

Abstract
Brain-derived neurotrophic factor (BDNF) is essential for maintaining energy and glucose balance within the central nervous system (CNS). Because the study of its metabolic actions has been limited to effects in neuronal cells, its role in other cell types within the brain remains poorly understood. Here we show that astrocytic BDNF signaling within the ventromedial hypothalamus (VMH) modulates neuronal activity in response to changes in energy status. This occurs via the truncated TrkB.T1 receptor. Accordingly, either fasting or central BDNF depletion enhances astrocytic synaptic glutamate clearance, thereby decreasing neuronal activity in mice. Importantly, selective depletion of TrkB.T1 in VMH astrocytes blunts the effects of energy status on excitatory transmission, as well as on responses to leptin, glucose and lipids. These effects are driven by increased astrocytic invasion of excitatory synapses, enhanced glutamate reuptake and decreased neuronal activity. We thus identify BDNF/TrkB.T1 signaling in VMH astrocytes as an essential mechanism that participates in energy and glucose homeostasis.
Introduction
Obesity and its associated metabolic disorders are pervasive and challenging to treat. A better understanding of the central neural circuitry controlling energy and glucose balance is necessary to devise effective strategies to tackle this debilitating disease. Metabolic circuits are well represented in the hypothalamus, which integrates caloric status signals originating in the periphery and responds by altering feeding behavior and energy expenditure to meet the nutritional demands of the animal. The ventromedial hypothalamus (VMH) is an energy balance-regulating center, containing primarily excitatory glutamatergic neurons that suppress feeding and promote glycemic control1. While brain derived-neurotrophic factor (BDNF) signaling in the VMH is known to play chief roles mediating these actions, the underlying mechanisms remain poorly defined.
BDNF signals via the TrkB receptor to regulate synaptic transmission and plasticity in many regions of the mature brain2. In the VMH, it is a requisite appetite-suppressing factor and its expression there is reduced during conditions of negative energy balance3,4. Accordingly, brain-wide or VMH-specific deletion of Bdnf in mice elicits hyperphagia, obesity, and metabolic dysfunction, and these alterations are associated with diminished excitatory drive onto anorexigenic VMH neurons4,5. Significant associations of mutations in Bdnf and Trkb with obesity susceptibility have also been reported in human populations, further attesting to the importance of this signaling pathway in mediating metabolic health6–8.
Previous investigations have focused exclusively on defining the effects of neuronal BDNF signaling on energy and glucose homeostasis. Although a limited number of studies show that cortical and hippocampal astrocytes only express TrkB.T1, the truncated isoform of the BDNF receptor, the effects of astrocytic BDNF/TrkB.T1 signaling on metabolic function have not been investigated. It is important to consider these effects as astrocytes actively participate in synaptic transmission via gliotransmitter signaling, synaptic glutamate clearance, and synapse remodeling. Furthermore, perisynaptic astrocyte processes (PAPs) are highly plastic, rapidly altering their synaptic coverage in response to diverse stimuli9–11. The proximity of PAPs to synapses influences the ability of the astrocytic glutamate transporters GLAST (EAAT1) and GLT-1 (EAAT2) to regulate the duration of glutamate events at the synapse and glutamate spillover to extrasynaptic receptors and neighboring synapses12. Despite these important roles, only a few studies have examined the function of astrocytes in feeding circuits and many of those focused on effects under pathological conditions, including dietary challenge.
Here, we investigated for the first time the physiological role of BDNF/TrkB.T1 signaling in VMH astrocytes regulating excitatory transmission and energy and glucose homeostasis. We show that VMH astrocytes are functionally dynamic and regulated by energy status. Notably, we demonstrate an essential role of astrocytic TrkB.T1 signaling regulating PAP invasion of VMH synapses, synaptic glutamate reuptake, VMH neuronal activity, energy and glucose balance, and responses to leptin.
RESULTS
VMH neuronal activity is regulated by energy status and BDNF signaling
Behavioral and physiological responses to changes in caloric status are thought to be mediated by dynamic synaptic remodeling and concomitant changes in the activity of feeding circuits13,14. To investigate whether this plasticity occurs in the VMH, we examined the effects of energy status on the density of excitatory synapses in this region. For this, co-localization of excitatory presynaptic (VGLUT2) and postsynaptic (PSD95) markers was measured in VMH sections from fed and fasted wild type (WT) mice. Fasted WT animals exhibited a significant decrease in excitatory synapse density in the central and dorsomedial (dm/cVMH) but not in the ventrolateral (vlVMH) VMH compared to fed mice (Fig. 1a, b and c). Because fasting reduces levels of BDNF in the VMH4, we tested whether this neurotrophin regulates density of excitatory synapses in this region using BDNF2L/2L:CK-Cre mutant mice, which have global depletion of BDNF in the brain5. Similar to fasted WT mice, fed BDNF2L/2L:CK-Cre mice, displayed a significant reduction in excitatory synapse density in the dm/cVMH but not in the vlVMH compared to fed controls (Fig. 1d and e).
Figure 1: VMH neuronal activity is dynamically regulated by energy status and BDNF signaling.
