

Abstract
Tumor Necrosis Factor-α (TNFα) is a key mediator of immune and radiation-induced cytotoxicity, but many cancers, including head and neck squamous cell carcinomas (HNSCC), display TNF resistance due to activation of the canonical IKK-NF-κB/RELA pro-survival pathway. However, toxicities associated with direct targeting of the canonical pathway point to the need to identify mechanism(s) contributing to TNFα resistance and synthetic lethal targets to overcome such resistance in cancer cells. Here, RNAi screening for modulators of TNFα-NF-κB reporter activity and cell survival unexpectedly implicated the WEE1 and CDC2 G2/M checkpoint kinases. The IKKα/β-RELA and WEE1-CDC2 signaling pathways are activated by TNFα and form a complex in cell lines derived from both Human Papilloma Virus (−) and (+) subtypes of HNSCC. WEE1 inhibitor AZD1775 reduced IKK/RELA phosphorylation and the expression of NF-κB-dependent pro-survival proteins Cyclin D1 and BCL2. Combination of TNFα and AZD1775 enhanced caspase-mediated apoptosis in vitro, and combination treatment with radiation and AZD1775 potentiated inhibition of HNSCC tumor xenograft growth in vivo, which could be significantly attenuated by TNFα depletion. These data offer new insight into the interplay between NF-κB signaling and WEE1-mediated regulation of the G2/M cell cycle checkpoint in HNSCC.
Keywords: NF-κB, IKKα/β, TNFα, WEE1, CDC2/CDK1, AZD1775/Adavosertib, radiation, head and neck cancer
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, with over 600,000 cases and around 300,000 deaths per year (1), with the primary causes including alcohol consumption and smoking or infection with Human Papillomavirus (HPV). The current standard therapeutic options include surgery, radiotherapy, and chemotherapies, with the more recent approval of immune targeted therapies with anti-programmed cell death protein (PD-1) therapy (pembrolizumab) in advanced stage, recurrent, and/or metastatic HNSCC (1). The survival rate for HNSCC can be stratified based on HPV status of the tumor. HPV+ HNSCC has a 3-year overall survival (OS) rate of around 80%, whilst for HPV− HNSCC it is around 55% (1). Thus, despite advances and differences in response to standard treatment, there is still a need for investigation and development of better and less toxic therapeutics in both HPV− and + HNSCC.
Tumor necrosis factor-α (TNFα) is an important mediator of inflammatory, immune, and radiation-induced cytotoxic therapies in cancer (2,3,4). TNFα can induce cell death extrinsically via the TNF Receptor (TNFR), or intrinsically via induction of reactive oxygen species (ROS) and genotoxic DNA damage (5,6). However, the cytotoxic effects of TNFα and genotoxic therapies are often attenuated via activation of the Inhibitor-kappaB kinase α/β/γ complex (IKK), which promotes phosphorylation and degradation of inhibitor kappaB (IκB), and activation of the heterodimeric transcription factor NF-κB1/RELA. RELA phosphorylation at serine 536 (S536) by IKK induces NF-κB1/RELA-dependent transcription, including the expression of the G1 cell cyclin CCND1 and anti-apoptosis protein BCL2, that can enhance cell proliferation and resistance to caspase and mitochondrial-dependent cell death, respectively (6).
We and others have shown that the canonical IKK-NF-κB/RELA pathway is often aberrantly activated in head and neck squamous cell carcinomas (HNSCC) and promotes resistance to the effects of TNFα, immune mediated, and radiation-induced cytotoxicity (7). As part of the The Cancer Genome Atlas (TCGA), we found that HNSCC subsets harbor genomic alterations in components that mediate signaling between TNF receptor family complexes and the NF-κB and cell death pathways, and that the components affected differ between human papillomavirus (HPV) negative and positive cancers (8). Further, HPVE6 can induce NF-κB activation in cervical squamous cell carcinomas, contributing to proliferation and survival (9). However, the mechanism(s) orchestrating NF-κB activation and TNFα resistance in HNSCC and other cancers are incompletely understood.
The genotoxic effects of immune or other DNA damaging therapies may also be opposed by DNA repair mechanisms during the cell cycle checkpoints at G0/G1 or G2/M (10). In HNSCC and other cancers, the G0/G1 checkpoint is often disabled by mutation or HPV E6-mediated inactivation of the tumor suppressor protein TP53 (11). In cells with defective TP53, the DNA repair process relies on activation of the G2/M cell cycle checkpoint. WEE1 is a key kinase controlling the G2/M checkpoint via phosphorylation and inactivation of the cyclin-dependent kinase CDC2 (CDK1) at tyrosine 15 (Y15) (12,13). Activation of WEE1 and CDC2 (Y15) phosphorylation has been reported in many cancer types, including HNSCC (12–14). The WEE1/CDC2 axis promotes a G2/M checkpoint pause, enabling repair of DNA double strand breaks before entering mitosis. AZD1775 is a pyrazole-pyrimidine derivative and is a first-in-class, potent adenosine triphosphate (ATP)-competitive small-molecule inhibitor of WEE1/2 (12,14). AZD1775 has demonstrated anti-tumor effects in HNSCC and other cancers as a single agent and/or in combination with DNA-damaging agents in clinical trials (12–14). In preclinical studies, we found that immune-mediated cytotoxicity was enhanced by AZD1775, and this could be partially rescued by inhibition of TNFα and other death ligands, or by excess nucleotides, potentially implicating both TNFα signaling and G2/M DNA repair mechanisms (15). However, how TNFα-induced activation of the IKK/NF-κB pathway and WEE1/CDC2 G2/M checkpoint signalling are mechanistically linked is unclear.
In an RNAi screen using an HPV(−) HNSCC cell line expressing NF-κB promoter response elements linked to a β-lactamase reporter gene (16), we unexpectedly found that siRNAs targeting WEE1, CDC2, and other G2/M cell cycle components inhibited TNFα induced NF-κB activity and cell density. Here, we demonstrate that WEE1/CDC2 G2/M checkpoint and IKK/NF-κB signaling mechanistically interact and that WEE1 inhibition sensitizes both HPV(−) and (+) HNSCC cells to TNFα–induced cell death in vitro and the TNFα-related anti-tumor effects of radiation in HNSCC xenografts in vivo. Analysis of TCGA HNSCC data indicate that increased WEE1 is associated with worse prognosis in HPV− and better prognosis in HPV+ tumor subsets, enriched for distinct genomic signatures linked to inflammation and NF-κB. As we previously found that WEE1 inhibition can more selectively sensitize tumor cells than immune cells to cytotoxicity (15), WEE1 inhibition could potentially enhance the effects of therapies mediated by TNFα with less systemic immune suppression and toxicity than observed with direct interruption of IKK-NF-κB/RELA signaling (6).
Materials and Methods
HNSCC cell lines
A panel of HNSCC cell lines from the University of Michigan squamous cell carcinoma (UMSCC) series were obtained from Dr. T.E. Carey. These UMSCC cell lines were authenticated by genotyping with 9 markers as described previously (17). UMSCC cell lines were cultured in minimal essential medium (MEM) supplemented with 10% fetal calf serum, penicillin and streptomycin (100 µg/mL), and MEM non-essential amino acids (NEAA). Human primary oral keratinocytes (HOK) from oral gingival mucosa were purchased from Science Cell Research laboratories and used as a control cell line. The cells were cultured in serum-free Oral Keratinocyte Medium with supplements (Science Cell) for less than four passages.
Genome-wide RNAi screening using an NF-κB reporter line
A stable reporter line was established by transfecting UMSCC1 cells with a pLenti-based vector containing 6 repeated κB binding sites upstream of a b-lactamase reporter gene (pLenti-bsd-NFκB-bla; created using pLenti6/V5-DEST™ Gateway™ vector (V49610; Life Technologies) and the following NF-κB response element sequence 5’CGCGGGGACTTTCCGCTTGGGGACTTTCCGCTGGGGACTTTCCGCTGGGGACTTTCCGCTGGGGACTTTCCGCGGAGACTCTAGAGGGTATATAATGGAAGCTCGAATTCCAGCTTGGCATTCCGGTACTGTTGGTAAAAGA’3) and a blasticidin resistance gene for selection (16). The β-lactamase reporter enzyme can cleave a fluorescent FRET substrate (LiveBLAzer™ FRET-B/G Loading Kit with CCF4-AM; Life Technologies, cat. #K1095), which disrupts FRET and results in blue fluorescence (Supplementary Figure 1A–C). The blue:green fluorescence ratio indicates the activity of the NF-κB reporter: cell viability. Following stimulation with TNFa, the most stimulated and responsive cells exhibiting blue FRET activity were isolated by fluorescence-activated cell sorting (FACS) (Supplementary Figure 1D–F). Using this stable cell line, UMSCC1κB, a large-scale RNAi screening of the kinome, and druggable genome was conducted to identify potential targets that modulate NF-kB activity by the RNAi screening facility, National Center for Advancing Translational Sciences (NCATS), NIH. Each siRNA library containing three distinct siRNAs targeted to the different regions of the same mRNA were transfected individually into UM-SCC-1 kB cells in wells of 384-well plates for 56 hr followed by a 16 hour treatment with TNFα (20ng/ml). Relative NF-κB activity (blue/green, fluorescent ratio) and viability (green FRET activity) was then observed via β-lactamase FRET assay, and effects on viability were independently determined by Cell Titer-Glo (CTG; Promega) assay for cytotoxic siRNAs. Replicate screens were performed to validate the assay and ensure reproducibility. Kinome siRNA libraries each contains siRNAs targeting 709 kinase mRNAs and Druggable Genome targeting 9031 mRNAs (Ambion Silencer Select libraries V4, ThermoFisher Scientific, Waltham, Massachusetts, USA. For the RNAi screen, Redundant siRNA Activity (RSA) analysis method was used to integrate the results of multiple siRNAs tested for the same gene (18).
Co-immunoprecipitation assay (Co-IP)
Co-IP experiments were performed using Pierce™ Crosslink Magnetic IP/Co-IP Kit (Thermo Fisher Scientific, USA) following the manufacturer’s protocol, except whole-cell lysates were collected using lysis buffer (1% Triton X-100, 0.5% deoxycholate, 1% NP-40, 50 mM Tris, 100 mM NaCl, 2 mM MgCl2, 10% glycerol). For each IP, 50 µg protein was incubated with 5 µL antibody: WEE1 (1:500; sc-5285, Santa Cruz Biotechnology (SCBT)), CDC2 (9116, CST), IKKα (11930, CST), IKKβ (8943, CST), IKKγ (2685, CST), RELA (3039, CST), RELB (1:500; 4922, CST) . For input lysates, 20 µg protein was loaded and the input and immunoprecipitated proteins were then analyzed by SDS-PAGE and Western blot. Western blot was performed as described in the supplementary methods. Non-immune rabbit IgG or mouse IgG was included as the negative control.
