

Abstract
Highly identical segmental duplications (SDs) account for over 5% of the human genome and are enriched in the short arm of the chromosome 16. These SDs are susceptibility factors for recurrent chromosomal rearrangements mediated by non-allelic homologous recombination (NAHR). Chromosomal microarray analysis (CMA) has been widely used as the first-tier test for individuals with developmental disabilities and/or congenital anomalies and several genomic disorders involving the 16p-arm have been identified with this technique. However, the resolution of CMA and the limitations of short-reads whole genome sequencing (WGS) technology do not allow the full characterization of the most complex chromosomal rearrangements. Herein, we report on two unrelated patients with a de novo 16p13.11p11.2 triplication associated with a 16p11.2 duplication, detected by CMA. These patients share a similar phenotype including hypotonia, severe neurodevelopmental delay with profound speech impairment, hyperkinetic behavior, conductive hearing loss, and distinctive facial features. Short-reads WGS could not map precisely any of the rearrangement’s breakpoints that lie within SDs. We used optical genome mapping (OGM) to determine the relative orientation of the triplicated and duplicated segments as well as the genomic positions of the breakpoints, allowing us to propose a mechanism involving recombination between allelic SDs and a NAHR event. In conclusion, we report a new clinically recognizable genomic disorder. In addition, the mechanism of these complex chromosomal rearrangements involving SDs could be unraveled by OGM.
Subject terms: Cytogenetics, Genetics research
Introduction
Highly identical segmental duplications (SDs) account for over 5% of the human genome and are enriched in the short arm of the chromosome 16. These SDs are susceptibility factors for recurrent chromosomal rearrangements mediated by non-allelic homologous recombination (NAHR) which have been called “genomic disorders” [1]. Chromosomal microarray analysis (CMA) has been widely used as the first-tier test for individuals with developmental disabilities and/or congenital anomalies and several genomic disorders involving the 16p-arm have been described with this technique [2]. For example, the 16p11.2 proximal deletion is among the most commonly known genetic etiologies associated with autism spectrum disorder (ASD) [3]. Similarly, the recurrent deletion of a distal 200-kb region at 16p11.2, including the SH2B1 gene, confers an increased risk for developmental delay, intellectual disability, and obesity [4]. Interstitial triplications, unlike deletions and duplications, are much less frequent. The phenotype is usually more severe than duplications because of further increases in gene dosage. Herein, we report on two unrelated patients with a de novo 16p13.11p11.2 triplication associated with a 16p11.2 duplication between BP1–BP3 and BP1–BP2 in patient 1 and patient 2, respectively, sharing a similar phenotype including hypotonia, severe neurodevelopmental delay with profound speech impairment and hyperkinetic behavior, conductive hearing loss, and distinctive facial features. Complete genetic characterization of these events was unreliable by whole genome sequencing (WGS) because the breakpoints lie within SDs. Consequently, we used optical genome mapping (OGM) to fully characterize these chromosomal abnormalities. Thus, we propose a mechanism to explain these rearrangements associated with a new clinically recognizable syndrome.
Materials, subjects and methods
Patients
The two female patients from two unrelated families were referred to the medical genetics department of Necker-Enfants Malades hospital because of syndromic intellectual disability. Informed consent for genetic testing on each patient was obtained from the parents. The parents of patient 1 gave informed consent for publication of medical photographs.
Patient 1
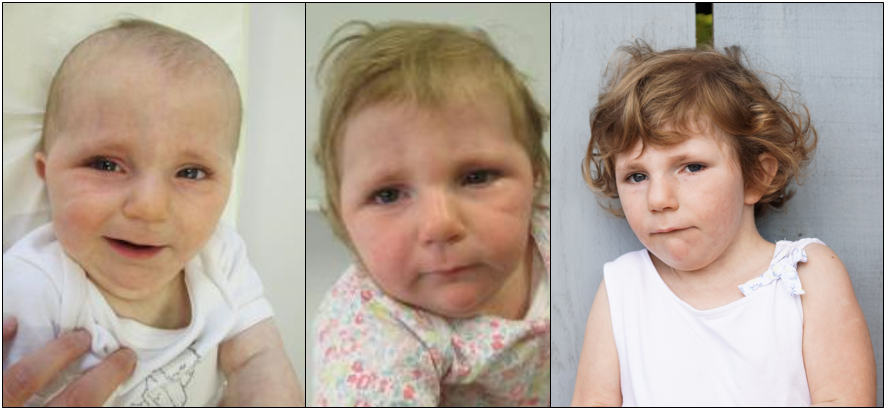
Patient 1 is the second child of an unrelated 33-year-old female and 36-year-old male. She has one healthy older brother. Her mother was diagnosed with hypothyroidism and treated with levothyroxine. She was born at full term by C-section after an uneventful pregnancy. At birth, at 38 weeks plus 4 days of gestation, weight was 3350 g (50th percentile), length 48 cm (16th p) and head circumference 34 cm (37th p). She had feeding difficulties (HP:0008872) and was fed through a nasogastric tube for several weeks. Club foot (HP:0001762), sacral dimple (HP:0000960), and cutis marmorata (HP:0000965) were noted. The magnetic resonance imaging (MRI) of the lumbar spine demonstrated a tethered cord (HP:0002144) at the level of L3–L4 and a filum terminale lipoma (HP:0012032) requiring surgery at 1 year and 3 months of age. In the first year of life, she presented with pharyngo-laryngomalacia (HP:0001601) and obstructive sleep apnea (HP:0002870). She also had a history of recurrent episodes of bronchitis (HP:0002837) and of bilateral seromucinous otitis (HP:0000389). She