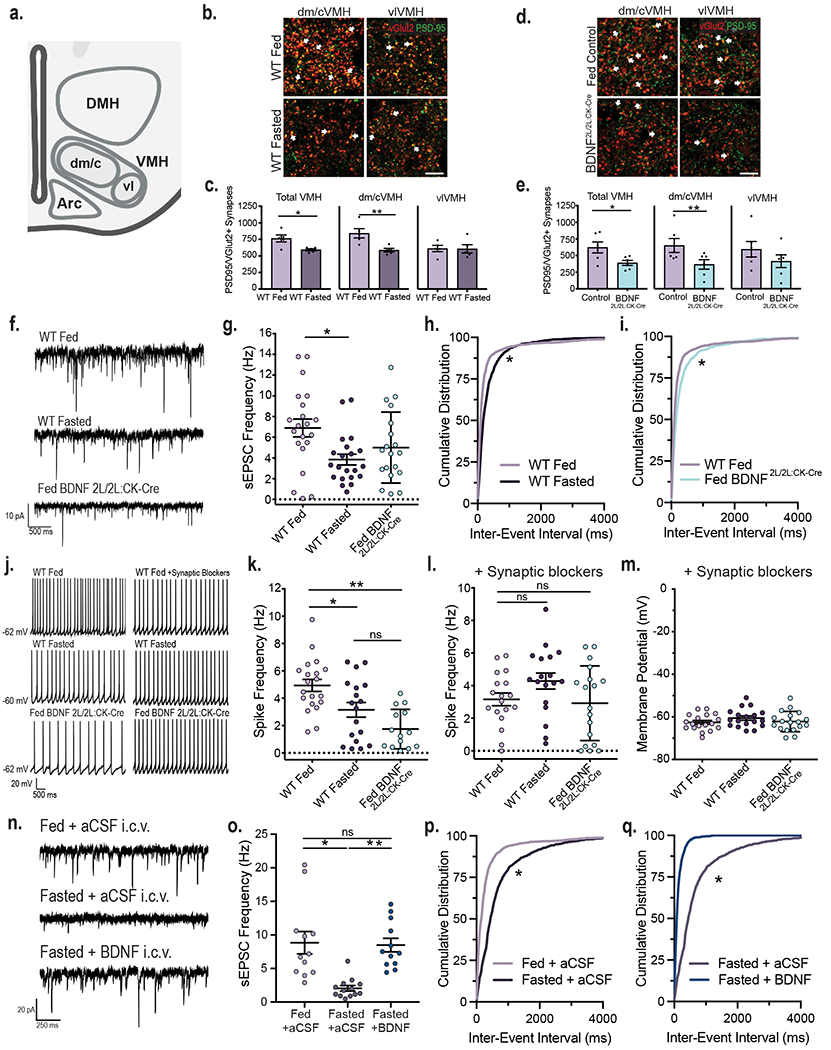
a, Diagram indicating the central/dorsomedial (dm/c) and ventrolateral (vl) regions of the VMH where excitatory synapse density was measured. Created with Biorender.com b, Representative images of VMH tissue from fed and fasted wild type (WT) mice co-immunolabeled with anti-vGlut2 and anti-PSD95. Arrows indicate PSD95 and vGlut2 co-localization (scale bar = 15 uM). c, Quantification of number of excitatory synapses in the VMH of fed and fasted WT mice (n = 5 mice). Student’s unpaired two-sided t-test, *, p = 0.01; **; p = 0.009. d, Representative images of VMH from fed control and BDNF2L/2L:CK-cre mice (scale bar 15 uM). e, Quantification of number of excitatory synapses in the VMH of fed control and BDNF2L/2L:CK-Cre mice (n = 6 mice). Student’s unpaired two-sided t-test, *, p = 0.02; **, p = 0.04. f, Representative traces of sEPSCs in VMH neurons. g, Frequency of sEPSCs in VMH neurons of fed (n = 21 cells) and fasted WT (n = 21 cells) and fed BDNF2L/2LCK:Cre mice (n = 19 cells) from 4 - 6 mice. Ordinary One-way ANOVA, p = 0.01. Tukey multiple comparisons, *, p = 0.01. h, Cumulative distributions of inter-event interval generated from 50 random events per recorded cell for fed and fasted WT (*, p < 0.001, KS = 0.227, Kolmogorov-Smirnov) and i, fed WT and BDNF2L/2LCK:Cre mice (*, p < 0.001, KS = 0.167, Kolmogorov-Smirnov, two-sided). j, Representative traces in current clamp showing VMH neuronal firing. k, Spike frequency of VMH neurons of fed (n = 20 cells) and fasted WT (n = 18 cells) and fed BDNF2L/2LCK:Cre mice (n = 14 cells) (4 – 6 mice per group). Ordinary One-way ANOVA, p < 0.0001. Tukey multiple comparisons, *, p = 0.019; **, p < 0.0001. l, Spike frequency and m, Membrane potential of VMH neurons of fed and fasted WT and fed BDNF2L/2LCK:Cre mice in the presence of 10 uM SR 95531, 50 uM NBQX and 10 uM CPP (n = 18 cells, 3 - 4 mice). Spike Frequency Ordinary One-way ANOVA, p = 0.1078. n, Representative traces of sEPSCs in VMH neurons in Fed WT + aCSF, Fasted WT + aCSF and Fasted WT + BDNF mice. o, Frequency of sEPSCs in VMH neurons in Fed WT + aCSF (n = 12 cells), Fasted WT + aCSF (n = 13 cells) and Fasted WT + BDNF (n = 12 cells) mice (4 mice per group). Ordinary One-way ANOVA, p = 0.0001. Tukey multiple comparisons, *, p < 0.0004; **, p = 0.0007. p, Cumulative distributions of inter-event interval generated from 50 random events per recorded cell for Fed WT + aCSF and Fasted WT + aCSF conditions (*, p < 0.0001, KS = 0.409, Kolmogorov-Smirnov, two-sided) and q, Fasted WT + aCSF and Fasted WT + BDNF conditions (*, p < 0.0001, KS = 0.591, Kolmogorov-Smirnov, two-sided). Data represented as mean +/− SEM.
To determine whether the observed synaptic remodeling was associated with changes in neuronal excitability, spontaneous excitatory postsynaptic currents (sEPSCs) were recorded from neurons in the dm/cVMH of fed and fasted WT animals and fed BDNF2L/2L:CK-Cre mice in the presence of the GABAA antagonist SR95531 to isolate excitatory events. In WT animals, sEPSC frequency was significantly decreased in the fasted compared to the fed state, corresponding to a rightward shift of the sEPSC interevent interval (Fig. 1f, g and h), whereas amplitude was similar in both groups (Extended Data Fig. 1a). No significant differences in sEPSC frequency or amplitude were observed in fed BDNF2L/2L:CK-Cre compared to fed and fasted control animals (Fig. 1f and g and Extended Data Fig. 1a). However, there was a significant rightward shift of the sEPSC interevent interval in BDNF2L/2L:CK-Cre mice compared to fed control animals, indicating that excitatory events are less frequent in fed BDNF mutants (Fig. 1i).