In vivo tumor studies
All in vivo experiments were approved by the NIDCD/NIH Animal Care and Use Committee under protocol #1322, in compliance with the Guide for the Care and Use of Laboratory Animal Resource National Research Council. Fifty-five 4 to 6-week-old female athymic nu/nu mice were obtained from Jackson laboratory and housed in a pathogen-free NIH animal facility. Two HPV− (UMSCC1 and UMSCC46) cell lines and one HPV+ cell line (UPCI:SCC090) were harvested and mixed with Matrigel (R & D systems) in a 70% cells:30% Matrigel solution. Each mouse was injected subcutaneously in the right thigh with 5×106 UMSCC46 cells. Tumor volumes were measured thrice weekly and mice were randomized into the respective groups when tumor volumes reached ~0.25 cm3. AZD1775 was given by oral gavage to mice as a suspension in 0.5% methyl cellulose. For UMSCC46 tumor xenografts, mice were divided into four groups; Control (0.5% Methylcellulose), AZD1775 (120 mg/kg), radiation (20 Gy total; 10 x 2 Gy) and combination (AZD1775 (120mg/kg) plus radiation (10 x 2 Gy)). For UPCI:SCC090 and UMSCC1 tumor xenografts, a lower dose of 60mg/kg AZD1775 was used in the drug alone and combination groups, based on additional activity and toxicity data and recommendations of the manufacturer. The treatments were conducted for 2 weeks in a 5-days-on, 2-days-off, 5-days-on schedule. For UMSCC1 tumor xenografts, an additional group of mice were intraperitoneally injected with mouse anti-TNFα antibody (200 μg per mouse) for 7 times. The injection was started 2 days before treatment, and then injected 3 time per week for the following 2 weeks with drug and radiation treatment. Mouse body weight, body condition, and tumor size was recorded two or three times a week for the indicated days. The tumor volume was calculated with the formula V=½ L*W2, and tumor growth was reported as the mean tumor volume +/− standard error. Kaplan-Meier survival analysis was performed using the GraphPad PRISM software (v6.05). p values for tumor growth were calculated using the Student’s t test. Survival statistics were performed using the Gehan-Breslow-Wilcoxon test.
Analyses of publicly available datasets
Analysis of gene and protein expression in HNSCC cases was performed using publicly available data from the following resources: TCGA (https://portal.gdc.cancer.gov/repository) (8,19), Clinical Proteomic Tumor Analysis Consortium (NCI/NIH) (CPTAC, https://cptac-data-portal.georgetown.edu) (20,21), DepMap Public 20Q2 release (https://depmap.org/portal/) (22), Project Achilles (23,24), and the Gene Expression Omnibus (GEO; GSE40774 (25) and GSE117973). Further analysis details are in the supplementary methods.
Statistical Analysis
Cell viability was analyzed by multiple linear regression of TNF addition and log-transformed concentration of AZD1775 to determine statistical significance. Multiple regression permits the simultaneous handling of many variables to isolate the effect of each factor adjusted for the presence of the remainder. The analyses and data visualization were conducted in R. The Bliss synergy analysis and synergy score was calculated using SynergyFinder (26). Kaplan-Meier survival curves comparing PFS and OS for patients based on high versus low expression of the genes from the WEE1 and NFκB axis. The patients were dichotomized into high and low expression groups for each of these genes. The survminer R package was used to find the optimal cut-point that corresponds to the most significant relation with survival. Patient overall survival data were downloaded from Firehose at the Broad Institute (https://confluence.broadinstitute.org/display/GDAC/Home/). Patient progression-free survival data were downloaded from Supplement Information of a pan-cancer clinical study (27). The clinical survival endpoints for patients were progression-free survival (PFS), and overall survival (OS). PFS is the time from the date of diagnosis until tumor progression or death, whichever occurs first. OS is the period from the date of diagnosis until the date of death from any cause. The PFS and OS curves were obtained using Kaplan-Meier method and were compared using the log-rank test. The Cox proportional hazards model was used to estimate Hazard Ratios (HRs) with 95% Confidence Intervals (CIs).
For differential gene expression analysis between normal and tumor tissue, and HPV− and HPV+ tumor tissue, Wilcoxon sum rank test was used to assess statistical significance. All graphs were prepared using the GraphPad Prism (GraphPad, USA) or R software (28) version 4.0.2. Error bars represent mean ± the standard error. Statistical significance was determined as follows: NS = not significant, *p<0.05, **p<0.01, ***p<0.001.
Additional methods for Western blot, siRNA experiments, cell cycle analysis, Annexin V assay, impedance assay, XTT viability assay and colony formation assay are presented in the Supplemental Materials and Methods.
Results
RNAi screening identifies G2/M checkpoint kinases as modulators of TNFα-induced NF-κB activity and survival in HNSCC.
HPV(−) HNSCC have poorer prognosis, and we have previously shown that HPV(−) HNSCC cell line resistance to TNFα is promoted by IKK-NF-κB (29). In order to investigate the mechanism(s) driving TNFα-dependent NF-κB signaling and TNFα resistance in HNSCC, we established UMSCC1κB, which is an HPV− HNSCC cell line (UMSCC1) stably expressing an NF-κB response element-β-lactamase reporter gene, to enable detection of NF-κB activity normalized for cell viability by flow cytometry or spectrophotometry (Supplementary Figure 1). Using this cell line, we performed an RNAi screen using libraries targeting the human kinome and druggable genome, to identify potential regulators of the NF-κB pathway and cell survival (16). Supporting the veracity of this assay, the top hits identified included components of the IKK/NF-κB pathway (e.g. CHUK (IKKα), IKBKB (IKKβ) and RELA; Figure 1A and B). Unexpectedly, we also found that depletion of key regulators of the G2/M checkpoint, namely WEE1 and CDC2, also significantly inhibited NF-κB activity and cell viability (Figure 1A and B). Similar effects were observed with depletion of the same components queried in replicate kinome and the druggable genome screens.
Figure 1. Kinome and genome-wide siRNA screen identifies G2/M kinases WEE1 and CDC2 as regulators of NF-κB signaling and survival in HNSCC cells.
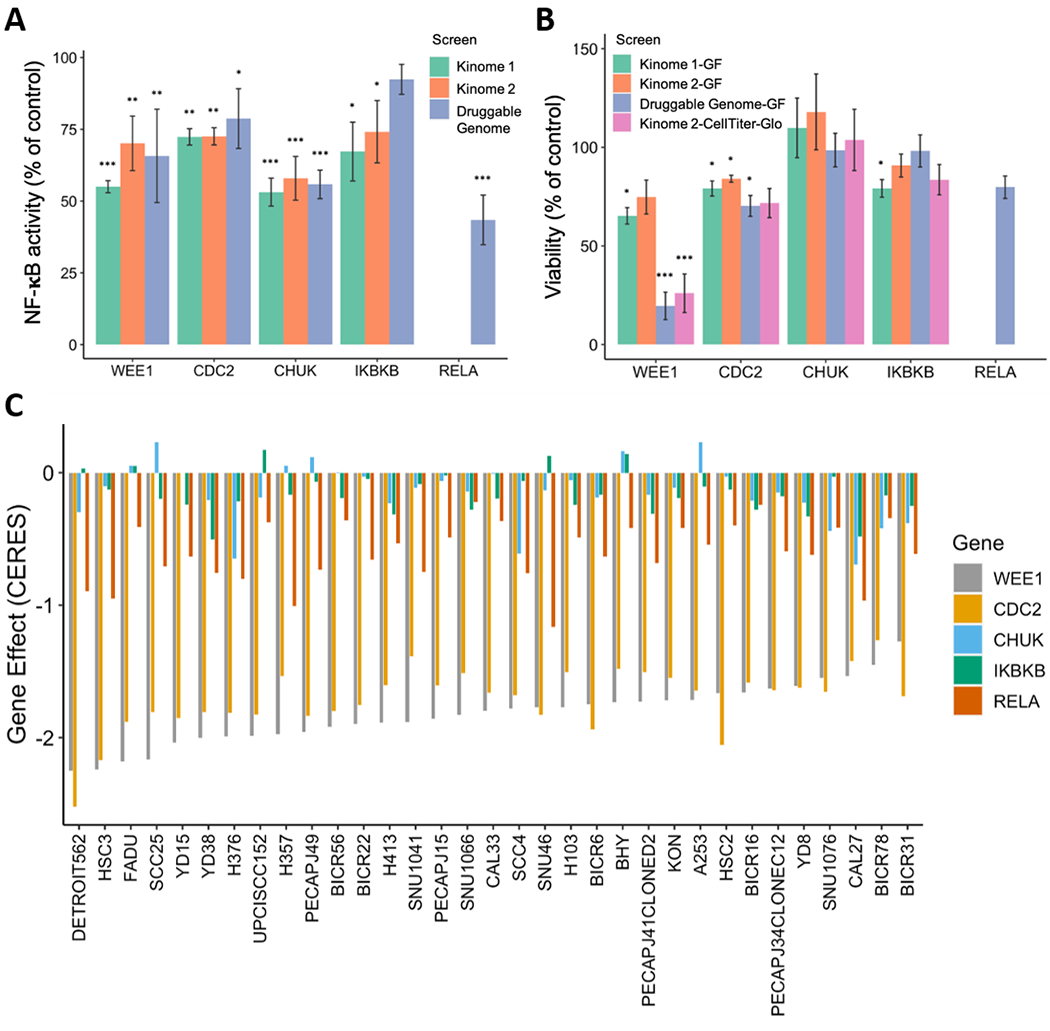
For siRNA screening, NF-κB activity and cell viability in the HPV− HNSCC line UMSCC1κB, stably expressing an NF-κB response element-β-lactamase reporter gene assessed by flow cytometry. Each bar represents a different RNAi screen. The β-lactamase reporter enzyme cleaves a fluorescent FRET substrate, which disrupts FRET and results in blue fluorescence. The blue:green fluorescence ratio indicates the activity of the NF-κB reporter. (A) NF-κB activity is shown as percentage of the negative control siRNA. (B) The change in cell viability is shown as percentage of siRNA control. For Kinome 1-GF, Kinome 2-GF and Druggable Genome-GF, viability is measured by green FRET activity, and significance was calculated by two-tailed Student’s t-tests compared to negative control. For Kinome 2-CellTiter-Glo, ATP-based Cell Titer-Glo assay was used to measure cell proliferation and cytotoxicity. Except for Kinome 1 and 2, in all the screens for NF-κB activity and viability, significance was analyzed by RSA analysis for gene hits. Error bars show standard error of the mean. *p < 0.05, **p < 0.01 and ***p < 0.001. (C) CERES-inferred gene dependency score in the CRISPR-Cas9 screen from project Achilles for WEE1, CDC2, CHUK/IKKα, IKBKB/IKKβ and RELA in 33 head and neck cell lines. The order is based on the gene effect of WEE1 (most dependent cell line (lowest value) to least dependent cell line (highest value)).
To broadly examine the dependence of HNSCC on these components for cell survival, we utilized the DepMap portal, which contains CRISPR and RNAi screening data in 33 head and neck cancer cell lines (32 HPV− and 1 HPV+; Supplementary Table 1). The effect of the depletion of a gene on cell viability is given a CERES dependency score; a lower CERES score indicates a higher likelihood that the gene of interest is essential in a given cell line, with a score of −1 comparable to the median of all pan-essential genes (30). We used this database to assess the dependance of a large panel of head and neck cancer cell lines on the expression of WEE1, CDC2, CHUK/IKKα, IKBKB/IKKβ and RELA (hereafter referred to as ‘genes from the WEE1 and NFκB axis’). The data demonstrates that both WEE1 and CDC2 are essential genes in these cancers, while inhibition of the NF-κB genes alone, for which there are functionally redundant IKK and NF-κB/REL subunits, have a more variable effect when inhibited in the absence of TNFα (Figure 1C).
WEE1 inhibition attenuates TNFα-induced WEE1/CDC2 and IKK/RELA activation in multiple HNSCC lines.
