

Abstract
PURPOSE
This phase I study aimed to define the recommended phase II dose (RP2D) of tebentafusp, a first-in-class T-cell receptor/anti-CD3 bispecific protein, using a three-week step-up dosing regimen, and to assess its safety, pharmacokinetics, pharmacodynamics, and preliminary clinical activity in patients with metastatic uveal melanoma (mUM).
METHODS
In this open-label, international, phase I/II study, HLA-A*02 or HLA-A*02:01+ patients with mUM received tebentafusp 20 μg once in week 1 and 30 μg once in week 2. Dose escalation (starting at 54 μg) began at week 3 in a standard 3 + 3 design to define RP2D. Expansion-phase patients were treated at the RP2D (20-30-68 μg). Blood and tumor samples were collected for pharmacokinetics/pharmacodynamics assessment, and treatment efficacy was evaluated for all patients with baseline efficacy data as of December 2017.
RESULTS
Between March 2016 and December 2017, 42 eligible patients who failed a median of two previous treatments were enrolled: 19 in the dose escalation cohort and 23 in an initial dose expansion cohort. Of the dose levels investigated, 68 μg was identified as the RP2D. Most frequent treatment-emergent adverse events regardless of attribution were pyrexia (91%), rash (83%), pruritus (83%), nausea (74%), fatigue (71%), and chills (69%). Toxicity attenuated following the first three doses. The overall response rate was 11.9% (95% CI, 4.0 to 25.6). With a median follow-up of 32.4 months, median overall survival was 25.5 months (range, 0.89-31.1 months) and 1-year overall survival rate was 67%. Treatment was associated with increased tumor T-cell infiltration and transient increases in serum inflammatory mediators.
CONCLUSION
Using a step-up dosing regimen of tebentafusp allowed a 36% increase in the RP2D compared with weekly fixed dosing, with a manageable side-effect profile and a signal of efficacy in mUM.
INTRODUCTION
Uveal melanoma (UM) is a rare malignancy that develops from melanocytes of the choroid, ciliary body, and iris, and is the most common primary eye cancer in adults.1 Incidence varies by geography, race, and age, but is < 10 cases per million.2,3 Up to 50% of patients with UM develop systemic metastases generally affecting the liver4-7 and, thereafter, the median overall survival (OS) historically is < 1 year.4,8 No previous therapy has been demonstrated in a randomized setting to improve OS in patients with metastatic UM (mUM), a diagnosis that is uniformly fatal and that represents a significant area of unmet clinical need.
CONTEXT
Key Objective
Tebentafusp showed promising activity in patients with metastatic uveal melanoma (mUM) in a first-in-human study, with greater responses observed at or exceeding the maximum tolerated dose. A step-up dosing regimen was developed in an effort to permit the safe administration of higher doses, with escalation of the final dose using a standard 3 + 3 design.
Knowledge Generated
This study identified a recommended phase II dose 36% higher than the maximum tolerated dose identified in the first-in-human trial. A 1-year survival of 67% and median overall survival of 25.5 months suggest clinically meaningful antitumor activity as monotherapy in patients with mUM.
Relevance
Data from this study in patients with previously treated mUM provided rationale for the dosing regimen used in the phase II portion of this trial (ClinicalTrials.gov identifier: NCT02570308) and the randomized controlled study of tebentafusp versus investigator's choice of therapy in patients with previously untreated mUM (ClinicalTrials.gov identifier: NCT03070392).
UM differs significantly from cutaneous melanoma (CM) in molecular and tumor-immune microenvironment features, as well as clinical response to treatment with immune checkpoint inhibitors.1,9 Despite the presence of intratumoral cytotoxic T cells in both diseases, the clinical activity of single-agent immunologic checkpoint blockade is markedly inferior in UM.8,10 The differential response is, in part, explained by the distinct immunologic microenvironment, the low mutational burden, and relatively low expression of programmed death-ligand 1 (PD-L1) in mUM when compared with cutaneous disease.11 Notwithstanding the stark differences between these two tumor types, phenotypic commonalities remain, such as expression of the melanoma associated antigen gp100.
Two single-arm phase II studies of combination checkpoint inhibitors nivolumab and ipilimumab, a standard of care in CM, have recently been reported: one a multicenter trial in first-line patients (GEM-1402) and the other a mixed-line single-institution study (PROSPER).12,13 Although both studies showed promising response rates (11.5% and 18%) and median OS (12.7 months and 19.1 months) after approximately 13 months of follow-up, there was no clear benefit on OS with 1-year OS rates matching recent meta-analyses (52%-56%),8,14 and treatment-related toxicity was greater than observed with single-agent checkpoint blockade.
Tebentafusp (formerly IMCgp100) is a first-in-class investigational bispecific fusion protein composed of a soluble affinity-enhanced HLA-A*02:01-restricted T-cell receptor that is specific for the gp100 peptide, YLEPGPVTA, fused to an anti-CD3 scFv.15-18 These ImmTAC (Immune mobilizing monoclonal T-cell receptors Against Cancer) molecules redirect and activate polyclonal T cells toward target cells presenting peptide-HLA complexes on their cell surface. In the first-in-human (FIH) multicenter phase I study of tebentafusp enrolling HLA-A*02-positive patients with advanced melanoma, including 19 with UM, 61 with CM, and four with other melanoma subtypes, a maximum tolerated dose (MTD) of 50 μg was identified when administered at a fixed once weekly dose. No differences in efficacy were observed when tebentafusp was administered using this weekly fixed-dose schedule or when using a daily dosing schedule. Three of 15 (20%) evaluable mUM patients achieved a partial response (PR), and 7 (47%) achieved stable disease.17 Greater responses were observed at dose levels at or exceeding the MTD, suggesting a dose-response relationship.19-21 Although dosing was limited by toxicities, including cytokine release syndrome (CRS), these toxicities were manageable and reversible within 24 hours, and most occurrences were limited to the first 2 weeks of treatment.19-21
These observations suggested that (1) a dosing regimen using doses of tebentafusp below the MTD for the first two doses may reduce the occurrence of severe toxicity as has been observed for other anti-CD3 bispecifics22; and (2) a dosing strategy that permits increased exposure above the previously identified MTD may permit an enhanced tumor response.
