

Abstract
The success of anti-cancer therapies is often limited by heterogeneity within and between tumors. While much attention has been devoted to understanding the intrinsic molecular diversity of tumor cells, the surrounding tissue microenvironment is also highly complex and co-evolves with tumor cells to drive clinical outcomes. Here, we propose that diverse types of solid tumors share common physical motifs that change in time and space, serving as universal regulators of malignancy. We use breast cancer and glioblastoma as instructive examples and highlight how invasion in both diseases is driven by appropriation of structural guidance cues, contact-dependent heterotypic interactions with stromal cells, and elevated interstitial fluid pressure and flow. We discuss how engineering strategies show increasing value for measuring and modeling these physical properties for mechanistic studies. Moreover, engineered systems offer great promise for developing and testing novel therapies that improve patient prognosis by normalizing the physical tumor microenvironment.
Keywords: Breast cancer, glioblastoma, physical microenvironment, mechanobiology, tumor invasion, engineering strategies
1. INTRODUCTION
Cancer was once viewed as a tumor cell-autonomous disease in which the accumulation of successive genetic mutations drives uncontrolled cell proliferation and resistance to death (1). In time, scientists and clinicians gradually began to appreciate the contribution of surrounding non-cancerous tissue, known as the stroma, to disease progression (2). Stephen Paget first proposed this “seed and soil” hypothesis in 1889, positing that a given tissue environment acts as “soil” to either promote or restrict the growth of tumor “seeds.” However, this theory was not widely embraced for another century as cancer became increasingly framed as a systems-level disease. Initially, the “microenvironment” was viewed solely in terms of biochemical interactions between tumor and stromal cells, including secreted cytokines and growth factors that drive disease progression (3, 4). It has now become clear that physical remodeling of the stroma also powerfully contributes to tumor progression (5, 6). However, uncovering the exact mechanisms by which physical properties of the tumor microenvironment mediate disease outcomes requires further investigation.
Developing tumors induce numerous physical changes in the stroma, including 1) aberrant tissue microarchitecture, 2) altered material properties such as stiffness, 3) increased solid stress, and 4) elevated interstitial fluid pressure (IFP) (5). Increased tissue stiffness is the most clinically accessible of these changes, with lesions that are firm, irregular, and palpable classically drawing the most suspicion. Tissue stiffening results from increased total cell density and extracellular matrix (ECM) deposition by both tumor and stromal cells. As a developing tumor expands, it also deforms and displaces existing structures within the peritumoral space, exerting tensile and compressive forces that generate solid stress within the surrounding tissue (7). Notably, increased solid stress disrupts normal tissue microarchitecture and leads to the collapse of blood and lymphatic vessels within the tumor and nearby stroma (8). As a result, blood flow and lymphatic drainage are compromised, leading to fluid accumulation and increased IFP. This elevated IFP in turn increases interstitial fluid flow (IFF) from the tumor into surrounding non-cancerous tissues where pressure is lower, facilitating the transport of tumor-secreted factors in the process (9). To identify how these changes impact the development, progression, and prognosis of cancer, it will be critical to integrate experimental approaches across the biological and physical sciences.
In this review, we discuss physical changes to the tissue microenvironment that drive cancer progression and highlight how bioengineering strategies can be used to fundamentally improve our understanding of these phenomena. Although many of the concepts we cover are relevant to all solid tumors, we compare breast and brain cancer as instructive examples. In particular, we highlight how physical aspects of the breast and brain stroma modulate invasion with a focus on how tumor cells exploit structural guidance cues, contact-dependent heterotypic cell-cell contact, and elevated IFP in both diseases. We then outline how specific bioengineering strategies have contributed to our ability to measure and model these mechanical properties for mechanistic studies.
2. BREAST CANCER AND GLIOBLASTOMA AS AN INSTRUCTIVE COMPARISON
Mechanistic discovery in cancer research has almost exclusively focused on dissecting the molecular events that drive the development of specific cancer types and subtypes (1). These efforts have deeply enriched our understanding of tumorigenesis and facilitated the development of targeted therapies. However, efforts to generalize findings have been frustrated by the diverse molecular backgrounds of each tumor and patient, yielding variable and unpredictable responses to treatment (10). For example, breast cancer originates from epithelial tissue, has favorable patient survival for early-stage disease, but poor prognosis when tumors metastasize (11). Glioblastoma (GBM), on the other hand, originates from neuroglial tissue, universally exhibits poor patient survival, and almost never metastasizes (12, 13). In contrast to molecular events, physical changes are often shared between solid tumors regardless of the tissue of origin. Both breast cancer and GBM exploit structural guidance cues, contact-dependent heterotypic cell-cell interactions, and dysregulated IFP and IFF to mediate tumor invasion. In breast cancer, invasion precedes entry into the vasculature and formation of distant metastases. In GBM, invasion produces diffuse tumor margins that make surgical resection difficult and recurrence more probable. Breast cancer cells align collagen fibers in the stroma to regulate their invasion, while GBM cells co-opt existing vascular structures to infiltrate the brain parenchyma (14–16). Invasion is further aided by direct cell-cell contact of breast cancer cells with myofibroblasts and GBM cells with neurons via heterotypic cadherin-based or microtube-dependent junctions, respectively (17, 18). Similarly, breast cancer and GBM cells must both overcome physical barriers provided by the myoepithelium and astrocytic processes that impede invasion (19, 20). Collapse and dysfunction of tumor-associated vessels elevate IFP and IFF for both tumor types, leading to the formation of chemotactic gradients that drive cancer cell migration out of the tumor bulk (21, 22). Moreover, increased IFP and IFF also impede delivery of chemotherapies to the tumor core (23). Thus, understanding and targeting these shared physical phenomena that collectively modulate invasion could offer a novel approach to restrict disease progression and improve patient prognosis across otherwise distinct tumor types.
3. REVIEW OF MECHANOTRANSDUCTION
Both tumor and stromal cells respond to physical cues via mechanotransduction, the process by which cells convert mechanical inputs into biochemical signals (24) (Figure 1). One of the most studied mechanisms by which cells sense physical cues is through the formation of focal adhesions and consequential remodeling of the cellular cytoskeleton (25). Focal adhesions are micron-scale, multi-protein complexes that include clusters of transmembrane integrin heterodimers (26). Outside the cell, integrins bind to ECM proteins such as collagen, fibronectin, and laminin. Inside the cell, scaffolding proteins such as talin, tensin, and vinculin connect integrins to the cytoskeleton. While the actin cytoskeleton is often emphasized in discussions of FA biology, FAs also coordinate transmission of mechanical force to microtubules and intermediate filaments (27–29). External mechanical forces transmitted to focal adhesions change the conformation of specific intracellular proteins (e.g., talin), thus altering binding kinetics and recruiting downstream signaling molecules such as focal adhesion kinase (FAK), paxillin, and Src that elicit biochemical responses. Similarly, adherens junctions are cell-cell adhesions formed between transmembrane cadherins that bind to the actin cytoskeleton via catenin proteins (30). In addition, forces may also be transmitted between cells through intermediate filament-based complexes, such as desmosomes (31, 32). Applied forces at cell-matrix or cell-cell adhesions can propagate through the cytoskeleton and act directly on the nucleus via linker of nucleoskeleton and cytoskeleton (LINC) complexes (33). The resulting deformation of the nucleus is thought to alter chromatin packing and transport of biomolecules via nuclear pores to either promote or inhibit transcription of mechanoresponsive genes (34, 35).
Figure 1.

Mechanisms of cell mechanotransduction. Both tumor and stromal cells use a variety of mechanisms to sense mechanical signals within their microenvironments. These include force transmission at cell-matrix and cell-cell adhesions, propagation of force through the cytoskeleton to the nucleus, activation of ion channels in response to membrane tension, transduction of fluid shear stress by surface-bound glycocalyx macromolecules, and conformational changes of caveolae in the plasma membrane.
In addition to cytoskeleton-dependent mechanisms, stretch-activated ion channels help mediate the cellular response to mechanical stimuli (36). These transmembrane channels, such as Piezo1, undergo a conformational change and open upon the application of membrane tension, transporting ions from the extracellular space to the cytosol (37). In turn, altered intracellular ion concentrations and membrane potentials elicit biochemical responses. Moreover, a layer of membrane-bound proteoglycans, known as the glycocalyx, deforms in response to fluid shear stress and is known to transduce these forces in endothelial and tumor cells (38, 39). Interestingly, extension or compression of the plasma membrane in and of itself can modulate cell signaling by unfolding or refolding small membrane invaginations known as caveolae (40). These conformational changes release otherwise sequestered biomolecules or enable intracellular docking of curvature-sensing proteins that initiate downstream signaling cascades (40–42). Moreover, compression is associated with cell volume reduction that, in turn, increases the effective concentration of intracellular molecules (43). This phenomenon, termed macromolecular crowding, slows diffusion within the cytosol by reducing unoccupied void space, altering biochemical reaction kinetics as a result (44).
4.1. THE PHYSICAL MICROENVIRONMENT OF BREAST CANCER
Overview of breast cancer progression
While breast cancer arises from genetic transformation of mammary epithelial cells (11), the properties of the surrounding stroma influence whether benign disease becomes malignant and invasive. Indeed, irradiated fibroblasts promote tumor formation from otherwise non-malignant epithelial cells, while implantation of malignant cells into normal embryos prevents tumor development (45, 46). The mammary duct, where over 80% of breast cancers originate, consists of an inner layer of polarized luminal epithelial cells and an outer layer of contractile myoepithelial cells. These epithelial layers are encased by a dense, collagen IV- and laminin-rich basement membrane (47). In early-stage disease, pre-malignant epithelial cells become highly proliferative and fill the central lumen of the mammary duct but stay confined within the basement membrane. As tumors progress, the basement membrane degrades, enabling transformed cells to migrate into the surrounding stroma (Figure 2). Subsequently, biochemical and physical properties of the stroma further promote or restrict tumor invasion and entry into nearby blood and lymphatic vessels. These processes are key rate-liming steps of metastasis, which accounts for up to 90% of breast cancer-related deaths (48).
Figure 2.

Schematic of invasive breast cancer. Breast cancer cells invade through the mammary gland basement membrane and along aligned collagen fibers. Both tumor cells and activated myofibroblasts contribute to fiber alignment and remodeling. Elevated interstitial fluid pressure due to dysfunctional vasculature drives interstitial fluid flow from the tumor into the nearby stroma, creating chemotactic gradients of soluble factors as a result.
Structural guidance cues in breast cancer
The breast stroma refers to connective tissue that surrounds mammary ducts and becomes altered in the context of cancer. It typically consists of adipocytes, fibroblasts, immune cells, as well as blood and lymphatic vessels embedded in a network of ECM molecules (47). In the 1980s, clinicians identified a population of α-smooth muscle actin (αSMA)-positive myofibroblasts that was contained in malignant but not healthy breast tissue (49). These cells, also termed cancer-associated fibroblasts (CAFs), develop from resident or recruited fibroblasts or mesenchymal progenitor cells when exposed to tumor-secreted transforming growth factor-β1 (TGFβ1) (50–52). CAFs deposit and remodel ECM components including collagen I, fibronectin, and hyaluronic acid (HA) that drive changes in tissue architecture and stiffness (53). Epidemiologically, dense breast tissue caused by increased deposition and structural remodeling of fibrotic ECM is one of the most significant risk factors for breast cancer and used to detect tumors clinically (54, 55). One hallmark of breast cancer-associated ECM remodeling is that collagen fibers adopt distinct patterns of orientation (56) (Figure 2). Densely aligned collagen fibers oriented perpendicular to the tumor border not only correlate with increased invasion, but also poor patient prognosis regardless of tumor grade, size, receptor status, or subtype (57). Thus, the physical organization of peritumoral collagen fibers can serve as a prognostic biomarker to help predict clinical outcomes for breast cancer patients.
Several mechanisms contribute to the correlation between collagen fiber alignment, tumor cell invasion, and patient prognosis. For example, local fiber alignment and crosslinking increase tissue stiffness, which drives tumor progression and malignancy via enhanced cytoskeletal tension and integrin signaling (58, 59). Similar changes increase the activation of resident fibroblasts into CAFs in response to soluble TGFβ1, further promoting tumor progression (60, 61). Independent of stiffness, aligned collagen fibers can promote tumor cell invasion by directing cellular protrusions to increase migration persistence in the direction of alignment (14). Importantly, ECM stiffness and fiber orientation are reciprocally linked with cell phenotype via a positive feedback loop (62, 63). As cells pull on and align nearby collagen fibers, collagen strain-stiffens, which induces a reciprocal tensile strain on the tumor cells, activating calcium ion channels and Rho signaling pathways to further enhance cell contractility and migration. This interplay between the material properties of collagen and cellular mechanosignaling increases not only tumor cell invasion but also activation of fibroblasts into CAFs (62, 64). Fiber alignment also impacts cellular confinement, which can independently drive tumor cell invasion (65–68). For example, cells confined in three-dimensional (3D) collagen microtracks form larger, vinculin-rich adhesions that correspond with higher cell contractility than cells in unconfined conditions. Thus, as tumor and stromal cells become more contractile while aligning dense peritumoral collagen, fiber reorganization may simultaneously confine cell polarity and increase tumor cell migration.