To explore whether WEE1/CDC2 G2/M checkpoint and IKK/NF-κB protein expression and signaling could be linked, we initially compared the protein expression level of various components of the two signaling pathways in normal human oral keratinocytes (HOK) and a panel of 7 HPV(−) HNSCC cell lines (Figure 2A). We observed increased protein expression of WEE1, phosphorylated CDC2 at tyrosine 15 (Y15), a canonical WEE1 substrate site, and CDC2 total protein in UMSCC1, UMSCC11A, UMSCC38 and UMSCC46 when compared to normal HOK, or UMSCC9, 22A or 74A (Figure 2A). Interestingly, phosphorylated IKKα/β, total IKKα/β, phosphorylated RELA and total RELA were also expressed at higher in cell lines expressing high WEE1/CDC2 protein (e.g. UMSCC1, 11A and 46), and lower levels in cell lines expressing low WEE1/CDC2 protein (HOK, UMSCC9 and UMSCC22A). This variation was not related to deficient or overexpressed mutant TP53 protein (31). These observations suggest that the WEE1/CDC2 and IKK/NF-κB signalling pathways could be linked in a subset of HPV(−) HNSCC cell lines. HNSCC can broadly be classified into HPV− and HPV+ subtypes, and these subtypes can differ substantially in terms of clinical response to standard and targeted therapies. We therefore examined expression of these proteins in two HPV− (UMSCC1 and UMSCC22A) and two HPV+ (UMSCC47 and UPCI:SCC090) HNSCC cell lines (Figure 2B (31). A similar trend in aberrant increases in WEE1/CDC2 and IKK-RELA protein and/or signal phosphorylation was observed in UMSCC1 and the two HPV+ lines relative to HOK. Next, to assess if these two pathways may functionally interact, we looked at the activation of WEE1/CDC2 and IKK/RELA signaling after TNFα treatment, an inducer of the IKK-RELA signal phosphorylation. As expected, TNFα treatment induced activation of the classical IKK/NF-κB pathway, as evidenced by increased phosphorylation of IKKα/β (S176/180) and RELA (S536). Strikingly, TNFα also induced the phosphorylation of CDC2 (Y15), a WEE1 substrate, in all cell lines tested (Figure 2C; lane 2 in each panel). These data suggest a potential for signal cross talk between these two signaling pathways. To assess this further, we looked at the impact of WEE1 inhibition on TNFα–induced CDC2, IKK and RELA phosphorylation. AZD1775, a well characterized and potent WEE1 inhibitor (12–15), ablated basal and TNFα-inducible CDC2 (Y15) phosphorylation. Interestingly, AZD1775 also inhibited TNFα induced IKKα/β phosphorylation and partially inhibited RELA phosphorylation, in both HPV− and HPV+ cell lines (Figure 2C; lane 3 and 4 in each panel)). The reduction of TNFα-induced phosphorylation after AZD1775 treatment was also observed but delayed (evident at 12 hours vs 6 hours) in an additional cell line UMSCC46,, which exhibits high basal CDC2 and RELA phosphorylation (Supplementary Figure 2).
Figure 2. WEE1/CDC2 and IKK/NF-κB signaling components are co-expressed in HNSCC and WEE1 inhibition attenuates TNFα-induced IKK/RELA activation.
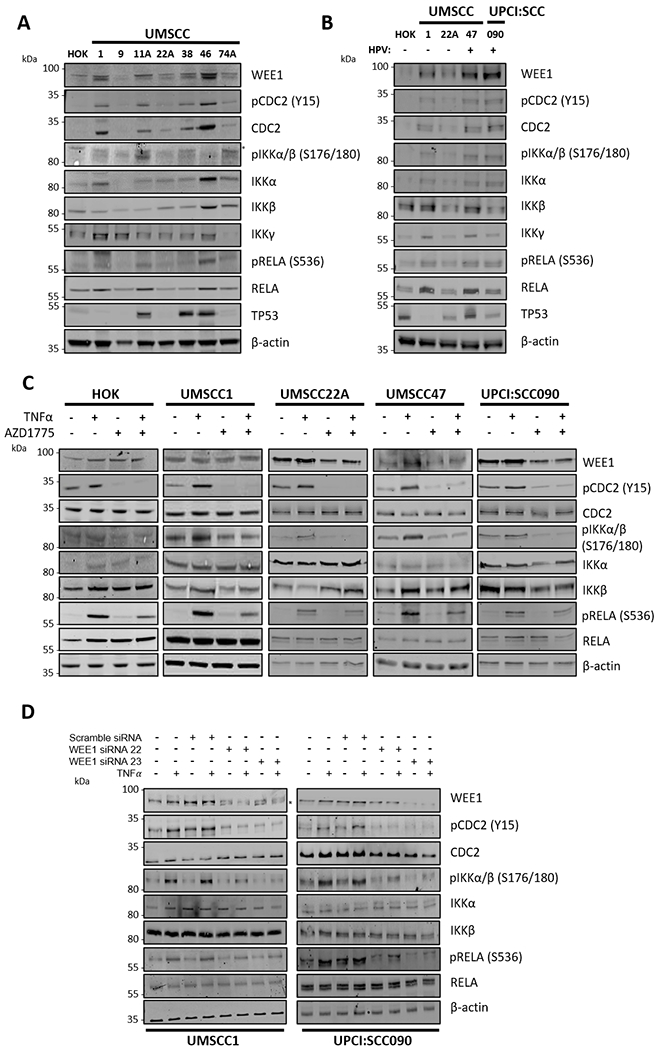
(A) Whole cell lysates of a panel of UMSCC cells and human oral keratinocytes (HOK) were collected for western blot analysis for the expression of WEE1, total and phosphorylated CDC2, total and phosphorylated IKKα/β, IKKγ, total and phosphorylated RELA and TP53. β-actin was used as the loading control. (B) Whole cell lysates of human oral keratinocytes (HOK), HPV− UMSCC1 and UMSCC22A and HPV+ UMSCC47 and UPCI:SCC090 were collected for western blot analysis for the expression of WEE1, total and phosphorylated CDC2, total and phosphorylated IKKα/β, total and phosphorylated RELA, and TP53. β-actin was used as the loading control. (C) HOK, UMSCC1, UMSCC22A, UMSCC47 and UPCI:SCC090 cells were treated with AZD1775 for six hours. 30 minutes before harvest, 10 ng/ml TNFα was added. Whole cell lysates were analyzed by western blot for the expression of WEE1, total and phosphorylated CDC2, total and phosphorylated IKKα/β, total and phosphorylated RELA. β-actin was used as the loading control. (D) UMSCC1 and UPCI:SCC090 cells were treated with scramble or two specific WEE1 siRNAs for 48 hours. 30 minutes before harvest, 10 ng/ml TNFα was added. Whole cell lysates were analyzed by western blot for the expression of WEE1, total and phosphorylated CDC2, total and phosphorylated IKKα/β, total and phosphorylated RELA. β-actin was used as the loading control. *P<0.05, **P<0.01 and ***P<0.001 (Wilcoxon rank sum test).
To confirm these data were not due to potential off target effects of AZD1775, we knocked down the expression of WEE1 with two distinct WEE1-specific siRNAs in both HPV− UMSCC1 and HPV+ UPCI:SCC090 cells. Both siRNAs reduced WEE1 expression by at least 50% in each cell line (Figure 2D). In line with the inhibitor data, WEE1 knockdown drastically reduced both basal and TNF-induced CDC2, as well as IKK phosphorylation, while partially inhibiting RELA phosphorylation (Figure 2D). Taken together, these data demonstrate that TNFα can activate the WEE1/CDC2 signaling pathway in addition to the classical IKK/NF-κB pathway, and that inhibition of WEE1 reciprocally reduces TNFα–induced IKK-NF-κB RELA subunit phosphorylation, in cell lines of either HPV status.
WEE1/CDC2 physically interact with the IKK/RELA complex in a WEE1 kinase dependent manner.
To investigate if WEE1/CDC2 and IKK/RELA physically interact, reciprocal co-immunoprecipitations (Co-IPs) were performed. Our Co-IP data demonstrated that WEE1, CDC2, and canonical pathway components IKKα, IKKβ, IKKγ and RELA can all form complexes in both HPV− (UMSCC1 and UMSCC22A) and HPV+ (UPCI:SCC090) cells, while immunoprecipitation with control IgG or the RELB and IB with NF-κB2 p100 or RELB subunits from the alternative IKKα-NF-κB2/RELB pathway could not (Figure 3A). Next, we wanted to see if treatment with TNFα could alter the formation of these complexes. Treatment with TNFα did not alter the basal complex formation between WEE1, CDC2, IKKα, IKKβ and RELA after immunoprecipitation with WEE1 or IKKβ (Figure 3B). Interestingly, however, addition of AZD1775 reduced complex formation between WEE1, IKKα, IKKβ and RELA; additionally, AZD1775 inhibited the interaction between CDC2 and IKKβ, but not WEE1 (Figure 3B). Together, these data demonstrate that WEE1/CDC2 physically interacts with the IKK/RELA complex and this may be disrupted by inhibition of WEE1 kinase activity.
Figure 3. WEE1/CDC2 physically interact with the IKK/RELA complex in a WEE1 kinase dependent manner.
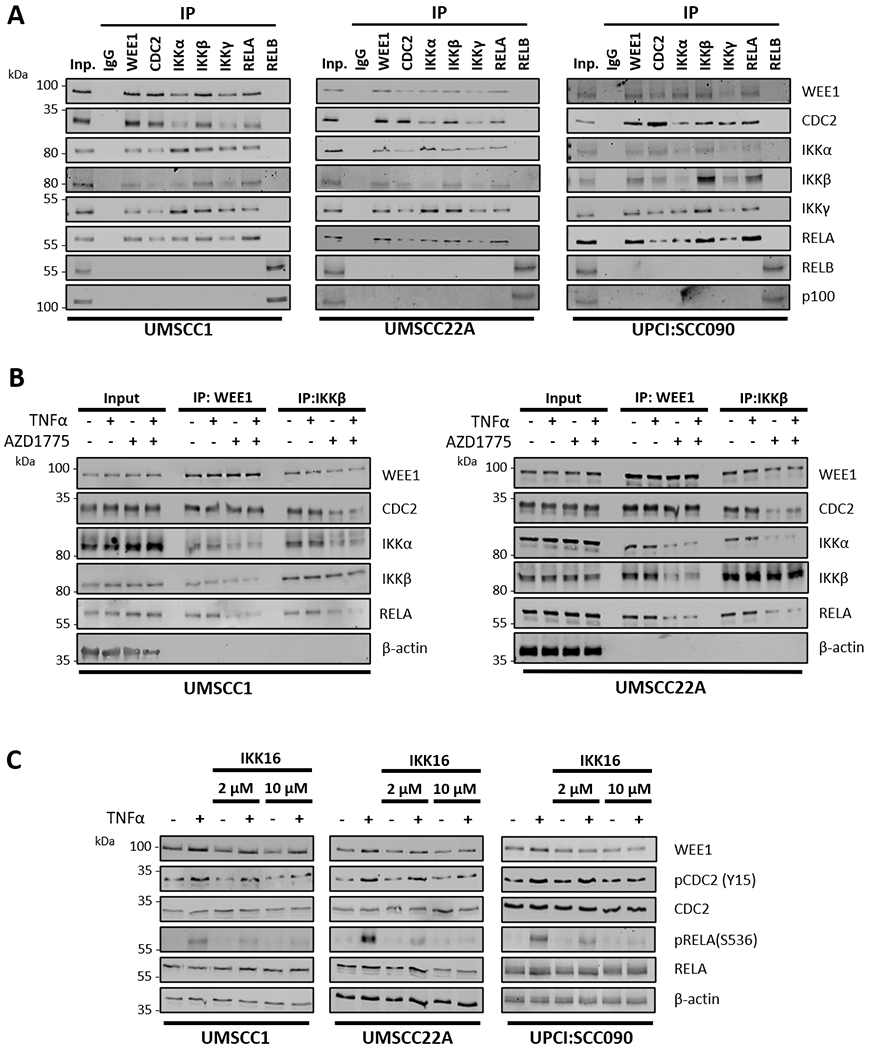
(A) Endogenous WEE1, CDC2, IKKα, IKKβ, IKKγ, RELA and RELB were immunoprecipitated from HPV− UMSCC1, HPV− UMSCC22A and HPV+ UPCI:SCC090 cells using the corresponding antibodies. Co-immunoprecipitated proteins were detected by western blot using the respective antibodies. p100 was detected as a positive control for the RELB IP. IgG was used as a specificity control. (B) UMSCC1 and UMSCC22A cells were treated with AZD1775 for six hours. 30 minutes before harvest, 10 ng/ml TNFα was added. Endogenous WEE1 and IKKβ were immunoprecipitated from UMSCC1 and UMSCC22A using the corresponding antibodies. Co-immunoprecipitated proteins were detected by western blot using the respective antibodies. β-actin was used as the loading control. (C) UMSCC1, UMSCC22A, UPCI:SCC090 cells were treated with 2 μM and 10 μM IKK16 for 6 hours. 30 minutes before harvest, 10 ng/ml TNFα was added. Whole cell lysates were analyzed by western blot for the expression of WEE1 and total and phosphorylated of CDC2, IKKα/β, and RELA. β-actin was used as the loading control.