Fasting also triggered a significant reduction in the firing rate of VMH neurons compared to the fed state in WT mice (Fig. 1j and k), consistent with previous studies indicating that VMH neurons are anorexigenic and thus, expected to be less active when energy stores are low15. Notably, whereas the firing rate in fed BDNF2L/2L:CK-Cre mice was significantly reduced compared to fed WT controls, it was similar to that of fasted controls (Fig. 1j and k).
Modifications in intrinsic excitability can also contribute to changes in neuronal activity. Thus, whole-cell recordings were performed in fed and fasted WT mice and fed BDNF2L/2L:CK-Cre mice in the presence of synaptic blockers for GABAA, AMPA, and NMDA receptors. Under these conditions, firing rates across all groups were no longer different (Fig. 1j and l), indicating that synaptic changes elicited by caloric status and diminished BDNF signaling drove the differences in neuronal activity observed. Accordingly, there were no significant differences in the number of action potentials evoked by increasing depolarizing current injections across groups, indicating that changes in intrinsic excitability did not contribute to changes in activity elicited by fasting or BDNF depletion (Extended Data Fig. 1c and d). Resting membrane potential was also similar between groups (Fig. 1m).
The data indicate that VMH neuronal activity is decreased when BDNF levels are low, as observed in fasted WT and fed BDNF2L/2L:CK-Cre mice (Fig. 1g, h and k). To determine whether diminished BDNF signaling in the fasted state is responsible for reductions in VMH neuronal excitability, we tested the sufficiency of BDNF reversing the effects of caloric status. For this, sEPSCs in VMH neurons were measured in fed WT animals that received intracerebroventricular (ICV) delivery of aCSF and in fasted WT mice with ICV delivery of aCSF or BDNF 2 hours prior. Fasted WT mice administered aCSF had a significant reduction in sEPSC frequency and a rightward shift in sEPSC interevent interval compared to fed WT animals administered aCSF (Fig. 1n, o and p). Strikingly, ICV administration of BDNF in fasted WT mice was sufficient to fully reverse VMH neuronal sEPSC frequency to the levels of fed mice treated with aCSF, inducing a large leftward shift in sEPSC interevent interval (Fig. 1n, o and q).
The cumulative data indicate that positive energy status and associated elevations in BDNF signaling augment the activity of VMH neurons by increasing the excitatory synaptic drive onto those cells, without affecting neuronal intrinsic excitability.
Energy status and BDNF regulate astrocytic glutamate clearance at VMH synapses
Extracellular glutamate is removed primarily by the astrocytic excitatory amino acid transporters GLT-1 and GLAST, limiting postsynaptic glutamate receptor activation and shaping excitatory neurotransmission16,17. We asked whether changes in glutamate clearance from VMH synapses might contribute to BDNF and energy status-dependent alterations in neuronal excitability. First, we examined the effects of feeding status and BDNF levels on protein levels of GLT-1 and GLAST in the VMH. Whereas GLAST levels were unaffected, there was a significant increase in GLT-1 expression in both fasted WT and fed BDNF2L/2L:CK-Cre animals compared to fed WT mice (Fig. 2a, b and c). Notably, GLT-1 expression in fasted WT and fed BDNF2L/2L:CK-Cre mice was similar. The findings show that feeding and BDNF negatively regulate GLT-1 expression in the VMH.
Figure 2: Energy status and BDNF regulate astrocytic glutamate uptake at VMH synapses.
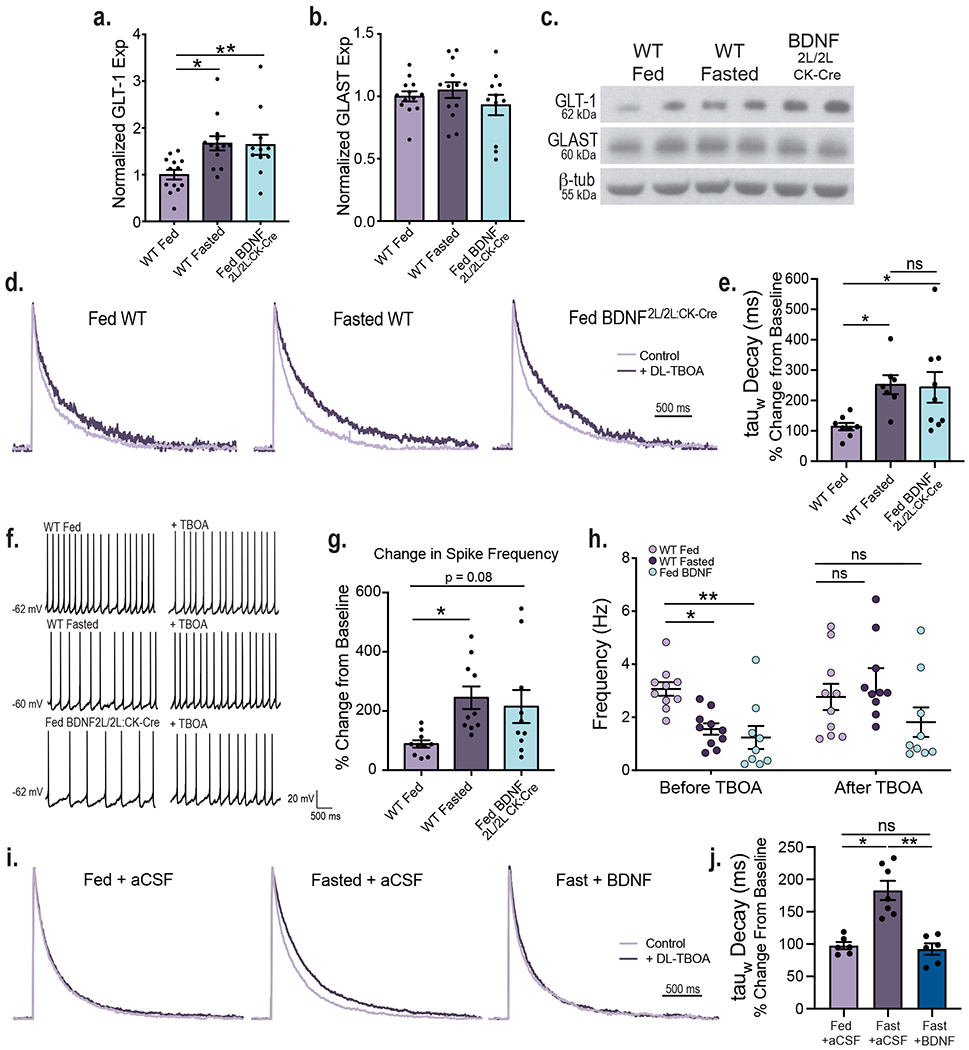
VMH GLT-1 (a) and GLAST (b) protein expression in fed (n = 13) and fasted WT (n =13) and fed BDNF2L/2L:CK-Cre mice (n = 11). GLT1 Ordinary One-way ANOVA, p = 0.006. GLAST Ordinary One-way ANOVA, p = 0.4. Tukey multiple comparisons, *, p = 0.01; **, p= 0.02. c, Representative western blots of GLT-1 and GLAST expression in VMH (Data collected from four experiments). d, Average amplitude-normalized NMDAR responses at baseline (light purple) and following application of 100 uM DL-TBOA (dark purple) in fed and fasted WT and fed BDNF2L/2L:CK-Cre mice. e, Effect of DL-TBOA on Decay of NMDAR responses (weighted tau) of fed (n = 8 cells) and fasted (n = 7 cells) WT and fed BDNF2L/2L:CK-Cre (n = 9 cells) mice ( 4 – 6 mice). Ordinary One-way ANOVA, p = 0.02. Tukey multiple comparisons, *, p = 0.04. f, Representative traces in current clamp showing VMH