was diagnosed with conductive deafness (HP:0000405). She has worn hearing aids since she was 2 years old. In early infancy, she showed developmental delay (HP:0012758) and hypotonia (HP:0001252). She was able to support her head at 6 months, and sat with assistance at 9 months. She developed relatively good eye contact. On examination at 10 months, growth parameters were length 67 cm (−2 SD), weight 7 kg (−2 SD), and head circumference 41 cm (−2.5 SD). A brain MRI performed at the age of 1 year and 6 months showed ventricular dilatation (HP:0002119), cerebral atrophy (HP:0002059), and myelination delay (HP:0012448). Reevaluation at 25 months of age confirmed significant psychomotor retardation. She could not walk without help and had no speech. Isolated growth hormone deficiency (HP:0032367) was diagnosed with normal pituitary imaging and growth hormone therapy was started at the age of 2 years and 6 months. At 4 years, growth parameters were weight 15 kg (−0.5 SD), height 95 cm (−2 SD), and head circumference 46 cm (−3 SD). She began to walk without help but still had no speech. Expressive language was more affected than the receptive. She presented with hyperkinetic behavior and sleep disturbance (HP:0002360) treated by melatonin. Facial features showed microcephaly (HP:0000252), enophtalmia (HP:0000490), retrognathia (HP:0000278), prominent chin with horizontal crease (HP:0011823), thin upper lip (HP:0000219), and long and smooth philtrum (HP:0000343, HP:0000319) (Supplementary Fig. 1). Due to chronic upper airway obstruction (HP:0002781), adenoidectomy and insertion of tympanostomy tube were performed at the age of 4 years and 7 months. MRI at the same age confirmed microcephaly and simplified bifrontal gyration pattern (HP:0002536). Clinical and genetics information regarding patient 1 is included in the Decipher database (accession number: 436458) [5].
Patient 2
Patient 2 was referred for a clinical genetic consultation at the Necker-Enfants Malades Hospital at the age of 2 years and 4 months due to hypotonia (HP:0001252) associated with developmental delay (HP:0012758). She is the only child of an unrelated 25-year-old female and 34-year-old male. She has one older half-brother from her mother’s side. She was born at full term after an uneventful pregnancy by vaginal delivery. At birth, at 37 weeks of gestation, her weight was 3785 kg (90th percentile), length 50 cm (68th p), and head circumference 39.5 cm (>99th p). An umbilical hernia (HP:0001537) was noted and she underwent surgery at the age of 4 years and 4 months. Conductive hearing loss (HP:0000405) was diagnosed at the age of 4 months in a context of chronic otitis media (HP:0000389). She began wearing hearing aids at the age of 10 months. Due to recurrent episodes of otitis, she underwent an adenoidectomy as well as an insertion of tympanostomy tube at the age of 22 months. In early infancy, she showed developmental delay and hypotonia. She was able to sit at 9 months. At physical examination at the age of 2 years and 4 months, growth parameters were length 89 cm (mean) and head circumference 48 cm (mean). She was able to stand and to sit with assistance and to crawl alone. Her speech was limited to only some simple words but she developed relatively good eye contact. Expressive language, especially verbal language, was more affected than the receptive. MRI at 2 years of age revealed normal inner ear, no major intra-cranial malformation but a slightly simplified bifrontal gyration (HP:0002536). Upon reevaluation at 4 years and 5 months, she still had hypotonia and developmental delay. She was not able to walk without help and her speech remained limited to a few words. Growth parameters were normal. At 6 years and 9 months, she began to walk by herself but she still was not able to speak more than a few isolated words. She also had hyperkinetic behavior. Facial features showed a large forehead (HP:0002003), enophthalmia (HP:0000490), prominent chin with horizontal crease (HP:0011823), and a long, smooth philtrum (HP:0000343, HP:0000319) as well as an indentation of the left helix (HP:0011262). Clinical and genetics information regarding patient 2 is included in the Decipher database (accession number: 436460).
Molecular cytogenetics
Agilent CGH Microarray 60 K (Agilent Technologies, Santa Clara, CA, USA) was used for genomic copy number (CN) analyses that was carried out according to manufacturers’ recommendations. Agilent CytoGenomics v5.0.2 software was used to analyze and report the data. Genomic positions are relative to human genome Build GRCh37/hg19. Using standard protocols, chromosomal rearrangement characterization and parental testing were performed by fluorescence in situ hybridization (FISH) with the BAC (Bacterial Artificial Chromosome) probes RP11–489O1 (accession number AC020621.4) at 16p13.11 (both patients and parents), RP11–152L13 at 16p12.1 (both patients and the parents of patient 1), and CTD-2515C15 at 16p11.2 (patient 1 and her parents) on chromosome preparations from leukocyte cultures.