IKK activity is required for TNFα-inducible CDC2 phosphorylation.
Our data thus far suggest a functional interaction between WEE1/CDC2 and the IKK/RELA complex. Based on these observations, we hypothesized that IKKα/β may also play a role in the TNFα-induced WEE1-CDC2 (Y15) phosphorylation. To test this, we stimulated UMSCC1, UMSCC22A and UPCI:SCC090 cells with TNFα with or without pre-treatment with IKK16, a small molecule inhibitor that inhibits IKKβ>α activity (32). Compared to basal and TNFα stimulated cells, IKK16 inhibited TNFα-induced phosphorylation of CDC2 (Y15) and IKK-dependent RELA (S536) in a dose dependent manner (Figure 3C). This suggests that IKKα/β is involved in TNFα-induced CDC2 (Y15) phosphorylation and thus may be involved in regulating the G2/M checkpoint in these cells.
AZD1775 inhibits TNFα–induced expression of NF-κB dependent cyclin and pro-survival proteins and cell proliferation.
We have previously shown that NF-κB activity promotes the proliferation and survival of HNSCC (7). We therefore examined the impact of WEE1 inhibition on the expression of TNFα and NF-κB inducible proteins Cyclin D1 (CCND1) and BCL2, which can promote cell proliferation, survival and resistance to TNFα–induced cell death. TNFα induced Cyclin D1 and BCL2 expression between 12-24 hours in both HPV− UMSCC1 and HPV+ UPCI:SCC090 cells. The addition of AZD1775 inhibited the induction of these proteins by 24 hours (Supplementary Figure 3A). Since the induction of BCL2 may inhibit TNFα-induced cell death, we examined if BCL2 siRNA knockdown affects cell growth in combination with TNFα. In HPV− UMSCC1 cells, BCL2 depletion alone resulted in ~40% reduction is cell growth (Supplementary Figure 3B and C). TNFα treatment alone resulted in a ~25% increase in cell growth; however, TNFα treatment in BCL2 depleted cells resulted in a further reduction in cell growth compared to BCL2 depletion alone (~75% decrease), suggesting that TNFα–dependent BCL2 expression contributes to resistance to TNFα–induced cell death (Supplementary Figure 3C). Similar results were observed in HPV+ UPCI:SCC090 cells (Supplementary Figure 3D–E). To examine the effects of WEE1 on cell growth, we performed similar experiments after AZD1775 treatment or WEE1 depletion (Supplementary Figure 3F–K). Inhibition or depletion of WEE1 significantly impaired cell growth, and this was exacerbated after the addition of TNFα. To further assess effect of AZD1775 on cell growth, we performed XTT cell viability experiments. Multiple linear regression analysis demonstrated that, in each cell line, AZD1775 and TNFα significantly reduced cell viability (Supplementary Table 2), with IC50 values ranging from 471 nM (HPV− UMSCC1) to 207 nM (HPV+ UPCI:SCC090) (Supplementary Figure 4A). To address the combinatorial effect of AZD1775 and TNFα, we treated cells with increasing dose of AZD1775 with or without 2 different doses of TNFα and performed Bliss synergy analysis as in our previous work (33). The addition of TNFα reduced the IC50 of AZD1775 in each cell in a dose dependent manner; Bliss synergy analysis demonstrated that in each cell line, the effects of TNFα on AZD1775 function was moderately synergistic (defined as a Bliss synergy score >0; Supplementary Figure 4B). Furthermore, colony formation in HNSCC cells was reduced after WEE1 inhibition and this was significantly enhanced in combination with TNFα (Supplementary Figure 4C–F). Similar results were observed after WEE1 depletion (Supplementary Figure 5A and B). Together, these data demonstrate that AZD1775 inhibits the TNFα–inducible expression of the NF-κB inducible proteins Cyclin D1 and BCL2 that contribute to proliferation, survival, and TNFα resistance in HPV− and HPV+ HNSCC cells.
Inhibition of WEE1 sensitizes HNSCC cells to TNFα in vitro.
Our above data suggests that WEE1 may play a role in the expression of TNFα– and NF-κB inducible targets involved in proliferation, survival and TNFα resistance; we therefore assessed the effect of WEE1 inhibition on cell cycle progression and cell death in the presence of TNFα. In HPV− UMSCC1 and HPV+ UMSCC47 cells, TNFα alone had minimal impact on cell cycle progression, whereas in HPV− UMSCC22A cells TNFα treatment resulted in a reduction of cells in S phase (Figure 4A and Supplementary Figure 6A). In contrast, HPV+ UPCI:SCC090 cells were more sensitive to TNFα alone, which induced significant accumulation of sub-G1 DNA particularly at later timepoints. AZD1775 treatment resulted in an increased proportion in S or G2/M phase in each cell line. At later time points, WEE1 inhibition increased cells with sub-G1 DNA, consistent with the induction of cell death and DNA fragmentation. Combination treatment of AZD1775 and TNFα reduced the accumulation of cells in S and G2 phase after AZD1775 treatment and significantly increased cells in sub-G1 DNA at later time points in all cell lines, particularly HPV− UMSCC22A and HPV+ UPCI:SCC090. When compared, UMSCC22A and UPCI:SCC090 were more sensitive to WEE1 inhibition than HPV− UMSCC1 and HPV+ UMSCC47 cells, but in all cell lines AZD1775 treatment significantly sensitized cells to TNFα (observed as an increase in sub-G1 DNA).
Figure 4. AZD1775 sensitizes HNSCC cells to TNFα-induced cell death.
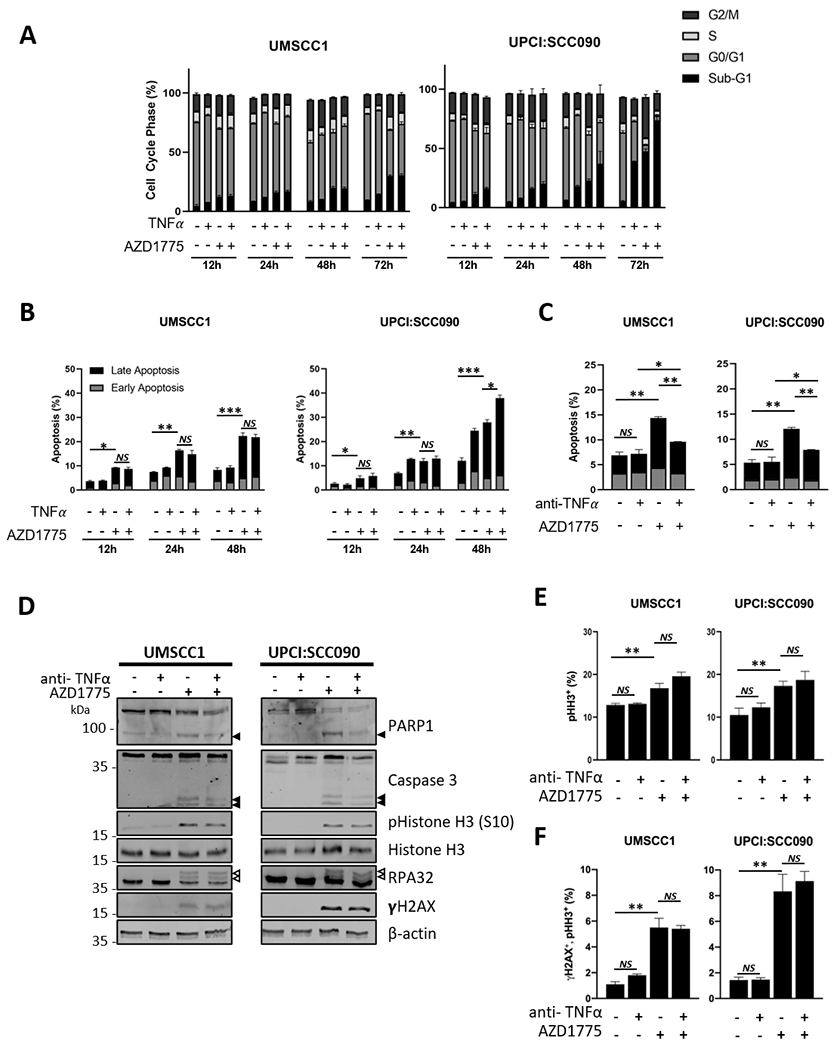
(A) HPV− UMSCC1 and HPV+ UPCI:SCC090 cells were treated with AZD1775 (500 nM for UMSCC1, 250 nM UPCI:SCC-090) and/or TNFα (10 ng/mL) for 12, 24 48 or 72 hours. Flow cytometric analysis of the cell cycle was performed using a FACS Canto flow cytometer. Data from 10,000 cells per sample analyzed using Flow-Jo analysis software. (B) UMSCC1 and UPCI:SCC090 cells were treated with AZD1775 as above for 12, 24 and 48 hours. Flow cytometric analysis of apoptosis was performed using Annexin V assay. Data from 10,000 cells per sample analyzed using Flow-Jo analysis software. Early apoptotic cells were defined as Annexin positive/PI negative and late apoptotic cells was defined as Annexin positive/PI positive. (C) UMSCC1 and UPCI:SCC090 cells were treated with AZD1775 for 20 hours after pre-treatment with a neutralizing TNFα or IgG control for 4 hours. Flow cytometric analysis of the cell cycle was performed using a FACS Canto flow cytometer. Data from 10,000 cells per sample analyzed using Flow-Jo analysis software. (D) UMSCC1 and UPCI:SCC090 cells were treated with AZD1775 for 20 hours after pre-treatment with a neutralizing TNFα or IgG control for 4 hours. Whole cell lysates were analyzed for PARP1 cleavage and Caspase 3 cleavage, phosphorylated and total Histone H3, RPA32 and γH2AX expression. β-actin was used as a loading control. (E) UMSCC1 and UPCI:SCC090 cells were treated with AZD1775 for 20 hours after pre-treatment with a neutralizing TNFα or IgG control for 4 hours. The expression of phosphorylated Histone H3 (S10) was analyzed by flow cytometry using a FACS Canto flow cytometer. The percent of positive cells is shown as a marker for mitosis. Data from 10,000 cells per sample analyzed using Flow-Jo analysis software. (F) UMSCC1 and UPCI:SCC090 cells were treated with AZD1775 for 20 hours after pre-treatment with a neutralizing TNFα or IgG control for 4 hours. The expression of phosphorylated Histone H3 (S10) and γH2AX was analyzed by flow cytometry using a FACS Canto flow cytometer. The percent of double positive cells is shown as a marker for DNA damage in mitosis. Data from 10,000 cells per sample analyzed using Flow-Jo analysis software. *P<0.05, **P<0.01 and ***P<0.001 (Student’s T Test). Error bars represent the mean ± standard deviation of a minimum of three biological repeats. *P<0.05, **P<0.01 and ***P<0.001 (Student’s T Test).