In addition to modulating migration speed, ECM architecture influences the mechanisms by which tumor cells invade. Invasion depends on a combination of mesenchymal migration, where cells degrade the surrounding ECM to create spaces to invade, and amoeboid migration, where cells squeeze through pre-existing gaps in their microenvironments (69). Studies with viscoelastic hydrogels indicate that breast cancer cells can also irreversibly widen ECM pores (70). Given that these studies also found tumor-associated tissue to be more plastic than normal tissue, this mechanism may allow breast cancer cells to create permanent microtracks that facilitate invasion by subsequent cells. Moreover, altered cell-cell adhesions and matrix-dependent confinement jointly regulate whether breast cancer cells invade individually or as collective strands (68). For tumor cells without cell-cell adhesions, dense ECM patterns confine migration paths to collective strands of moving particles, much like the molecules of an active fluid. In less confined regions of matrix, however, cells decondense or individualize into a pattern of gas-like single-cell dissemination. Collectively, this suggests that tumor-associated ECM can act as a deformable scaffold for leader cells to mediate migration of follower cells and confine invasion to collective strands even for tumor cells who have lost the strong cell-cell adhesions characteristic of epithelia.
Direct heterotypic cell-cell contact in breast cancer
In addition to structural guidance cues provided by the ECM, direct interactions between breast cancer cells and non-cancerous stromal cells influence tumor invasion. For example, normal fibroblasts have been shown to restrict tumor progression, while CAFs mediate the opposite effect (71). In collagen-based coculture studies, CAFs promote collective invasion of carcinoma cells in a manner that requires direct cell-cell contact (72). Indeed, CAFs form heterotypic cadherin-based adhesions with carcinoma cells, generate intercellular forces, and physically pull on tumor cells to guide and facilitate collective invasion (17, 73) (Figure 2). Forces generated by CAFs can further promote tumor cell invasion by mechanically deforming the basement membrane to enable cancer cell migration independent of proteolysis (73). While these studies underscore the role of physical interactions between CAFs and tumor cells, similar mechanisms can promote migration of otherwise non-invasive cells. Indeed, fibroblast-like adipose stromal cells isolated from obese breast tissue express CAF markers and are more contractile than their lean counterparts, enabling more effective invasion of premalignant mammary epithelial cells (74, 75). These mechanisms likely contribute to the clinical observation that obesity is associated with a worse clinical prognosis for breast cancer patients.
Similar to CAFs, heterotypic interactions with myoepithelial cells regulate invasion, but often in the opposite direction. For example, direct contact between breast cancer cells and normal myoepithelial cells promotes their self-assembly into non-invasive, growth-arrested acini (76). This anti-tumorigenic effect depends on the formation of heterotypic cell-cell adhesions between both cell types that direct cell sorting and morphogenesis. Additionally, normal myoepithelial cells can act as a dynamic barrier to the invasion of tumor cells (19). Myoepithelial cells localize to the surface of mammary duct organoids, restrain protrusions of migrating cancer cells, and even recapture cells that escape from the organoid bulk. However, given sufficient exposure to tumor-secreted signals, myoepithelial cells lose their anti-invasive properties and ability to direct assembly of transformed epithelial cells into growth-arrested acini (77). Indeed, disruption of normal myoepithelial adhesion or contractility results in a patchier surface layer on mammary duct organoids with frequent gaps that are unable to restrain invasive cancer cells (19). These findings are clinically relevant as upregulation of αvβ6 integrins in myoepithelial cells—used as a surrogate marker for a cancer-associated phenotype—strongly correlates with tumor progression and reduced median time to recurrence in a cohort of breast cancer patients (78).
Elevated interstitial fluid pressure and flow in breast cancer
Elevated IFP in human breast tumors compared to adjacent non-cancerous tissue was first documented in 1994 for women undergoing biopsy (79). Subsequent studies in mice indicated that rapidly expanding tumors generate sufficient solid stress to collapse nearby blood and lymphatic vessels (80). Coupled with vessel dysfunction and increased deposition of hygroscopic HA, fluid readily accumulates around developing tumors. This creates a net pressure gradient that drives interstitial fluid into the surrounding stroma (23, 81). Historically, the resulting IFF was thought to promote disease progression primarily by impeding delivery of cytotoxic therapies to the tumor core. However, more recent studies found that IFF can independently induce tumor malignancy by generating gradients of autocrine factors including CCL19 and CCL21 that stimulate tumor cell migration in the direction of flow via activation of CCR7 (21) (Figure 2). Somewhat surprisingly, these effects are most pronounced at low tumor cell densities, with increasing cell densities hypothesized to flatten local chemotactic gradients due to simultaneous secretion of the same cues by neighboring cells (82).
Interestingly, subsequent experiments demonstrated that IFF can also induce upstream tumor cell migration (i.e., against the direction of fluid flow) (82). This phenomenon, termed rheotaxis, depended on asymmetric tension generated on cellular adhesions by fluid drag forces. The application of higher tension on the upstream edges of cells resulted in local activation of FAK, polarized recruitment of FA complexes, formation of actin-rich membrane protrusions, and finally directed migration against fluid flow (83). The rheotactic response of breast cancer cells may also be controlled by upregulating the mesenchymal markers vimentin and Snail without losing expression of the epithelial marker E-cadherin, as might be expected for complete epithelial-mesenchymal transition (84). These studies indicate that IFF can direct downstream chemotactic migration of cancer cells away from the tumor during early-stage disease when cell densities are low. Subsequently, upstream rheotactic migration of cancer cells against transmural fluid flow radiating from intact capillaries could mediate homing to the nearby stromal vasculature. In addition to influencing tumor cell migration directly, elevated IFF can modulate stromal cells in the peritumoral stroma to create a pro-invasive environment. Fluid flow levels similar to those generated by tumor-associated IFP, for example, promote fibroblast-to-myofibroblast conversion and collagen alignment in a manner that depends on autocrine TGFβ1 secretion and integrin α1β1 signaling (85). These changes are functionally relevant as protease- and contractility-dependent remodeling and alignment of collagen by fibroblasts in response to fluid flow enhanced tumor cell migration via similar mechanisms (86). Hence, fluid flux out of the tumor bulk and into the adjacent stroma synergistically promotes invasion by influencing tumor cells directly and by reinforcing the structural guidance cues described above. Moreover, elevated IFF also induces the formation of new lymphatic vessels and increases the secretion of chemotactic ligands by existing lymphatic vessels—providing additional stimuli to drive cancer cell migration toward the stromal vasculature and thus metastatic spread via the circulation (21, 87).
4.2. THE PHYSICAL MICROENVIRONMENT OF GLIOBLASTOMA
Overview of glioblastoma progression
GBM develops from the genetic transformation of neural stem cells and glial progenitor cells including astrocytes and oligodendrocytes (88, 89). While GBM tends to be quite heterogeneous within and between patients, tumors are often classified in terms of mutation (e.g., isocitrate dehydrogenase, IDH), amplification (e.g., epidermal growth factor receptor, EGFR and platelet derived growth factor receptor alpha, PDGFRA), or promoter methylation (e.g., MGMT) of specific genes (90, 91). Next-generation sequencing has also produced a broad classification scheme enabling stratification of GBM into transcriptional subtypes (classical, mesenchymal, proneural), although single-cell sequencing has painted a more nuanced picture where bulk tumors are composed of distinct cell populations that represent different subtypes (90, 92). Functionally, GBM exhibits highly malignant growth and rapid invasion, where bulk tumor expansion precedes rapid cellular infiltration into neighboring healthy brain tissue. In early-stage disease, tumor expansion caused by highly proliferative tumor cells leads to compression of intratumoral blood vessels, which contributes to the development of a hypoxic, necrotic tumor core (Figure 3). Hypoxic signaling then induces pseudopallisading infiltration in which tumor cells slowly invade from the tumor bulk into the brain parenchyma and subsequently shift to a faster invasion scheme along white matter tracts. Finally, the tumor cells eventually reach the brain perivascular space and rapidly invade along blood vessels via vascular co-option (93).
Figure 3.
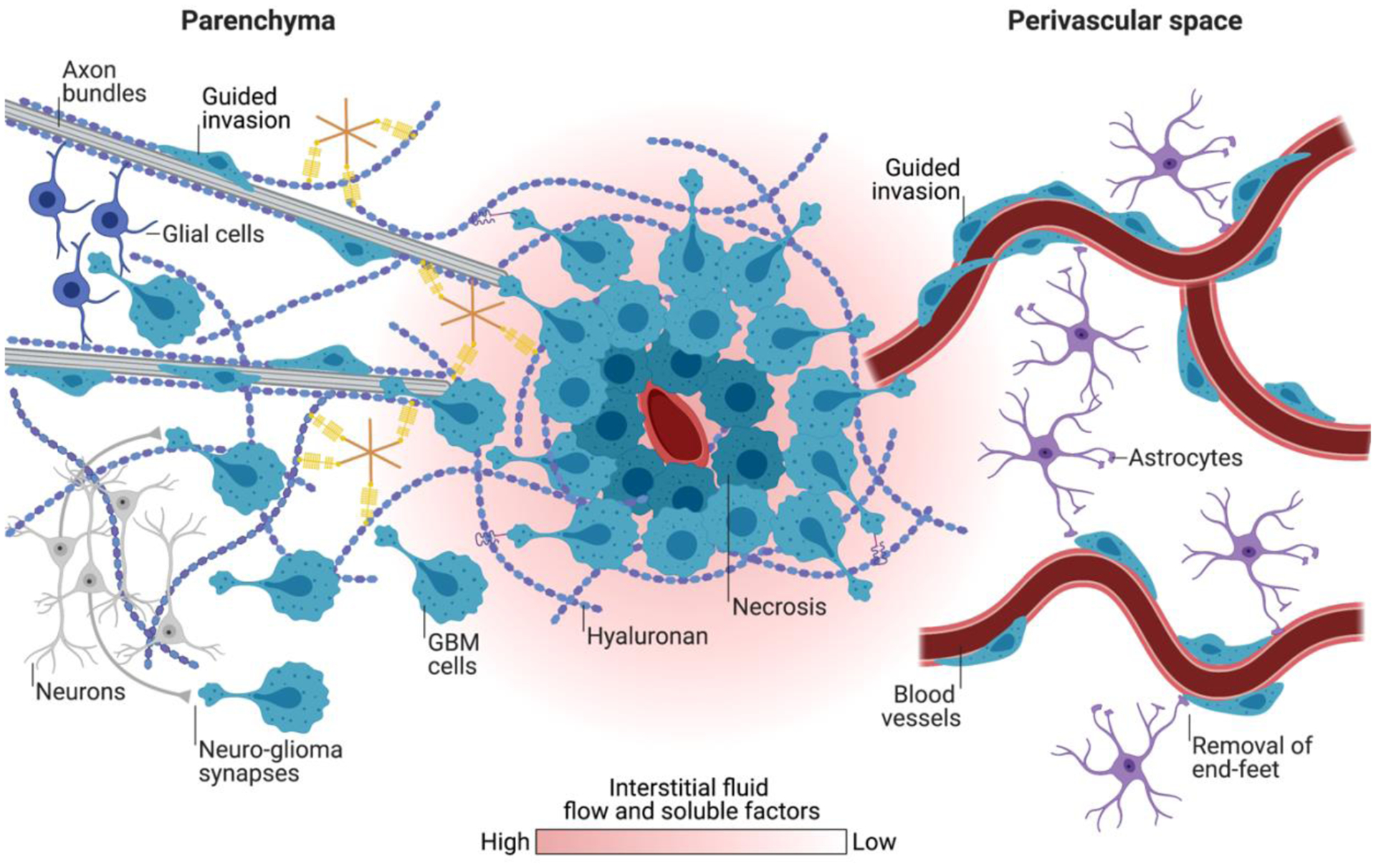
Schematic of invasive GBM. GBM cells invade the parenchyma and perivascular space of the brain after escaping the tumor core. The brain parenchyma contains a nanoporous matrix including hyaluronic acid, as well as numerous cellular protrusions which physically hinder and confine invasion. Alternatively, the linear structures of white matter tracts in the parenchyma and blood vessels in the perivascular space encourage rapid invasion of GBM cells along these guidance cues.
Structural guidance cues in glioblastoma
While breast cancer invasion is largely driven by structural guidance cues provided by the ECM, GBM invasion is guided by the unique architecture and structural features of the brain parenchyma and vasculature. Many in vivo studies using GBM mouse models report that tumor cells associated with blood vessels and white matter tracts achieve higher net displacements than cells not associated with these structures, contributing to the idea that these cues promote infiltration (20, 94–96). How vascular structures and white matter tracts facilitate invasion may be understood in terms of the contrast between these structures and the surrounding parenchyma. The grey matter of the brain parenchyma consists of an isotropic polysaccharide- and glycoprotein-rich ECM (e.g., HA, tenascin-c, aggrecan, osteopontin) and densely packed cells (e.g., neuronal cell bodies, glia), which collectively pose significant steric barriers to invasion (15, 16) (Figure 3). By contrast, white matter tracts (i.e., axons) are surrounded by a linearized ECM and space that more easily accommodates tumor cell infiltration (97). Similarly, blood vessels are embedded within the subarachnoid space, which lowers physical resistance for GBM invasion and provides a linear geometry that preferentially directs cell migration (15).