neuronal firing at baseline and following application of DL-TBOA. g, Percent change in neuronal firing rate (Hz) in response to bath DL-TBOA (100 uM) application (n = 10 cells, 4 – 5 mice). Ordinary One-way ANOVA, p = 0.0227. Tukey multiple comparisons, *, p = 0.0255. h, Spike frequency of VMH neurons of fed (n =10 cells) and fasted (n = 10 cells) WT and fed BDNF2L/2L:CK-Cre (n = 9 cells) mice (4 – 5 mice per group) before and after bath application of 100 uM DL-TBOA. Two-way ANOVA; Treatment, p = 0.044; Genotype, p = 0.006; Interaction, p = 0.041. Bonferroni multiple comparisons, *, p = 0.024; **, p = 0.006. i, Average amplitude-normalized NMDAR responses at baseline (light purple) and following application of 100 uM DL-TBOA (dark purple) in Fed WT + aCSF, Fasted WT + aCSF and Fasted WT + BDNF mice. j, Effect of DL-TBOA on Decay of NMDAR responses (weighted tau) (Fed WT + aCSF n = 6 cells; Fasted WT + aCSF n = 7 cells; Fasted WT + BDNF n = 6 cells; from 4 mice per group) . Ordinary One-way ANOVA, p < 0.0001. Tukey multiple comparisons, *, p = 0.0001; **, p < 0.0001. Data represented as mean +/− SEM.
Next, we investigated whether the observed changes in GLT-1 expression had functional consequences. The rate of glutamate uptake and resulting synaptic glutamate availability alter the duration and kinetics of NMDA receptor (NMDAR) activation. Decreased glutamate clearance leads to a prolonged NMDAR response, as indicated by a prolonged decay of NMDAR-mediated currents18. To obtain a functional readout of the degree of glutamate uptake occurring at VMH synapses, we measured stimulus-invoked NMDAR currents before and after bath application of the glutamate transporter inhibitor DL-TBOA in fed and fasted WT and fed BDNF2L/2L:CK-Cre mice. DL-TBOA application will have a larger impact in conditions where glutamate transporter activity is elevated, leading to prolonged NMDAR responses. Fasted WT and fed BDNF2L/2L:CK-Cre animals exhibited a significantly greater increase in NMDAR current decay time after bath application of DL-TBOA compared to fed controls, consistent with their increased levels of GLT-1 in the VMH, indicating an enhancement in glutamate clearance in these states (Fig. 2d and e). Notably, the effect of DL-TBOA on NMDAR decay kinetics was similar in fasted WT compared to fed BDNF2L/2L:CK-Cre mice. It is important to note that the baseline kinetics of NMDAR receptors as measured by current amplitude, charge transfer and decay were similar across experimental groups (Extended Data Fig. 2). Therefore, observed differences in NMDAR kinetics in response to DL-TBOA are due to alterations in synaptic glutamate uptake.
We interrogated the role of enhanced synaptic glutamate clearance in reducing neuronal activity in the VMH of fasted WT and fed BDNF2L/2L:CK-Cre mice. For this, the firing rate of VMH neurons was recorded before and after bath application of DL-TBOA, with no other drugs present in the bath. Following glutamate transporter blockade with DL-TBOA, firing rate was 88.5%, 244% and 215% that of baseline (no DL-TBOA) in fed WT, fasted WT and fed BDNF2L/2L:CK-Cre mice, respectively (Fig. 2f, g and h). Notably, neuronal firing rates across all experimental groups were no longer significantly different following application of DL-TBOA, indicating that changes in astrocytic glutamate clearance were largely responsible for differences in VMH neuronal activity elicited by caloric status and BDNF signaling.
To determine whether BDNF is the critical signal mediating the effects of energy status, we examined the consequence of ICV BDNF delivery in fasted WT mice on glutamate uptake at VMH synapses. We found that DL-TBOA had a significantly greater effect on NMDAR decay kinetics in fasted WT mice compared to fed WT animals administered aCSF, indicating increased glutamate clearance in the fasted state (Fig. 2i and j). Notably, ICV BDNF delivery was sufficient to fully reverse the effects of fasting on glutamate clearance (Fig. 2i and j). Accordingly, the effects of DL-TBOA on NMDAR decay kinetics were similar in fasted WT mice treated with BDNF compared to fed WT mice treated with aCSF. The baseline kinetics of NMDAR responses were similar across groups (Extended Data Fig. 3).
In total, the results indicate that inhibition of astrocytic glutamate clearance at VMH synapses is a chief mechanism driven by positive energy balance and concomitant increases in BDNF signaling to augment the activity of anorexigenic neurons.