To optimize response through increase in exposure and mitigate treatment toxicity in mUM, we conducted a phase I/II trial using a three-week step-up dosing regimen with the objective of allowing the safe administration of higher, and potentially more effective, doses of tebentafusp. We now report the safety and efficacy results of the phase I dose escalation (DE) portion of the trial together with 23 patients enrolled on an initial dose expansion portion of the study, as well as initial biomarker assessment to explore biologic effects in an mUM population.
METHODS
Study Design and Participants
This open-label, international, phase I/II study (ClinicalTrials.gov identifier: NCT02570308) was composed of a phase I DE and an initial expansion cohort that was subsequently expanded into a full phase II study and was carried out in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol was approved by the relevant ethical bodies at each participating site, and patients provided written informed consent before being screened for enrollment. A trial safety committee that comprised study investigators and representatives from the sponsor convened at regular intervals to review the safety of trial participants and dose-limiting toxicities (DLTs), and to determine the MTD and recommended phase II dose (RP2D).
Eligible patients were age ≥ 18 years, HLA-A*02- or HLA-A*02:01-positive (eligibility was changed to mandate HLA-A*02:01 as per an amendment dated September 7, 2016), had histologically or cytologically confirmed diagnosis of mUM, had received any number of prior lines of therapy, had an Eastern Cooperative Oncology Group performance score of ≤ 1,23 and had adequate organ function. In the DE portion of the study, patients with high tumor burden, defined as liver replacement of > 60% hepatic organ volume with tumor, were excluded because of concerns for hepatic toxicity. Other key exclusions included patients with symptomatic or untreated CNS metastases, a history of severe hypersensitivity reactions to other biologic drugs or monoclonal antibodies, or out-of-range laboratory values.
Tebentafusp was administered intravenously on a weekly basis, with each cycle consisting of four doses. To minimize the risk of CRS and early skin toxicity, which had been observed to ameliorate after the initial three doses in the completed FIH study, we used a three-week step-up dosing regimen where all patients received sub-MTD doses of 20 μg tebentafusp at cycle 1 day 1 and 30 μg at cycle 1 day 8. Interpatient DE began at cycle 1 day 15 (C1D15; Fig 1). Inpatient administration and monitoring for at least 16 hours following the first three doses was mandated to enable prompt treatment of CRS if needed. A standard 3 + 3 design was used to define the MTD and RP2D. Three to six patients were enrolled in sequentially enrolling dose cohorts. Patients treated in cohort 1 received tebentafusp 54 μg at C1D15 and thereafter once weekly; dosing was increased in subsequent cohorts until an MTD and/or RP2D was defined. Patients in the expansion cohort were treated at the RP2D. Treatment continued until confirmed disease progression as per immune-related response criteria or intolerable toxicity.
FIG 1.
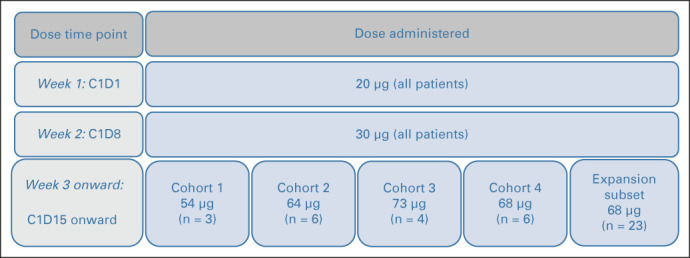
Dose escalation trial design. C1D1, cycle 1 day 1; C1D8, cycle 1 day 8; C1D15, cycle 1 day 15.
Outcomes and Assessments
Treatment efficacy was assessed using RECIST v1.1 and Kaplan-Meier survival analysis. We further defined an exploratory category of minor response where the sum of the longest diameters of target lesions is reduced by 10%-29%. OS was measured from the start of treatment to time of death. Patients alive at the time of the analysis were censored on the last date they were known to be alive. Treatment-emergent adverse events (TEAEs) were coded according to Common Terminology Criteria for Adverse Events v4.03. Incidence of DLT were defined as adverse events (AEs) of grade 3 or higher occurring in the first 4-week cycle during the DE portion of the study. AEs of special interest included rash, CRS, and elevation in liver function tests (LFTs).
Safety/toxicity, pharmacodynamic, pharmacokinetic (PK), and biomarker data from 42 patients (19 patients treated in the DE portion of the study and 23 patients treated in an expansion cohort by December 31, 2017) were included for analysis. Detailed methods for PK and pharmacodynamic assessments can be found in the Data Supplement (online only).
Statistical Analysis
Data were analyzed and reported on the basis of all patient data up to the data cutoff date of June 26, 2019. CIs for overall response rate (ORR) and related end points were calculated using the exact methods. Time-to-event estimates of OS were calculated via Kaplan-Meier methods, and the median and 95% CIs calculated by the method of Brookmeyer and Crowley. Hazard ratios were derived from a Cox proportional hazards regression model.
The analysis set includes all patients assigned to treatment who received at least one dose of tebentafusp in the DE (n = 19) portion of the study and the 23 dose expansion patients treated by December 31, 2017. Efficacy parameters for the 23 patients enrolled in the expansion phase, including imaging and response data, were assessed by investigators. This initial gated expansion cohort subsequently expanded into the formal phase II portion of the trial.
RESULTS
Nineteen patients were enrolled in the DE portion of the study between March 2016 and September 2016 and were treated across four dose levels defined by the dose administered at C1D15 and beyond (Fig 1). The 23 expansion patients were first treated between October 2016 and December 2017. The median age was 55 years (range, 34-73 years) for DE and 61 years (range, 45-79 years) for expansion, with a near even split between sexes (Table 1). Both groups of subjects had similar drug exposure, as well as baseline clinical characteristics, including demographics, prior treatment history, extent of disease, and prognostic factors. The DE patients were, however, characterized by a lower prevalence of largest liver metastasis size > 3 cm (5/19, 25%) when compared with the expansion patients (14/23, 61%).
TABLE 1.