As GBM cells invade brain tissue, they alternate between a variety of motility strategies or “modes” depending on the specific constraints of the local microenvironment. While these modes differ in their speed and molecular mechanisms, all involve adhesion and transmission of force between the ECM and cytoskeleton through specialized molecular machinery. The HA adhesion receptor CD44 is particularly important for GBM cells to navigate through the brain parenchyma and can serve as a predictor for disease outcomes (98, 99). GBM cells use CD44 to engage and transduce mechanical signals from HA, which in turn facilitate migration through ECM pores (100). For example, CD44-dependent “microtentacles,” analogous to those sometimes seen in circulating tumor cells, mechanically couple the actin and microtubule cytoskeleton to HA through a molecular motor-clutch system that includes the actin-binding protein IQGAP1 and the microtubule-binding protein CLIP170 (101). Elsewhere, it has been reported that GBM cells extend pseudopodia-like protrusions to navigate through the brain parenchyma. These protrusions depend on the Rho GTPases Rac1 and Cdc42, which direct actin polymerization, and Lck-I, which supports FA generation through regulation of paxillin phosphorylation (94, 102).
Within the perivascular space, GBM cells adopt a unipolar morphology with prominent anterior processes to linearly invade along blood vessels in a saltatory fashion (95). As a result, vascular architecture influences GBM invasion dynamics. For example, during invasion, cell division often takes place near vascular branch points and GBM cells that interact with multiple vessels are more invasive (95, 96). Interestingly, GBM cell migration is also driven by engagement with less common ECM components, including collagen IV and laminin on the abluminal surface of vessels (103). Interactions with vascular ECM often involve integrins, which are typically upregulated by invasive GBM cells. Yet, integrin suppression only partially inhibited vascular migration in preclinical studies and yielded disappointing results in clinical trials (103, 104). The residual invasion following integrin suppression could be due to physical guidance cues provided by vessels or cell adaptation to other adhesive systems such as formins, which have been implicated in GBM migration on laminin-coated tracks (103, 105). Additionally, vascular engagement by tumor cells leads to local vascular deformation through an actomyosin driven motor-clutch invasion mechanism to propagate tumor growth (106). However, it is not clear whether these invasion-driven deformations also contribute to vascular dysfunction. In summary, GBM cells adopt unique morphologies driven by ECM adhesion proteins and cytoskeletal components to invade along vessels and within the brain parenchyma.
Direct heterotypic cell-cell contact in glioblastoma
Direct cell-cell contact between GBM cells and brain stromal cells including astrocytes, endothelial cells, and neurons also supports GBM growth and invasion. While the vasculature is a permissible route for invasion overall, astrocytic processes that physically maintain the integrity of the blood brain barrier hinder tumor cell migration. To overcome this obstacle, perivascular GBM cells physically displace astrocytes from the vasculature (Figure 3), which concurrently contributes to vessel dysfunction and fluid accumulation (20). Remarkably, a single perivascular GBM cell is sufficient to disrupt the vasculature by displacing astrocytic end-feet (20). Moreover, removal of astrocytic end-feet interferes with vascular tone regulation (i.e., vasodilation or vasoconstriction), resulting in vessels that are unable to respond to vasoactive molecules released by astrocytes. GBM cells can thus usurp this vasomodulatory role by mobilizing calcium-activated potassium ion channels to constrict vessels, increase perivascular space, and enable greater perivascular invasion (20). However, some instances of GBM-induced vascular modulation have also been reported to induce vessel dilation (96).
Furthermore, vessel architecture and barrier function are disrupted due to direct contact between GBM and endothelial cells. Notably, GBM vascular co-option is associated with loss of endothelial cell tight junction proteins including zonula occludens-1 and claudins, which otherwise maintain vascular integrity (20, 107). Additionally, direct contact with endothelial cells supports growth and self-renewal of GBM stem-like cells. For example, endothelial cells that express Notch ligands are often found adjacent to Nestin- and Notch- receptor-positive cancer stem-like cells (108). Knocking down the Notch ligands JAG or DLL in endothelial cells decreases the presence of CD133-positive GBM stem-like cells adjacent to blood vessels in vivo. This corroborates previous findings that the microvasculature supports the maintenance of a stem-like state for GBM (109).
Physical and biochemical interactions with neurons have also been recently reported to promote GBM progression. Indeed, direct GBM contact with the subventricular zone of the neural stem cell niche is associated with decreased patient survival (110). Two recently published studies reported that neuron-to-glioma synaptogenesis, mediated by the transmembrane glutamate receptor AMPAR, regulates tumor growth and invasion. Here, electrochemical cell communication was mediated by AMPAR-dependent neuro-glioma synapses where GBM cells were often found on the postsynaptic side of these junctions (111). Elsewhere, neuro-glioma synapses have been observed to connect with microtubes on GBM cells. Thus, targeting neuro-glioma synapses by genetic and pharmacological perturbation of AMPAR-mediated calcium activation attenuated disease progression in GBM mouse models (18).
Elevated interstitial fluid pressure and flow in glioblastoma
Similar to most solid tumors, elevated IFP contributes to invasion in GBM (112). Vascular abnormalities including loss of barrier function in GBM is one of the primary causes of elevated IFP, along with an impaired fluid drainage system. The compromised GBM-associated vasculature permits fluid accumulation and immune cell infiltration in the interstitial space surrounding the tumor. Subsequent immune cell secretion of pro-invasive growth factors such as TGFβ1, epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF), together with elevated IFP and IFF, amplify invasion (113–115). Similar to the observations in breast cancer described previously, the combination of IFF and cytokine secretion can create chemotactic gradients used by GBM cells to invade healthy brain tissue (116).
Despite its role in regulating invasion, the biological mechanisms through which IFF drives GBM invasion remain underexplored. CXCR4-CXCL12- and CD44-based mechanisms are known means by which GBM cells migrate in response to flow. Specifically, the presence of IFF enhances GBM cell motility by activating CXCR4, which responds to autocrine CXCL12 gradients (116). However, this response is not universal, as specific subpopulations of GBM cells respond to flow by CD44-dependent mechanisms instead (113). These results were reproduced in a mouse model of GBM where IFF induced by convection-enhanced delivery increased tumor cell invasion and the population of CXCR4-positive cells, whereas administration of a CXCR4 antagonist abrogated these effects (117). Additionally, irradiation led to an increase in CXCR4-positive cells, which could then respond to IFF-induced gradients of CXCL12 at the tumor border to potentiate invasion (117). As a result, how candidate therapies influence IFP and IFF must be considered moving forward given their implications for tumor invasion.
Elevated IFP in GBM also leads to cerebral edema and frustrates successful delivery of therapeutic agents to the tumor core. Efforts to improve drug delivery by decreasing IFP include normalizing the tumor vasculature with anti-angiogenic agents that inhibit VEGF signaling (118). For example, administration of anti-VEGF monoclonal antibodies reduced IFP and inhibited tumor growth in a xenograft model of GBM (119). Similarly, in recurrent GBM, anti-VEGF therapies reduced edema and resulted in improved survival when delivered in combination with other chemotherapies in a phase II clinical trial (120, 121). Thus, normalizing the peritumoral vasculature increases uptake of cytotoxic drugs while reducing cerebral edema. However, anti-angiogenic therapies can also produce undesirable compensatory effects, such as elevated secretion of other pro-angiogenic factors, including basic fibroblast growth factor (bFGF) and stromal cell-derived factor 1 alpha (SDF1α), expansion of treatment-resistant GBM progenitor cells, and even increased tumor cell invasion (120, 122). Thus, more research is needed to understand how treatment-induced vascular modulation translates into clinical outcomes.
5.1. ENGINEERING STRATEGIES TO MEASURE PHYSICAL ASPECTS OF THE TUMOR MICROENVIRONMENT
Established methods from the physical sciences have been adapted to quantify and improve our understanding of the physical tumor microenvironment. For example, atomic force microscopy (AFM), first developed to characterize nanoscale materials in the 1980s, has been applied to measure the viscoelastic properties of cancer cells, stromal cells, and ECM components (123). Likewise, the bulk mechanical properties of tumors have been assessed via extensional and shear rheology (70, 124). Beyond pre-existing technologies, new tools to probe cell and tissue mechanics have been engineered with specific biological questions in mind. For an overview of these strategies, see Table 1.
Table 1.
Example systems used to measure physical traits of the tumor microenvironment.
| Category | Measurement systems | Physical traits | References |
|---|---|---|---|
| Cell mechanics | Micropipette aspiration, real-time deformability cytometry, nanoindentation, optical tweezers | Generalized cell material properties | (124–126, 128–134) |
| Subcellular mechanics | Atomic force microscopy, directed micropipette aspiration | Organelle cell material properties | (123, 127–129) |
| Substrate mechanics | Atomic force microscopy, (micro)-rheology, nanoindentation, optical tweezers | ECM material properties | (14, 51, 63, 64, 70, 73, 74, 124) |
| Cell adhesion forces | Spinning disc assays, single-cell force spectroscopy, dual micropipette aspiration, magnetic tweezers | Cell-substrate adhesion forces, cell-cell adhesion forces | (17, 144–151) |
| Cell traction forces | Traction force microscopy, microfabricated post-array-detectors, optical tweezers | Cell-generated traction forces, strain stiffening | (17, 51, 63, 136–141, 143) |
| Flows, pressures, and stresses | Pressure transducer systems, contrast-enhanced imaging, deformable microdroplets, molecular tension sensors | Fluid flow, fluid pressure, and solid stress | (8, 9, 152–157, 159) |
Measuring the material properties of cells
The material properties of cells can be measured by assessing cell deformation in response to forces applied with an external object (e.g., a cantilever or piston) or by exposing cells to fluid shear stress and pressure. Micropipette aspiration, where cells in suspension are partially aspirated into narrow glass pipettes, is perhaps the most established of these methods (125). After aspiration, the extent of cell distension into the pipette is measured with a microscope and used to calculate the viscoelastic properties given the applied suction pressure. In practice, micropipette aspiration is sensitive enough to distinguish low-grade from high-grade tumor cells due to their differential mechanics acquired over malignant transformation (126). These methods are complementary, as micropipette aspiration integrates whole cell mechanics while AFM provides higher resolution measurements that can detect differences in subcellular mechanics (127). Recent advances in micropipette aspiration have focused on improving the speed at which cells can be sampled and spatially directing aspiration to measure the mechanical properties of specific organelles such as the nucleus (128, 129). Similar in principle, real-time deformability cytometry (RT-DC) assesses cell mechanics by monitoring the shapes of individual cells flowing through pressurized channels in response to shear stress (130, 131). RT-DC is higher throughput (100s to 1000s of cells per minute) than micropipette aspiration and simultaneously measures relative cell size and granularity like conventional flow cytometry. In practice, RT-DC has been applied to distinguish malignant from normal blood cells in samples collected from patients with leukemia based on relative differences in cell deformability (132).
The development of advanced imaging systems has enabled optics-based measurements of cell mechanical properties. For example, “optical tweezers” and related technologies mechanically manipulate particles and cells by taking advantage of small forces generated by light refracting and changing momentum as it passes through an optical interface, such as between extracellular fluid and the cytoplasm (133). By measuring the deformation of a given cell along the applied axis of light, the relative elasticity can be inferred. Studies using these approaches found that tumor cell deformability correlated with invasive potential and that malignant cells altered their cellular mechanics in response to substrate mechanics while non-malignant cells did not (134, 135). Similar principles have been applied to measure ECM mechanical properties via optics-based microrheology (124). By optically pulling on spherical beads embedded in a substrate of interest, microscale viscoelastic properties can be calculated. For collagen substrates composed of fine, uniform fiber networks, this approach yielded comparable results to conventional shear rheology. However, for substrates with large pores and thick fibers, microscale elasticities were often much larger than bulk elasticities given that individual fiber mechanics dominate at the microscale. Independent studies using a similar setup measured the degree to which collagen fibers stiffen when elongated by contractile tumor cells (63). When both beads and tumor cells were embedded in collagen gels, bead-conjugated fibers located near cells resisted optical deformation more than those far away from cells or those located near non-contractile control cells. These results indicate that tumor-cell-generated traction forces led to local ECM stiffening in the immediate vicinity of a given cell.
