

Abstract
zVAD-fmk is a widely used pan-caspase inhibitor that blocks apoptosis but has undesirable side-effects, including autophagy. In this issue, Needs et al. proposes that zVAD-fmk induces autophagy by inhibiting the N-glycanase NGLY1 rather than caspases. NGLY1 is essential for the ERAD response and patients with inactivating mutations in NGLY1 present with neurodevelopmental defects and organ dysfunction. The ability of NGLY1 to inhibit basal levels of autophagy may contribute to this pathology. This study demonstrates possible cross talk between protein turnover and autophagy while also underscoring the importance of specificity when using chemical tools to interrogate these pathways.
Keywords: NLGY1, zVAD-fmk, autophagy, apoptosis, caspase
Graphical Abstract
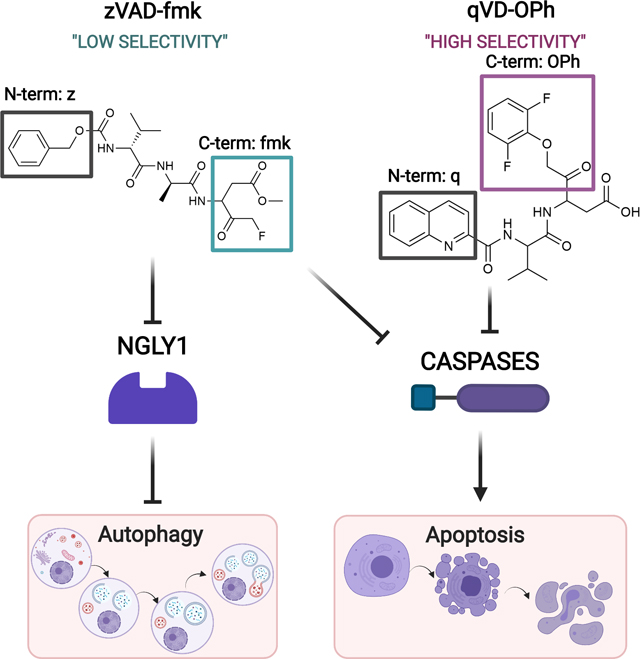
zVAD-fmk is a pan-caspase inhibitor that blocks apoptosis and also induces autophagy in some contexts. Needs et al. propose that zVAD-fmk induces autophagy by inhibiting the N-glycanase NGLY1. A different pan-caspase inhbitior, qVD-OPh, with a more highly selective warhead, does not inhibit NGLY1. This report underscores the importance of specificity when using chemical inhibitors to interrogate apoptosis and autophagy pathways.
Does caspase inhibition induce autophagy?
Apoptosis is a physiological cell death process that is mediated by the caspase family of aspartate-specific cysteine proteases. As essential initiators and executioners of apoptosis, caspase inhibition is widely used as a research tool to investigate the apoptotic cascade [1]. This has led to the development of an array of specific and broad-spectrum caspase inhibitors. These inhibitors comprise mono- to tetra-peptides that mimic the target cleavage sequence of the caspase substrate, which is broadly defined as a DXXD motif [2]. They contain an alkoxy group at the N-terminus to increase hydrophobicity and cell permeability, while the C-terminus is conjugated to either chloromethyl ketone (cmk), fluoromethyl ketone (fmk), or aldehyde (CHO) to allow for irreversible competitive inhibition. They form covalent bonds with the thiol group of the cysteine that comprises the active site of caspases. This prevents substrates, including the caspase itself, from binding to the active site inhibiting apoptosis.
The most commonly used pan-caspase inhibitor is zVAD-fmk, but it has poor stability and toxicity. It is able to inhibit other enzymes not involved in apoptosis that can impact lysosomal degradation and ER stress pathways, among others. In 2004, Lenardo and colleagues reported the observation that treatment of cells with zVAD-fmk led to the induction of cellular autophagy [3]. Autophagy is a catabolic system that delivers dysfunctional cytoplasmic components to lysosomes for demolition or recycling. Autophagy can be induced by a range of signals including endoplasmic reticulum (ER) stress, hypoxia, and nutrient starvation. It is a tightly regulated process that involves the sequential activation of a number of highly conserved autophagy-related genes (ATG) to execute autophagosome formation, maturation, and fusion with lysosomes [4]. Induction of autophagy by zVAD-fmk was initially proposed to be the result of inhibition of caspase-8-mediated cleavage of receptor-interacting protein kinase1 (RIPK1) [3]. However, it is now understood that inhibition of caspase-8 induces an alternate form of cell death known as RIPK1-dependent necrosis or necroptosis in response to TNF stimulation [5]. While there is evidence showing that blocking autophagy increases sensitivity to necroptosis [6], until now, it has remained unclear how caspase inhibition could induce autophagy. In this issue, Needs et al. provide evidence that zVAD-fmk induces autophagy, not by inhibition of a particular caspase, but by inhibiting NGLY1, a protein traditionally known for its role in the ER-associated degradation (ERAD) response [7].