Consistent with the above data, TNFα alone did not induce significant levels of apoptosis in HPV− UMSCC1 cells in an Annexin V apoptosis assay but did induce significant apoptosis in HPV+ UPCI:SCC090 at later time points (Figure 4B). AZD1775 induced significant levels of apoptosis in all cell lines, and this was further enhanced in combination with TNFα (Figure 4B and Supplementary Figure 6B). To investigate the mechanism of cell death, western blot analysis was used to look for caspase activation. AZD1775 treatment induced the cleavage of caspase 3 and its substrate PARP1, and this was enhanced when combined with TNFα (Supplementary Figure 7A and 8A). Treatment with the pan-caspase inhibitor Z-VAD-FMK and/or the RIP kinase inhibitor Necrostatin-1 confirmed that AZD1775 alone, and in combination with TNFα, primarily induced caspase-dependent apoptosis (Supplementary Figure 7B).
As a critical regulator of the intra-S and G2/M checkpoints, WEE1 activation prevents cells from undergoing unscheduled DNA replication or allowing cells with DNA damage from entering mitosis (10). Therefore, inhibition of WEE1 results in unscheduled DNA replication, activation of the DNA damage response (DDR) and DNA damage in mitotic cells. To confirm if the addition of TNFα to AZD1775 modulated the number of mitotic cells or the induction of DNA damage, we looked at the expression of phospho-Histone H3 (Ser 10, a marker of mitosis; (34)), phospho-RPA32 (a marker of replication stress; (35)) and γH2AX (a marker of dsDNA breaks; (36)). As expected, treatment with AZD1775 induced phospho-Histone H3 (Ser 10), phospho-RPA32 (indicated by the arrows as slower migrating bands) and γH2AX expression, indicating the an increase in mitotic cells and the induction of DNA damage (Supplementary Figure 7A and 8A). Co-treatment of AZD1775 and TNFα, however, did not significantly alter the induction of mitosis or the DDR when compared to AZD1775 treatment alone. This was confirmed using flow cytometry to identify the percentage of mitotic cells (Supplementary Figure 7C and 8B) and the percentage of mitotic cells containing DNA damage (a marker of mitotic catastrophe; Supplementary Figure 7D and 8C). These data suggest that the addition of TNFα did not further exacerbate mitotic induction and DNA damage induced by WEE1 inhibition alone, as no significant increase in the induction of mitosis or DNA damage markers was observed. In corroboration with the WEE1 inhibitor data, WEE1 depletion resulted in a significant increase in sub-G1 DNA 48h post-treatment and the addition of TNFα for 24 hours increased this further, particularly in HPV+ UPCI:SCC090 cells (Supplementary Figure 9A). Also, depletion of WEE1 with siRNA led to similar results for PARP1, Caspase 3 and pHistoneH3, although the effect of TNF was more muted, likely due to significant effect of WEE1 depletion alone on the induction of apoptosis in these cells (Supplementary Figure 9B and C). Representative flow cytometry histograms for pharmacologic and siRNA inhibition are shown in Supplementary Figure 10.
Next, we wanted to investigate if the effects of WEE1 inhibition on proliferation and apoptosis were TNF-dependent. Pre-treatment of cells with a neutralizing TNFα antibody partially restored colony formation after AZD1775 treatment (Supplementary Figure 11). Furthermore, the neutralizing TNFα antibody also reduced AZD1775-induced apoptosis, without altering the induction of mitosis or the DDR (Figure 4C–F, Supplementary Figure 6C–F). Together, these data suggest that WEE1 inhibition or depletion sensitizes both HPV− and HPV+ HNSCC cells to TNFα–induced, caspase-dependent cell death, without impacting the induction of mitosis or the DDR. Furthermore, the effects of WEE1 inhibition on cell proliferation and apoptosis appear to be partially TNF-dependent.
AZD1775 plus radiation delays the growth of xenograft tumors, which can be reduced by TNFα depletion.
Radiation induces TNFα as well as direct DNA damage that mediate the cytotoxic and antitumor effects in SCC and other cancers (4). We examined the activity of AZD1775 alone and in combination with radiation, in athymic nude mice bearing the relatively radiation-sensitive HPV− UMSCC46 cells, HPV+ UPCI:SCC090 cells, and radiation-resistant HPV− UMSCC1 xenograft models (Supplemental Figure 12) (37,38). UMSCC46 tumors grew rapidly in both the control group and the AZD1775 monotherapy group (Figure 5A). Although radiation delayed tumor growth (p <0.05), this was further delayed (Figure 5A; p </= 0.0001) and survival was prolonged (Figure 5B, p = 0.0069) when AZD1775 and radiation were combined. Nude mice bearing UPCI:SCC090 xenografts also showed tumor growth delay in response to radiation alone and prolongation in combination with AZD1775 (Figure 5C, p </=0.036), but more than half of tumor bearing mice in the combination group were cured, demonstrating durable responses and survival more than 100d (Figure 5D, p = 0.029). Thus, AZD1775, when given in combination with radiation, significantly suppresses growth of the HPV− UMSCC46 and HPV+ UPCI:SCC090 derived tumors and can eliminate the HPV+ UPCI:SCC090 cell line derived tumors in ~50% mice, enhancing survival, consistent with the relatively greater TNFα sensitivity of the latter cell line.
Figure 5. AZD1775 significantly delayed the tumor growth in nude mice when combined with radiation.
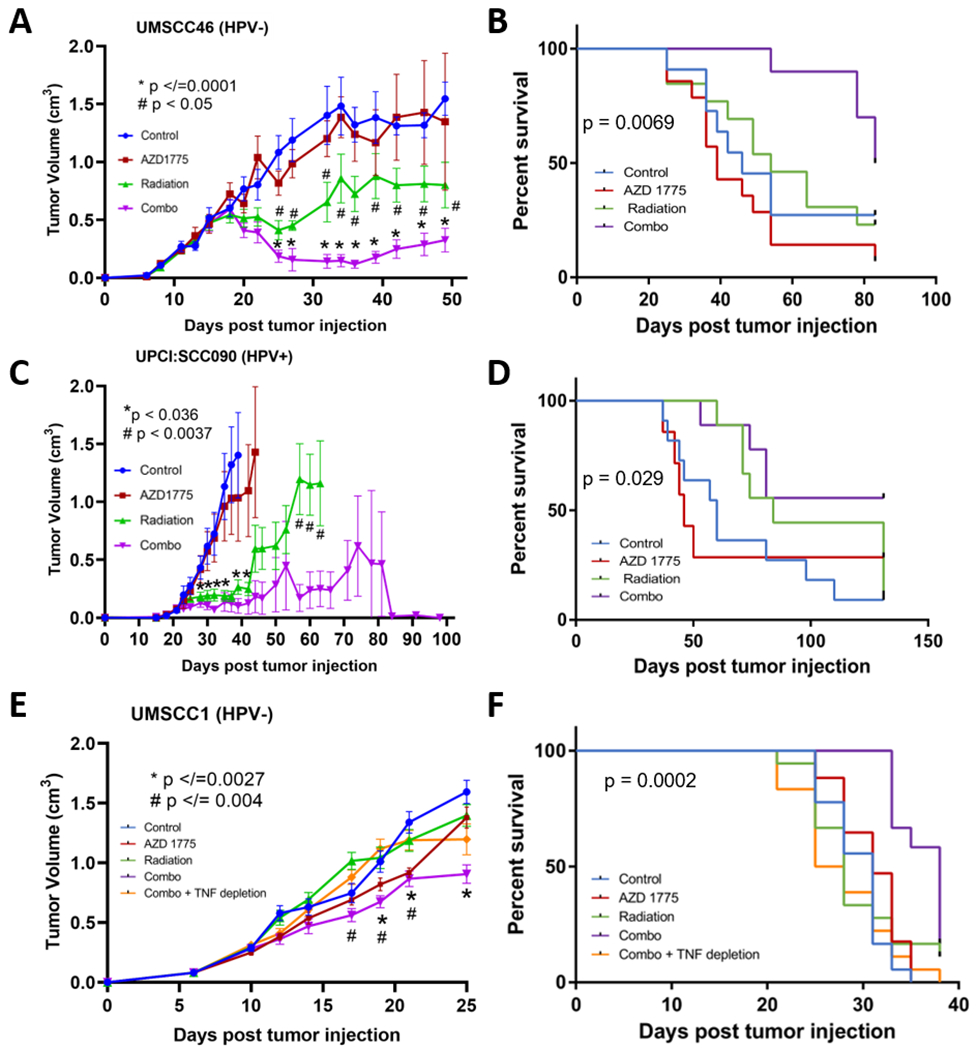
(A) Nude mice were injected with HPV− UMSCC46 cells ( and randomized into four groups: control (blue), treated with AZD1775 alone (120mg/kg daily, red), radiation alone (2Gy daily, green), or in combination with the same doses (purple). Each group contained 13-14 mice and the experimental treatments were carried 5 days a week for two weeks as shown in Supplemental Figure 7. Tumor growth was measured 2-3 times a week and is reported as the mean volume (cm3) with standard error of the mean. p values for the tumor growth were calculated using Student’s t-test. * indicates the p value of the difference between the control and the combination group, and # indicates a significant difference between the radiation group and the combination group. (B) Survival curves for the 4 groups of mice bearing UMSCC46. The p value represents the significant difference between the combination group and all other groups calculated using Gehan-Breslow-Wilcoxon test. (C) Mice injected with HPV+ UPCI:SCC090 cells were randomized into four groups and treated as described in (A and Supplemental Figure 7), except using 60mg/kg AZD1775. Each group consisted of 7-11 mice, and tumor growth was measured and reported as in (A). The significant difference between the control versus combination groups and control versus radiation groups are indicated as *, and # indicates a significant difference between the radiation versus the combination groups. (D) Survival curves for the four groups of mice bearing UPCI:SCC090 tumors. The p value shows the significant difference between the combination group and all other groups calculated using the Gehan-Breslow-Wilcoxon test. (E) Mice injected with HPV− UMSCC1 cells were randomized into five groups and treated with the same dosage regimen as showing (C). One additional group in which anti-TNFα antibody (200 μg) was intraperitoneally injected for seven days (referred to as Combo + TNF depletion, Supplemental Figure 7) was included. Each group consisted of 17-18 mice. Tumor growth is reported as the mean volume (cm3) with standard error of the mean. p values for the tumor growth were calculated using the Student’s t- test. * indicates the p value of the difference between the control and the combination group, and # indicates a significant difference between the combination group and the combination plus TNFα depletion group. (F) Survival curves for the four groups of mice bearing UMSCC1 tumors. The p value shows the significant difference between the combination group and all other groups calculated using the Gehan-Breslow-Wilcoxon test.