Measuring cell-matrix traction forces
Traction force microscopy (TFM) is widely used to estimate the forces that adherent cells generate against their substrates. In this method, cells are cultured on top of substrates that contain fluorescent tracer particles (136). Cell-generated forces produce net displacement of these tracers relative to their relaxed reference positions. If the mechanical properties of the substrate material are known, the stress distribution can then be inferred from the net bead displacements by solving an inverse problem. TFM was first established to examine the traction stresses generated by migrating fibroblasts on two-dimensional (2D) collagen-coated polyacrylamide gels and has since been applied to interrogate diverse cell-substrate interactions in applications ranging from tissue morphogenesis to tumor invasion (136, 137). In a similar manner, culturing cells on elastomeric micropost arrays enables direct measurement of 2D cellular traction stresses by measuring the deformation of the underlying posts (138, 139). Enabled by the improved axial resolution of modern confocal microscopes, the more recent development of 2.5D and 3D TFM permits traction forces to be measured in all three spatial dimensions by cells seeded on or within 3D substrates, respectively (140, 141). Newer methods such as optical coherence microscopy (OCM)-based TFM enable label- and tracer-free measurements of cell-generated traction forces by directly imaging collagen fiber deformation (142). Given that OCM uses near-infrared light, this method is also well suited for time-lapse analysis of biological samples (e.g., those with large volumes embedded in scattering media). In addition, the use of astigmatic imaging, which infers 3D spatial information within a few micrometers of a given focal plane, has enabled 2.5D and 3D TFM to be performed without collecting multi-image stacks (143). Thus, adopting advanced imaging modalities has greatly improved both the spatial and temporal resolutions of traditional TFM methods.
Measuring cell adhesion forces
Early methods for measuring cell adhesion forces involved culturing cells on spinning discs and tracking cell detachment due to the applied rotational forces (144). While this approach proved extraordinarily valuable for semi-quantitative population-level measurements, it does not yield absolute single-cell measurements of adhesion. To this end, AFM-based single-cell force spectroscopy has emerged to measure adhesion forces in the sub-nN range (145–147). Here, a modified AFM cantilever is functionalized and coupled to a cell of interest. The conjugated cell is then lowered onto an opposing cell or substrate until adhesion occurs, the cantilever is retracted, and the force required for separation is recorded (148). Similarly, in dual pipette assays, cells are held in place by micropipette-generated suction and brought into contact to facilitate adhesion (149, 150). The cell doublet is then pulled in opposing directions and the suction pressure is increased in a stepwise fashion until the cells detach from each other instead of from the pipettes. In addition, magnetic tweezers have been used to detach micron-scale magnetic beads conjugated with E-cadherin from epithelial cells to examine the role of cell-cell adhesion strength in tissue dynamics (151). In the context of tumor invasion, researchers extended this platform to demonstrate that CAFs bind and pull cancer cells to enhance their migration via the formation of heterotypic cadherin-based adhesions (17).
Measuring fluid pressure and solid stress
While many strategies have been developed to measure microscale mechanical properties and cell-generated traction and adhesion forces, relatively few methods exist to measure tissue-scale changes in IFP and solid stress associated with tumor development (5). Elevated IFP within tumors relative to normal tissue has been quantified by direct insertion of wick-in-needle, fiberoptic, or piezoelectric pressure transducer systems (152–154). However, these techniques are invasive, and there is some debate about the extent to which their readings reflect pressure contributed by free interstitial fluid, bound interstitial fluid, or artifacts of solid stress. More recently, fluid pressure has been non-invasively inferred by imaging IFF (driven by relative differences in pressure) via contrast-enhanced imaging modalities (155). Monitoring solid stress within tumors has proven even more challenging. Injection of deformable fluorescent microdroplets into tissues and ectopic expression of molecular tension sensors have enabled local stresses to be calculated from microdroplet deformation and changes in sensor fluorescence, respectively (156, 157). However, these methods measure forces at sub-cellular or cellular scales and are subjected to the optical limitations of confocal microscopy. To measure tissue-level solid stress, researchers have developed protocols to embed resected tumors in agarose and monitor tissue displacement following tumor slicing as a metric of stress dissipation (158, 159). As this technique is destructive, the evolution of tumor solid stress over the course of disease progression cannot be monitored longitudinally. Moving forward, improved strategies to non-invasively measure tissue-level mechanics in real time will help reveal new mechanisms of tumor pathophysiology and identify physical targets for clinical intervention.
5.2. ENGINEERING STRATEGIES TO MODEL PHYSICAL ASPECTS OF THE TUMOR MICROENVIRONMENT
While cell and tissue engineering strategies were initially developed with an eye toward regenerative medicine, these approaches are similarly valuable for cancer research (160, 161). Indeed, tumor engineering publications increased 24-fold from 2000 to 2020 and have isolated specific physical characteristics of the tumor microenvironment for mechanistic studies (162, 163). Key design considerations for these systems include the ability to independently tune mechanical properties, define ECM composition and architecture, integrate multiple cell types, and visualize outcomes in real-time with live-cell imaging techniques. For an overview of these strategies, see Table 2.
Table 2.
Example systems used to model physical traits of the tumor microenvironment.
| Model system | Physical traits | References |
|---|---|---|
| Advanced biomaterials | Material properties, cleavable crosslinks, ligand presentation, microarchitecture | (64, 70, 160, 165–168, 172–186, 203) |
| Protein micropatterns | Geometric constraints, heterotypic cell-cell contacts | (191–196) |
| Microfabricated devices | Structural guidance cues, material properties, confinement | (14, 67, 84, 198, 199, 202, 204, 206, 207) |
| Tumor-on-chips, microfluidics | Heterotypic cell-cell contacts, interstitial fluid flow, pharmacodynamics | (85, 128, 198, 205, 208–210, 212) |
| Pressure-driven flow systems | Interstitial fluid flow, heterotypic cell-cell contacts | (21, 22, 39, 82–84, 86, 113, 212) |
Advanced biomaterials to mimic ECM mechanics and microarchitecture
Natural biomaterials, such as solubilized collagen and reconstituted basement membrane, were among the first substrates to enable 3D culture of cells. Studies with these hydrogels enabled key insights into how ECM-derived cues regulate cell behaviors ranging from morphogenesis to malignant transformation (164, 165). However, the physical and biochemical properties of natural biomaterials cannot be easily decoupled, making it difficult to interrogate how physical motifs in the microenvironment regulate cell phenotype (166). For example, altering the stiffness of collagen matrices by modulating collagen concentration simultaneously changes the density of adhesion ligands, the pore size between collagen fibers, as well as fiber length and thickness, all of which can independently affect cell behavior (64, 167). Moreover, tissue-derived matrices, including those derived from epithelial basement membranes, often include growth factors and cytokines and suffer from batch-to-batch variability (168, 169). Synthetic or semi-synthetic biomaterials can address these limitations by independently tuning of parameters such as ligand density, microarchitecture, and stiffness.
Synthetic biomaterials are often functionalized with bioactive components to recapitulate specific properties of the ECM (170, 171). For example, biocompatible, photopolymerizable polyethylene glycol (PEG)-based hydrogels have been modified with integrin-binding adhesion peptides and enzymatically cleavable crosslinks to enable cell-ECM interactions and proteolytic remodeling of hydrogels following cell encapsulation (172, 173). The stiffness of such PEG-based systems depends on the degree of photoinducible crosslinking and can be patterned to explore how substrate mechanical gradients impact tumor cell migration. For example, tumor cells that started in regions of low stiffness migrated rapidly and reversed directions upon encountering regions of higher stiffness, while cells that started in regions of high stiffness migrated more slowly but readily invaded into regions of lower stiffness (174). These results indicate that ECM stiffnesses above a certain threshold restrict cell migration. However, it should be noted that the response to stiffness varies as a function of ECM remodeling, as cell migration depended on cleaving crosslinks in stiff but not soft PEG-based substrates (175). PEG-based systems have also increased our understanding of how adhesion ligand density affects encapsulated cell behavior (176). While increasing the density of RGD adhesion ligands at a constant stiffness prompted metastatic carcinoma cells to self-assemble into structures that resembled healthy acini (177), increasing adhesion ligand density in the presence of TGFβ1 stimulated epithelial-mesenchymal transition. This discrepancy reveals the complex and often unexpected interplay between physical and biochemical cues on cell behavior. Importantly, the material properties of synthetic PEG-based platforms can also be adjusted in vivo, enabling direct comparison with in vitro findings. For example, application of transdermal light to subcutaneous PEG-maleimide scaffolds freed previously unavailable RGD adhesion sites from photolabile cages, prompting inflammation and vascularization of the implanted material (178, 179).
Given that tumors and tumor-associated stroma often stiffen over the course of disease progression, dynamic biomaterials that can alter their physical properties on demand represent promising platforms to research the consequences of evolving tumor mechanics (58). Methacrylated HA hydrogels coated with collagen can be dynamically stiffened from 100 to 3000 Pa in response to stepwise doses of ultraviolet irradiation. Pre-malignant epithelial cells cultured on soft substrates that were later stiffened to pathological levels resisted adopting malignant phenotypes when compared to cells directly seeded onto pre-stiffened substrates, indicating that cells respond to both the timing and degree of matrix stiffening (180). Using methacrylated HA to adjust substrate stiffness is particularly attractive for studies of GBM invasion since HA is an essential component of the native brain ECM (181, 182). For example, GBM cells plated on stiff 5 kPa HA spread, adhered, and migrated more than those on soft 150 Pa HA. Interestingly, CD44-HA adhesions alone could facilitate migration of GBM cells (183). Introducing RGD ligands to enable integrin-based adhesions only increased the rate of invasion when CD44 was inhibited. These findings indicate that CD44-based mechanisms result in greater GBM invasion speeds while integrin-based mechanisms facilitate a slower, alternative mode of migration. Finally, HA-based platforms can also be leveraged to examine other aspects of the tumor microenvironment such as the induction of angiogenesis. Studies with RGD-functionalized tunable HA hydrogels, for example, suggested that substrate stiffness and ECM degradation are both essential for robust endothelial cell sprouting and vascular tube formation (184, 185).
In addition to photopolymerization, dynamic substrate stiffening has been accomplished with interpenetrating networks (IPNs) of alginate and Matrigel that contain liposomes filled with soluble calcium (186). Near infrared irradiation released calcium from these liposomes, creating ionic crosslinks within the alginate and increasing the elastic moduli of the hydrogels from 150 to 1200 Pa. This on-demand stiffening promoted encapsulated breast cancer cells to downregulate epithelial markers, adopt more protrusive morphologies, and form into large, disorganized clusters. IPNs of alginate and Matrigel also expanded our understanding of how viscoelastic substrate properties influence tumor cell invasion (70). IPNs with high plasticity (i.e., non-reversible deformation in response to cell-generated forces) induced tumor cells to extend invadopodia-like protrusions and create permanent pores in the surrounding alginate-Matrigel network that were large enough for cells to squeeze through in a proteolysis-independent manner. Consequently, these materials revealed a new mechanism of tumor cell migration under confined conditions, an insight that would not have been possible with conventional approaches.
While the described synthetic or semi-synthetic biomaterials offer many advantages over natural biomaterials, most of them polymerize into nanoporous structures that lack the fibrous microarchitecture characteristic of native ECM (187). These ECM microarchitectural cues (i.e., interfibrillar pore size, fiber alignment, length, and thickness) influence malignancy independently of bulk material properties (64, 167). Thus, there is a need to develop platforms that combine the tractable nature of synthetic biomaterials while also recapitulating the fibrillar architecture of native ECM. One approach is to form fibers by electrospinning synthetic biomaterials into fine, nanoscale filaments (188). For example, reducing the stiffness of spun methacrylated dextran (DexMA) fibers encouraged spreading, proliferation, and FA formation by encapsulated cells. The authors determined that cells exerted traction forces to recruit fibers and increase the local concentration of RGD peptides to elicit these effects, a phenomenon that was hindered when fibers became too stiff to easily remodel. Increasing the bulk stiffness of similar scaffolds inhibited myofibroblast differentiation while increasing the concentration of spun fibers promoted fibroblast-to-myofibroblast conversion upon exposure to TGFβ1 (189). Notably, both studies contradict previous findings on traditional substrates, where increased stiffness promotes cell spreading, proliferation, and FA assembly, as well as myofibroblast differentiation (58, 61). These discrepancies indicate that changes to bulk substrate mechanics do not necessarily influence cell behavior in the same manner as similar changes to microscale fiber mechanics. Moreover, matrices composed of spun DexMA fibers have also been leveraged to discover new mechanisms of tumor cell migration via live imaging studies (190). Breast cancer cells intermittently stopped migrating in these matrices to pull fibers in front of them toward their cell bodies, storing elastic energy in the process. Eventually, adhesions at the trailing edge of a given cell failed and the tumor cell ricocheted forward as the recruited fibers returned to their starting positions. Despite the stationary period, this mode of tumor cell migration was ultimately faster and resulted in greater net displacement than continuous mesenchymal migration.