Whole genome sequencing (WGS)
WGS analysis was performed only on patient 1. Libraries were prepared using the TruSeq DNA PCR-free Library Preparation Kit (Illumina) according to the manufacturer’s instructions, with 2.1 or 2.2 μg DNA input sheared to a mean target size of 550 bp using the Covaris E220 sonicator. A pool of libraires was sequenced on the NovaSeq6000 (Paired-End reads 150 bases +150 bases). A total of ~420 million of passing filter paired-end reads were targeted per library to achieve a minimum mean depth coverage of at least 30X. The samples were analyzed by a bioinformatic pipeline using alignment and variant calling programs widely distributed in the community. Sequences in fastq format were aligned with the reference sequence of the human genome hg19 using the bwa program [6]. In order to interpret and characterize precisely the structural variants (SVs) in patient 1, three bioinformatics approaches were used: (1) Wisecondor (WisecondorX version 1.0) which is a depth of coverage method with data normalization, segmentation and aberration calling steps; (2) Canvas (canvas-1.34.0.1201+master), a control free depth of coverage method; and (3) Manta (manta-1.5.0.Centos6) which detects read-pair aberration combined with split-read evidence [7–9]. SVs were then annotated and scored according to the number of callers having detected the variant, by giving a greater weight to Wisecondor which has higher specificity. They are then prioritized and presented in a web interface, named PolyCyto (‛in-house’ software). Allelic frequencies for SNVs along the chromosome 16 were called by GATK Haplotypecaller (version 4) and plotted [10].
Optical genome mapping (OGM)
Ultra high molecular weight DNA was isolated from both patients using the SP Blood and Cell Culture DNA Isolation Kit (SKU 80030, Bionano Genomics®, San Diego, CA, USA) and fluorescently labeled with the enzyme DLE-1 (Bionano Genomics) using the DLS DNA Labeling Kit (SKU 80005, Bionano Genomics ®, San Diego, USA). Labeled DNA was loaded on Saphyr chip and imaged on the Saphyr instrument, for collection of 1300 Gb of molecules >150 kb [11, 12]. For all samples, a minimum of 320 Gb of data was acquired. First, an automatic whole genome de novo assembly was performed for each sample. De novo genome assemblies and variant calling were performed via Bionano Access software (v1.4.3) using the Bionano Tools version 1.4.3 for assembly and variant calling using pipeline version 7981 and RefAligner version 7989. The Bionano tools version includes a CN analysis tool that is performed as part of the de novo assembly pipeline. The CN analysis tool analyzes an input molecule-to-reference alignment, normalizes this raw molecule coverage profile using control data, and segments the genome based on detected changes in the underlying CN state. CNV calls are output and annotated with confidence scores (set at 0.66). Variants were called against GRCh38/hg38 human genome reference using default filter setting except for translocations (Ins: 0, Del:0, Inv: 0.01, Dup: −1, Trans: −1) and only rare structural variation larger than 5 kb and absent from Bionano control sample database were considered. In order to increase the contiguity of the assembled optical maps, the assembly did not perform any break at SDs (no -SplitSegDup). A targeted approach was performed to further refine the assembly at the 16p13.11p11.2 reference sequence. Briefly, molecules larger than 700 kb were subsampled from the original molecules file and aligned against 16p13.11p11.2. All aligned molecules covering this locus were merged into highly accurate consensus maps using stringent merging threshold to avoid merging non-allelic homologous regions.
Results
Identification of 16p13.11p11.2 triplication and 16p11.2 duplication in both patients
According to CMA probes, patients 1 and 2 harbor a 16p13.11p11.2 heterozygous triplication which is approximately 13 Mb in size and flanked by SDs: NC_000016.9:g.(15,154,747_15,492,317)_(28,335,137_28,543,103)trip (Fig. 1A, B). According to the Database of Genomic Variants, this triplication has not been previously reported [13]. The centromeric SD (BP1) is located at the genomic position chr16:28.300.000-28.500.000 (GRCh37/hg19). The telomeric SD (unnamed distal SD, udSD) is located at the genomic position chr16:14.800.000-15.500.000 and is composed of two main duplicons which are in an inverted orientation (chr16:14.865.000-15.000.000 and chr16:15.255.000-15.450.000, respectively). This triplicated segment encompasses 179 genes, including 100 protein-coding genes (Supplementary Fig. 2 and Supplementary Table 1). Dual-color BAC FISH analysis using the probe pair RP11–489O1 (located proximal to udSD) and RP11–152L13 (just distal to BP1) (Supplementary Fig. 2) localized the triplication on one of the chromosomes 16, excluding a chromosomal insertion. In addition, this approach revealed three repeats on the rearranged chromosome 16, with the middle repeat inverted (Fig. 2A, B). FISH analyses of the parental samples demonstrated that the triplication occurred de novo (data not shown). The CMA also showed that both patient harbor a 16p11.2 heterozygous duplication, albeit with different sizes. In detail, a duplication between 330 and 700 kb in size, NC_000016.9:g.(28,335,138_28,543,163)_(28,876,705_29,042,119)dup, was identified in patient 1, whereas a duplication between 30 and 500 kb, NC_000016.9:g.(28,335,138_28,543,103)_(28,574,419_28,837,449)dup, was detected in patient 2. These duplications are flanked by SDs: in both patients, the telomeric SD is BP1, whereas the centromeric SD is BP3 (chr16:29,000,000-29,700,000) for patient 1 and BP2 (chr16:28,600,00-28,800,000) for patient 2. For patient 1, dual-color FISH using the CTD-2515C15 and RP11–489O1 probes illustrated that the distal BP1–BP3 interval was located between the two distal udSD-BP1 repeats (Fig. 2C, D), although with this technique it was not possible to establish the orientation of the BP1–BP3 repeats. FISH analyses of the parental samples demonstrated that the duplication occurred de novo for patient 1 (data not shown). For patient 2, the duplication could not be confirmed by FISH analysis owing to the poor quality sample with insufficient metaphases.