Since we hypothesized that the action of AZD1775 in combination with radiation treatment could be via TNFα-mediated mechanisms; we therefore examined the effects of the combination without or with depletion of endogenous mouse TNFα in the HPV− UMSCC1 xenograft mouse model (Figure 5E–F). Murine anti-TNFα antibody was administered through seven intraperitoneal injections (over three weeks) to one group of mice that received the combination treatment. Consistent with the results from previous models, significant although reduced anti-tumor effects were observed in the combination group vs control (Figure 5E; p </=0.0027). However, TNFα depletion significantly abrogated this anti-tumor effect (Figure 5F; p =0.004) and survival benefit (Fig; p= 0.0002) of the combination therapy. These data support the hypothesis that the anti-tumor effect of combination therapy of AZD1775 with radiation is at least partially TNFα dependent. Therefore, our in vivo data suggest that WEE1 inhibition enhances radiation effects upon tumor growth in both HPV− and HPV+ xenografts.
WEE1, CDC2 and IKK-RELA expression in HNSCC tumors.
To investigate the possible clinical correlatives of our data, we examined the mRNA expression of the genes from the WEE1 and NFκB axis in the HNSCC TCGA database (19). Whilst WEE1 and CHUK/IKKα expression was significantly lower in HNSCC tumors, CDC2 expression was significantly higher (Figure 6A). We then analyzed the expression of the genes from the WEE1 and NFκB axis by HPV status (Figure 6B). Interestingly, although WEE1 expression was lower in HNSCC cases combined, mean WEE1 expression was significantly higher in HPV+ HNSCC compared to HPV− HNSCC (but not compared to normal tissue). CDC2 expression was higher with either HPV status, but highest in HPV+ HNSCC cases. Additionally, the expression of IKBKB/IKKβ was also higher in HPV+ HNSCC, whereas CHUK/IKKα and RELA expression were comparable to normal tissue. Overall, variation in mRNA expression of the genes from the WEE1 and NFκB axis significantly correlated with copy number (Figure 6C). These data were further confirmed using two separate databases from the GEO repository (Supplementary Figure 13A and B). We next analyzed the protein expression of these genes in HNSCC from CPTAC (20), which includes data from HPV− HNSCC. In corroboration with the gene expression analysis, we observed decreased WEE1 expression and significantly increased CDC2 protein expression in HNSCC tumors (Figure 6D).
Figure 6. Expression and copy number correlation analysis of the genes from the WEE1 and NFκB axis from TCGA HNSCCs.
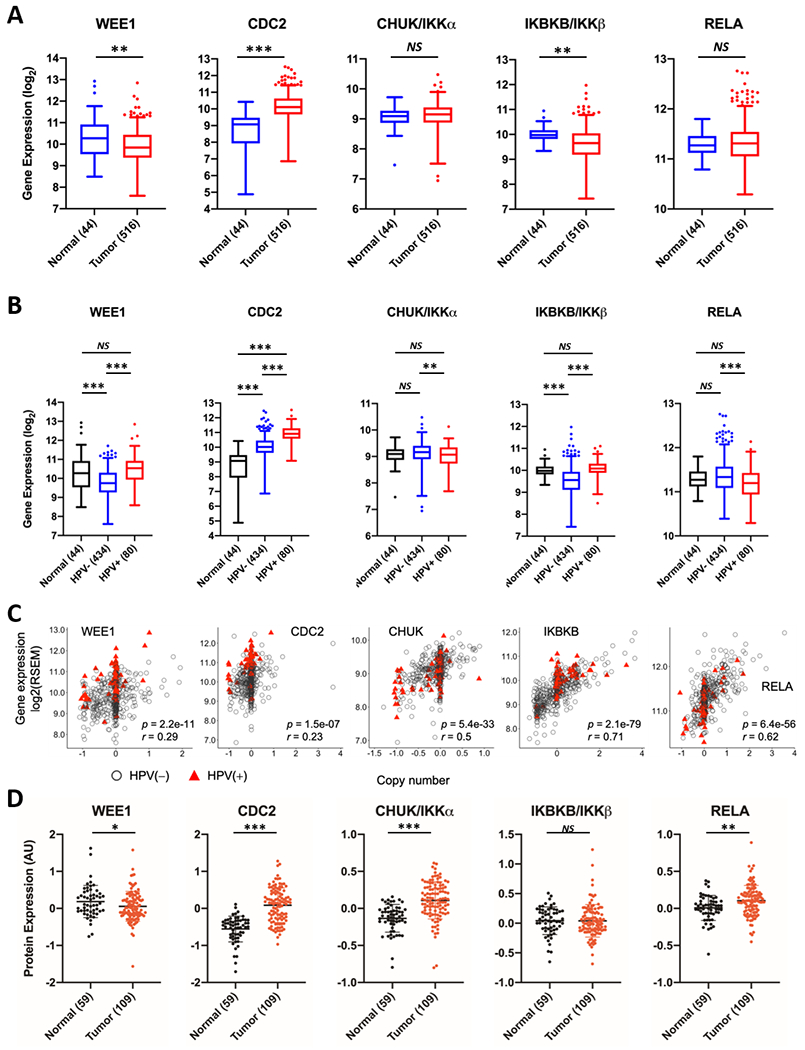
WEE1, CDC2, IKKα, IKKβ and RELA are expressed differentially between tumor vs. normal (A) and HPV(−) vs. HPV(+) vs. normal (B). Gene expression depicted in boxplots was quantified from RNA-Seq data with RSEM and log2 transformed. Statistical significance of the differential expression was assessed with Wilcoxon rank-sum test. *p < 0.05, **p < 0.01 and ***p < 0.001. (C) Correlation of gene copy number with mRNA expression in HNSCCs based on HPV status. Scatterplots for the genes from the WEE1 and NFκB axis showing significant CNV and gene expression correlation. CNVs were presented in X axis, as one-copy loss (−1), diploid (0), one-copy gain (0.58), and amplification (larger than 1). mRNA expression calculated by RSEM is presented in the y axis as the log2 scale. Pearson correlation tests were conducted for 508 HNSCC tumor specimens, including 428 HPV− (gray circles), and 80 HPV(+) samples (red triangles). p values give the significance of the Pearson correlation between mRNA expression and CNV. r values represent Pearson correlation coefficients. (D) Scatter dot plot data of protein expression of WEE1, CDC2, IKKα, IKKβ and RELA taken from the HNSCC dataset from the Clinical Proteomic Tumor Analysis Consortium (CPTAC). The log ratio of the values compared to the reference is shown in the y-axis. Brackets indicate the number of samples in each group; black indicates normal samples, red indicates tumor samples.
Genomic and survival analysis reveals distinct prognosis and associated inflammatory and NF-κB signatures associated with WEE1 expression and HPV status in HNSCC patients.
To investigate the prognostic relevance of the genes from the WEE1 and NFκB axis, we performed survival analysis using the HNSCC TCGA dataset. Interestingly, we observed that the expression of these genes had a differential relationship to survival depending on HPV status. For WEE1, CDC2 and CHUK/IKKα, high expression is significantly associated with worse progression-free and overall survival in HPV− HNSCC, but better progression-free and overall survival in HPV+ HNSCC (Figure 7A and Supplementary Figure 14). We investigated this further using individual patient data, stratified by WEE1 expression and HPV status, and examined potential associations with previously identified genomic signatures associated with SCC in the pan-squamous TCGA study (Figure 7B and C) (19). HPV− tumors that display low WEE1 expression and better prognosis are significantly associated with an expression profile enriched for NF-κB-related inflammatory, immune checkpoint, and Myeloid Derived Suppressor Cell signatures (MDSC-INF, Cluster C1 and 2; p = 6.2E-04, Fisher’s exact test), and the expression of EMT markers (ZEB1, p = 9.7E-04; ZEB2, p = 2.4E-05 and VIM, p = 0.0038; mRNA C6. p = 1.7E-06, Fisher’s exact test). In HPV+ tumors with better prognosis, high WEE1 expression is significantly associated with copy number loss/deletion and/or mutations in TNF-NF-κB pathway components TRAF3 and CYLD (p = 0.030, Fisher’s exact test), previously identified to be associated with greater survival and NF-κB pathway activation in HPV+ tumors (39,40). These data suggest that the expression of WEE1 has a differential relationship with survival dependent on HPV status and subtypes with distinct genomic signatures related to NF-κB, inflammation, and cell survival.
Figure 7. The association between distinct molecular subtypes/genomic alterations and WEE1 expression underlies different survival outcome based on HPV status.
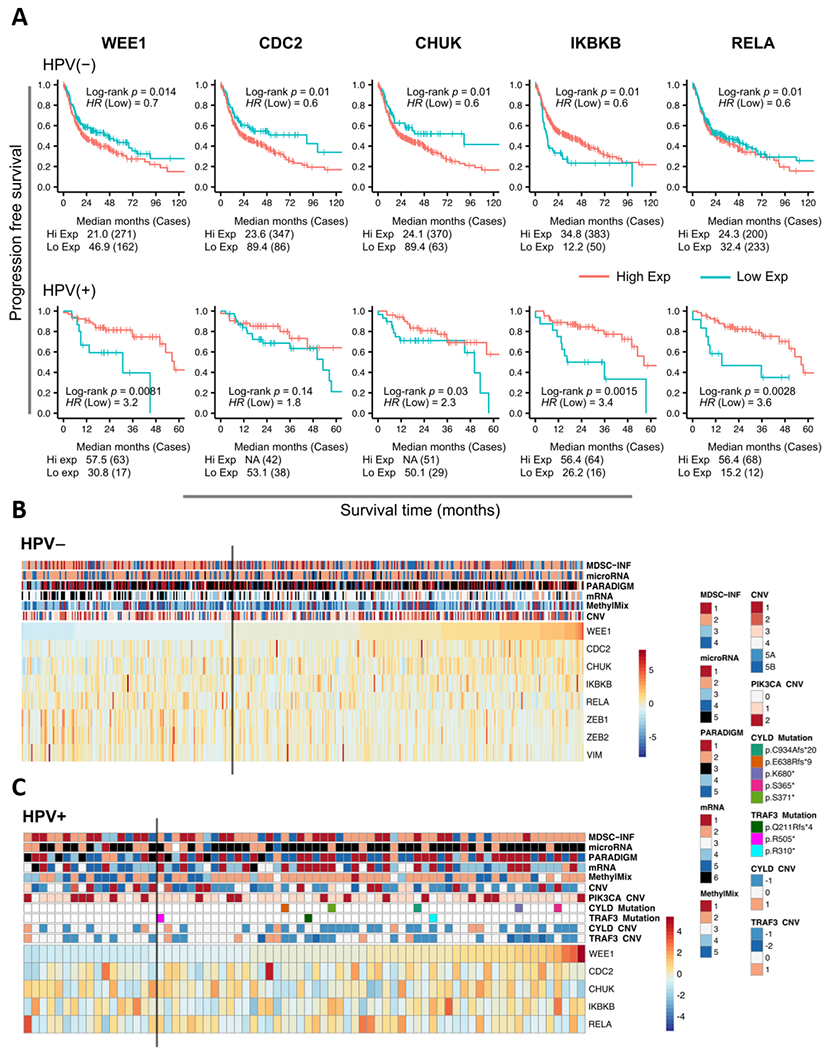
(A) Progression free survival outcome for HPV− and HPV+ HNSCC patients in the TCGA HNSCCs based on the expression of the genes from the WEE1 and NFκB axis by Kaplan-Meier analysis. Red indicates high expression, cyan indicates low expression. p values were determined using a log rank test. Plots were truncated at 10 years for HPV(−) patients and five years for HPV(+) patients, but the analyses were conducted using all of the data. (B) and (C) Heatmap display of the expression of the genes from the WEE1 and NFκB axis and the corresponding molecular subtypes or genomic events in HPV(−) (B) and HPV(+) (C) TCGA cohort. Samples are arranged in columns and ordered by WEE1 expression. The annotation tracks are labeled with the copy number variations, DNA mutations and other molecular features identified from the previous TCGA study (23). The gray vertical bar indicates the cut point for dichotomization of WEE1 expression in the association analysis.