zVAD-fmk: not just a caspase inhibitor
NGLY1 is a well-conserved cytosolic enzyme known to catalyze the removal of asparagine-linked glycans from glycoproteins. The primary function of NGLY1 is non-lysosomal clearance of misfolded proteins from the ER, thereby playing an essential role in protein surveillance [8]. Studies focusing on NGLY1 gained particular attention when Need et al. reported a patient harboring an inactivating NGLY1 mutation [9]. Individuals with NGLY1-deficiency display a broad spectrum of clinical presentations including inability to produce tears, neurodevelopmental disorders, and multi-organ dysfunction. Causative factors for these clinical features can only be partly attributed to the dysregulation of the NGLY1-ERAD substrates because reactivation of ERAD in NGLY1-deficient cells does not fully rescue these pathologies [10]. Thus, there are likely additional consequences of NGLY1 inactivation that contribute to the clinical complications in these patients.
It was known that zVAD-fmk could potently inhibit NGLY1 and this has been implicated in the disruption of activation of the transcription factor nuclear respiratory factor 1 (NRF1). NRF1 is the chief NGLY1 substrate that mediates proteasome recovery. Inhibition of NGLY1 inactivates NRF1 leading to dysregulated proteasome machinery promoting ER stress [11], impaired mitochondrial biogenesis, and inflammatory response [12]. Needs et al. demonstrate that inhibition of NGLY1 by zVAD-fmk also promotes autophagosome formation and upregulation of ATG proteins [7]. Like caspases, NLGY1 has an active site cysteine and it cleaves glycans after an aspartate [13]. This mechanism likely explains the ability of zVAD-fmk to target NLGY1. However, the same effect was not seen when a different pan-caspase inhibitor, qVD-OPh, was used. qVD-OPh is a more potent inhibitor of caspases that contains an N-terminal quinolyl group and a C-terminal phenoxy moiety. The smaller size of the peptide motif and the phenoxy moiety lead to enhanced cellular permeability, substrate access, and stability compared to zVAD-fmk. However, despite the similar positions of aspartate in the P1 position (immediately before the cleavage site) in zVAD-fmk and qVD-OPh, the latter had no activity against NLGY1. Similarly, an aldehyde-based inhibitor Ac-VAD-CHO had only 60% efficacy against NLGY1 compared to zVAD-fmk [13]. Thus, the position and presence of the fluoromethylketone warhead is essential for optimal NLGY1 inhibition and induction of autophagy. Based on this data, it appears likely that zVAD-fmk induces autophagy through inhibition of NLGY1 rather than through direct inhibition of a specific caspase [7].
NGLY1: a role beyond misfolded protein clearance?
It may seem counterintuitive that a protein that is essential for the degradation of misfolded proteins inhibits autophagy, a process that also plays a crucial role in protein clearance. A major difference between ER-activated autophagy and ERAD is the half-life of the degraded protein, where autophagy specifically clears long-lived proteins. These results may indicate crosstalk between ERAD and autophagy that is regulated by NGLY1. Inhibition of NGLY1 is sufficient to induce autophagy independent of traditional stress signals. This may suggest that the accumulation of N-glycosylated proteins in the absence of NGLY1 is sufficient to trigger autophagy. It is likely that this process is mediated by the regulation of transcription or stability of autophagy genes since positive regulators of autophagy like ATG3, ATG4B, and ATG7 were enriched upon inhibition of NGLY1 [7]. However, it is yet unclear if NGLY1 plays a direct role in the autophagy process.
Mice engineered to overexpress ATG5 have increased life spans [14]. In contrast, NGLY1 deletion in mice results in embryonic or perinatal lethality depending on the genetic background [10]. Therefore, induction of autophagy by NGLY1-deficiency does explain the associated lethality. Endo-β-N-acetylglucosaminidase (ENGASE) is a protein that is responsible for processing of N-glycans released by NGLY1. Further deletion of Engase in Ngly1−/− mice restores ERAD and rescues the embryonic lethality. However, the mice retain symptoms like weight loss and bent spines that are similar to those of NGLY1-deficient patients [3]. It remains to be determined if increased autophagy contributes to these symptoms. Interestingly, increased accumulation of N-glycoproteins was detected in Engase−/−/Ngly1−/− cells, which, as discussed, may serve as a trigger for autophagy [14]. Counter to this argument, an inducer of autophagy called Dactosilib has been shown to induce the degradation of NGLY1-dependent substrates in NGLY1-deficienct cells [15]. To further understand the role of autophagy in NGLY1-deficiency it will be important to investigate the impact of inhibition or induction of autophagy on the clinical complications of this disease. In turn, this may provide new avenues for drug discovery.
