting capabilities through antagonistic antibodies or single-chain variable fragments (scFv) (57,58). Pre-clinical testing of directly fused TF-Dox compounds revealed activity at the plasma membrane and cytoplasm, in strong contrast to unconjugated single-agent Dox’s activity in the nucleus, and this discrepancy was among factors that ultimately halted further development (59–61). An engineered diphtheria toxin mutant CRM107 fused to TF promoted a tumor response in 9 of 15 evaluable brain-tumor patients during a phase 1 trial (24), but ultimately failed due to seizure toxicity of unclear etiology (25). TFR1-targeting nanoparticles successfully delivered siRNA to human melanoma patients but failed to evolve into a clinically applicable therapy (26). Recent advances in nanotechnology provide new opportunities to optimize the TFR1-targeted treatment paradigm. In addition, previous efforts have not assessed efficacy against DLBCL, a disease in which Dox is still considered the most active drug as part of frontline chemoimmunotherapy.
Our CNDs are low-cost, non-toxic, eco-friendly nanoparticles with advantageous properties for therapeutic development (33,36,39,40,62,63). In this study, we developed and tested the anti-tumor efficacy of a novel chemotherapeutic nanocarrier compound comprising holo-TF and Dox linked to CNDs (Fig. 2A). Our results suggest this is a viable therapeutic strategy for targeting TFR1 in DLBCL. Cell viability assessments revealed DLBCL cell lines are dramatically more sensitive to CDT than single-agent Dox (Fig. 3A–B), with onset of apoptosis and DNA damage occurring at lower doses (Fig. 3C–D). Overall, we find that in cell lines, baseline sensitivity to Dox appears most closely associated with observed CDT efficacy rather than the absolute level of TFR1 expression. This is consistent with well-described rapid recycling of TFR1 back to the cell surface after each round of endocytosis (48–50), and so while raw TFR1-expression may differ across cell lines, rapid CDT uptake occurs nonetheless (Supplementary Fig. 4D). Regardless, gain-and-loss of function experiments demonstrate CDT activity is ultimately mediated by TFR1 (Fig. 4A–D) and facilitated cellular entry via TF-TFR1 interaction (Fig. 4E) with subsequent cytoplasmic separation of Dox, either alone or still bound to CND, from TF (Fig. 4F). This enabled strong entry and activity of Dox in the nucleus. We establish the safety of CNDs in NSG mice and identify a working dose of our CDT (Supplementary Fig. 7–8).
When compared with Dox MTD in a high-TFR1 expressing DLBCL PDX model, we find CDT has similar anti-tumor efficacy and an improved toxicity profile (Supplementary Fig. 8). To more accurately explore clinical relevance, we substituted Dox with CDT in frontline R-CHOP, creating R-nanoCHOP, administered in clinically standard 21-day cycles. Compared with R-CHOP, R-nanoCHOP had similar anti-lymphoma efficacy with diminished toxicity (Fig. 5A–B). Strikingly, we see a significant improvement in the OS of R-nanoCHOP treated mice compared to those treated with R-CHOP (Fig. 5C). While histology showed similar effects of both treatment modalities, it is important to note that this level of minimal toxicity occurred in R-nanoCHOP treated mice on average two cycles after it was seen in R-CHOP treated mice. We found that CDT has similar activity against and binding to human and murine cells, addressing potential caveats regarding possible lack of cross-species reactivity (Fig. 6B–D). This shows R-nanoCHOP delayed non-malignant organ toxicity substantially longer than R-CHOP (Fig. 6A, Supplementary Fig. 9). Specifically, we observed indication of diminished negative effects in the bone marrow with R-nanoCHOP, despite known high TFR1 expression on cells of the erythrocyte lineage. Additionally, in spite of additional dosing with R-nanoCHOP, no increase in cardiotoxicity was observed. In this proof-of-principle work, we believe we have demonstrated a biodistribution of CDT that results in highly promising anti-lymphoma activity with reduced toxicities to non-malignant tissues compared to Dox. Underlying germline mutations found in immuno-incompetent NSG mice limit their potential for a full panel of clinically relevant toxicity analyses (64), a potential limitation of our study. Indeed, the NSG mice tolerated at most only two cycles of R-CHOP compared to six cycles routinely given to human patients. Further study in additional model systems therefore will be needed as this approach moves forward. We first plan development of optimized therapeutic molecules based on the proof-of-principle established here. We currently are synthesizing CDTsc, a version in which holo-TF is replaced with an anti-TF scFv. We expect the resulting substantial decrease in molecular weight (>50% overall) will improve pharmacokinetic properties and allow increased dosing, while preserving strong anti-lymphoma activities. This system is also highly adaptable to conjugation of additional antineoplastic compounds and optimization with modifications that enhance delivery inside cells such as linkers specifically cleaved in lysosomes as are used in approved antibody-drug conjugates (65). All of these approaches we have under active investigation.