Patient Baseline Characteristics

At the time of data cutoff, the median follow-up was 35 months (range, 32-39 months) for the DE subjects and 21 months (range, 18-29 months) for the expansion subjects, with seven patients still receiving study treatment at the time of analysis. The median follow-up for all subjects was 32.4 months (range, 18-39 months). The median number of cycles of tebentafusp started and completed were 7.5 (range, 1-41) and 6 (range, 0-39), respectively. Median time from initial diagnosis to development of metastatic disease was 3.6 years (range, 0-20 years) for the DE group and 4.4 years (range, 1-22 years) for the expansion group. Eleven of the 19 (58%) DE and 12 of the 23 (52%) expansion subjects had a baseline lactate dehydrogenase (LDH) above the upper limit of normal (ULN). Almost all patients had at least one prior anticancer therapy in the metastatic setting, including 18 of the 19 (95%) patients enrolled in the DE portion (median 2; range, 0-6) and all patients in the expansion portion of the study (median 2; range, 1-5).
During the DE portion of the study, four dose levels (54 μg, 64 μg, 73 μg, and 68 μg) were investigated. Three DLTs consisting of hepatic events were observed in one of six patients treated at 64 μg (grade 3 transaminase elevation with concurrent mild increase in bilirubin) and in two of four patients treated at 73 μg (grade 3 or 4 transaminase elevations). Each of these events resolved within 1 week without the administration of corticosteroids. All patients experiencing a DLT reinitiated and tolerated therapy with a dose level reduction. Dose level 3 (73 μg) was deemed not tolerable because of transaminase elevations, and an intermediate dose level (68 μg) was enrolled. Six patients were treated at this dose level with no DLTs or significant elevations of hepatic transaminases observed, and 68 μg was identified as the RP2D.
As of the data cutoff, all patients (N = 42) had experienced at least one TEAE and 30 (71%) patients experienced a grade 3 or higher TEAE. The most frequently reported TEAEs regardless of causality were pyrexia (86%), nausea (74%), chills (69%), pruritus (67%), and fatigue (62%; Data Supplement). The most frequently reported grade ≥ 3 TEAEs were abdominal pain (12%), hypotension (9%), fatigue (9%), and hypophosphatemia (9%). A total of 16 (38%) patients experienced at least one serious adverse event, with the most frequently reported being abdominal pain (7%), AST increased (7%), ALT increased (5%), hypotension (5%), hyperbilirubinemia (5%), and hypophosphatemia (5%). Increases in transaminases from baseline were observed in 9 (21%) patients, including four patients with grade 3 or 4 events; these events generally occurred early in the treatment course in the setting of CRS. Chronic elevations in LFTs were mostly associated with disease progression. Two (4%) patients experienced an AE that led to study drug discontinuation (CRS and abdominal pain). No treatment-related deaths occurred.
In total, 38 (90%) patients experienced CRS of any grade as defined using 2019 CRS grading criteria,24 which was applied retrospectively. CRS was grade 1 or 2 in severity in 98% of cases and was generally reversible with intravenous fluids and, in some cases, short courses of corticosteroids. One patient (2%) experienced grade 4 CRS and recovered from the event but did not continue treatment because of protocol toxicity management criteria. Only one patient received supplemental oxygen, and no use of anti–interleukin (IL)-6 antibodies, such as tocilizumab, was required.
Skin toxicity was among the most frequent AEs observed with tebentafusp and is believed to be an on-target (response to gp100 protein in skin melanocytes), off-tumor effect. In a post hoc analysis, we developed a composite list of skin toxicity comprising all unique MedDRA preferred terms under the system organ class of Skin and Subcutaneous Tissue Disorders (Data Supplement). Skin toxicity composite terms groups into the following categories: rash (83%), pruritus (83%), dry skin (64%), pigment change (57%), erythema (57%), edema (57%), and other changes (43%). Skin toxicity was grade 1 or 2 in severity in 67% of cases and occurred approximately 1-2 days after one or more of the first few doses. No cases of Stevens-Johnson syndrome or toxic epidermal necrosis have been reported. Symptomatic patients were managed with antihistamine and topical corticosteroid therapy. AEs including rash, hypotension, pruritus, and pyrexia reduced in incidence and severity with repeated dosing (Fig 2).
FIG 2.
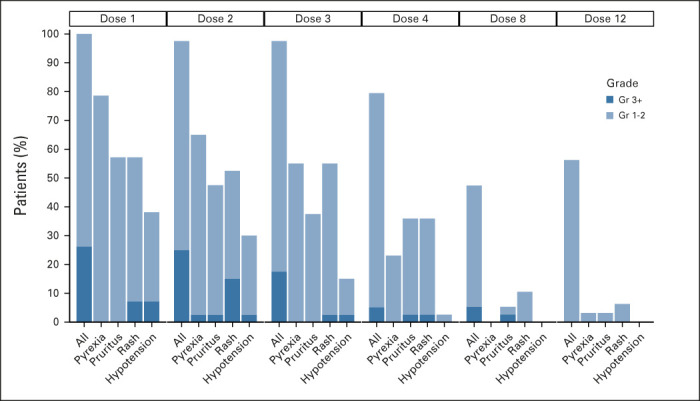
Occurrence and severity of AEs attenuate with repeated weekly dosing. Percentage of treated patients experiencing any grade or Gr 3+ of select treatment-related AEs occurring within 7 days of the first four doses (cycle 1), dose 8 (end cycle 2), and dose 12 (end cycle 3). Rash is a composite term for a list of skin toxicities of any grade (Data supplement). AE, adverse event; Gr, grade.
Confirmed PR was achieved in five of the 42 evaluable patients, resulting in an ORR of 11.9% (95% CI, 4.0 to 25.6). Three of the responses occurred in the DE portion of the study, all within cohort four (68 μg). Fifty-five percent of patients achieved some degree of tumor reduction in target lesions (Fig 3). Median time to response was 7.4 months (range, 3.7-9.2 months), median duration of response was 8.5 months (range, 3.7-8.5 months), and median PFS was 4.6 months (range, 0.7-25.9 months; Data Supplement). The 24-week disease control rate was 33%. The 1-year OS rate was 67% (95% CI, 52 to 81), with a median OS of 25.5 months (range, 0.89-31.1 months; Fig 4A). Of patients with LDH > ULN at baseline, 48% (11/23) had OS > 12 months (Data Supplement). In a post hoc analysis, patients experiencing skin toxicity of any grade within 7 days of first dose (n = 24) survived longer than those who did not have rash (HR 0.23; 95% CI, 0.10 to 0.54; Fig 4B), with a 1-year OS rate of 83% (95% CI, 68 to 98) versus 44% (95% CI, 21 to 67), respectively.