TrkB.T1 signaling in VMH astrocytes is required for body weight control
We sought to ascertain whether BDNF acts directly on VMH astrocytes to regulate neuronal activity and energy and glucose balance control. BDNF signals through TrkB.FL and activates the MAP kinase, PI3 kinase, and phospholipase Cγ pathways to regulate synaptic plasticity and neuronal function2. The TrkB locus also generates a truncated form of the receptor, TrkB.T1, that lacks the intracellular tyrosine kinase domain and binds BDNF with the same affinity as TrkB.FL19. However, our understanding of its biological functions is limited. Interestingly, cortical and hippocampal astrocytes exclusively express TrkB.T1 and its activation by BDNF regulates astrocyte Ca2+ influx and cell morphology via Rho GTPase activity20–22. Our measurements in whole WT VMH tissue showed protein expression of both TrkB.FL and TrkB.T1, with TrkB.T1 expression levels being significantly higher (Fig. 3a and b). Astrocytes were isolated from acute WT VMH tissue using magnetic beads conjugated to the astrocyte-specific cell surface marker ACSA-2 to determine which TrkB isoforms they express. Validation studies show that this approach efficiently isolates astrocytes from other cell types in dissociated tissue (Extended Data Fig. 4a and b). Whereas isolated VMH astrocytes express TrkB.T1 mRNA, TrkB.FL mRNA was undetectable in this cell population (Fig. 3c). Moreover, the flowthrough fraction, consisting primarily of neurons as indicated in Extended Data Fig. 4a and b, contained TrkB.FL mRNA as well as TrkB.T1 mRNA at lower levels (Fig. 3c).
Figure 3: TrkB.T1 signaling in VMH astrocytes is essential for the regulation of energy balance.
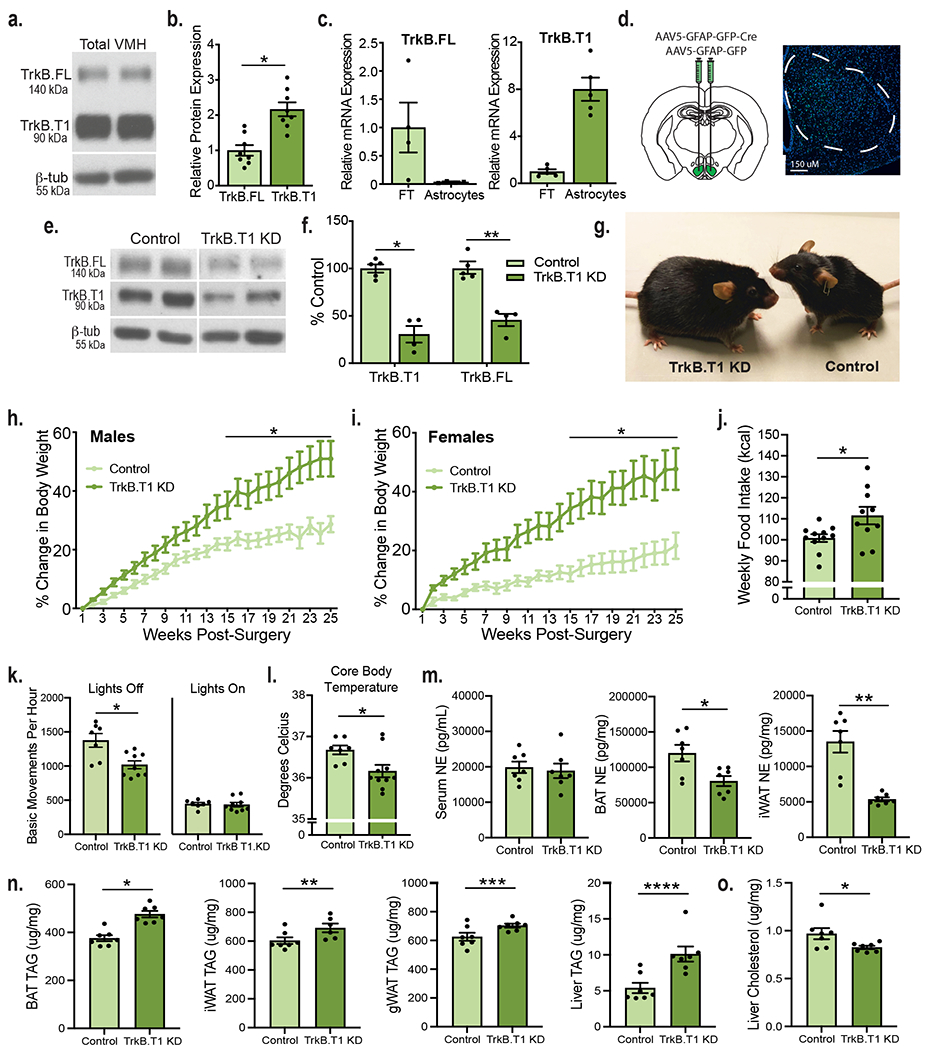
a, Representative western blot showing TrkB expression in the adult WT VMH. b, Quantification of TrkB FL and TrkB.T1 protein and in the WT VMH (n = 8). Students unpaired two-sided t-test, *, p = 0.0003. c, Expression of TrkB.FL (n = 4) and TrkB.T1 (n = 5) mRNA in astrocytes isolated from adult WT VMH compared to flowthrough. d, Diagram showing experimental approach for depleting TrkB.T1 in adult VMH astrocytes bilaterally and AAV5-GFAP-driven GFP signal throughout the VMH. e, Representative western blot showing knockdown of TrkB.FL and TrkB.T1 in mice delivered AAV5-GFAP-GFP (control) or AAV5-GFAP-GFPCre (TrkB.T1 KD) 5 weeks post-surgery. f, Representative quantification of viral knock down of TrkB.T1 and TrkB.FL 5 weeks post-surgery (Control n = 5 and TrkB.T1 KD, n = 4). Student’s unpaired two-sided t-test, *, p = 0.0001; **, p = 0.0004. g, Representative image of TrkB.T1 KD and control males 25 weeks post-surgery. h, Percentage body weight gain in TrkB.T1 KD (n = 10) and control males (n = 12) fed a chow diet. Two-way RM ANOVA: Genotype, p = 0.003; Time, p <0.0001; Genotype x Time Interaction, p < 0.0001; Subjects (matching), p < 0.0001. Bonferroni multiple comparisons, *, p < 0.05. i, Percentage body weight gain in TrkB.T1 KD (n = 12) and control females (n = 8) fed a chow diet. Two-way RM ANOVA: Genotype, p = 0.005; Time, p < 0.0001; Genotype x Time Interaction, p < 0.0001; Subjects (matching), p < 0.0001. Bonferroni multiple comparisons, *, p < 0.01. j, Weekly food intake (kcal) weeks 3 - 6 post-surgery (Control, n = 11 and TrkB.T1 KD, n = 10). Students unpaired two-sided t-test, *, p = 0.02. k, Basic movements per hour of TrkB.T1 KD (n = 9) and control (n = 7) males during 6 days of measurements during the light and dark cycles. Students unpaired two-sided t-test, *, p = 0.005. l, Rectal body temperature of TrkB.T1 KD (n = 10) and control males (n = 7). Students unpaired two-sided t-test, *, p = 0.02. m, Norepinephrine levels (pg/mL) in TrkB.T1 KD and control males (n = 7). Student’s unpaired two-sided t-test, *, p < 0.01; **, p = 0.0002. n, Triglyceride levels (ug/mg) in TrkB.T1 KD (n = 7) and control males (n = 6). Student’s unpaired two-sided t-test, *, p = 0.0001; **, p = 0.03; ***; p = 0.02; ****, p = 0.002. o, Cholesterol levels (ug/mg) in the liver of TrkB.T1 KD and control males (n = 7). Student’s unpaired two-sided t-test, *, p = 0.03. Data represented as mean +/− SEM.