Fig. 1. Identification of triplication and duplication events on the short arm of chromosome 16 using CMA.

A, B CMA profiles for patients 1 and 2. Identification of a 16p13.11p11.2 triplication between udSD and BP1 for both patients (log2 ratio at 1). Identification of a 16p11.2 duplication (log2 ratio at 0.58) between BP1 and BP3 for patient 1 and between BP1 and BP2 for patient 2. C Schematic representation of the short arm of chromosome 16. The centromere is represented by a black circle. The centromere and the telomere are indicated by “cen” and “tel”, respectively. The SDs are indicated by hatched rectangles. Their genomic positions (according to human genome Build GRCh37/Hg19) are shown on the left. The six SDs of interest (udSD and BP1 to BP5) are indicated on the right [18]. The major genes of the region are indicated in the gray rectangles.
Fig. 2. FISH results and schematic representation of the short arm of chromosome 16.

FISH results (A–C) and schematic representation of the short arm of chromosome 16 for patient 1 (D). A, B, D Hybridization with BAC RP11–489O1 (red) and RP11–152L13 (green) revealed two signals of different size for each probe on the rearranged chromosome 16 (white arrow) in patient 1 (A) and 2 (B): the distal green signal is larger than the proximal one, and the proximal red signal is larger than the distal one. The hybridization pattern is consistent with the presence of three udSD-BP1 repeats, of which the middle one is inverted, as shown by the scheme in D (black-dotted arrows). C, D Hybridization with BAC CTD-2515C15 (blue) revealed two signals of similar size on the rearranged chromosome 16 (white arrow) for patient 1 (C), which is consistent with the presence of two BP1–BP3 repeats. In addition, FISH analysis revealed that the distal blue signal was located between the red signals, thus clarifying that BP1–BP3 repeats are not side by side. However, the orientation of these repeats could not be specified (D).
Whole genome sequencing was insufficient to precisely map chromosomal rearrangements
To further investigate the complex chromosomal rearrangement, we performed WGS analysis in patient 1. Out of three bioinformatics approaches we explored (Wisecondor, Canvas, and Manta), only Wisecondor revealed a genomic gain at the 16p13.11p11.2 region with a size of 14.16 Mb. Initially, three regions were detected which were grouped into one according to their proximity. Among those three segments, two had a mean log2 ratio (observed CN/expected CN) greater than 0.7 (indicating a triplication) and one of them had a ratio of 0.43 (indicating a duplication) (Supplementary Fig. 3A). The genomic gain at the 16p13.11p11.2 region is clearly detected as a heterozygous triplication (NC_000016.9:g.14,890,000-28,350,000trip), followed by a heterozygous duplication (NC_000016.9:g.28,480,001-29,050,000dup). In contrast the Canvas approach revealed a genomic gain at the 16p13.11p12.2 region which was 6.32 Mb in size (NC_000016.9:g.16:15,430,637-21,746,874trip). This segment and the one found by Wisecondor were not considered as a single event because they do not meet the criterion of 60% overlap of their reciprocal length. The Manta approach did not identify any genomic gains or losses in the 16p-arm. We also used WGS dataset to simulate a SNP array. The allelic frequencies analysis showed four bands corresponding to 25%, 50, 75 and 100% (the genomic positions with 0% of alternative allele were not plotted because they were not called). There was no loss of heterozygosity distal to the rearrangements (Supplementary Fig. 3B). While the WGS analysis allowed us to confirm the genomic gains and simulate a SNP array, the precise orientations and organization of the duplicated and triplicated segments were still unclear.
Deciphering complex chromosomal rearrangements with optical genome mapping
To overcome limitations of CMA and WGS, we performed OGM, a technique that enables constitutional chromosomal aberration detection. In both patients the analysis confirmed the 16p13.11p11.2 triplication (number of copies = 4), which is ~ 13 Mb in size (udSD-BP1 region, NC_000016.10:g.15,322,000_28,469,000trip or NC_000016.10:g.15,322,000_28,398,000trip) (Fig. 3A, E). The proximal breakpoints of the triplication are slightly different between the two patients but are located at the same SD (BP1). In patient 1 we detected a 16p11.2 duplication (number of copies = 3), which is ~ 550 kb in size and encompassing the SH2B1 gene (BP1–BP3 region, NC_000016.10:g.28,469,000_29,039,000dup). In patient 2, a 16p11.2 duplication, which is ~300 kb in size (BP1–BP2 region, NC_000016.10:g.28,398,000_28,716,000dup), was identified. Given the large size of these chromosomal rearrangements, the de novo assembly pipeline was complemented by a targeted assembly of this region. Thus, a thorough analysis of the optical maps covering the junction points between two adjacent rearranged segments allowed us to unravel the organization of the region (Fig. 3B, C, F, G). Our OGM analysis revealed in both patients that the three copies of udSD-BP1 region are in a direct-inverted-direct orientation and the two copies of either BP1–BP3 or BP1–BP2 regions are in a direct orientation. On patient 1’s rearranged chromosome 16, the segments are organized as follows from telomere to centromere: udSD-BP1 region (direct)—BP1–BP3 region (direct)—udSD-BP1 region (inverted)—udSD-BP1 region (direct)—BP1–BP3 region (direct) (Fig. 3D). For patient 2, the organization of these segments is identical, except that the duplication involves the BP1–BP2 region, instead of the BP1–BP3 region (Fig. 3H).