FIG 3.
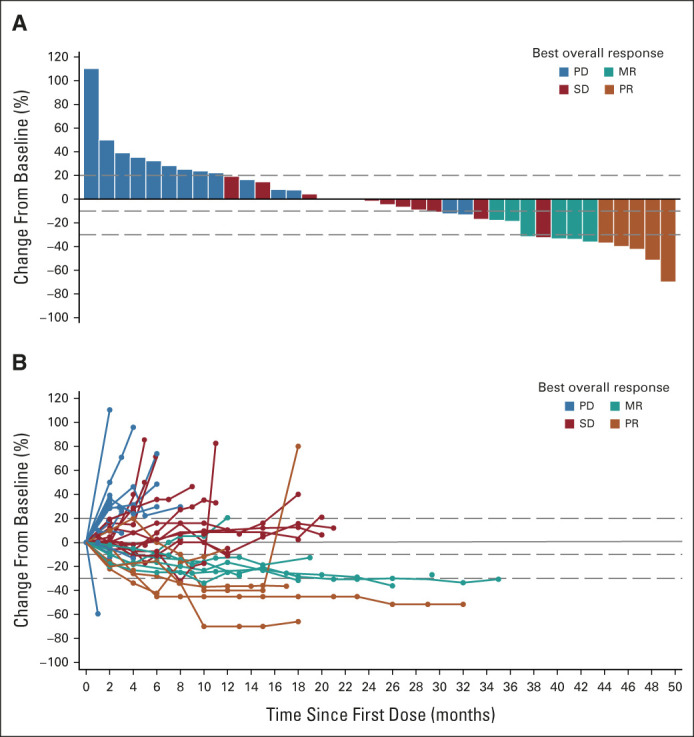
Clinical activity of tebentafusp. (A) Waterfall plot showing the best change in tumor size (n = 38). (B) Spider plot showing individual patients and the time frame (n = 40). For both (A) and (B), tumor size is measured as the sum of longest diameters of the target lesions according to RECISTv1.1 by investigator opinion. Best percent change in target lesion size is the maximum percent reduction from baseline or the minimum percent increase from baseline (in the absence of a reduction). Reference lines at 20%, –10%, and –30% mark target lesion response criteria for PD, MR, and PR, respectively. Only patients with baseline (A) or at least one evaluable postbaseline target lesion scans (A and B) are included. MR, minor response; PD, disease progression; PR, partial response; SD, stable disease.
FIG 4.

OS for tebentafusp-treated metastatic uveal melanoma patients. (A) Kaplan-Meier plot of OS (N = 42). Events are deaths because of any cause. Patients not known to have died at the time of analysis are censored. Median (95% CI) is 25.5 (12.4 to 31.1) months. (B) Kaplan-Meier plot of OS by rash (any grade) within 7 days (N = 42). Hazard ratio (95% CI) for rash: 0.235 (0.101 to 0.544). Rash is a composite term for a list of skin toxicities of any grade (Data supplement). OS, overall survival.
PK profiles were analyzed using noncompartmental analysis and included PK sampling on cycle 1 day 1 and C1D15. Tebentafusp serum levels were detectable at all dose levels administered (Data Supplement). No accumulation was observed with repeated dosing at therapeutic doses beyond day 15. Peak concentrations were observed at the end of infusion. Overall exposure as measured by Cmax and area under the curve increased with dose but was not powered to test proportionality (Data Supplement). A terminal half-life of approximately 6-8 hours was estimated across the dose range, and distribution was primarily limited to the central blood volume (Vss approximately 6-7 L).
Levels of 11 markers of immune modulation were measured in serum samples taken pretreatment and at 8 hours and 12-24 hours following the first and third doses of tebentafusp. In response to the first dose of 20 μg, all except one patient exhibited a treatment-induced increase (Fig 5A; Data Supplement). Across the 11 analytes measured, the greatest fold increases relative to baseline were calculated for CXCL10, CXCL11, IL-2, IL-6, IL-10, and interferon (IFN)γ (median approximately 20-80-fold), with approximately 60%-95% of the patients with a > 10-fold increase. Temporal analysis showed increases in serum markers to be transient, reaching maximal levels 8-24 hours postdose, with the majority returning to baseline levels before the next dose (Fig 5B). Notable exceptions to this were CXCL10 and CXCL11, which remained elevated relative to baseline levels. Analysis of mean fold change in serum markers showed that the IFNγ-induced chemokines CXCL10, CXCL11, and CXCL9, as well as IFNγ, IL-10, and IL-6 have the greatest treatment induced increase. This increase was also seen in response to the third dose (Data Supplement), and these increases in serum chemokines coincided with lymphocyte migration from the blood (Fig 5C).
FIG 5.

Tebentafusp induced a pharmacodynamic response in multiple peripheral immune markers and increased CD3+, CD8+, or CD4+ cells in the TME. (A) Maximal postdose (log2) fold change, relative to baseline concentration, in response to first dose in serum markers (n = 41). (B) Temporal profile of post first and third dose fold-change response 12-24 hours following first dose (D1) versus third dose (D15); data points represent mean ± SEM, N = 42. (C) Temporal profile of blood lymphocytes post first and third dose expressed as a fold change from baseline. Data points represent mean ± SEM, n = 32. (D) Number of CD3+, CD8+, or CD4+ cells per mm2 tumor in paired pretreatment and on-treatment biopsies (taken cycle 1 day 16) from up to six patients; line per patient. (E) Representative immunohistochemistry images from patient 2 in (D) from pretreatment and on-treatment biopsies for CD3+ (total T cells), CD4+ (T cells and monocytes), and CD8+ cells (T cells and NK cell subset). HGF, hepatocyte growth factor; IFN, interferon; IL, interleukin; NK, natural killer; TME, tumor microenvironment; TNF, tumor necrosis factor.