Targeting the autophagy pathway for therapeutic benefit has been a long held goal of the scientific community. In early clinical studies, when used in combination with other drugs, autophagy inhibition has shown some efficacy against cancer [16], while induction of autophagy may be beneficial in neurodegenerative diseases. The paper by Needs et al underscores the complex nature of interpreting the functional outcomes of inhibiting or inducing autophagy while also highlighting how important it is to consider the specificity of chemical inhibitors of caspases when interrogating apoptotic pathways.
Acknowledgements
The LBH lab is funded by NIH/NIGMS R01GM121389 (LBH), NIH/NCI R21CA256606 (LBH) NIH/NIDDK F32DK121479 (BB) and CPRIT #RP210027 (DS). The graphical abstract was generated using Biorender
Abbreviations
- ATG
autophagy-related gene
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ENGASE
endo-β-N-acetylglucosaminidase
- NGLY1
N-Glycanase 1
- NRF1
nuclear respiratory factor 1
- qVD-OPh
quinoline-Val-Aspdifluorophenoxymethylketone
- RIPK1
receptor-interacting protein kinase1
- TNF
tumor necrosis factor
- zVAD-fmk
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl] fluoromethylketone
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Poreba M, Strozyk A, Salvesen GS & Drag M. (2013) Caspase substrates and inhibitors, Cold Spring Harb Perspect Biol. 5, a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT & Nicholson DW (1997) A Combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B: FUNCTIONAL RELATIONSHIPS ESTABLISHED FOR KEY MEDIATORS OF APOPTOSIS *, Journal of Biological Chemistry. 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- 3.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH & Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8, Science. 304, 1500–2. [DOI] [PubMed] [Google Scholar]
- 4.He C. & Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy, Annual review of genetics. 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linkermann A. & Green DR (2014) Necroptosis, New England Journal of Medicine. 370, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kist M. & Vucic D. (2021) Cell death pathways: intricate connections and disease implications, EMBO J. 40, e106700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Needs SH BM, Grotzke JE, Kramer HB & Allman SA (2021) NGLY1 knockdown or pharmacological inhibition induces cellular autophagy. , FEBS J. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki T, Huang C. & Fujihira H. (2016) The cytoplasmic peptide:N-glycanase (NGLY1) - Structure, expression and cellular functions, Gene. 577, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH & Goldstein DB (2012) Clinical application of exome sequencing in undiagnosed genetic conditions, J Med Genet. 49, 353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujihira H, Masahara-Negishi Y, Tamura M, Huang C, Harada Y, Wakana S, Takakura D, Kawasaki N, Taniguchi N, Kondoh G, Yamashita T, Funakoshi Y. & Suzuki T. (2017) Lethality of mice bearing a knockout of the Ngly1-gene is partially rescued by the additional deletion of the Engase gene, PLoS genetics. 13, e1006696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomlin FM, Gerling-Driessen UIM, Liu YC, Flynn RA, Vangala JR, Lentz CS, Clauder-Muenster S, Jakob P, Mueller WF, Ordonez-Rueda D, Paulsen M, Matsui N, Foley D, Rafalko A, Suzuki T, Bogyo M, Steinmetz LM, Radhakrishnan SK & Bertozzi CR (2017) Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity, ACS Cent Sci. 3, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang K, Huang R, Fujihira H, Suzuki T. & Yan N. (2018) N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1, Journal of Experimental Medicine. 215, 2600–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misaghi S, Pacold ME, Blom D, Ploegh HL & Korbel GA (2004) Using a small molecule inhibitor of peptide: N-glycanase to probe its role in glycoprotein turnover, Chem Biol. 11, 1677–87. [DOI] [PubMed] [Google Scholar]
- 14.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, Jung S. & Jung YK (2013) Overexpression of Atg5 in mice activates autophagy and extends lifespan, Nature communications. 4, 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller WF, Jakob P, Sun H, Clauder-Munster S, Ghidelli-Disse S, Ordonez D, Boesche M, Bantscheff M, Collier P, Haase B, Benes V, Paulsen M, Sehr P, Lewis J, Drewes G. & Steinmetz LM (2020) Loss of N-Glycanase 1 Alters Transcriptional and Translational Regulation in K562 Cell Lines, G3 (Bethesda). 10, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy JMM, Towers CG & Thorburn A. (2017) Targeting autophagy in cancer, Nat Rev Cancer. 17, 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]