Fig. 3. Schematic representation of the chromosomal rearrangements identified by optical genome mapping.

A, E Detection of the 16p13.11p11.2 triplication (Copy Number = 4, udSD-BP1 region) for both patients and detection of the 16p11.2 duplication (Copy Number = 3, BP1–BP3 region for patient 1 and BP1–BP2 region for patient 2). B, C, F, G Optical maps spanning the junctions between adjacent segments. The patients’ optical maps are indicated in blue, the genome reference hg38’s are indicated in green. The molecules used to assemble the optical maps are indicated in yellow. B, F Optical maps spanning the junction between either the BP1–BP3 region (patient 1) or BP1–BP2 region (patient 2) and the second copy of udSD-BP1 region. On the rearranged chromosome, BP1 in an inverted orientation is located directly proximal to BP3 or BP2 (for patient 1 and 2, respectively) proving that the middle udSD-BP1 repeat has an inverted orientation. C, G Optical maps spanning the junction between the two proximal udSD-BP1 repeats. The junction is located in the distal udSD which is ~700 kb in size. The inverted sequences allow us to prove that the two proximal udSd-BP1 have inverted orientations. D, H Schematic representation of the rearrangement of the chromosome 16 for both patients. The three udSD-BP1 repeats have a direct-inverted-direct orientation. The two BP1–BP3 or BP1–BP2 repeats (for patient 1 and 2, respectively) have a direct orientation and the distal repeat is located between the two distal udSD-BP1 repeats.
Discussion
In this study, we identified and molecularly characterized two patients with a 16p13.11p11.2 triplication which is ~13 Mb in size. In addition, we found that patient 1 harbors a 16p11.2 duplication involving the SH2B1 gene between BP1 and BP3, while a different 16p11.2 duplication between BP1 and BP2, not involving the SH2B1 gene, was detected in patient 2. In order to determine the mechanisms underlying the chromosomal rearrangements in the two patients, three combined technical approaches (FISH, WGS, OGM) were employed. Full genetic characterization of these types of complex rearrangement events is challenging, especially since the breakpoints lie within SDs. These repeat sequences, which correspond to blocks of DNA ranging from 1 to 400 kb in length with at least 90% of identity, represent 5% of the genome [14, 15]. NAHR between directly oriented SDs results in recurrent deletion and duplication of the intervening sequence. Approximately 10% of chromosome 16 consists of SDs which is among the most enriched compared to the other autosomes [16]. They are particularly clustered along the p arm of the chromosome. Due to this density of SDs, chromosome 16p shows a high frequency of genomic rearrangements such as at the BP2–BP3 and BP4–BP5 loci [4, 17]. These recurrent CNVs mediated by NAHR between directly oriented SDs are associated with neurodevelopmental disorders with highly variable expressivity and incomplete penetrance. Triplications involving neurosusceptibility loci are far less frequent. For example, several patients with a triplication of the 22q11.2 or 1q21 regions have been reported [18, 19]. A NAHR mechanism has also been proposed to explain these genomic imbalances. It is worth noting that complex rearrangements have been increasingly unraveled due to the greater resolution of the advanced genome technologies such as cases of duplications containing an embedded triplicated and inverted segment at the MECP2 and PLP1 loci [20]. Replication-based mechanisms (RBM), involving a low-processivity polymerase, have been proposed to explain the formation of these complex chromosomal anomalies [21]. Regarding large intrachromosomal triplications visible by conventional karyotyping, few case reports involving the chromosomes 3, 7, 13, 15, and 21 have been reported [22–26]. Proposed mechanisms include U-type exchanges associated in some cases with NAHR. Finally, interstitial duplications/triplications in conjunction with uniparental isodisomy, generated by a microhomology-mediated break-induced replication-based mechanism, were found in patients with syndromic intellectual disability [27, 28].
In the present patients, CMA detected a 16p13.11p11.2 triplication which is ~13 Mb in size. In addition, the recurrent 16p11.2 duplication involving the SH2B1 gene between BP1 and BP3 was found in patient 1. Another 16p11.2 duplication not involving the SH2B1 gene, between BP1–BP2, was detected in patient 2. The exact localization of the breakpoints was not possible by short-reads WGS because the detection of a junction-spanning sequence read or read-pair within large highly identical SDs is highly unlikely [29]. Thus, the relative orientation of the triplicated/duplicated segments as well as the exact genomic positions of the breakpoints at the molecular level could not be determined by short-reads-based technologies [30]. Currently, OGM has emerged as a promising tool for characterizing all classes of SV, even the most complex ones. Unlike CMA or long read sequencing, this technique leverages molecules of several hundred of kilobase pairs to provide information about location and orientation of segments involved in chromosomal rearrangements, regardless of the presence of repetitive DNA at the junctions. As shown in our study, this technology offers a more complete and comprehensive understanding of the chromosomal abnormalities found in our two patients. Our data demonstrated that the two copies of the 16p11.2 region (BP1–BP3 for patient 1 and BP1–BP2 for patient 2) are directly oriented and located between the two distal triplicated segments and at the boundary of the proximal triplicated segment (Fig. 3). According to SNP data from WGS (i.e., the presence of four bands of SNPs at BAF 0.25, 0.5, 0.75 and 1), we can deduce that the chromosomal rearrangement occurred during meiosis and not in the postzygotic stage during the mitotic divisions [27, 28]. The generation of the triplicated segment requires the involvement of three chromatids and, thus, the formation of a transient dicentric chromosome resulting from a recombination between two sister chromatids. This recombination may have occurred during pre-meiotic S-phase to correct a double-strand break or, alternatively, during the pachytene stage of the first meiotic prophase. The event is located at the distal SD (udSD) for both patients and could have been facilitated by homologous sequences in an inverted orientation located in the same SD. It is well known that SDs predispose to NAHR as well as RBM events [21]. Both hypotheses can be supported by our data. The event between BP1–BP3 or BP1–BP2 for patient 1 and 2, respectively, is likely a NAHR event leading to the observed rearrangements (trip-dup) (Fig. 4). It is possible that the triplication and duplication may have resulted from either two simultaneous events or a two-steps process. The mechanism proposed is almost similar to cases previously reported involving the chromosomes 7 and 15 [26, 31, 32].