At baseline, none of the biomarkers assessed were associated with tumor shrinkage; however, below median levels of hepatocyte growth factor, IFNγ, IL-6, and CCL2 were associated with longer OS (Data Supplement), consistent with the hypothesis that systemic inflammation is associated with worse prognosis. On-treatment changes on tebentafusp were not associated with OS but below median increase (< 6 fold) in IL-10 was associated with greater tumor shrinkage, which is consistent with an immunosuppressive role for IL-10.
Preliminary assessment of changes in immune cells in the tumor microenvironment following tebentafusp treatment was determined in paired pre- and cycle 1 day 16 tumor biopsies available from six subjects (one from the DE portion of the study and five from expansion phase). Immunohistochemistry analyses revealed that most on-treatment biopsy samples had a greater presence of CD3+ T cells (median 1,989; range, 553-3,269) per mm2 tumor compared with baseline (median 520; range, 0-1,266; median fold change 3.8; Fig 5D). An increase in detection of CD4 (median 4,319; range, 585-8,102), which marks T cells and inflammatory monocytes, and CD8 (median 631; range, 183-1,791), marking T cells and natural killer cells, was also evident in all but one patient after treatment with tebentafusp, with a median fold change of 3.7 and 2.5, respectively. Notably, an increase in CD8+ cells on-treatment was observed even in patients with relatively few intratumoral T cells before treatment (patient 5; 21 cells per mm2 pretreatment to 522 cells per mm2 on-treatment; Fig 5E).
DISCUSSION
In this study, we demonstrate a 36% increase in the RP2D of tebentafusp when using a 3-week step-up dosing regimen compared with the MTD of 50 μg when using a fixed weekly dosing schedule.17 We observed promising preliminary clinical benefit of tebentafusp in a previously treated mUM patient population with a 1-year OS rate of 67% and median OS of 25.5 months, a benefit that has recently been confirmed in an open-label, phase III trial of tebentafusp administered using this dosing regimen versus investigator's choice of therapy in previously untreated, HLA-A*02:01 positive patients with mUM.25 Even patients with elevated LDH at baseline, a known negative prognostic factor, had improved OS benefit compared with that reported in a recent meta-analysis, with half of the patients with LDH > ULN at baseline in this study surviving beyond 1 year.14
Although inpatient hospitalization was mandated for the first three weekly doses of tebentafusp on this study for AE management, because of the attenuation of toxicities observed over time, subsequent doses were generally administered as an outpatient infusion. The AEs observed were consistent with the proposed mechanism of action of tebentafusp, including dermatologic events because of the drug-induced proximity of T cells to gp100+ melanocytes as well as cytokine-mediated AEs, such as fever and CRS.26 By grouping skin toxicity composite terms, we observed that patients with skin toxicity within 7 days of treatment survived longer than those who did not develop rash in this time frame. This finding led to the inclusion of OS in patients developing a rash within 1 week of tebentafusp treatment as a coprimary end point in the phase III trial.25 Although the development of rash in the phase III trial did appear to be associated with longer survival, patients who developed rash had more favorable baseline characteristics, indicating that rash is not an independent predictor of outcome.25,27
Hepatotoxity was more commonly observed in this study compared with the FIH trial, consistent with nearly all patients having liver metastasis. Similar to other treatment-related AEs on study, most LFT elevations were mild and occurred early on-treatment, which may be attributed to T-cell redirection into liver lesions, whereas chronic LFT elevations were temporally associated with disease progression.
In this study, we observed clear pharmacodynamic responses to treatment, with rapid peripheral immune activation and induction of IFNγ pathway–related markers evident 8-24 hours after dosing. High levels of serum chemokines, CXCL9, CXCL10, CXCL11, and CCL2, and trafficking of lymphocytes from the blood are consistent with increased presence of immune cells in on-treatment biopsies, which was not correlated with the number of tumor-infiltrating lymphocytes at baseline. In contrast to the FIH study, on-treatment changes in serum biomarkers were not associated longer OS, possibly related to either the smaller cohort or different patient population in this study.
We have previously hypothesized that the changes induced within the tumor microenvironment by tebentafusp may increase the efficacy of immunologic checkpoint blockade. Consistent with this hypothesis, we observed that, of 24 patients treated with immune checkpoint inhibitors post-tebentafusp, one achieved a complete response and three achieved a PR, for an ORR of 16.7%.28 Notably, post-treatment biopsy specimens demonstrated increased expression of PD-L1 and programmed cell death protein-1. In this regard, combination of tebentafusp with anti–programmed cell death protein-1/PD-L1 antibody may potentially increase the efficacy of tebentafusp, and further investigation on this combination is warranted.
In conclusion, we have identified a novel treatment regimen of tebentafusp that permits the administration doses nearly 40% greater than what is possible with fixed weekly dosing and that has been used in the phase III trial of tebentafusp in treatment-naive, HLA-A*02:01-positive patients with mUM.25 The spectrum of on-target AEs and peripheral and intratumoral pharmacodynamic effects observed confirm our predicted mechanism of action.
ACKNOWLEDGMENT
The investigators are grateful to the patients, their family members, and the study teams at participating sites for their support of this trial. They acknowledge everybody who contributed to the study including data acquisition, analysis, interpretation, or writing, in particular Mohammed Dar, Koustubh Ranade, David Berman, Kate Lawrence, Revashnee Naidoo, Emma Leach, and Michelle L. McCully.