Microfabricated models of the tumor microenvironment
How tumor cells respond to the geometric and mechanical constraints of their microenvironments has been extensively studied with protein micropatterns (191, 192). Typically, protein micropatterns are printed onto glass or polyacrylamide (PA) substrates using photolithography approaches that employ either an elastomeric polydimethylsiloxane (PDMS)-based or deep UV irradiation-based technique (191, 193). The former uses a PDMS template with defined geometric features to print proteins onto a given substrate, which determines the features, location, and resolution of the resulting micropattern (192). The latter approach relies on deep UV exposure through a photomask that permits targeted irradiation of a given substrate coated with a photosensitive protein repellent, thus enabling protein adsorption to irradiated areas (191). Both techniques are able to achieve a patterning resolution on the order of 1–10 μm (189, 190). In one example, fibronectin micropatterns were used to demonstrate that imposed interfacial geometry can guide tumor cells toward a stem-like state via altered integrin α5β1 signaling (195). Recent advances in micropatterning technologies have also enabled real-time patterning to examine how cells adapt to dynamic presentation of adhesion sites within their microenvironments. In this study, real-time patterning was achieved using pulsed lasers that oxidized a PEG-based substrate to create new adhesion sites via subsequent protein adsorption onto oxidized regions (196).
While micropatterning has been used to present proteins in controlled 2D spatial configurations, other microfabrication methods have been implemented to model 3D aspects of the ECM microarchitecture. For example, injecting collagen into narrow microfabricated PDMS channels aligns the resulting fibers in the direction of the channel (197, 198). As the channel width increases, the fibers lose their aligned orientation and default to a randomly organized architecture. Studies with this platform demonstrated that collagen fiber orientation enhances tumor cell migration efficiency, directional persistence, and distance traveled (197). One alternative method includes casting collagen into PDMS molds that are stretched during polymerization to orient the resulting fibers, an approach that affected the migratory and morphogenic behavior of pre-seeded tumor and stromal cells (199). To more accurately mimic the 3D environment of developing tumors, a coaxial rotating cylinder platform was used to encase breast cancer spheroids with both perpendicularly and tangentially aligned collagen fibers (200). This method leveraged the nucleation and elongation phases of collagen polymerization to produce a system where the spheroids contacted two common fiber orientations found in breast cancer. In particular, fiber alignment in this system was driven by horizontal laminar Couette flow in the nucleation stage of polymerization, while gravitational forces guided fiber growth during elongation. In agreement with previous observations, breast cancer spheroids preferentially invaded along perpendicularly rather than tangentially aligned collagen fibers.
Numerous engineered model systems have leveraged microfabricated channels with defined dimensions to examine confined tumor cell migration in vitro (201). Notably, PDMS can be functionalized with cell adhesion proteins and is optically transparent, permitting real-time imaging that enables mechanistic studies of confined migration dynamics. Studies using these platforms found that tumor cells polarize their cytoskeleton and physically soften to achieve faster migration speeds when confined (202, 203). Furthermore, these microchannels can be modified to introduce additional physical constraints that tumor cells may encounter during invasion. These include physical gradients, where cells migrate from a wider into a narrower channel, or bifurcations, where cells are presented with a channel that splits into two channels of different widths (201, 204). In addition, microfabricated platforms have been used to examine nuclear deformation during confined tumor cell migration (205). Here, nuclear deformation compromised nuclear envelope integrity, leading to envelope rupture and subsequent DNA damage (206). Interestingly, follow-up studies found that mechanical deformation of the nucleus also damaged DNA without nuclear envelope rupture (207). These findings suggest that mechanically induced DNA damage contributes to the genomic instability commonly associated with metastatic tumor cells.
Many engineered systems take a reductionist approach to modeling cancer, restricted to studying tumor cells in isolation. While this approach enables the fundamental behavior of tumor cells to be studied without confounding factors, its application to studying tumor-stroma interactions is limited. Microfluidic-based tumor-on-chip systems address this challenge by introducing other stromal compartments such as blood vessels, immune cells, and fibroblasts. These platforms enable direct heterotypic cell-cell interactions to be studied by integrating multicellular compartments on a single platform. In addition to stromal cells, other aspects of the tumor microenvironment such as pressure gradients, specific ECM components, and defined mechanical properties can also be incorporated (208). Using these methods, an intravasation model consisting of tumor cells, endothelial cells, and macrophages demonstrated that impaired endothelial barrier function facilitates rapid tumor cell intravasation via direct heterotypic cell-cell interactions (209). Here, macrophage secretion of tumor necrosis factor alpha (TNF-α) disrupted endothelial cell tight junctions, which subsequently enhanced tumor cell transmigration by increasing tumor-endothelial cell interactions. Moreover, such tumor-on-chip systems are a promising method to systematically evaluate the efficacy and toxicity of candidate therapies (208). Interestingly, by integrating tumor cells and cardiac muscle cells on a single culture platform, researchers were able to more accurately predict both direct and off-target effects of tyrosine kinase inhibitors (210).
PDMS-based systems have also been used to model IFP and IFF in vitro. For example, IFP was simulated in a PDMS-based system by varying the volume of media between two reservoirs connected by a channel seeded with tumor cells. This setup established a pressure differential of 1.2 mmHg and generated fluid flow with a velocity of 1 μms−1. The authors found that IFF alters expression of mesenchymal genes which influences the invasion phenotype of tumor cells (211). Alternatively, transwell-based assays, where tumor cells are seeded on top of a porous membrane, have been used to study IFF-mediated invasion (22). In this setup, tumor cells were encapsulated in a hydrogel on top of the transwell membrane and a pressure head was introduced to establish fluid flow through the gel. More recently, a modified version of this system was established to stimulate a monolayer of lymphatic endothelial cells (LECs) with both transmural (perpendicular) and luminal (shear) flow (212). In this system, LECs simultaneously experienced transmural flow via an applied pressure head and luminal flow induced by a peristaltic pump. Moreover, this system enabled real-time monitoring of tumor cell transmigration across the LEC monolayer as a model of intravasation. Ultimately, the authors found that transmural and luminal flow synergistically upregulated LEC expression of CCL21 to promote breast cancer cell transmigration (212).
6. CLINICAL TRANSLATION AND FUTURE PERSPECTIVES
While the impact of the physical microenvironment on tumor progression is now well established, targeting these phenomena in the clinic remains challenging. For example, cilengitide, a peptide inhibitor of integrins αvβ3 and αvβ5, initially demonstrated anti-invasive effects in pre-clinical tumor models and phase I/II clinical trials (213, 214), but failed to improve patient survival in a phase III clinical trial (215). Perhaps the most successful example of targeting physical tissue properties thus far is the use of anti-angiogenic agents, which were shown to normalize tumor vasculature, reduce IFP, and improve drug penetration in pre-clinical studies (23, 216). Bevacizumab, a humanized anti-VEGF monoclonal IgG1 antibody, was the first anti-angiogenic agent approved for clinical use by the Food and Drug Administration (FDA) in 2004 (217). While still recommended for numerous cancer types including GBM, the FDA revoked bevacizumab’s approval for advanced breast cancer in 2011 (218). This decision resulted from a failure to improve survival while exposing recipients to adverse hemodynamic effects (219–221). Indeed, the efficacy of bevacizumab is highly variable and it is unclear whether patients who respond to treatment benefit from vascular normalization or secondary, angiogenesis-independent effects such as immunomodulation (222, 223). Ultimately, while bevacizumab is often considered the most successful therapy that targets the tumor microenvironment to date, it has not become a universal treatment for solid tumors as researchers once hoped.
Anti-fibrotic agents that target the ECM comprise another class of emerging therapies directed toward the physical tumor microenvironment (224). These therapies tend to either 1) enzymatically degrade specific ECM components, 2) normalize or deplete ECM remodeling cells such as CAFs, or 3) interfere with the ability of cells to sense aberrant ECM by inhibiting mechanosignaling pathways (225). Despite success in pre-clinical studies, therapies that directly modulate the ECM such as PEGPH20, a modified hyaluronidase, or simtuzumab, an inhibitor of the collagen crosslinker LOXL2, have largely failed clinical trials due to a lack of therapeutic benefit and high incidence of adverse events (226–228). Strikingly, therapies that deplete CAFs worsened outcomes in pre-clinical studies (229). These negative outcomes underscore the complexity of tumor-stroma interactions, which can either promote or restrict disease progression. Therapies that interfere with pro-fibrotic or mechanosignaling pathways offer an alternative to target tumor-associated ECM. Indeed, multiple inhibitors for FAK, Rho-associated kinase (ROCK), and TGFβ are currently under investigation (230, 231). For example, the use of galunisertib, a small molecule inhibitor of pro-fibrotic TGFβ1, improved overall survival for patients with unresectable pancreatic cancer and advanced liver cancer in phase I/II clinical trials (232, 233). Collectively, these findings reveal the clinical potential of targeting the physical tumor microenvironment, but also emphasize our incomplete understanding of these phenomena which impedes successful translation of candidate therapies from bench to bedside.
Bioengineers are well positioned to address outstanding questions in cancer mechanobiology given their multi-disciplinary training and ability to “translate” between basic scientists, technicians, oncologists, and other healthcare professionals. Moving forward, interrogating physical parameters with more representative models of cancer, such as patient-derived organoids, will be critical to further our understanding of how the tumor microenvironment interfaces with the molecular heterogeneity of human disease (234, 235). In particular, combining patient-derived tissues with humanized mouse models or organ-on-chip systems will enable better screening of candidate therapies in the pre-clinical setting and facilitate clinical translation to improve patient prognosis (236–238). Moreover, integrating spatial transcriptomics with high resolution mapping of tissue mechanics in these studies will reveal how molecular and physical heterogeneity synergize to coordinate disease behavior. As we move toward adaptive strategies that aim to control tumor growth and prevent genetic selection of therapy resistance (239, 240), we must also consider how the physical microenvironment evolves in response to treatment to collectively regulate clinical outcomes.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge support from the National Institutes of Health (R01CA227136, R01CA260443, and R01GM122375 to SK and U54CA210184 to CF), the National Science Foundation (NNCI-2025233 to CF and a Graduate Research Fellowship to GFB), and the Howard Hughes Medical Institute (Gilliam Fellowship for Advanced Studies to KA). Figures created with BioRender.com.