CDT’s successful delivery of Dox to DLBCL tumors in vivo with diminished treatment-limiting off-tumor toxicities demonstrates a proof of principle for targeting TFR1 in this disease. We believe these studies provide compelling rationale for development of TFR1-targeting therapies for DLBCL, perhaps using high TFR1 tumor expression as a biomarker in the design of relevant clinical trials. We postulate the use of optimized reagents based on the CDT concept in place of Dox could improve DLBCL patient outcomes in frontline or rel/ref settings.
Supplementary Material
Significance.
Targeted nanoparticle delivery of doxorubicin chemotherapy via the TRF1 receptor presents a new opportunity against high-risk DLBCL tumors using potency and precision.
Acknowledgements
We would like to thank Dr. Yu Zhang (J. Rosenblatt Lab, University of Miami) for her input in flow cytometry experimental technique and Ashanti Osteen (Cancer Modeling Shared Resource, Sylvester Comprehensive Cancer Center, University of Miami) for help with histological preparation of the slides. We would like to thank the Sandeep Dave Lab (Duke University) for kindly providing access to their DLBCL RNA-seq dataset. A.D.N. was supported in part by NIH Medical Scientist Training Program award T32GM112601. R.M.L., P.Y.L. and B.C.L.B.F. would like to thank the support from the National Science Foundation under the award numbers 1809060 and 2041413. J.H.S. and A.A. would like to thank the support from the Sylvester Comprehensive Cancer Center at the University of Miami Miller School of Medicine.
Footnotes
Conflicts of interest
A.A, P.Y.L, R.M.L, and J.H.S. have a patent pending with regards to this work.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34 [DOI] [PubMed] [Google Scholar]
- 2.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program 2011;2011:498–505 [DOI] [PubMed] [Google Scholar]
- 3.Bonadonna G, Monfardini S, De Lena M, Fossati-Bellani F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br Med J 1969;3:503–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology 2010;115:155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep 2017;7:44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers C. The role of iron in doxorubicin-induced cardiomyopathy. Semin Oncol 1998;25:10–4 [PubMed] [Google Scholar]
- 7.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 2006;121:144–58 [DOI] [PubMed] [Google Scholar]
- 8.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer 2013;13:342–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol 2006;121:159–76 [DOI] [PubMed] [Google Scholar]
- 10.Yang DC, Wang F, Elliott RL, Head JF. Expression of transferrin receptor and ferritin H-chain mRNA are associated with clinical and histopathological prognostic indicators in breast cancer. Anticancer Res 2001;21:541–9 [PubMed] [Google Scholar]
- 11.Prior R, Reifenberger G, Wechsler W. Transferrin receptor expression in tumours of the human nervous system: relation to tumour type, grading and tumour growth fraction. Virchows Arch A Pathol Anat Histopathol 1990;416:491–6 [DOI] [PubMed] [Google Scholar]
- 12.Kondo K, Noguchi M, Mukai K, Matsuno Y, Sato Y, Shimosato Y, et al. Transferrin receptor expression in adenocarcinoma of the lung as a histopathologic indicator of prognosis. Chest 1990;97:1367–71 [DOI] [PubMed] [Google Scholar]
- 13.Seymour GJ, Walsh MD, Lavin MF, Strutton G, Gardiner RA. Transferrin receptor expression by human bladder transitional cell carcinomas. Urol Res 1987;15:341–4 [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Zhang J, Dai R, Xu J, Feng H. Transferrin receptor-1 and VEGF are prognostic factors for osteosarcoma. J Orthop Surg Res 2019;14:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakurai K, Sohda T, Ueda S, Tanaka T, Hirano G, Yokoyama K, et al. Immunohistochemical demonstration of transferrin receptor 1 and 2 in human hepatocellular carcinoma tissue. Hepatogastroenterology 2014;61:426–30 [PubMed] [Google Scholar]
- 16.Rosager AM, Sorensen MD, Dahlrot RH, Hansen S, Schonberg DL, Rich JN, et al. Transferrin receptor-1 and ferritin heavy and light chains in astrocytic brain tumors: Expression and prognostic value. PLoS One 2017;12:e0182954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR, et al. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat 2010;119:283–93 [DOI] [PubMed] [Google Scholar]