Richard D. Carvajal
Consulting or Advisory Role: Merck, Aura Biosciences, Castle Biosciences, Immunocore, PureTech, Sorrento Therapeutics, Chimeron Bio, Rgenix, InxMed, Pierre Fabre, TriSalus Life Sciences, Iovance Biotherapeutics, Oncosec, Regeneron, Genzyme, Amgen (Inst), Astellas Pharma (Inst), AstraZeneca (Inst), Bristol Myers Squibb/Medarex (Inst), Corvus Pharmaceuticals (Inst), Ideya (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Pfizer (Inst), Plexxikon (Inst), Roche/Genentech (Inst)
Speakers' Bureau: Bristol Myers Squibb/Medarex
Research Funding: Amgen (Inst), Astellas Pharma (Inst), AstraZeneca (Inst), Bayer (Inst), Bellicum Pharmaceuticals (Inst), Bristol Myers Squibb (Inst), Corvus Pharmaceuticals (Inst), Lilly (Inst), Immunocore (Inst), Incyte (Inst), Macrogenics (Inst), Merck (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Pfizer (Inst), Plexxikon (Inst), Roche/Genentech (Inst), Array BioPharma (Inst), IDEAYA Biosciences (Inst), Regeneron (Inst)
Paul Nathan
Consulting or Advisory Role: AstraZeneca, Bristol Myers Squibb, MSD, Immunocore, Pfizer, Pierre Fabre, Novartis, GlaxoSmithKline, Ipsen, 4SC, Merck
Speakers' Bureau: Bristol Myers Squibb, Novartis, MSD, Merck
Travel, Accommodations, Expenses: Bristol Myers Squibb, MSD
Joseph J. Sacco
Honoraria: Immunocore, Pierre Fabre, Novartis
Consulting or Advisory Role: Immunocore (Inst), Delcath Systems, MSD, BMS, Immunocore
Research Funding: AstraZeneca (Inst), Immunocore (Inst), Replimune (Inst), BMS (Inst)
Travel, Accommodations, Expenses: BMS, MSD
Marlana Orloff
Consulting or Advisory Role: Immunocore, TriSalus Life Sciences, IDEAYA Biosciences
Speakers' Bureau: Bristol Myers Squibb
Research Funding: Bristol Myers Squibb (Inst), Immunocore (Inst), Delcath Systems (Inst), Plexxikon (Inst), IDEAYA Biosciences (Inst), Linnaeus Therapeutics (Inst)
Leonel F. Hernandez-Aya
Consulting or Advisory Role: Massive Bio, Bristol Myers Squibb, Castle Biosciences
Speakers' Bureau: Sanofi/Regeneron
Research Funding: Bristol Myers Squibb (Inst), Regeneron (Inst), Immunocore (Inst), Merck (Inst), Polynoma (Inst), Corvus Pharmaceuticals (Inst), Roche/Genentech (Inst), Merck Serono (Inst), Amgen (Inst), MedImmune (Inst), Takeda (Inst), Moderna Therapeutics (Inst), Foghorn Therapeutics (Inst)
Travel, Accommodations, Expenses: Sanofi/Regeneron, Bristol Myers Squibb, Castle Biosciences
Jason J. Luke
Stock and Other Ownership Interests: Actym Therapeutics, Mavu Pharmaceutical, Pyxis, Alphamab, Tempest Therapeutics, Kanaph Therapeutics, Onc.AI, Arch Oncology
Consulting or Advisory Role: Bristol Myers Squibb, Merck, EMD Serono, Novartis, 7 Hills Pharma, Janssen, Reflexion Medical, Tempest Therapeutics, Alphamab, Spring Bank, AbbVie, Astellas Pharma, Bayer, Incyte, Mersana, Partner Therapeutics, Synlogic, Eisai, Werewolf Therapeutics, Ribon Therapeutics, Checkmate Pharmaceuticals, CStone Pharmaceuticals, Nektar, Regeneron, Rubius Therapeutics, Tesaro, Xilio Therapeutics, Xencor, Alnylam, Crown Bioscience, Flame Biosciences, Genentech, Kadmon, KSQ Therapeutics, Immunocore, Inzen Therapeutics, Pfizer, Silicon Therapeutics, TRex Bio
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Incyte (Inst), Corvus Pharmaceuticals (Inst), AbbVie (Inst), Macrogenics (Inst), Xencor (Inst), Array BioPharma, Agios, Astellas Pharma, EMD Serono, Immatics, Kadmon, Moderna Therapeutics, Nektar, Spring bank, Trishula Therapeutics, KAHR Medical, Fstar, Genmab, Ikena Oncology, Numab, Replimune, Rubius Therapeutics, Synlogic, Takeda, Tizona Therapeutics Inc, Trishula Therapeutics, BioNTech, Scholar Rock
Patents, Royalties, Other Intellectual Property: Serial #15/612,657 (Cancer Immunotherapy), Serial #PCT/US18/36052 (Microbiome Biomarkers for Anti–PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Array BioPharma, EMD Serono, Janssen, Merck, Novartis, Reflexion Medical, Mersana, Pyxis, Xilio Therapeutics
Marcus O. Butler
Honoraria: Roche, Merck, Bristol Myers Squibb, Novartis
Consulting or Advisory Role: Merck, Bristol Myers Squibb, Novartis, Immunovaccine, Immunocore, Adaptimmune, EMD Serono, GlaxoSmithKline, Genzyme, Sanofi, LaRoche Posay, Sun Pharma, Instil Bio, Iovance Biotherapeutics, Pfizer
Research Funding: Merck, Takara Bio
Expert Testimony: Merck
Sarah Stanhope
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Laura Collins
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Cheryl McAlpine
Employment: Immunocore, Adaptimmune
Chris Holland
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore, Amgen, MacroGenics
Patents, Royalties, Other Intellectual Property: Coinvolvement in a patent for the compositions and methods for treating diffuse large B-cell lymphoma
Shaad E. Abdullah
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Patents, Royalties, Other Intellectual Property: AU2019270277A1—Treatment of Cancer, WO2020225552A1—Combination of monalizumab, durvalumab, chemotherapy, and bevacizumab or cetuximab for the treatment of colorectal cancer
Takami Sato
Consulting or Advisory Role: Immunocore, Castle Biosciences
Research Funding: Immunocore (Inst), Verastem (Inst)
Travel, Accommodations, Expenses: Castle Biosciences
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the ASCO Annual Meeting, June 2-6, 2017, Chicago, IL.
SUPPORT
Funded by Immunocore Ltd.