Discussion
Current treatment options for HNSCC include radio- chemo- and immune checkpoint therapy, which are critical inducers of the DNA damage response (DDR) and innate and adaptive immune T cell responses that can produce cytotoxic TNF-family death ligands. DNA damage and genotoxic stress can induce activation of ataxia-telangiectasia-related (ATR) protein kinase or ataxia-telangiectasia mutated (ATM) protein kinase (12,41,42). ATR is the upstream kinase responsible for the phosphorylation and activation of checkpoint kinase 1 (Chk1), which phosphorylates WEE1 and Cdc25C. WEE1 then phosphorylates CDC2 at tyrosine 15, inactivating it and releasing Cyclin B (CCNB), resulting in cell cycle arrest at G2. This allows the repair of DNA, preventing cells with extensively damaged chromosomal DNA from undergoing mitosis. Additionally, DNA damage-induced ATM (43,44) and TNFα-induced kinase TAK1 can activate the canonical IKKα/β kinases and NF-κB1/RELA (45,46), to promote expression of several target pro-survival proteins that contribute to proliferation and resistance to cytotoxicity (6,46). Here, we performed an RNAi screen to identify new modulators of TNFα-induced NF-κB activity and cell cytotoxicity, which unexpectedly led to the discovery of a relationship and interaction between TNFα-induced activation of IKKα/β and RELA in the core canonical NF-κB pathway, and the key G2/M checkpoint kinases WEE1 and CDC2.
Pharmacologic or genetic WEE1 inhibition partially attenuated the signaling effects of TNFα, and we showed that the effects on WEE1-mediated CDC2 phosphorylation were greater than those upon phosphorylation of IKK and RELA, and subsequent functional activity of NF-κB. That siRNA depletion and CRISPR knockout of WEE1 and CDC2 in the reporter and DEPMAP panel demonstrated greater dependency with TNFα or alone than depletion of individual IKKs or RELA, may reflect the fact that there are multiple IKK, NF-κB and REL subunits with functional overlap, and co-activating pathways, that may limit effective cytotoxicity of specific inhibitors (7). Consistent with this, our mechanistic data demonstrated an interaction between WEE1/CDC2 and the canonical IKK/RELA, but not non-canonical RELB and p100 subunits, which was at least partially dependent on WEE1 kinase activity. Notably, WEE1 inhibitor AZD1775 partially decreased the TNFα-inducible phosphorylation of canonical TAK1 kinase phospho-acceptor sites on IKKα/β and phosphorylation of IKK-dependent S536 site on RELA (6,45,46), and also disrupted the WEE1-CDC2/IKKα/β/RELA complex. Together, these observations suggest that AZD1775 disrupts signaling within and between these pathways, and the stability of the interaction between the components of the complex. Of additional interest, we note that in each cell line, TNFα treatment resulted in a small, but reproducible increase in WEE1 protein expression, while IKK inhibitor negated this effect. We are currently investigating if the effect on WEE1 expression could be due to the transcriptional effects of TNF-induced NF-κB signalling, or via protein stabilization of WEE1 via the interaction with the IKK/RELA complex or other proteins activated via the TNFR complex (46).
Consistent with the latter possibility, our data also demonstrated that the TNFα-induced CDC2 phosphorylation is at least partially dependent on IKK activity, suggesting that crosstalk between WEE1 and IKK kinase activity may be bidirectional. Intriguingly, we have previously observed that antagonists of Inhibitors of Apoptosis (IAP)s, involved in forming ubiquitin scaffolds between the TNFR and canonical IKK-NF-κB pathway, can also modulate TNFα resistance, cell cycle, and survival in HNSCC (37,38). Together, these observations are consistent with established mechanisms by which TNFα and the TNFR recruits and stabilizes complexes containing IKKα/β and RELA with other kinases that converge to interact with the canonical IKK/NF-κB pathway. Further studies may reveal how processes downstream of the TNFα/TNFR1 receptor mediate the recruitment and signaling between these complexes.
There is evidence for broader clinical relevance of cross talk between IKK/NF-κB, WEE1/CDC2, and other G2/M checkpoint kinases in therapeutic resistance to TNF family death ligands or genotoxic therapies in HNSCC and other cancers. We previously reported that resistance of murine oral carcinomas to immune checkpoint therapy and cytotoxic T lymphocytes (CTL) could be overcome by AZD1775, and this sensitization could be abrogated by neutralization of death ligands TNFα, TRAIL or FASL, or provision of excess nucleotides, known to promote DNA repair (15). Importantly, WEE1 inhibition by AZD1775 potently sensitized these HNSCC to immune checkpoint therapy in vivo and CTL in vitro, without significant immunosuppression, CTL or hepatotoxicity that has been observed with IKK, proteasome, or other inhibitors of the NF-κB pathway (6,46). Consistent with this, the interaction with WEE1/CDC2 appears to be specific to the canonical IKKα/β/RELA complex important in cell survival, and not the alternative IKKα/RELB pathway implicated in immunity, as the complex included IKKα/β and RELA, but not RELB (Figure 3A). Thus, WEE1 inhibitors merit investigation for their potential to enhance therapies mediated by TNFα without the systemic immune suppression and hepatotoxicity previously observed with direct interruption of IKK-NF-κB/RELA signaling (6,46).
Analogous to the interaction and role of WEE1/CDC2 in modulating TNFα-induced IKK/NF-κB pro-survival signaling, a previous study demonstrated that Aurora A and B, that are also involved in the regulation of mitosis, can form a complex with IKKα/β (47). In that study, the authors show that inhibition of Aurora kinases inhibits pro-survival NF-κB signaling induced by TRAIL, resembling our observations between WEE1 and TNFα-induced, pro-survival NF-κB signaling. Concurrently, our lead author and collaborators have recently extended the findings in the current study, demonstrating that the DNA damaging agent cisplatin can also co-activate WEE1/CDC2 as well as RELA/BCL2 activation in osteosarcoma cells (48). Cisplatin- induced CDC2 and RELA phosphorylation and resistance in osteosarcomas could also be attenuated by AZD1775. Together, these studies highlight the combinatorial potential for inhibitors of G2/M checkpoint and their interactions with the IKK/NF-κB axis to overcome resistance to immune and genotoxic therapies. Furthermore, our data suggests that the cell cycle and IKK/NF-κB signalling may be functionally linked, and this may have consequences for cancer therapy. Indeed, several studies have demonstrated that NF-κB signalling may be differentially regulated at different stages of the cell cycle (49,50). Further studies are required to investigate if this occurs in HNSCC cells and if other cell cycle regulators may also be involved in NF-κB in these cancers.
Our data suggests that the inhibition of WEE1 is detrimental to both HPV− and HPV+ HNSCC cells at least in part by reducing NF-κB activity and sensitizing cells to TNF-induced cell death. AZD1775 treatment alone had a modest impact on tumor growth in vivo in each cell model tested. However, combination treatment of AZD1775 with radiation, a critical inducer of TNFα (4), significantly suppressed tumor growth and prolonged survival in vivo in three HNSCC xenograft mouse models, including HPV− and HPV+ xenografts. Interestingly, both our in vitro and in vivo data demonstrate that the inhibition of proliferation and induction of apoptosis upon WEE1 inhibition was at least partially dependent on TNFα, as depletion of TNFα with a neutralizing antibody significantly reduced the cytotoxic effects of AZD1775. This is similar to a recent study that reported that both Chk1 and WEE1 inhibition resulted in TNF-dependent apoptosis in acute myeloid leukemia (AML; (51)), suggesting that inhibition of WEE1 may induce its cytotoxic effects in a TNF-dependent manner in diverse cancer types.
Several previous studies have investigated the impact of HPV status on the sensitivity to AZD1775 (13,52–55). Early studies demonstrated that the sensitivity of HPV− HNSCC to AZD1775 appears to be dependent on TP53 mutation – treatment of HNSCC cells expressing high-risk TP53 mutations with AZD1775 sensitized them to cisplatin treatment, promoting mitotic arrest and senescence (13,52). Subsequent studies demonstrated that HPV+ HNSCC are also highly sensitive to AZD1775, which was also sensitive to radiation and the combinatory treatments in vivo in our study (55,56). The combination treatment exhibited durable anti-tumor effects, with more than half of the tumor bearing mice cured by the combination therapy. Interestingly, we found that although the UPCI:SCC090 cell line was highly sensitive to AZD1775 in vitro, the UPCI:SCC090 tumor model was less sensitive to single agent treatment in vivo. A recent study demonstrated that expression of HPV16 E6 and E7 in the wild type TP53 cell line UMSCC74A sensitized them to AZD1775 by inducing a FOXM1-CDK signaling axis, driving aberrant mitosis and DNA damage (56). This study, and another recent study (57), have also demonstrated higher expression of WEE1 expression in HPV+ HNSCC, in accordance with our data in HNSCC cell lines and from the TCGA.
We observed potentially interesting relationships between expression of WEE1 and other previously defined immune and NF-κB gene signatures that co-clustered in subsets of HPV− and + tumors, with prognosis of patients from the TCGA dataset, who were treated primarily with standard surgery, radiation and chemotherapy. Intriguingly, increased expression of WEE1 was associated with worse survival in HPV− but better survival in HPV+ HNSCC. In HPV− tumors, the larger subset with worse survival and increased WEE1 expression is associated with previously defined clusters lacking immune signatures, which have previously been associated with a lack of response to immune checkpoint blockade (ICB). In contrast, a smaller subset associated with better survival and lower WEE1 expression was significantly enriched for expression of previously defined NF-κB/immune inflamed/checkpoint and MDSC mRNA signatures, that overlapped with a smaller cluster enriched for mRNAs and microRNAs associated with EMT (19). Conversely, in HPV+ tumors, TCGA data showed that high WEE1 expression in HPV+ HNSCC is linked to better survival and CNA/mutations in TRAF3 and/or CYLD, which are suppressors of NF-κB activation, and whose loss was previously associated with increased NF-κB-related inflammatory signatures and better prognosis (39,40).
While the therapeutic implications for WEE1 as a target may or may not be extrapolated from the relationship of WEE1 expression to prognosis, these relationships suggest that combinations of WEE1 inhibitors with TNFα induction by radiation or ICB merits investigation. The poorer prognosis HPV− subset with higher WEE1 expression and immune deficient signatures could warrant examining combination of WEE1 inhibition with radiation, to enhance induction of genotoxic and TNFα-mediated cytotoxicity. Further studies would be needed to determine if the subset associated with lower expression of WEE1 and immune activated and checkpoint signatures would be more sensitive or resistant to a combination of WEE1 inhibitor with TNFα induced by ICB, which we observed in a syngeneic Murine Oral Cancer model (15). The HPV+ tumors that express high levels of WEE1 and inflammatory signatures, may merit investigation of WEE1 inhibitors and ICB, while the non-inflamed tumors may be responsive to a combination with radiation. Overall, HPV+ HNSCC, will likely respond better to WEE1 inhibition (56) and therefore may enable reduction in the intensity of chemo-radiotherapy, or offer ICB as a possible substitute for the more toxic chemotherapies currently used. Taken together, our experimental and bioinformatic analyses suggest that expression of WEE1/CDC2 G2/M checkpoint and related NF-κB pro-survival and inflammatory signatures could provide additional candidate biomarkers and targets to help elucidate and investigate combination therapies in distinct HNSCC subtypes.
Supplementary Material
Implications.