Having determined that VMH astrocytes only express TrkB.T1, we examined its necessity in the regulation of metabolic function. Mice with selective deletion of TrkB.T1 in VMH astrocytes (TrkB.T1 KD) and controls were generated by bilateral stereotaxic delivery of AAV5-GFAP-GFP-Cre or AAV5-GFAP-GFP, respectively, to the VMH of floxed TrkB mice23 (Fig. 3d). The use of this AAV5 serotype, the GFAP promoter and optimized stereotaxic coordinates mediates astrocyte-specific transduction covering the VMH while avoiding nearby hypothalamic nuclei (Fig. 3d; Extended Data Fig. 4d, e, f and g). Immunolabeling of TrkB.T1 in tissue obtained from a floxed TrkB mouse injected unilaterally with AAV-GFAP-GFP-Cre shows a decrease in TrkB.T1 protein, 5 weeks after viral delivery compared to the un-injected side (Extended Data Fig. 4c). Furthermore, western blot analysis indicated a 70% reduction in TrkB.T1 protein content in VMH of TrkB.T1 KD mice compared to controls (Fig. 3e and f).
Bilateral knockdown of TrkB.T1 in adult VMH astrocytes led to a significant increase in weight gain in both male and female mice fed a chow diet (Fig. 3g, h and i and Extended Data Fig. 5a and b). At twenty-five weeks post-surgery, male TrkB.T1 KD mice exhibited a 50.9% increase in weight gain compared to a 28.6% increase in control animals (Fig. 3h). Female TrkB.T1 KD exhibited a 48.8% increase in weight gain compared to a 21.6% increase in controls (Fig. 3i). To rule out non-specific effects of the viral vectors, the effects of delivering AAV5-GFAP-GFP-Cre or AAV5-GFAP-GFP to the VMH of wild type C57Bl6 mice were compared to those elicited by delivering AAV5-GFAP-Cre to the VMH of floxed TrkB mice. As shown in Extended Data Fig. 5c, AAV-GFAP-GFP-Cre delivery to the VMH of floxed TrkB mice elicited a significant increase in percentage body weight gain compared to both, WT mice delivered AAV-GFAP-GFP-Cre or AAV-GFAP-GFP. Moreover, body weight gain in WT mice delivered AAV-GFAP-GFP-Cre or AAV-GFAP-GFP was not significantly different, ruling out non-specific effects of the viral vectors.
We discovered that knockdown of TrkB.T1 in VMH astrocytes also produced a significant decrease in TrkB.FL expression in the VMH (Fig. 3e and f), the primary isoform expressed by neurons but not by astrocytes (Fig. 3c). Because TrkB.FL expression is regulated by neuronal activity in other brain regions24–26, it is possible that putative changes in neuronal excitatory drive in astrocytic TrkB.T1 KD mice caused this deficit. We sought to determine whether reduced neuronal TrkB.FL expression contributed to the excessive weight gain observed in TrkB.T1 KD mice. For this, mice with selective deletion of TrkB in VMH neurons (Neuronal TrkB KD) and controls were generated by stereotaxic delivery of AAV2-CMV-Cre-GFP or AAV2-CMV-GFP bilaterally into the VMH of floxed TrkB mice (Extended Data Fig. 6a). The use of this serotype AAV2 viral vector mediates neuronal-specific transduction within the VMH (Extended Data Fig. 6b) and reduced both TrkB.FL and TrkB.T1 expression (Extended Data Fig. 6c–d), as expected as neurons contain both isoforms27,28.
In contrast to TrkB.T1 KD mice, male neuronal TrkB KD mice exhibited similar levels of weight gain and total body weight as controls (Extended Data Fig. 7a and b), showing that TrkB.FL and TrkB.T1 signaling in VMH neurons is not required for the regulation of energy balance. Importantly, the data indicate that the decrease in TrkB.FL observed in TrkB.T1 KD mice is unlikely to contribute significantly to their excessive weight gain. The findings identify VMH astrocytes as a critical cellular substrate for the effects of BDNF facilitating energy balance control.
TrkB.T1 in VMH astrocytes regulates feeding and energy expenditure.