CLINICAL TRIAL INFORMATION
AUTHOR CONTRIBUTIONS
Conception and design: Richard D. Carvajal, Joseph J. Sacco, Cheryl McAlpine, Chris Holland, Shaad E. Abdullah, Takami Sato
Provision of study materials or patients: Richard D. Carvajal, Paul Nathan, Joseph J. Sacco, Marlana Orloff, Leonel F. Hernandez-Aya, Jessica Yang, Jason J. Luke, Marcus O. Butler, Takami Sato
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I Study of Safety, Tolerability, and Efficacy of Tebentafusp Using a Step-Up Dosing Regimen and Expansion in Patients With Metastatic Uveal Melanoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Richard D. Carvajal
Consulting or Advisory Role: Merck, Aura Biosciences, Castle Biosciences, Immunocore, PureTech, Sorrento Therapeutics, Chimeron Bio, Rgenix, InxMed, Pierre Fabre, TriSalus Life Sciences, Iovance Biotherapeutics, Oncosec, Regeneron, Genzyme, Amgen (Inst), Astellas Pharma (Inst), AstraZeneca (Inst), Bristol Myers Squibb/Medarex (Inst), Corvus Pharmaceuticals (Inst), Ideya (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Pfizer (Inst), Plexxikon (Inst), Roche/Genentech (Inst)
Speakers' Bureau: Bristol Myers Squibb/Medarex
Research Funding: Amgen (Inst), Astellas Pharma (Inst), AstraZeneca (Inst), Bayer (Inst), Bellicum Pharmaceuticals (Inst), Bristol Myers Squibb (Inst), Corvus Pharmaceuticals (Inst), Lilly (Inst), Immunocore (Inst), Incyte (Inst), Macrogenics (Inst), Merck (Inst), Mirati Therapeutics (Inst), Novartis (Inst), Pfizer (Inst), Plexxikon (Inst), Roche/Genentech (Inst), Array BioPharma (Inst), IDEAYA Biosciences (Inst), Regeneron (Inst)
Paul Nathan
Consulting or Advisory Role: AstraZeneca, Bristol Myers Squibb, MSD, Immunocore, Pfizer, Pierre Fabre, Novartis, GlaxoSmithKline, Ipsen, 4SC, Merck
Speakers' Bureau: Bristol Myers Squibb, Novartis, MSD, Merck
Travel, Accommodations, Expenses: Bristol Myers Squibb, MSD
Joseph J. Sacco
Honoraria: Immunocore, Pierre Fabre, Novartis
Consulting or Advisory Role: Immunocore (Inst), Delcath Systems, MSD, BMS, Immunocore
Research Funding: AstraZeneca (Inst), Immunocore (Inst), Replimune (Inst), BMS (Inst)
Travel, Accommodations, Expenses: BMS, MSD
Marlana Orloff
Consulting or Advisory Role: Immunocore, TriSalus Life Sciences, IDEAYA Biosciences
Speakers' Bureau: Bristol Myers Squibb
Research Funding: Bristol Myers Squibb (Inst), Immunocore (Inst), Delcath Systems (Inst), Plexxikon (Inst), IDEAYA Biosciences (Inst), Linnaeus Therapeutics (Inst)
Leonel F. Hernandez-Aya
Consulting or Advisory Role: Massive Bio, Bristol Myers Squibb, Castle Biosciences
Speakers' Bureau: Sanofi/Regeneron
Research Funding: Bristol Myers Squibb (Inst), Regeneron (Inst), Immunocore (Inst), Merck (Inst), Polynoma (Inst), Corvus Pharmaceuticals (Inst), Roche/Genentech (Inst), Merck Serono (Inst), Amgen (Inst), MedImmune (Inst), Takeda (Inst), Moderna Therapeutics (Inst), Foghorn Therapeutics (Inst)
Travel, Accommodations, Expenses: Sanofi/Regeneron, Bristol Myers Squibb, Castle Biosciences
Jason J. Luke
Stock and Other Ownership Interests: Actym Therapeutics, Mavu Pharmaceutical, Pyxis, Alphamab, Tempest Therapeutics, Kanaph Therapeutics, Onc.AI, Arch Oncology
Consulting or Advisory Role: Bristol Myers Squibb, Merck, EMD Serono, Novartis, 7 Hills Pharma, Janssen, Reflexion Medical, Tempest Therapeutics, Alphamab, Spring Bank, AbbVie, Astellas Pharma, Bayer, Incyte, Mersana, Partner Therapeutics, Synlogic, Eisai, Werewolf Therapeutics, Ribon Therapeutics, Checkmate Pharmaceuticals, CStone Pharmaceuticals, Nektar, Regeneron, Rubius Therapeutics, Tesaro, Xilio Therapeutics, Xencor, Alnylam, Crown Bioscience, Flame Biosciences, Genentech, Kadmon, KSQ Therapeutics, Immunocore, Inzen Therapeutics, Pfizer, Silicon Therapeutics, TRex Bio
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Incyte (Inst), Corvus Pharmaceuticals (Inst), AbbVie (Inst), Macrogenics (Inst), Xencor (Inst), Array BioPharma, Agios, Astellas Pharma, EMD Serono, Immatics, Kadmon, Moderna Therapeutics, Nektar, Spring bank, Trishula Therapeutics, KAHR Medical, Fstar, Genmab, Ikena Oncology, Numab, Replimune, Rubius Therapeutics, Synlogic, Takeda, Tizona Therapeutics Inc, Trishula Therapeutics, BioNTech, Scholar Rock
Patents, Royalties, Other Intellectual Property: Serial #15/612,657 (Cancer Immunotherapy), Serial #PCT/US18/36052 (Microbiome Biomarkers for Anti–PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Array BioPharma, EMD Serono, Janssen, Merck, Novartis, Reflexion Medical, Mersana, Pyxis, Xilio Therapeutics
Marcus O. Butler
Honoraria: Roche, Merck, Bristol Myers Squibb, Novartis
Consulting or Advisory Role: Merck, Bristol Myers Squibb, Novartis, Immunovaccine, Immunocore, Adaptimmune, EMD Serono, GlaxoSmithKline, Genzyme, Sanofi, LaRoche Posay, Sun Pharma, Instil Bio, Iovance Biotherapeutics, Pfizer
Research Funding: Merck, Takara Bio
Expert Testimony: Merck
Sarah Stanhope
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Laura Collins
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Cheryl McAlpine
Employment: Immunocore, Adaptimmune
Chris Holland
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore, Amgen, MacroGenics
Patents, Royalties, Other Intellectual Property: Coinvolvement in a patent for the compositions and methods for treating diffuse large B-cell lymphoma
Shaad E. Abdullah
Employment: Immunocore
Stock and Other Ownership Interests: Immunocore
Patents, Royalties, Other Intellectual Property: AU2019270277A1—Treatment of Cancer, WO2020225552A1—Combination of monalizumab, durvalumab, chemotherapy, and bevacizumab or cetuximab for the treatment of colorectal cancer
Takami Sato
Consulting or Advisory Role: Immunocore, Castle Biosciences
Research Funding: Immunocore (Inst), Verastem (Inst)
Travel, Accommodations, Expenses: Castle Biosciences
No other potential conflicts of interest were reported.