- 18.Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 2017;36:4089–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, et al. Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med 2001;7:723–34 [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, et al. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther 2002;1:337–46 [PubMed] [Google Scholar]
- 21.Yu W, Pirollo KF, Rait A, Yu B, Xiang LM, Huang WQ, et al. A sterically stabilized immunolipoplex for systemic administration of a therapeutic gene. Gene Ther 2004;11:1434–40 [DOI] [PubMed] [Google Scholar]
- 22.Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther 2013;21:1096–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pirollo KF, Nemunaitis J, Leung PK, Nunan R, Adams J, Chang EH. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol Ther 2016;24:1697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med 1997;3:1362–8 [DOI] [PubMed] [Google Scholar]
- 25.Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol 2003;65:3–13 [DOI] [PubMed] [Google Scholar]
- 26.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010;464:1067–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S, Skromne I, Peng Z, Dallman J, Al-Youbi AO, Bashammakh AS, et al. “Dark” carbon dots specifically “light-up” calcified zebrafish bones. J Mater Chem B 2016;4:7398–405 [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Lu Y-C, Huang H, Feng J-J, Chen J-R, Wang A-J. Facile synthesis of water-soluble and biocompatible fluorescent nitrogen-doped carbon dots for cell imaging. Analyst 2014;139:1692–6 [DOI] [PubMed] [Google Scholar]
- 29.Ma J, Guo B, Cao X, Lin Y, Yao B, Li F, et al. One-pot fabrication of hollow cross-linked fluorescent carbon nitride nanoparticles and their application in the detection of mercuric ions. Talanta 2015;143:205–11 [DOI] [PubMed] [Google Scholar]
- 30.Zhou J, Yang Y, Zhang C-y. A low-temperature solid-phase method to synthesize highly fluorescent carbon nitride dots with tunable emission. Chemical Communications 2013;49:8605–7 [DOI] [PubMed] [Google Scholar]
- 31.Ge J, Jia Q, Liu W, Guo L, Liu Q, Lan M, et al. Red-Emissive Carbon Dots for Fluorescent, Photoacoustic, and Thermal Theranostics in Living Mice. Adv Mater 2015;27:4169–77 [DOI] [PubMed] [Google Scholar]
- 32.Jiang K, Sun S, Zhang L, Lu Y, Wu A, Cai C, et al. Red, green, and blue luminescence by carbon dots: full-color emission tuning and multicolor cellular imaging. Angew Chem Int Ed Engl 2015;54:5360–3 [DOI] [PubMed] [Google Scholar]
- 33.Peng Z, Miyanji EH, Zhou Y, Pardo J, Hettiarachchi SD, Li S, et al. Carbon dots: promising biomaterials for bone-specific imaging and drug delivery. Nanoscale 2017;9:17533–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Jiang W, Qiu L, Jiang X, Zuo D, Wang D, et al. One pot synthesis of highly luminescent polyethylene glycol anchored carbon dots functionalized with a nuclear localization signal peptide for cell nucleus imaging. Nanoscale 2015;7:6104–13 [DOI] [PubMed] [Google Scholar]
- 35.Huang X, Zhang F, Zhu L, Choi KY, Guo N, Guo J, et al. Effect of injection routes on the biodistribution, clearance, and tumor uptake of carbon dots. ACS Nano 2013;7:5684–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruan S, Qian J, Shen S, Zhu J, Jiang X, He Q, et al. A simple one-step method to prepare fluorescent carbon dots and their potential application in non-invasive glioma imaging. Nanoscale 2014;6:10040–7 [DOI] [PubMed] [Google Scholar]
- 37.Yang ST, Cao L, Luo PG, Lu F, Wang X, Wang H, et al. Carbon dots for optical imaging in vivo. J Am Chem Soc 2009;131:11308–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng M, Ruan S, Liu S, Sun T, Qu D, Zhao H, et al. Self-Targeting Fluorescent Carbon Dots for Diagnosis of Brain Cancer Cells. ACS Nano 2015;9:11455–61 [DOI] [PubMed] [Google Scholar]
- 39.Liyanage PY, Graham RM, Pandey RR, Chusuei CC, Mintz KJ, Zhou Y, et al. Carbon Nitride Dots: A Selective Bioimaging Nanomaterial. Bioconjug Chem 2019;30:111–23 [DOI] [PubMed] [Google Scholar]
- 40.Liyanage PY, Zhou Y, Al-Youbi AO, Bashammakh AS, El-Shahawi MS, Vanni S, et al. Pediatric glioblastoma target-specific efficient delivery of gemcitabine across the blood–brain barrier via carbon nitride dots. Nanoscale 2020 [DOI] [PubMed] [Google Scholar]