References
- 1.Vogelstein B, Kinzler KW. 1993. The multistep nature of cancer. Trends in Genetics. 9(4):138–41 [DOI] [PubMed] [Google Scholar]
- 2.Fidler IJ, Poste G. 2008. The “seed and soil” hypothesis revisited. The Lancet Oncology. 9(8):808. [DOI] [PubMed] [Google Scholar]
- 3.Kenny PA, Lee GY, Bissell MJ. 2007. Targeting the tumor microenvironment. Front Biosci. 12:3468–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joyce JA. 2005. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 7(6):513–20 [DOI] [PubMed] [Google Scholar]
- 5.Nia HT, Munn LL, Jain RK. 2020. Physical traits of cancer. Science. 370(6516): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butcher DT, Alliston T, Weaver VM. 2009. A tense situation: forcing tumour progression. Nat Rev Cancer. 9(2):108–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seano G, Nia HT, Emblem KE, Datta M, Ren J, et al. 2019. Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nature Biomedical Engineering 2019 3:3. 3(3):230–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. 2013. Coevolution of Solid Stress and Interstitial Fluid Pressure in Tumors During Progression: Implications for Vascular Collapse. Cancer Res. 73(13):3833–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, et al. 2012. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. PNAS. 109(38):15101–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagogo-Jack I, Shaw AT. 2018. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 15(2):81–94 [DOI] [PubMed] [Google Scholar]
- 11.Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, et al. 2019. Breast cancer. Nature Reviews Disease Primers. 5(1):1–31 [DOI] [PubMed] [Google Scholar]
- 12.Weller M, Wick W, Aldape K, Brada M, Berger M, et al. 2015. Glioma. Nat Rev Dis Primers. 1(1):1–18 [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Yuan F, Qi Y, Liu B, Chen Q. 2021. Circulating Tumor Cells for Glioma. Front Oncol. 11:607150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, et al. 2014. 3D Collagen Alignment Limits Protrusions to Enhance Breast Cancer Cell Persistence. Biophysical Journal. 107(11):2546–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuddapah VA, Robel S, Watkins S, Sontheimer H. 2014. A neurocentric perspective on glioma invasion. Nature Reviews Neuroscience. 15(7):455–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolf KJ, Lee S, Kumar S. 2018. A 3D topographical model of parenchymal infiltration and perivascular invasion in glioblastoma. APL Bioengineering. 2(3):031903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, et al. 2017. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nature Cell Biology. 19(3):224–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, et al. 2019. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 573(7775):532–38 [DOI] [PubMed] [Google Scholar]
- 19.Sirka OK, Shamir ER, Ewald AJ. 2018. Myoepithelial cells are a dynamic barrier to epithelial dissemination. Journal of Cell Biology. 217(10):3368–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H. 2014. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nature Communications. 5(1):1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. 2007. Autologous Chemotaxis as a Mechanism of Tumor Cell Homing to Lymphatics via Interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell. 11(6):526–38 [DOI] [PubMed] [Google Scholar]
- 22.Munson JM, Bellamkonda RV, Swartz MA. 2013. Interstitial Flow in a 3D Microenvironment Increases Glioma Invasion by a CXCR4-Dependent Mechanism. Cancer Res. 73(5):1536–46 [DOI] [PubMed] [Google Scholar]
- 23.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. 2004. Vascular Normalization by Vascular Endothelial Growth Factor Receptor 2 Blockade Induces a Pressure Gradient Across the Vasculature and Improves Drug Penetration in Tumors. Cancer Res. 64(11):3731–36 [DOI] [PubMed] [Google Scholar]
- 24.Hoffman BD, Crocker JC. 2009. Cell Mechanics: Dissecting the Physical Responses of Cells to Force. Annu. Rev. Biomed. Eng 11(1):259–88 [DOI] [PubMed] [Google Scholar]
- 25.DuFort CC, Paszek MJ, Weaver VM. 2011. Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol. 12(5):308–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Kamm RD, Lee RT. 2004. Cell mechanics and mechanotransduction: pathways, probes, and physiology. American Journal of Physiology-Cell Physiology. 287(1):C1–11 [DOI] [PubMed] [Google Scholar]
- 27.Seetharaman S, Etienne-Manneville S. 2019. Microtubules at focal adhesions – a double-edged sword. Journal of Cell Science. 132(19): [DOI] [PubMed] [Google Scholar]
- 28.Leube RE, Moch M, Windoffer R. 2015. Intermediate filaments and the regulation of focal adhesion. Curr Opin Cell Biol. 32:13–20 [DOI] [PubMed] [Google Scholar]
- 29.Seetharaman S, Vianay B, Roca V, Farrugia AJ, De Pascalis C, et al. 2021. Microtubules tune mechanosensitive cell responses. Nat Mater [DOI] [PubMed] [Google Scholar]
- 30.Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, et al. 2012. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell–cell contacts upon externally applied stretch. PNAS. 109(31):12568–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broussard JA, Yang R, Huang C, Nathamgari SSP, Beese AM, et al. 2017. The desmoplakin–intermediate filament linkage regulates cell mechanics. MBoC. 28(23):3156–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price AJ, Cost A-L, Ungewiß H, Waschke J, Dunn AR, Grashoff C. 2018. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat Commun. 9(1):5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaalouk DE, Lammerding J. 2009. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 10(1):63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirby TJ, Lammerding J. 2018. Emerging views of the nucleus as a cellular mechanosensor. Nat Cell Biol. 20(4):373–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, et al. 2017. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell. 171(6):1397–1410.e14 [DOI] [PubMed] [Google Scholar]
- 36.Wang JH-C, Thampatty BP. 2006. An Introductory Review of Cell Mechanobiology. Biomech Model Mechanobiol. 5(1):1–16 [DOI] [PubMed] [Google Scholar]
- 37.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, et al. 2010. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 330(6000):55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. 2003. Mechanotransduction and flow across the endothelial glycocalyx. PNAS. 100(13):7988–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qazi H, Palomino R, Shi Z-D, Munn LL, Tarbell JM. 2013. Cancer cell glycocalyx mediates mechanotransduction and flow-regulated invasion. Integr Biol. 5(11):1334–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Roux A-L, Quiroga X, Walani N, Arroyo M, Roca-Cusachs P. 2019. The plasma membrane as a mechanochemical transducer. Philosophical Transactions of the Royal Society B: Biological Sciences. 374(1779):20180221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, et al. 2011. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell. 144(3):402–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, et al. 2004. BAR Domains as Sensors of Membrane Curvature: The Amphiphysin BAR Structure. Science. 303(5657):495–99 [DOI] [PubMed] [Google Scholar]
- 43.Miermont A, Waharte F, Hu S, McClean MN, Bottani S, et al. 2013. Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. PNAS. 110(14):5725–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, et al. 2018. mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell. 174(2):338–349.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barcellos-Hoff MH, Ravani SA. 2000. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 60(5):1254–60 [PubMed] [Google Scholar]
- 46.Dolberg DS, Bissell MJ. 1984. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature. 309(5968):552–56 [DOI] [PubMed] [Google Scholar]
- 47.Place AE, Jin Huh S, Polyak K. 2011. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Research. 13(6):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seyfried TN, Huysentruyt LC. 2013. On the Origin of Cancer Metastasis. Crit Rev Oncog. 18(1–2):43–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sappino AP, Skalli O, Jackson B, Schürch W, Gabbiani G. 1988. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer. 41(5):707–12 [DOI] [PubMed] [Google Scholar]
- 50.Rønnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. 1995. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 95(2):859–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandler EM, Seo BR, Califano JP, Eguiluz RCA, Lee JS, et al. 2012. Implanted adipose progenitor cells as physicochemical regulators of breast cancer. PNAS. 109(25):9786–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, et al. 2011. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 19(2):257–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, et al. 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer. 20(3):174–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyd NF, Lockwood GA, Byng JW, Tritchler DL, Yaffe MJ. 1998. Mammographic densities and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 7(12):1133–44 [PubMed] [Google Scholar]
- 55.Li T, Sun L, Miller N, Nicklee T, Woo J, et al. 2005. The Association of Measured Breast Tissue Characteristics with Mammographic Density and Other Risk Factors for Breast Cancer. Cancer Epidemiol Biomarkers Prev. 14(2):343–49 [DOI] [PubMed] [Google Scholar]
- 56.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. 2006. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Medicine. 4(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, et al. 2011. Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma. The American Journal of Pathology. 178(3):1221–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, et al. 2005. Tensional homeostasis and the malignant phenotype. Cancer Cell. 8(3):241–54 [DOI] [PubMed] [Google Scholar]
- 59.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, et al. 2009. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 139(5):891–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arora PD, Narani N, McCulloch CA. 1999. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Pathol. 154(3):871–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C. 2003. α-Smooth Muscle Actin Is Crucial for Focal Adhesion Maturation in Myofibroblasts. Mol Biol Cell. 14(6):2508–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahmadzadeh H, Webster MR, Behera R, Valencia AMJ, Wirtz D, et al. 2017. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. PNAS. 114(9):E1617–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han YL, Ronceray P, Xu G, Malandrino A, Kamm RD, et al. 2018. Cell contraction induces long-ranged stress stiffening in the extracellular matrix. PNAS. 115(16):4075–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seo BR, Chen X, Ling L, Song YH, Shimpi AA, et al. 2020. Collagen microarchitecture mechanically controls myofibroblast differentiation. PNAS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pathak A, Kumar S. 2013. Transforming potential and matrix stiffness co-regulate confinement sensitivity of tumor cell migration. Integr Biol (Camb). 5(8):1067–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu Y-C, Chu T, Hall MS, Fu D-J, Shi Q, et al. 2019. Physical confinement induces malignant transformation in mammary epithelial cells. Biomaterials. 217:119307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mosier JA, Rahman-Zaman A, Zanotelli MR, VanderBurgh JA, Bordeleau F, et al. 2019. Extent of Cell Confinement in Microtracks Affects Speed and Results in Differential Matrix Strains. Biophysical Journal. 117(9):1692–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ilina O, Gritsenko PG, Syga S, Lippoldt J, La Porta CAM, et al. 2020. Cell–cell adhesion and 3D matrix confinement determine jamming transitions in breast cancer invasion. Nature Cell Biology. 22(9):1103–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Talkenberger K, Cavalcanti-Adam EA, Voss-Böhme A, Deutsch A. 2017. Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments. Sci Rep. 7:9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wisdom KM, Adebowale K, Chang J, Lee JY, Nam S, et al. 2018. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nature Communications. 9(1):4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaukonen R, Mai A, Georgiadou M, Saari M, De Franceschi N, et al. 2016. Normal stroma suppresses cancer cell proliferation via mechanosensitive regulation of JMJD1a-mediated transcription. Nature Communications. 7(1):12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, et al. 2007. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nature Cell Biology. 9(12):1392–1400 [DOI] [PubMed] [Google Scholar]
- 73.Glentis A, Oertle P, Mariani P, Chikina A, El Marjou F, et al. 2017. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun. 8(1):924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seo BR, Bhardwaj P, Choi S, Gonzalez J, Eguiluz RCA, et al. 2015. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Science Translational Medicine. 7(301):301ra130–301ra130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ling L, Mulligan JA, Ouyang Y, Shimpi AA, Williams RM, et al. 2020. Obesity-Associated Adipose Stromal Cells Promote Breast Cancer Invasion through Direct Cell Contact and ECM Remodeling. Advanced Functional Materials. n/a(n/a):1910650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Runswick SK, O’Hare MJ, Jones L, Streuli CH, Garrod DR. 2001. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nature Cell Biology. 3(9):823–30 [DOI] [PubMed] [Google Scholar]
- 77.Gudjonsson T, Rønnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. 2002. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. Journal of Cell Science. 115(1):39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allen MD, Thomas GJ, Clark S, Dawoud MM, Vallath S, et al. 2014. Altered Microenvironment Promotes Progression of Preinvasive Breast Cancer: Myoepithelial Expression of αvβ6 Integrin in DCIS Identifies High-risk Patients and Predicts Recurrence. Clin Cancer Res. 20(2):344–57 [DOI] [PubMed] [Google Scholar]
- 79.Nathanson SD, Nelson L. 1994. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Annals of Surgical Oncology. 1(4):333–38 [DOI] [PubMed] [Google Scholar]
- 80.Padera TP, Stoll BR, Tooredman JB, Capen D, Tomaso E di, Jain RK. 2004. Cancer cells compress intratumour vessels. Nature. 427(6976):695–695 [DOI] [PubMed] [Google Scholar]
- 81.Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, et al. 2000. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol. 156(2):529–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polacheck WJ, Charest JL, Kamm RD. 2011. Interstitial flow influences direction of tumor cell migration through competing mechanisms. PNAS. 108(27):11115–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polacheck WJ, German AE, Mammoto A, Ingber DE, Kamm RD. 2014. Mechanotransduction of fluid stresses governs 3D cell migration. PNAS. 111(7):2447–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Piotrowski-Daspit AS, Tien J, Nelson CM. 2016. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integrative Biology. 8(3):319–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ng CP, Hinz B, Swartz MA. 2005. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. Journal of Cell Science. 118(20):4731–39 [DOI] [PubMed] [Google Scholar]
- 86.Shieh AC, Rozansky HA, Hinz B, Swartz MA. 2011. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 71(3):790–800 [DOI] [PubMed] [Google Scholar]