Fig. 4. Proposed mechanism to explain the chromosomal rearrangements observed in the two patients.

The SDs are represented by colored rectangles. The SH2B1 gene is represented by a yellow star. A recombination at a distal SD (udSD) between sister chromatids would have allowed the formation of a transient dicentric chromosome 16 for both patients. This event may have occurred during either pre-meiotic S-phase to correct a double-strand break (DSB) or the pachytene stage of the first meiotic prophase. The acentric fragment is lost. A NAHR between the SDs BP1–BP3 for patient 1 and the SDs BP1–BP2 for patient 2 could lead to the formation of 3 copies of udSD-BP1 region in a direct-inverted-direct orientation and two copies of the regions BP1–BP3 or BP1–BP2 for patient 1 and 2, respectively, in a direct orientation. The positions and orientations of the triplicated and duplicated segments are represented by black-dotted arrows and black arrows, respectively. The dicentric segment is presumed to be lost.
The two patients reported here share similar clinical features that make this condition clinically recognizable. They present with profound psychomotor delay, hypotonia, severe cognitive impairment, behavioral abnormalities and recurrent episodes of otitis media with hearing loss. They also display a distinct facial gestalt characterized by enophtalmia, prominent chin, and a long and smooth philtrum (Supplementary Fig. 1). To our knowledge, no patient with a similar triplication has been reported. Only three patients harboring a duplication of bands p11 to p13 detected by karyotyping but not characterized by CMA were described [33, 34]. Some of the clinical features of these individuals resemble those in our patients, such as psychomotor retardation, lack of speech, deeply set eyes and long philtrum. The phenotype of the two cases with overlapping triplications reported in the DECIPHER database (i.e., cases 263405 and 363860) is not well specified. In addition, the triplicated segment encompasses the 16p13.11 recurrent microduplication locus, which is generally 1.5 Mb in size [35–39]. The miR-484 (MicroRNA 484) and the NDE1 gene located at this locus seem to be involved in the neurodevelopmental phenotype of these patients [35]. However, given the controversial data regarding the clinical impact of this duplication and the lack of characteristic features, it is difficult to determine the contribution of this region in the phenotype of our two patients. Furthermore, in the present patients the triplicated segment encompasses 179 genes, including 100 protein-coding genes among which 24 are disease-associated genes (Supplementary Table 1) [5]. According to GTEx v.8, 18 protein-coding genes (namely, ARL6IP1, BMERB1, CACNG3, CCP110, CRYM, GPRC5B, GTF3C1, HS3ST4, LCMT1, NDE1, NPIPA5, PRKCB, SBK1, SLC5A11, SMG1, TNRC6A, UBFD1, and ZP2) are highly and specifically expressed in the brain or known to be involved in neurodevelopmental disorders, which suggest that they might play a role in the onset of the neurodevelopmental phenotype described here (Supplementary Table 1).
Although there is no evidence for triplosensitivity for any of the triplicated genes according to ClinGen, 13 out of 100 protein-coding genes (namely, CACNG3, GTF3C1, IL21R, MARF1, MYH11, PLK1, PRKCB, RBBP6, SMG1, TNRC6A, UBFD1, XPO6, and XYLT1) have a pLI (probability of being loss of function intolerant) ≥0.9, which indicates that they are likely intolerant to loss of function variants (Supplementary Table 1) [40, 41]. It is worth noting that a haploinsufficiency gene might be sensitive to overdosage as well, and several examples have been reported in the literature [42]. Therefore, by intersecting the protein-coding, brain-expressed, and haploinsufficient gene lists, we support CACNG3, GTF3C1, PRKCB, SMG1, TNRC6A, and UBFD1 as the best candidates. SMG1 (SMG1 nonsense-mediated mRNA decay-associated PI3K-related kinase) is involved in cell growth, in nonsense-mediated mRNA decay, genotoxic and oxidative stress pathways, and TNF-induced apoptosis. PRKCB (Protein kinase C beta) is a member of PKC family whose members phosphorylate a wide variety of protein targets and are known to be involved in diverse cellular signaling pathways. CACNG3 (calcium channel, voltage-dependent, gamma-3 subunit) encodes a type 1 transmembrane AMPA receptor regulatory protein which regulate trafficking and channel gating of the AMPA receptors. TNRC6A (trinucleotide repeat-containing gene 6A) encodes a component of a cytoplasmic ribonucleoprotein complex involved in regulating mRNA silencing, stability, and translation. Pathogenic variants in this gene have been reported in patients presenting with myoclonic epilepsy (OMIM 618074). GTF3C1 (General Transcription Factor IIIC Subunit 1) is a component of RNA polymerase III that binds B box promoter elements. UBFD1 may play a role as NF-kappa-B regulator [43].