We investigated mechanisms leading to increased body weight in TrkB.T1 KD mice. Because both male and female TrkB.T1 KD mutants exhibited energy balance dysregulation, we focused our studies on male mice. All measurements were performed at 3-6 weeks post-viral delivery to the VMH when weights of TrkB.T1 KD and controls are not yet significantly different to avoid secondary effects of obesity as confounding factors. TrkB.T1 KD mice exhibited a significant increase in chow intake compared to controls (Fig. 3j). They also showed a significant reduction in activity during the dark cycle (Fig. 3k, Extended Data Fig. 5d) as well as reduced core body temperature (Fig. 3l), indicating that decreased thermogenesis and energy expenditure also contributed to obesity in TrkB.T1 KD mice. Of note, mice with neuronal-specific depletion of TrkB in the VMH exhibited similar food intake, body temperature and locomotor activity as control animals, indicating that astrocyte TrkB.T1 is the critical cellular substrate of BDNF in regulating energy balance (Extended Data Fig. 7c, d and e).
The VMH plays chief roles regulating sympathetic output to metabolic tissues in the periphery. Notably, the dmVMH regulates sympathetic norepinephrine (NE) release onto brown (BAT) and white adipose tissue (WAT) fat pads via connections with the hindbrain, to influence thermogenesis and lipid mobilization29,30. We asked whether alterations in sympathetic tone may play a role eliciting thermogenesis dysregulation in TrkB.T1 KD mutant mice. We found that NE levels were significantly reduced in thermogenic BAT and inguinal WAT (iWAT) tissue as well as in gonadal WAT (gWAT) (Fig. 3m, Extended Data Fig. 5d). In contrast, NE content was normal in serum, liver and muscle in mutant compared to control mice (Fig. 3m and Extended Data Fig. 5e and f). These results suggest that impaired sympathetic output to adipose tissues may contribute to reduced thermogenesis in TrkB.T1 KD animals. Sympathetic output was also measured in neuronal TrkB KD mutants at 5-weeks post-surgery. These mice displayed a small but significant decrease in BAT NE content and normal NE content in iWAT, gWAT, liver, muscle and serum (Extended Data Fig. 7g).
Finally, we measured lipid levels in BAT, iWAT, gWAT and liver in TrkB.T1 KD mutants and controls considering that the VMH and sympathetic tone influence lipid metabolism31. These studies were performed 5 weeks post-surgery, when TrkB.T1 KD mutants exhibit similar body weights to controls to identify weight-independent alterations. BAT, iWAT, gWAT and livers of mutants contained significantly higher levels of triglycerides in TrkB.T1 KD mutants compared to controls (Fig. 3n). Additionally, hepatic levels of cholesterol were significantly reduced in the mutants (Fig. 3o).
The results indicate that astrocytic TrkB.T1 in the VMH is essential for the regulation of food intake, energy expenditure, and sympathetic output in the periphery, mediating energy and lipid balance control.
Glycemic control requires intact TrkB function in VMH astrocytes and neurons
The VMH is a glucose-sensing brain region mediating glycemic control. To determine whether astrocytic TrkB.T1 in the VMH is required for those functions, TrkB.T1 KD mice and controls were subjected to glucose tolerance tests (GTT) at 7 weeks post-surgery, when their body weights are not significantly different (Extended Data Fig. 5a). Whereas fasting levels of blood glucose were not different between groups (Fig. 4a, time point 0), TrkB.T1 KD animals exhibited impaired responses to a glucose challenge (Fig. 4a and b). To determine whether this alteration was associated with abnormal neuronal responses of VMH neurons to the glucose load, VMH neuronal activity was measured in fasted mice 60 minutes following glucose administration using c-fos expression as a surrogate for neuronal activity. TrkB.T1 KD mice showed a 42.3% decrease in VMH c-fos+ cells compared to controls, indicating decreased neuronal activity during a glucose challenge (Fig. 4c and d). Additionally, normal fasted levels of serum insulin and unaffected performance in the insulin tolerance test (ITT) suggest that insulin sensitivity and the counterregulatory response to hypoglycemia are normal in TrkB.T1 KD mutants (Fig. 4e, f and g).
Figure 4: Depletion of TrkB.T1 from VMH astrocytes of adult mice leads to impaired glycemic control and leptin resistance.
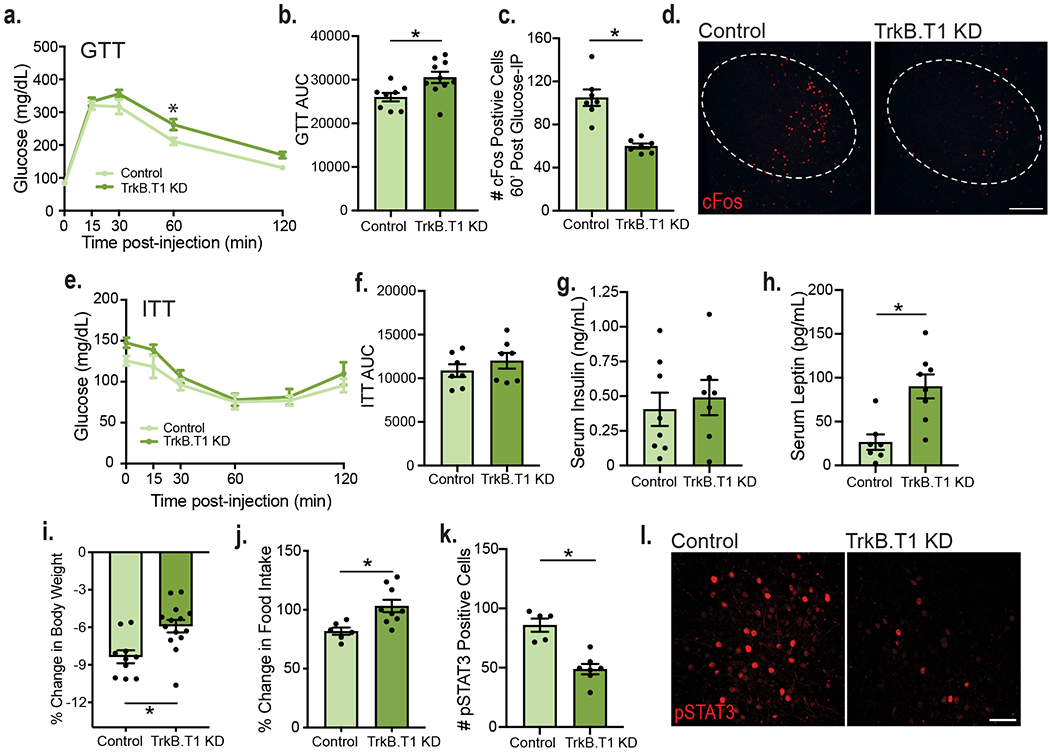