- 87.Boardman KC, Swartz MA. 2003. Interstitial Flow as a Guide for Lymphangiogenesis. Circulation Research. 92(7):801–8 [DOI] [PubMed] [Google Scholar]
- 88.D’Alessio A, Proietti G, Sica G, Scicchitano BM. 2019. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers. 11(4):E469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zong H, Verhaak RGW, Canoll P. 2012. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 12(4):383–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, et al. 2010. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17(1):98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sakthikumar S, Sakthikumar S, Roy A, Haseeb L, Pettersson ME, et al. 2020. Whole-genome sequencing of glioblastoma reveals enrichment of non-coding constraint mutations in known and novel genes. Genome Biology. 21(1):1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, et al. 2019. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 178(4):835–849.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seano G, Jain RK. 2020. Vessel co-option in glioblastoma: emerging insights and opportunities. Angiogenesis. 23(1):9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hirata E, Yukinaga H, Kamioka Y, Arakawa Y, Miyamoto S, et al. 2012. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family gtpases in glioblastoma cell invasion. Journal of Cell Science. 125(4):858–68 [DOI] [PubMed] [Google Scholar]
- 95.Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P. 2006. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia. 53(8):799–808 [DOI] [PubMed] [Google Scholar]
- 96.Winkler F, Kienast Y, Fuhrmann M, Von Baumgarten L, Burgold S, et al. 2009. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 57(12):1306–15 [DOI] [PubMed] [Google Scholar]
- 97.Louis DN. 2006. Molecular Pathology of Malignant Gliomas. Annual Review of Pathology: Mechanisms of Disease. 1(1):97–117 [DOI] [PubMed] [Google Scholar]
- 98.Klank RL, Decker Grunke SA, Bangasser BL, Forster CL, Price MA, et al. 2017. Biphasic Dependence of Glioma Survival and Cell Migration on CD44 Expression Level. Cell Reports. 18(1):23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pietras A, Katz AM, Ekström EJ, Wee B, Halliday JJ, et al. 2014. Osteopontin-CD44 Signaling in the Glioma Perivascular Niche Enhances Cancer Stem Cell Phenotypes and Promotes Aggressive Tumor Growth. Cell Stem Cell. 14(3):357–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim Y, Kumar S. 2014. CD44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Molecular Cancer Research. 12(10):1416–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolf KJ, Shukla P, Springer K, Lee S, Coombes JD, et al. 2020. A mode of cell adhesion and migration facilitated by CD44-dependent microtentacles. Proceedings of the National Academy of Sciences of the United States of America. 117(21):11432–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zepecki JP, Snyder KM, Moreno MM, Fajardo E, Fiser A, et al. 2019. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene. 38(10):1734–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gritsenko PG, Friedl P. 2018. Adaptive adhesion systems mediate glioma cell invasion in complex environments. Journal of Cell Science. 131(15): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weller M, Nabors LB, Gorlia T, Leske H, Rushing E, et al. 2016. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget. 7(12):15018–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Monzo P, Chong YK, Guetta-Terrier C, Krishnasamy A, Sathe SR, et al. 2016. Mechanical confinement triggers glioma linear migration dependent on formin FHOD3. Molecular Biology of the Cell. 27(8):1246–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu CJ, Shamsan GA, Akkin T, Odde DJ. 2019. Glioma Cell Migration Dynamics in Brain Tissue Assessed by Multimodal Optical Imaging. Biophysical Journal. 117(7):1179–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ishihara H, Kubota H, Lindberg RLP, Leppert D, Gloor SM, et al. 2008. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor β2 involves matrix metalloproteinases and tight junction proteins. Journal of Neuropathology and Experimental Neurology. 67(5):435–48 [DOI] [PubMed] [Google Scholar]
- 108.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, et al. 2011. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Research. 71(18):6061–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, et al. 2007. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell. 11(1):69–82 [DOI] [PubMed] [Google Scholar]
- 110.Mistry AM, Dewan MC, White-Dzuro GA, Brinson PR, Weaver KD, et al. 2017. Decreased survival in glioblastomas is specific to contact with the ventricular-subventricular zone, not subgranular zone or corpus callosum. Journal of Neuro-Oncology. 132(2):341–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, et al. 2019. Electrical and synaptic integration of glioma into neural circuits. Nature. 573(7775):539–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boucher Y, Salehi H, Witwer B, Harsh GR, Jain RK. 1997. Interstitial fluid pressure in intracranial tumours in patients and in rodents. British Journal of Cancer. 75(6):829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kingsmore KM, Logsdon DK, Floyd DH, Peirce SM, Purow BW, Munson JM. 2016. Interstitial flow differentially increases patient-derived glioblastoma stem cell invasion via CXCR4, CXCL12, and CD44-mediated mechanisms. Integr Biol. 8(12):1246–60 [DOI] [PubMed] [Google Scholar]
- 114.Akins EA, Aghi MK, Kumar S. 2020. Incorporating Tumor-Associated Macrophages into Engineered Models of Glioma. iScience. 23(12):101770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heldin C-H, Rubin K, Pietras K, Ostman A. 2004. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 4(10):806–13 [DOI] [PubMed] [Google Scholar]
- 116.Munson JM, Bellamkonda RV., Swartz MA. 2013. Interstitial flow in a 3d microenvironment increases glioma invasion by a cxcr4-dependent mechanism. Cancer Research. 73(5):1536–46 [DOI] [PubMed] [Google Scholar]
- 117.Cornelison RC, Brennan CE, Kingsmore KM, Munson JM. 2018. Convective forces increase CXCR4-dependent glioblastoma cell invasion in GL261 murine model. Scientific Reports. 8(1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gerstner ER, Duda DG, di Tomaso E, Ryg PA, Loeffler JS, et al. 2009. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 6(4):229–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee CG, Heijn M, Di Tomaso E, Griffon-Etienne G, Ancukiewicz M, et al. 2000. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Research. 60(19):5565–70 [PubMed] [Google Scholar]
- 120.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DGG, et al. 2007. AZD2171, a Pan-VEGF Receptor Tyrosine Kinase Inhibitor, Normalizes Tumor Vasculature and Alleviates Edema in Glioblastoma Patients. Cancer Cell. 11(1):83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vredenburgh JJ, Desjardins A, Herndon JE, Dowell JM, Reardon DA, et al. 2007. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clinical Cancer Research. 13(4):1253–59 [DOI] [PubMed] [Google Scholar]
- 122.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, et al. 2009. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell. 15(3):220–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Deng X, Xiong F, Li X, Xiang B, Li Z, et al. 2018. Application of atomic force microscopy in cancer research. J Nanobiotechnol. 16(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Staunton JR, Vieira W, Fung KL, Lake R, Devine A, Tanner K. 2016. Mechanical Properties of the Tumor Stromal Microenvironment Probed In Vitro and Ex Vivo by In Situ -Calibrated Optical Trap-Based Active Microrheology. Cel. Mol. Bioeng 9(3):398–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hochmuth RM. 2000. Micropipette aspiration of living cells. J Biomech. 33(1):15–22 [DOI] [PubMed] [Google Scholar]
- 126.Pachenari M, Seyedpour SM, Janmaleki M, Shayan SB, Taranejoo S, Hosseinkhani H. 2014. Mechanical properties of cancer cytoskeleton depend on actin filaments to microtubules content: Investigating different grades of colon cancer cell lines. Journal of Biomechanics. 47(2):373–79 [DOI] [PubMed] [Google Scholar]
- 127.Calzado-Martín A, Encinar M, Tamayo J, Calleja M, San Paulo A. 2016. Effect of Actin Organization on the Stiffness of Living Breast Cancer Cells Revealed by Peak-Force Modulation Atomic Force Microscopy. ACS Nano. 10(3):3365–74 [DOI] [PubMed] [Google Scholar]
- 128.Davidson PM, Fedorchak GR, Mondésert-Deveraux S, Bell ES, Isermann P, et al. 2019. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab Chip. 19(21):3652–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Irianto J, Xia Y, Pfeifer CR, Greenberg RA, Discher DE. 2017. As a Nucleus Enters a Small Pore, Chromatin Stretches and Maintains Integrity, Even with DNA Breaks. Biophys J. 112(3):446–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tse HTK, Gossett DR, Moon YS, Masaeli M, Sohsman M, et al. 2013. Quantitative diagnosis of malignant pleural effusions by single-cell mechanophenotyping. Sci Transl Med. 5(212):212ra163. [DOI] [PubMed] [Google Scholar]
- 131.Otto O, Rosendahl P, Mietke A, Golfier S, Herold C, et al. 2015. Real-time deformability cytometry: on-the-fly cell mechanical phenotyping. Nat Methods. 12(3):199–202 [DOI] [PubMed] [Google Scholar]
- 132.Toepfner N, Herold C, Otto O, Rosendahl P, Jacobi A, et al. 2018. Detection of human disease conditions by single-cell morpho-rheological phenotyping of blood. eLife. 7:e29213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, et al. 2005. Optical Deformability as an Inherent Cell Marker for Testing Malignant Transformation and Metastatic Competence. Biophysical Journal. 88(5):3689–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yousafzai MS, Coceano G, Bonin S, Niemela J, Scoles G, Cojoc D. 2017. Investigating the effect of cell substrate on cancer cell stiffness by optical tweezers. Journal of Biomechanics. 60:266–69 [DOI] [PubMed] [Google Scholar]
- 135.Wullkopf L, West A-KV, Leijnse N, Cox TR, Madsen CD, et al. 2018. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. MBoC. 29(20):2378–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Plotnikov SV, Sabass B, Schwarz US, Waterman CM. 2014. High-Resolution Traction Force Microscopy. Methods Cell Biol. 123:367–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dembo M, Wang YL. 1999. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J. 76(4):2307–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A. 100(4):1484–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rabodzey A, Alcaide P, Luscinskas FW, Ladoux B. 2008. Mechanical forces induced by the transendothelial migration of human neutrophils. Biophys J. 95(3):1428–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maskarinec SA, Franck C, Tirrell DA, Ravichandran G. 2009. Quantifying cellular traction forces in three dimensions. PNAS. 106(52):22108–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS. 2010. Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nat Methods. 7(12):969–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mulligan JA, Ling L, Leartprapun N, Fischbach C, Adie SG. 2021. Computational 4D-OCM for label-free imaging of collective cell invasion and force-mediated deformations in collagen. Sci Rep. 11(1):2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li D, Colin-York H, Barbieri L, Javanmardi Y, Guo Y, et al. 2021. Astigmatic traction force microscopy (aTFM). Nat Commun. 12(1):2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aj G, D P, B D. 1997. Quantification of cell adhesion using a spinning disc device and application to surface-reactive materials. Biomaterials. 18(16): [DOI] [PubMed] [Google Scholar]
- 145.Benoit M, Gabriel D, Gerisch G, Gaub HE. 2000. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat Cell Biol. 2(6):313–17 [DOI] [PubMed] [Google Scholar]
- 146.Panorchan P, Thompson MS, Davis KJ, Tseng Y, Konstantopoulos K, Wirtz D. 2006. Single-molecule analysis of cadherin-mediated cell-cell adhesion. J Cell Sci. 119(Pt 1):66–74 [DOI] [PubMed] [Google Scholar]
- 147.Selhuber-Unkel C, López-García M, Kessler H, Spatz JP. 2008. Cooperativity in Adhesion Cluster Formation during Initial Cell Adhesion. Biophysical Journal. 95(11):5424–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Friedrichs J, Legate KR, Schubert R, Bharadwaj M, Werner C, et al. 2013. A practical guide to quantify cell adhesion using single-cell force spectroscopy. Methods. 60(2):169–78 [DOI] [PubMed] [Google Scholar]
- 149.Chu Y-S, Thomas WA, Eder O, Pincet F, Perez E, et al. 2004. Force measurements in E-cadherin–mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. Journal of Cell Biology. 167(6):1183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maître J-L, Berthoumieux H, Krens SFG, Salbreux G, Jülicher F, et al. 2012. Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science. 338(6104):253–56 [DOI] [PubMed] [Google Scholar]
- 151.Bazellières E, Conte V, Elosegui-Artola A, Serra-Picamal X, Bintanel-Morcillo M, et al. 2015. Control of cell–cell forces and collective cell dynamics by the intercellular adhesome. Nat Cell Biol. 17(4):409–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Stohrer M, Boucher Y, Stangassinger M, Jain RK. 2000. Oncotic Pressure in Solid Tumors Is Elevated. Cancer Res. 60(15):4251–55 [PubMed] [Google Scholar]
- 153.Ozerdem U 2009. Measuring interstitial fluid pressure with fiberoptic pressure transducers. Microvasc Res. 77(2):226–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.DuFort CC, DelGiorno KE, Carlson MA, Osgood RJ, Zhao C, et al. 2016. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophysical Journal. 110(9):2106–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kingsmore KM, Vaccari A, Abler D, Cui SX, Epstein FH, et al. 2018. MRI analysis to map interstitial flow in the brain tumor microenvironment. APL Bioengineering. 2(3):031905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Campàs O, Mammoto T, Hasso S, Sperling RA, O’Connell D, et al. 2014. Quantifying cell-generated mechanical forces within living embryonic tissues. Nat Methods. 11(2):183–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, et al. 2010. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 466(7303):263–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, et al. 2012. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proceedings of the National Academy of Sciences. 109(38):15101–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nia HT, Liu H, Seano G, Datta M, Jones D, et al. 2016. Solid stress and elastic energy as measures of tumour mechanopathology. Nat Biomed Eng. 1: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, et al. 2007. Engineering tumors with 3D scaffolds. Nature Methods. 4(10):855–60 [DOI] [PubMed] [Google Scholar]
- 161.Hutmacher DW. 2010. Biomaterials offer cancer research the third dimension. Nat Mater. 9(2):90–93 [DOI] [PubMed] [Google Scholar]
- 162.Tan ML, Ling L, Fischbach C. 2021. Engineering strategies to capture the biological and biophysical tumor microenvironment in vitro. Advanced Drug Delivery Reviews. 176:113852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tavakol DN, Fleischer S, Vunjak-Novakovic G. 2021. Harnessing organs-on-a-chip to model tissue regeneration. Cell Stem Cell. 28(6):993–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Petersen OW, Rønnov-Jessen L, Howlett AR, Bissell MJ. 1992. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. PNAS. 89(19):9064–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, et al. 1997. Reversion of the Malignant Phenotype of Human Breast Cells in Three-Dimensional Culture and In Vivo by Integrin Blocking Antibodies. Journal of Cell Biology. 137(1):231–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Beri P, Matte BF, Fattet L, Kim D, Yang J, Engler AJ. 2018. Biomaterials to model and measure epithelial cancers. Nat Rev Mater. 3(11):418–30 [PMC free article] [PubMed] [Google Scholar]