Furthermore, the triplicated segment also encompasses several microRNAs and long non-coding RNAs (lncRNAs) including MIR3180-2 and MIR3180-3. Recently, Shi et al. showed that these microRNAs were frequently co-expressed with known Alzheimer’s disease risk protein-coding genes [37]. Thus, we speculate that the cumulative effects of many dysregulated genes and lncRNAs/miRNAs contribute to the clinical syndrome described in this study.
Finally patient 1 also harbors a duplication of a ∼550 kb region, flanked by SDs, at the 16p11.2 locus. Recurrent deletions of this region that include the SH2B1 gene predispose to severe early-onset obesity, developmental delay, intellectual disability, schizophrenia, and ASD [4, 44]. Conversely, 16p11.2 microduplication carriers have lower body mass index, reduced head circumference, mild cognitive impairment, and ASD [44, 45]. Recently, Sadler et al. suggested that this CNV may also contribute to scoliosis pathogenesis [46]. The clinical impact of this 16p11.2 duplication in patient 1 is difficult to assess since it is associated with a large chromosomal rearrangement. Regarding patient 2, the 16p11.2 microduplication between BP1–BP2 does not encompass the SH2B1 gene and has never been reported in the literature in patients with neurodevelopmental disorder. The severity of the phenotype of our two patients may essentially be explained due to the gene dosage effect of the triplication. Additional patients will be necessary to improve the genotype-phenotype correlation.
Conclusion
We report the first two patients with a 16p13.1p11.2 triplication associated with a 16p11.2 duplication between either BP1–BP3 or BP1–BP2. This recognizable genomic disorder is characterized by hypotonia, severe neurodevelopmental delay with profound speech impairment, hyperkinetic behavior, hearing loss, and distinctive facial features. Unlike CMA and short-read based NGS technologies, OGM was the only technology allowing us to fully characterize these complex chromosomal rearrangements involving SDs and to propose a mechanism that explains its formation.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the patients and their parents for their participation in this study. We thank Bionano Genomics for their technical assistances. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on 01/10/22.
Author contributions
RN and VM contributed to synthetizing and reviewing data, and writing of the manuscript. MR, AG, KSP, EO, PN, CBF, SR and AH contributed to generation of data and provided feedback on the manuscript. VM and VC provided project supervision and reviewed the manuscript.
Funding
This work was supported by State funding from the Agence Nationale de la Recherche under “Investissements d’avenir” program (ANR-10-IAHU-01) and the MSDAvenir fund (DEVO-DECODE project) and was also supported by the Fondation pour la Recherche Médicale (grants number M2R201906008521 to Romain NICOLLE and DEQ20160334938 to Vincent CANTAGREL).
Data availability
All data generated or analyzed during this study are included in the published article (and its Supplementary Information files).
Competing interests
The authors declare no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individuals included in this study.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01094-x.
References
- 1.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 4.Bachmann-Gagescu R, Mefford HC, Cowan C, Glew GM, Hing AV, Wallace S, et al. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet Med J Am Coll Med Genet. 2010;12:641–7. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 5.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–33. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straver R, Sistermans EA, Holstege H, Visser A, Oudejans CBM, Reinders MJT. WISECONDOR: detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2014;42:e31. doi: 10.1093/nar/gkt992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roller E, Ivakhno S, Lee S, Royce T, Tanner S. Canvas: versatile and scalable detection of copy number variants. Bioinformatics. 2016;32:2375–7. doi: 10.1093/bioinformatics/btw163. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 10.Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Genomics. 2017 doi: 10.1101/201178. [DOI] [Google Scholar]
- 11.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol. 2012;30:771–6. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barseghyan H, Tang W, Wang RT, Almalvez M, Segura E, Bramble MS, et al. Next-generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017;9:90. doi: 10.1186/s13073-017-0479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–92. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 15.Cheung J, Estivill X, Khaja R, MacDonald JR, Lau K, Tsui L-C, et al. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin J, Han C, Gordon LA, Terry A, Prabhakar S, She X, et al. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004;432:988–94. doi: 10.1038/nature03187. [DOI] [PubMed] [Google Scholar]
- 17.Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49:660–8. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yobb TM, Somerville MJ, Willatt L, Firth HV, Harrison K, MacKenzie J, et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am J Hum Genet. 2005;76:865–76. doi: 10.1086/429841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Dijck A, van der Werf IM, Reyniers E, Scheers S, Azage M, Siefkas K, et al. Five patients with a chromosome 1q21.1 triplication show macrocephaly, increased weight and facial similarities. Eur J Med Genet. 2015;58:503–8. doi: 10.1016/j.ejmg.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Carvalho CMB, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;43:1074–81. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–38. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vialard F, Mignon-Ravix C, Parain D, Depetris D, Portnoï M-F, Moirot H, et al. Mechanism of intrachromosomal triplications 15q11-q13: a new clinical report. Am J Med Genet A. 2003;118A:229–34. doi: 10.1002/ajmg.a.10164. [DOI] [PubMed] [Google Scholar]