Inhibiting WEE1 and IKK-RELA crosstalk could potentially enhance the effects of therapies mediated by TNFα with less systemic immune suppression and toxicity than observed with direct interruption of IKK-NF-κB/RELA signaling.
Acknowledgments
We thank Dr. Nicole Schmitt, MD, (Integrative Therapeutics Program, Head and Neck Surgery Branch, NIDCD/NIH), and Professor Cheng-Ming Chiang, PhD, (UT Southwestern Medical Center, Dallas, Texas) for reading this manuscript and providing helpful suggestions.
Funding information
This project is supported by NIDCD intramural projects Z01-DC-00016, 73, 74 (to C. Van Waes), and ZIA-DC000087 (to C. Allen). Zhengbo Hu was supported by the Nature Sciences Foundation of Guangdong, China (2017A030307012, 2018A0303070013).
Abbreviations
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papillomavirus
- RNAi
RNA interfere
- TNFα
Tumor necrosis factor-α
- ROS
oxygen species
- IKK
Inhibitor-kappaB kinase
- IκB
inhibitor kappaB
- CDK1/CDC2
cyclin-dependent kinase 1
- AZD1775/Adavosertib
AstraZeneca Drug 1775
- HOK
normal human oral keratinocytes
- UMSCC
University of Michigan squamous cell carcinoma
- MEM
minimal essential medium
- NEAA
non-essential amino acids
- IP
co-immunoprecipitation
- IB
immunoblot
- IF
Immunofluorescence
- qRT-PCR
Real-time quantitative polymerase chain reaction
- RTCA
Real-Time Cell Analysis
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
- OD
optical density
- SD
standard deviation
- ATM
ataxia-telangectasia mutated protein kinase
- ATR
ataxia-telangiectasia-related protein kinase
- Chk1
checkpoint kinase 1
- CCNB
Cyclin B
Footnotes
Disclosure of Potential Conflict of Interest: All authors declare having no conflict of interest.
Ethics Approval
In vivo experiments were approved by the NIDCD/NIH Animal Care and Use Committee under protocol #1322-16, in compliance with the Guide for the Care and Use of Laboratory Animal, National Research Council.
Competing interests
The authors declare that they have no competing interests. AZD1775 for in vivo studies was provided though a Cooperative Research and Development Agreement between NIDCD and AstraZeneca.
Availability of supporting data
The data used, analyzed and published in the current study are available from the corresponding author on request.
Reference
- 1.Chow LQM. Head and Neck Cancer. The New England Journal of Medicine. 2020;382:60–72. [DOI] [PubMed] [Google Scholar]
- 2.Morgan EL, Chen Z, Waes CV. Regulation of NFκB Signalling by Ubiquitination: A Potential Therapeutic Target in Head and Neck Squamous Cell Carcinoma? Cancers. 2020;12:2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urban JL, Shepard HM, Rothstein JL, Sugarman BJ, Schreiber H. Tumor necrosis factor: a potent effector molecule for tumor cell killing by activated macrophages. Proc National Acad Sci. 1986;83:5233–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc National Acad Sci. 1989;86:10104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ventura J-J, Cogswell P, Flavell RA, Baldwin AS, Davis RJ. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Gene Dev. 2004;18:2905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durand JK, Baldwin AS. Chapter Three Targeting IKK and NF-κB for Therapy. Adv Protein Chem Str. 2017;107:77–115. [DOI] [PubMed] [Google Scholar]
- 7.Allen CT, Ricker JL, Chen Z, Waes CV. Role of activated nuclear factor-κB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–71. [DOI] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan EL, Macdonald A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFκB-IL-6 signalling axis. PLoS Pathogens. 2019;15:e1007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benada J, Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomol. 2015;5:1912–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. [DOI] [PubMed] [Google Scholar]
- 12.Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle. 2013;12:3348–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osman AA, Monroe MM, Alves MVO, Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High-Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol Cancer Ther. 2015;14:608–19. 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33:3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, Moore E, Berman R, Clavijo PE, Saleh A, Chen Z, et al. WEE1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. Oncoimmunology. 2018;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleh AD, Cornelius S, Cheng H, Martin S, Ormanoglu P, Yang X, et al. Abstract 3400: Integrated functional RNAi screening and structural genomics identifies inverse co-modulators of TP53 family and NF-kB transitional activation as potential therapeutic targets in head and neck squamous cell carcinoma. Mol Cell Biol. 2014;3400–3400 [Google Scholar]
- 17.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.König R, Chiang C, Tu BP, Yan SF, DeJesus PD, Romero A, et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods. 2007;4:847–9. [DOI] [PubMed] [Google Scholar]
- 19.Campbell JD, Yau C, Bowlby R, Liu Y, Brennan K, Fan H, et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Reports. 2018;23:194–212.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, et al. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J Proteome Res. 2015;14:2707–13. [DOI] [PubMed] [Google Scholar]
- 21.Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD, Rodland KK, et al. Connecting Genomic Alterations to Cancer Biology with Proteomics: The NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013;3:1108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6:914–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keck MK, Zuo Z, Khattri A, Stricker TP, Brown CD, Imanguli M, et al. Integrative Analysis of Head and Neck Cancer Identifies Two Biologically Distinct HPV and Three Non-HPV Subtypes. Clin Cancer Res. 2015;21:870–81. 34 [DOI] [PubMed] [Google Scholar]
- 26.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic acids research. 2020;48:W488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Team RC. R: A language and environment for statistical computing. R foundation for Statistical Computing; 2015. [Google Scholar]
- 29.Duffey DC, Crowl-Bancroft CV, Chen Z, Ondrey FG, Nejad-Sattari M, Dong G, et al. Inhibition of transcription factor nuclear factor-kappaB by a mutant inhibitor-kappaBalpha attenuates resistance of human head and neck squamous cell carcinoma to TNF-alpha caspase-mediated cell death. British Journal of Cancer. 2000;83:1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy-number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng H, Yang X, Si H, Saleh AD, Xiao W, Coupar J, et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Reports. 2018;25:1332–1345.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awasthee N, Rai V, Chava S, Nallasamy P, Kunnumakkara AB, Bishayee A, et al. Targeting IκappaB kinases for cancer therapy. Semin Cancer Biol. 2018;56:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan EL, Patterson MR, Barba-Moreno D, Scarth JA, Wilson A, Macdonald A. The deubiquitinase (DUB) USP13 promotes Mcl-1 stabilisation in cervical cancer. Oncogene. 2021;40:2112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulson JR, Taylor SS. Phosphorylation of histones 1 and 3 and nonhistone high mobility group 14 by an endogenous kinase in HeLa metaphase chromosomes. J Biol Chem. 1982;257:6064–72. [PubMed] [Google Scholar]
- 35.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. Sites of UV-induced Phosphorylation of the p34 Subunit of Replication Protein A from HeLa Cells*. J Biol Chem. 1997;272:23896–904. [DOI] [PubMed] [Google Scholar]
- 36.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139*. J Biol Chem. 1998;273:5858–68. [DOI] [PubMed] [Google Scholar]
- 37.Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Research. 2016;76:5442–54. 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao R, An Y, Ye W, Derakhshan A, Cheng H, Yang X, et al. Dual Antagonist of cIAP/XIAP ASTX660 Sensitizes HPV− and HPV+ Head and Neck Cancers to TNFα, TRAIL, and Radiation Therapy. Clinical Cancer Research. 2019;25:6463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajek M, Sewell A, Kaech S, Burtness B, Yarbrough WG, Issaeva N. TRAF3/CYLD mutations identify a distinct subset of human papillomavirus-associated head and neck squamous cell carcinoma. Cancer. 2017;123:1778–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Chen T, Yang X, Cheng H, Späth SS, Clavijo PE, et al. Attenuated TRAF3 Fosters Activation of Alternative NF-κB and Reduced Expression of Antiviral Interferon, TP53, and RB to Promote HPV-Positive Head and Neck Cancers. Cancer Research. 2018;78:4613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc National Acad Sci. 2000;97:10389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. [DOI] [PubMed] [Google Scholar]
- 43.Sakamoto K, Hikiba Y, Nakagawa H, Hirata Y, Hayakawa Y, Kinoshita H, et al. Promotion of DNA repair by nuclear IKKβ phosphorylation of ATM in response to genotoxic stimuli. Oncogene. 2013;32:1854–62. [DOI] [PubMed] [Google Scholar]
- 44.Colomer C, Margalef P, Villanueva A, Vert A, Pecharroman I, Solé L, et al. IKKα Kinase Regulates the DNA Damage Response and Drives Chemo-resistance in Cancer. Mol Cell. 2019;75:669–682.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B, et al. TAK1 Is Recruited to the Tumor Necrosis Factor-α (TNF-α) Receptor 1 Complex in a Receptor-interacting Protein (RIP)-dependent Manner and Cooperates with MEKK3 Leading to NF-κB Activation*. J Biol Chem. 2005;280:43056–63. [DOI] [PubMed] [Google Scholar]
- 46.Häcker H, Karin M. Regulation and Function of IKK and IKK-Related Kinases. Sci Stke. 2006;2006:re13–re13. [DOI] [PubMed] [Google Scholar]
- 47.Mazzera L, Lombardi G, Abeltino M, Ricca M, Donofrio G, Giuliani N, et al. Aurora and IKK kinases cooperatively interact to protect multiple myeloma cells from Apo2L/TRAIL. Blood. 2013;122:2641–53. [DOI] [PubMed] [Google Scholar]
- 48.Hu Z, Li L, Lan W, Wei X, Wen X, Wu P, et al. Enrichment of Wee1/CDC2 and NF-κB Signaling Pathway Constituents Mutually Contributes to CDDP Resistance in Human Osteosarcoma. Cancer Res Treat. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ledoux AC, Perkins ND. NF-κB and the cell cycle. Biochem Soc T. 2014;42:76–81. 36 [DOI] [PubMed] [Google Scholar]
- 50.Ankers JM, Awais R, Jones NA, Boyd J, Ryan S, Adamson AD, et al. Dynamic NF-κB and E2F interactions control the priority and timing of inflammatory signalling and cell proliferation. Elife. 2016;5:e10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding H, Vincelette ND, McGehee CD, Kohorst MA, Koh BD, Venkatachalam A, et al. CDK2-Mediated Upregulation of TNFα as a Mechanism of Selective Cytotoxicity in Acute Leukemia. Cancer Res. 2021;81:2666–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Méndez E, Rodriguez CP, Kao M, Raju SC, Diab A, Harbison RA, et al. A Phase I Clinical Trial of AZD1775 in Combination With Neoadjuvant Weekly Docetaxel and Cisplatin Before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2018;24:2740–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanaka N, Patel AA, Tang L, Silver NL, Lindemann A, Takahashi H, et al. Replication Stress Leading to Apoptosis within the S-phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin Cancer Res. 2017;23:6541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diab A, Kao M, Kehrli K, Kim HY, Sidorova J, Méndez E. Multiple defects sensitize p53-deficient head and neck cancer cells to the WEE1 kinase inhibition. Mol Cancer Res. 2019;17: 1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M, et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clin Cancer Res. 2015;21:4831–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diab A, Gem H, Swanger J, Kim HY, Smith K, Zou G, et al. FOXM1 drives HPV+ HNSCC sensitivity to WEE1 inhibition. Proc National Acad Sci. 2020;117:28287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindemann A, Patel AA, Tang L, Tanaka N, Gleber-Netto FO, Bartels MD, et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated With Excessive DNA Damage and Replication Stress. Mol Cancer Ther. 2021;20:1257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used, analyzed and published in the current study are available from the corresponding author on request.