- 167.Velez DO, Tsui B, Goshia T, Chute CL, Han A, et al. 2017. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nature Communications. 8(1):1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu Z, Vunjak-Novakovic G. 2016. Modeling tumor microenvironments using custom-designed biomaterial scaffolds. Current Opinion in Chemical Engineering. 11:94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hansen K, Kiemele L, Maller O, O’Brien J, Shankar A, et al. 2009. An in-solution ultrasonication-assisted digestion method for improved extracellular matrix proteome coverage. Molecular & cellular proteomics : MCP. 8(7): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lutolf MP, Hubbell JA. 2005. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 23(1):47–55 [DOI] [PubMed] [Google Scholar]
- 171.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, et al. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. PNAS. 100(9):5413–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. 2009. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Advanced Materials. 21(48):5005–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Raeber GP, Lutolf MP, Hubbell JA. 2005. Molecularly Engineered PEG Hydrogels: A Novel Model System for Proteolytically Mediated Cell Migration. Biophysical Journal. 89(2):1374–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Singh SP, Schwartz MP, Lee JY, Fairbanks BD, Anseth KS. 2014. A peptide functionalized poly(ethylene glycol) (PEG) hydrogel for investigating the influence of biochemical and biophysical matrix properties on tumor cell migration. Biomaterials Science. 2(7):1024–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ehrbar M, Sala A, Lienemann P, Ranga A, Mosiewicz K, et al. 2011. Elucidating the Role of Matrix Stiffness in 3D Cell Migration and Remodeling. Biophysical Journal. 100(2):284–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Moon JJ, Saik JE, Poche RA, Leslie-Barbick JE, Lee S-H, et al. 2010. Biomimetic hydrogels with pro-angiogenic properties. Biomaterials. 31(14):3840–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gill BJ, Gibbons DL, Roudsari LC, Saik JE, Rizvi ZH, et al. 2012. A Synthetic Matrix with Independently Tunable Biochemistry and Mechanical Properties to Study Epithelial Morphogenesis and EMT in a Lung Adenocarcinoma Model. Cancer Res. 72(22):6013–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, et al. 2012. Maleimide Cross-Linked Bioactive PEG Hydrogel Exhibits Improved Reaction Kinetics and Cross-Linking for Cell Encapsulation and In Situ Delivery. Advanced Materials. 24(1):64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lee TT, García JR, Paez JI, Singh A, Phelps EA, et al. 2015. Light-triggered in vivo activation of adhesive peptides regulates cell adhesion, inflammation and vascularization of biomaterials. Nature Mater. 14(3):352–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ondeck MG, Kumar A, Placone JK, Plunkett CM, Matte BF, et al. 2019. Dynamically stiffened matrix promotes malignant transformation of mammary epithelial cells via collective mechanical signaling. PNAS. 116(9):3502–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ananthanarayanan B, Kim Y, Kumar S. 2011. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials. 32(31):7913–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wolf KJ, Chen J, Coombes J, Aghi MK, Kumar S. 2019. Dissecting and rebuilding the glioblastoma microenvironment with engineered materials. Nat Rev Mater. 4(10):651–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kim Y, Kumar S. 2014. CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol Cancer Res. 12(10):1416–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hanjaya-Putra D, Wong KT, Hirotsu K, Khetan S, Burdick JA, Gerecht S. 2012. Spatial control of cell-mediated degradation to regulate vasculogenesis and angiogenesis in hyaluronan hydrogels. Biomaterials. 33(26):6123–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shen Y-I, Abaci HE, Krupski Y, Weng L-C, Burdick JA, Gerecht S. 2014. Hyaluronic acid hydrogel stiffness and oxygen tension affect cancer cell fate and endothelial sprouting. Biomaterials Science. 2(5):655–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Allen SC, Widman JA, Datta A, Suggs LJ. 2020. Dynamic extracellular matrix stiffening induces a phenotypic transformation and a migratory shift in epithelial cells. Integrative Biology. 12(6):161–74 [DOI] [PubMed] [Google Scholar]
- 187.Shimpi AA, Fischbach C. 2021. Engineered ECM models: Opportunities to advance understanding of tumor heterogeneity. Curr Opin Cell Biol. 72:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, et al. 2015. Cell-mediated fibre recruitment drives extracellular matrix mechanosensing in engineered fibrillar microenvironments. Nature Mater. 14(12):1262–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Matera DL, DiLillo KM, Smith MR, Davidson CD, Parikh R, et al. 2020. Microengineered 3D pulmonary interstitial mimetics highlight a critical role for matrix degradation in myofibroblast differentiation. Science Advances. 6(37):eabb5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang WY, Davidson CD, Lin D, Baker BM. 2019. Actomyosin contractility-dependent matrix stretch and recoil induces rapid cell migration. Nat Commun. 10(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Azioune A, Storch M, Bornens M, Théry M, Piel M. 2009. Simple and rapid process for single cell micro-patterning. Lab Chip. 9(11):1640–42 [DOI] [PubMed] [Google Scholar]
- 192.Théry M 2010. Micropatterning as a tool to decipher cell morphogenesis and functions. Journal of Cell Science. 123(24):4201–13 [DOI] [PubMed] [Google Scholar]
- 193.Vignaud T, Ennomani H, Théry M. 2014. Chapter 6 - Polyacrylamide Hydrogel Micropatterning. In Methods in Cell Biology, ed Piel M, Théry M. 120:93–116. Academic Press; [DOI] [PubMed] [Google Scholar]
- 194.Tabdanov ED, Puram VV, Win Z, Alamgir A, Alford PW, Provenzano PP. 2018. Bimodal sensing of guidance cues in mechanically distinct microenvironments. Nat Commun. 9(1):4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lee J, Abdeen AA, Wycislo KL, Fan TM, Kilian KA. 2016. Interfacial geometry dictates cancer cell tumorigenicity. Nature Mater. 15(8):856–62 [DOI] [PubMed] [Google Scholar]
- 196.Vignaud T, Galland R, Tseng Q, Blanchoin L, Colombelli J, Théry M. 2012. Reprogramming cell shape with laser nano-patterning. Journal of Cell Science. 125(9):2134–40 [DOI] [PubMed] [Google Scholar]
- 197.Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, et al. 2014. 3D Collagen Alignment Limits Protrusions to Enhance Breast Cancer Cell Persistence. Biophys J. 107(11):2546–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lee P, Lin R, Moon J, Lee LP. 2006. Microfluidic alignment of collagen fibers for in vitro cell culture. Biomed Microdevices. 8(1):35–41 [DOI] [PubMed] [Google Scholar]
- 199.Brownfield DG, Venugopalan G, Lo A, Mori H, Tanner K, et al. 2013. Patterned Collagen Fibers Orient Branching Mammary Epithelium through Distinct Signaling Modules. Current Biology. 23(8):703–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Su C-Y, Burchett A, Dunworth M, Choi JS, Ewald AJ, et al. 2021. Engineering a 3D collective cancer invasion model with control over collagen fiber alignment. Biomaterials. 275:120922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Paul CD, Hung W-C, Wirtz D, Konstantopoulos K. 2016. Engineered Models of Confined Cell Migration. Annual Review of Biomedical Engineering. 18(1):159–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rianna C, Radmacher M, Kumar S. 2020. Direct evidence that tumor cells soften when navigating confined spaces. MBoC. 31(16):1726–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pathak A, Kumar S. 2012. Independent regulation of tumor cell migration by matrix stiffness and confinement. PNAS. 109(26):10334–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pathak A, Kumar S. 2012. Independent regulation of tumor cell migration by matrix stiffness and confinement. PNAS. 109(26):10334–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Davidson PM, Sliz J, Isermann P, Denais C, Lammerding J. 2015. Design of a microfluidic device to quantify dynamic intra-nuclear deformation during cell migration through confining environments. Integr. Biol 7(12):1534–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Denais CM, Gilbert RM, Isermann P, McGregor AL, Lindert M te et al. 2016. Nuclear envelope rupture and repair during cancer cell migration. Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shah P, Hobson CM, Cheng S, Colville MJ, Paszek MJ, et al. 2021. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Current Biology. 31(4):753–765.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sung KE, Beebe DJ. 2014. Microfluidic 3D models of cancer. Advanced Drug Delivery Reviews. 79–80:68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. 2012. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proceedings of the National Academy of Sciences. 109(34):13515–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chramiec A, Teles D, Yeager K, Marturano-Kruik A, Pak J, et al. 2020. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab Chip. 20(23):4357–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Piotrowski-Daspit AS, Tien J, Nelson CM. 2016. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integr Biol (Camb). 8(3):319–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pisano M, Triacca V, Barbee KA, Swartz MA. 2015. An in vitro model of the tumor-lymphatic microenvironment with simultaneous transendothelial and luminal flows reveals mechanisms of flow enhanced invasion. Integr Biol. 7(5):525–33 [DOI] [PubMed] [Google Scholar]
- 213.Bäuerle T, Komljenovic D, Merz M, Berger MR, Goodman SL, Semmler W. 2011. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using VCT, MRI and DCE-MRI in a longitudinal in vivo study. Int J Cancer. 128(10):2453–62 [DOI] [PubMed] [Google Scholar]
- 214.Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, et al. 2010. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 28(16):2712–18 [DOI] [PubMed] [Google Scholar]
- 215.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, et al. 2014. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 15(10):1100–1108 [DOI] [PubMed] [Google Scholar]
- 216.Zhang Q, Bindokas V, Shen J, Fan H, Hoffman RM, Xing HR. 2011. Time-Course Imaging of Therapeutic Functional Tumor Vascular Normalization by Antiangiogenic Agents. Mol Cancer Ther. 10(7):1173–84 [DOI] [PubMed] [Google Scholar]
- 217.Ferrara N, Hillan KJ, Gerber H-P, Novotny W. 2004. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 3(5):391–400 [DOI] [PubMed] [Google Scholar]
- 218.Aalders KC, Tryfonidis K, Senkus E, Cardoso F. 2017. Anti-angiogenic treatment in breast cancer: Facts, successes, failures and future perspectives. Cancer Treatment Reviews. 53:98–110 [DOI] [PubMed] [Google Scholar]
- 219.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, et al. 2007. Paclitaxel plus Bevacizumab versus Paclitaxel Alone for Metastatic Breast Cancer. New England Journal of Medicine. 357(26):2666–76 [DOI] [PubMed] [Google Scholar]
- 220.Robert NJ, Diéras V, Glaspy J, Brufsky AM, Bondarenko I, et al. 2011. RIBBON-1: Randomized, Double-Blind, Placebo-Controlled, Phase III Trial of Chemotherapy With or Without Bevacizumab for First-Line Treatment of Human Epidermal Growth Factor Receptor 2–Negative, Locally Recurrent or Metastatic Breast Cancer. JCO. 29(10):1252–60 [DOI] [PubMed] [Google Scholar]
- 221.Brufsky AM, Hurvitz S, Perez E, Swamy R, Valero V, et al. 2011. RIBBON-2: A Randomized, Double-Blind, Placebo-Controlled, Phase III Trial Evaluating the Efficacy and Safety of Bevacizumab in Combination With Chemotherapy for Second-Line Treatment of Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer. JCO. 29(32):4286–93 [DOI] [PubMed] [Google Scholar]
- 222.Martino EC, Misso G, Pastina P, Costantini S, Vanni F, et al. 2016. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discovery. 2(1):1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yang J, Yan J, Liu B. 2018. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Frontiers in Immunology. 9:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bejarano L, Jordāo MJC, Joyce JA. 2021. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 11(4):933–59 [DOI] [PubMed] [Google Scholar]
- 225.Hauge A, Rofstad EK. 2020. Antifibrotic therapy to normalize the tumor microenvironment. J Transl Med. 18(1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Van Cutsem E, Tempero MA, Sigal D, Oh D-Y, Fazio N, et al. 2020. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol. 38(27):3185–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Benson AB, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, et al. 2017. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist. 22(3):241–e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hecht JR, Benson AB, Vyushkov D, Yang Y, Bendell J, Verma U. 2017. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist. 22(3):243–e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, et al. 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 25(6):719–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Fennell DA, Baas P, Taylor P, Nowak AK, Gilligan D, et al. 2019. Maintenance Defactinib Versus Placebo After First-Line Chemotherapy in Patients With Merlin-Stratified Pleural Mesothelioma: COMMAND-A Double-Blind, Randomized, Phase II Study. J Clin Oncol. 37(10):790–98 [DOI] [PubMed] [Google Scholar]
- 231.McLeod R, Kumar R, Papadatos-Pastos D, Mateo J, Brown JS, et al. 2020. First-in-Human Study of AT13148, a Dual ROCK-AKT Inhibitor in Patients with Solid Tumors. Clin Cancer Res. 26(18):4777–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, et al. 2018. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 119(10):1208–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, et al. 2019. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clinical and Translational Gastroenterology. 10(7):e00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, et al. 2019. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Science Translational Medicine. 11(513):eaay2574. [DOI] [PubMed] [Google Scholar]
- 235.Driehuis E, Kretzschmar K, Clevers H. 2020. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc. 15(10):3380–3409 [DOI] [PubMed] [Google Scholar]
- 236.Tian H, Lyu Y, Yang Y-G, Hu Z. 2020. Humanized Rodent Models for Cancer Research. Frontiers in Oncology. 10:1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sontheimer-Phelps A, Hassell BA, Ingber DE. 2019. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer. 19(2):65–81 [DOI] [PubMed] [Google Scholar]
- 238.Lai BFL, Lu RXZ, Hu Y, Huyer LD, Dou W, et al. 2020. Recapitulating Pancreatic Tumor Microenvironment through Synergistic Use of Patient Organoids and Organ-on-a-Chip Vasculature. Advanced Functional Materials. 30(48):2000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.West J, You L, Zhang J, Gatenby RA, Brown JS, et al. 2020. Towards Multidrug Adaptive Therapy. Cancer Res. 80(7):1578–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gatenby RA, Brown JS. 2020. Integrating evolutionary dynamics into cancer therapy. Nat Rev Clin Oncol. 17(11):675–86 [DOI] [PubMed] [Google Scholar]