REFERENCES
- 1.Milam RW, Daniels AB: Uveal melanoma, in Riker AI. (ed): Melanoma: A Modern Multidisciplinary Approach. Cham, Switzerland, Springer International Publishing, 2018, pp 273-312 [Google Scholar]
- 2.Stang A, Parkin DM, Ferlay J, et al. : International uveal melanoma incidence trends in view of a decreasing proportion of morphological verification. Int J Cancer 114:114-123, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Virgili G, Gatta G, Ciccolallo L, et al. : Incidence of uveal melanoma in Europe. Ophthalmology 114:2309-2315, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Carvajal RD, Schwartz GK, Tezel T, et al. : Metastatic disease from uveal melanoma: Treatment options and future prospects. Br J Ophthalmol 101:38-44, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kujala E, Makitie T, Kivela T: Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci 44:4651-4659, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Lorenzo D, Piulats JM, Ochoa M, et al. : Clinical predictors of survival in metastatic uveal melanoma. Jpn J Ophthalmol 63:197-209, 2019 [DOI] [PubMed] [Google Scholar]
- 7.Weis E, Salopek TG, McKinnon JG, et al. : Management of uveal melanoma: A consensus-based provincial clinical practice guideline. Curr Oncol 23:e57-e64, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rantala ES, Hernberg M, Kivela TT: Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res 29:561-568, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coupland SE, Lake SL, Zeschnigk M, et al. : Molecular pathology of uveal melanoma. Eye (Lond) 27:230-242, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buder K, Gesierich A, Gelbrich G, et al. : Systemic treatment of metastatic uveal melanoma: Review of literature and future perspectives. Cancer Med 2:674-686, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Kooij MK, Speetjens FM, van der Burg SH, et al. : Uveal versus cutaneous melanoma; same origin, very distinct tumor types. Cancers (Basel) 11:845, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelster MS, Gruschkus SK, Bassett R, et al. : Nivolumab and ipilimumab in metastatic uveal melanoma: Results from a single-arm phase II study. J Clin Oncol 39:599-607, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piulats JM, Espinosa E, de la Cruz Merino L, et al. : Nivolumab plus ipilimumab for treatment-naive metastatic uveal melanoma: An open-label, multicenter, phase II trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J Clin Oncol 39:586-598, 2021 [DOI] [PubMed] [Google Scholar]
- 14.Khoja L, Atenafu EG, Suciu S, et al. : Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An International Rare Cancers Initiative (IRCI) ocular melanoma study. Ann Oncol 30:1370-1380, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Bossi G, Buisson S, Oates J, et al. : ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol Immunother 63:437-448, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liddy N, Bossi G, Adams KJ, et al. : Monoclonal TCR-redirected tumor cell killing. Nat Med 18:980-987, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Middleton MR, McAlpine C, Woodcock VK, et al. : Tebentafusp, A TCR/anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res 26:5869-5878, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boudousquie C, Bossi G, Hurst JM, et al. : Polyfunctional response by ImmTAC (IMCgp100) redirected CD8(+) and CD4(+) T cells. Immunology 152:425-438, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carvajal RD, Sato T, Shoushtari AN, et al. : Safety, efficacy and biology of the gp100 TCR-based bispecific T cell redirector, IMCgp100 in advanced uveal melanoma in two phase 1 trials. Journal for ImmunoTherapy of Cancer 5, 2017 (abstr P208)
- 20.Middleton M, Corrie P, Sznol M, et al. : A phase I/IIa study of IMCgp100: Partial and complete durable responses with a novel first-in-class immunotherapy for advanced melanoma. Cancer Res 75, 2015. (abstr CT106) [Google Scholar]
- 21.Middleton MR, Steven NM, Evans TJ, et al. : Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific t cell redirector with solid tumour activity: Results from the FIH study in melanoma. J Clin Oncol 34, 2016. (abstr 3016) [Google Scholar]
- 22.Saber H, Del Valle P, Ricks TK, et al. : An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharmacol 90:144-152, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Oken MM, Creech RH, Tormey DC, et al. : Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649-655, 1982 [PubMed] [Google Scholar]
- 24.Lee DW, Santomasso BD, Locke FL, et al. : ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 25:625-638, 2019 [DOI] [PubMed] [Google Scholar]
- 25.Nathan P, Hassel JC, Rutkowski P, et al. : Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med 385:1196-1206, 2021 [DOI] [PubMed] [Google Scholar]
- 26.Shoushtari A, Middleton M, Stevens N, et al. : Cytokine release syndrome (CRS) following treatment with tebentafusp, a novel bispecific TCR-anti-CD3 directed against gp100, in patients with advanced melanoma. Journal for ImmunoTherapy of Cancer 7, 2019 (abstr 283) [Google Scholar]
- 27.Hassel JC, Rutkowski P, Baurain J-F, et al. : Co-primary endpoint of overall survival for tebentafusp (tebe)-induced rash in a phase 3 randomized trial comparing tebe versus investigator’s choice (IC) in first-line metastatic uveal melanoma. J Clin Oncol 39, 2021. (abstr 9527) [Google Scholar]
- 28.Yang J, Orloff MM, Sacco JJ, et al. : Resensitization of uveal melanoma (UM) to immune checkpoint inhibition (ICI) by IMCgp100 (IMC). J Clin Oncol 37, 2019. (abstr 9592) [Google Scholar]