- 41.Hettiarachchi SD, Graham RM, Mintz KJ, Zhou Y, Vanni S, Peng Z, et al. Triple conjugated carbon dots as a nano-drug delivery model for glioblastoma brain tumors. Nanoscale 2019;11:6192–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One 2013;8:e74250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359:2313–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171:481–94 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Binaschi M, Bigioni M, Cipollone A, Rossi C, Goso C, Maggi CA, et al. Anthracyclines: selected new developments. Curr Med Chem Anticancer Agents 2001;1:113–30 [DOI] [PubMed] [Google Scholar]
- 46.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 2013;65:157–70 [DOI] [PubMed] [Google Scholar]
- 47.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 2006;10:839–50 [DOI] [PubMed] [Google Scholar]
- 48.Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol 1993;121:1257–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ciechanover A, Schwartz AL, Dautry-Varsat A, Lodish HF. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J Biol Chem 1983;258:9681–9 [PubMed] [Google Scholar]
- 50.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol 2004;5:121–32 [DOI] [PubMed] [Google Scholar]
- 51.Mohammad RM, Al-Katib A, Aboukameel A, Doerge DR, Sarkar F, Kucuk O. Genistein sensitizes diffuse large cell lymphoma to CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy. Mol Cancer Ther 2003;2:1361–8 [PubMed] [Google Scholar]
- 52.Mohammad RM, Wall NR, Dutcher JA, Al-Katib AM. The addition of bryostatin 1 to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy improves response in a CHOP-resistant human diffuse large cell lymphoma xenograft model. Clin Cancer Res 2000;6:4950–6 [PubMed] [Google Scholar]
- 53.Al-Katib A, Arnold AA, Aboukameel A, Sosin A, Smith P, Mohamed AN, et al. I-kappa-kinase-2 (IKK-2) inhibition potentiates vincristine cytotoxicity in non-Hodgkin’s lymphoma. Mol Cancer 2010;9:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019;380:45–56 [DOI] [PubMed] [Google Scholar]
- 55.Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol 2019;37:2105–19 [DOI] [PubMed] [Google Scholar]
- 56.Habeshaw JA, Lister TA, Stansfeld AG, Greaves MF. Correlation of transferrin receptor expression with histological class and outcome in non-Hodgkin lymphoma. Lancet 1983;1:498–501 [DOI] [PubMed] [Google Scholar]
- 57.Brooks D, Taylor C, Dos Santos B, Linden H, Houghton A, Hecht TT, et al. Phase Ia trial of murine immunoglobulin A antitransferrin receptor antibody 42/6. Clin Cancer Res 1995;1:1259–65 [PubMed] [Google Scholar]
- 58.Crepin R, Goenaga AL, Jullienne B, Bougherara H, Legay C, Benihoud K, et al. Development of human single-chain antibodies to the transferrin receptor that effectively antagonize the growth of leukemias and lymphomas. Cancer Res 2010;70:5497–506 [DOI] [PubMed] [Google Scholar]
- 59.Barabas K, Sizensky JA, Faulk WP. Transferrin conjugates of adriamycin are cytotoxic without intercalating nuclear DNA. J Biol Chem 1992;267:9437–42 [PubMed] [Google Scholar]
- 60.Kratz F, Beyer U, Roth T, Tarasova N, Collery P, Lechenault F, et al. Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J Pharm Sci 1998;87:338–46 [DOI] [PubMed] [Google Scholar]
- 61.Lai BT, Gao JP, Lanks KW. Mechanism of action and spectrum of cell lines sensitive to a doxorubicin-transferrin conjugate. Cancer Chemother Pharmacol 1998;41:155–60 [DOI] [PubMed] [Google Scholar]
- 62.Bao X, Yuan Y, Chen J, Zhang B, Li D, Zhou D, et al. In vivo theranostics with near-infrared-emitting carbon dots-highly efficient photothermal therapy based on passive targeting after intravenous administration. Light Sci Appl 2018;7:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng T, Ai X, An G, Yang P, Zhao Y. Charge-Convertible Carbon Dots for Imaging-Guided Drug Delivery with Enhanced in Vivo Cancer Therapeutic Efficiency. ACS Nano 2016;10:4410–20 [DOI] [PubMed] [Google Scholar]
- 64.Favreau-Lessard AJ, Blaszyk H, Jones MA, Sawyer DB, Pinz IM. Systemic and cardiac susceptibility of immune compromised mice to doxorubicin. Cardiooncology 2019;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 2010;363:1812–21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.