- 23.Rivera H, Bobadilla L, Rolon A, Kunz J, Crolla JA. Intrachromosomal triplication of distal 7p. J Med Genet. 1998;35:78–80. doi: 10.1136/jmg.35.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Expósito I, Bafalliu JA, Santos M, Fuster C, Puche-Mira A, Guillén-Navarro E. Intrachromosomal partial triplication of chromosome 13 secondary to a paternal duplication with mild phenotypic effect. Am J Med Genet A. 2008;146A:1190–4. doi: 10.1002/ajmg.a.32187. [DOI] [PubMed] [Google Scholar]
- 25.Ounap K, Ilus T, Bartsch O. A girl with inverted triplication of chromosome 3q25.3 –> q29 and multiple congenital anomalies consistent with 3q duplication syndrome. Am J Med Genet A. 2005;134:434–8. doi: 10.1002/ajmg.a.30134. [DOI] [PubMed] [Google Scholar]
- 26.Castronovo C, Crippa M, Bestetti I, Rusconi D, Russo S, Larizza L, et al. Complex de novo chromosomal rearrangement at 15q11-q13 involving an intrachromosomal triplication in a patient with a severe neuropsychological phenotype: clinical report and review of the literature. Am J Med Genet A. 2015;167A:221–30. doi: 10.1002/ajmg.a.36815. [DOI] [PubMed] [Google Scholar]
- 27.Sahoo T, Wang J-C, Elnaggar MM, Sanchez-Lara P, Ross LP, Mahon LW, et al. Concurrent triplication and uniparental isodisomy: evidence for microhomology-mediated break-induced replication model for genomic rearrangements. Eur J Hum Genet. 2015;23:61–6. doi: 10.1038/ejhg.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohmoto T, Okamoto N, Naruto T, Murata C, Ouchi Y, Fujita N, et al. A case with concurrent duplication, triplication, and uniparental isodisomy at 1q42.12-qter supporting microhomology-mediated break-induced replication model for replicative rearrangements. Mol Cytogenet. 2017;10:15. doi: 10.1186/s13039-017-0316-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuttle X, Itsara A, Shendure J, Eichler EE. Resolving genomic disorder-associated breakpoints within segmental DNA duplications using massively parallel sequencing. Nat Protoc. 2014;9:1496–513. doi: 10.1038/nprot.2014.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alkan C, Sajjadian S, Eichler EE. Limitations of next-generation genome sequence assembly. Nat Methods. 2011;8:61–5. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ungaro P, Christian SL, Fantes JA, Mutirangura A, Black S, Reynolds J, et al. Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J Med Genet. 2001;38:26–34. doi: 10.1136/jmg.38.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al Dhaibani MA, Allingham-Hawkins D, El-Hattab AW. De novo chromosome 7q36.1q36.2 triplication in a child with developmental delay, growth failure, distinctive facial features, and multiple congenital anomalies: a case report. BMC Med Genet. 2017;18:118. doi: 10.1186/s12881-017-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen MM, Lerner C, Balkin NE. Duplication of 16p from insertion of 16p into 16q with subsequent duplication due to crossing over within the inserted segment. Am J Med Genet. 1983;14:89–96. doi: 10.1002/ajmg.1320140114. [DOI] [PubMed] [Google Scholar]
- 34.Behjati F, Shafaghati Y, Firouzabadi SG, Kahrizi K, Bagherizadeh I, Najmabadi H, et al. M-banding characterization of a 16p11.2p13.1 tandem duplication in a child with autism, neurodevelopmental delay and dysmorphism. Eur J Med Genet. 2008;51:608–14. doi: 10.1016/j.ejmg.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Allach El Khattabi L, Heide S, Caberg J-H, Andrieux J, Doco Fenzy M, Vincent-Delorme C, et al. 16p13.11 microduplication in 45 new patients: refined clinical significance and genotype–phenotype correlations. J Med Genet. 2020;57:301–7. doi: 10.1136/jmedgenet-2018-105389. [DOI] [PubMed] [Google Scholar]
- 36.Tropeano M, Ahn JW, Dobson RJB, Breen G, Rucker J, Dixit A, et al. Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS ONE. 2013;8:e61365. doi: 10.1371/journal.pone.0061365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuang S-Q, Guo D-C, Prakash SK, McDonald M-LN, Johnson RJ, Wang M, et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011;7:e1002118. doi: 10.1371/journal.pgen.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hannes FD, Sharp AJ, Mefford HC, de Ravel T, Ruivenkamp CA, Breuning MH, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46:223–32. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ullmann R, Turner G, Kirchhoff M, Chen W, Tonge B, Rosenberg C, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28:674–82. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]
- 40.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372:2235–42. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carmona-Mora P, Walz K. Retinoic acid induced 1, RAI1: a dosage sensitive gene related to neurobehavioral alterations including autistic behavior. Curr Genomics. 2010;11:607–17. doi: 10.2174/138920210793360952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online Mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–98. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loviglio MN, Leleu M, Männik K, Passeggeri M, Giannuzzi G, van der Werf I, et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol Psychiatry. 2017;22:836–49. doi: 10.1038/mp.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loviglio MN, Arbogast T, Jønch AE, Collins SC, Popadin K, Bonnet CS, et al. The immune signaling adaptor LAT contributes to the neuroanatomical phenotype of 16p11.2 BP2-BP3 CNVs. Am J Hum Genet. 2017;101:564–77. doi: 10.1016/j.ajhg.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadler B, Haller G, Antunes L, Bledsoe X, Morcuende J, Giampietro P, et al. Distal chromosome 16p11.2 duplications containing SH2B1 in patients with scoliosis. J Med Genet. 2019;56:427–33. doi: 10.1136/jmedgenet-2018-105877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the published article (and its Supplementary Information files).