a, Glucose tolerance test (GTT) in TrkB.T1 KD (n = 10) and control males (n = 8). Two-way RM ANOVA; Time, p < 0.0001; Genotype, p = 0.0296; Time x Genotype Interaction, p = 0.0796; Subject, p = 0.0002. Bonferroni multiple comparisons, *, p = 0.01. b, Area under the curve for the GTT (Control, n = 8 and TrkB.T1 KD, n = 10). Students unpaired two-sided t-test, *, p = 0.01. c, Quantification of c-fos+ cells within the VMH of fasted (16 hours) TrkB.T1 KD mutant and control animals 60 min. following glucose administration (n = 7). Student’s unpaired two-sided t-test, *, p = 0.0001. d, Representative images of VMH c-fos expression 60 min. following glucose administration in fasted mice. Scale bar is 250 uM. e, Insulin tolerance test (ITT) in TrkB.T1 KD and control males (n = 7). Two-way RM ANOVA; Time, p < 0.0001; Genotype, p = 0.192; Time x Genotype Interaction, p = 0.626; Subject, p = 0.0001. Bonferroni multiple comparisons, NS. f, Area under the curve for the ITT (n = 7). Student’s unpaired two-sided t-test, NS g, Serum levels of insulin of fasted TrkB.T1 KD (n = 7) and control males (n = 8). h, Serum levels of leptin (pg/mL) in fed TrkB.T1 KD (n = 8) and control animals (n = 7). Students unpaired two-sided t-test, *, p = 0.002. i, Percent body weight change after 3 consecutive days of twice daily 3 ug/g leptin administration in TrkB.T1 KD (n = 14) and control males (n = 10). Students unpaired two-sided t-test, *, p = 0.003. j, Percent change in food intake from baseline (3-day vehicle administration) following 3 days of leptin administration (Control n = 6, TrkB.T1 KD n = 9). Student’s unpaired two-sided t-test, *, p = 0.009. k, Quantification of pSTAT3+ cells in TrkB.T1 mutant (n = 7) and control (n = 5) males 5 weeks post-surgery and 45 minutes post 5 ug/g leptin IP administration. Student’s t-test, *, p = 0.0003. h, Representative image of pSTAT3 expression in VMH 45 minutes post 5 ug/g leptin IP administration. Scale bar is 50 uM. Data represented as mean +/− SEM.
To determine whether the impaired glycemic control observed in TrkB.T1 KD mice was influenced by the homeostatic loss of neuronal TrkB observed in these mice, neuronal TrkB KD mice and controls were subjected to GTTs at 7-weeks post-surgery. Similar to TrkB.T1 KD mice, neuronal TrkB KD animals displayed normal fasting levels of glucose and normal responses in the ITT, while exhibiting impaired responses to a glucose challenge (Extended Data Fig. 7h–k). The data indicate that neuronal TrkB signaling mediates glycemic control and contributes to the glucose balance impairments observed in TrkB.T1 KD mice.
Astrocytic TrkB.T1 in the VMH mediates effects of leptin on metabolic function
Leptin receptor-containing cells in the VMH mediate some of the effects of this adipokine on energy and glucose balance control32–36. Notably, leptin administration elevates levels of BDNF in the VMH37. Thus, we asked whether effects of leptin on energy balance regulation required intact TrkB.T1 function in VMH astrocytes. TrkB.T1 KD mice displayed a 240% increase in serum leptin levels compared to controls at 5 weeks post-surgery, a time-point when mutant animals are not significantly heavier than controls (Fig. 4h and Extended Data Fig. 5a). Neuronal TrkB KD animals displayed a smaller but significant 100% increase in serum leptin levels 5 weeks post-surgery (Extended Data Fig. 7f). To determine whether the excessive weight gain observed in TrkB.T1 KD but not in neuronal TrkB KD mutants might be linked to diminished responses to leptin, the effects of three days of systemic administration of leptin were examined following a 3-day saline injection habituation period (Extended Data Fig. 5g). We found that control males lost significantly more weight than TrkB.T1 KD animals following chronic leptin treatment (Fig. 4i). Furthermore, whereas leptin delivery decreased food intake in controls, it did not affect feeding in TrkB.T1 KD mice (Fig. 4j). Using phosphorylated STAT3 (pSTAT3) as a surrogate for leptin receptor activation, we also observed a significant decrease in the density of pSTAT3+ cells in the dm/cVMH of TrkB.T1 KD mice compared to controls following leptin administration (Fig. 4k and l). The results show that BDNF/TrkB.T1 signaling in VMH astrocytes is requisite for effects of leptin mediating energy balance control.
Depletion of TrkB.T1 from VMH astrocytes decreases neuronal activity
To identify cellular mechanisms underlying the effects of astrocytic TrkB.T1 on metabolic function, neuronal activity was examined in TrkB.T1 KD and control mice 5 weeks post-surgery. Whole cell recordings were performed in fed and fasted mice to inform the necessity of astrocytic TrkB.T1 in mediating the effects of caloric status on VMH neuronal activity. sEPSC frequency was decreased in fasted compared to fed controls, with a rightward shift in sEPSC interevent interval (Fig. 5a–c). Notably, both fed and fasted TrkB.T1 KD mice exhibited a two-fold decrease in sEPSC frequency compared to fed control animals (Fig. 5a, b and d). In contrast to control animals, energy status did not impact sEPSC frequency or interevent interval in TrkB.T1 KD mice (Fig. 5a–c). Furthermore, there were no significant differences between fasted control animals and fed or fasted TrkB.T1 KD animals, suggesting that BDNF signaling in VMH astrocytes is required for elevations in VMH excitatory neurotransmission induced by feeding (Fig. 5b). Finally, there was a small but significant effect of genotype in sEPSC amplitude but the post hoc analysis did not reveal significant differences in amplitude in TrkB.T1 KD mice compared to controls (Fig. 5d).
Figure 5: Depletion of TrkB.T1 from VMH astrocytes of adult mice decreases neuronal activity.
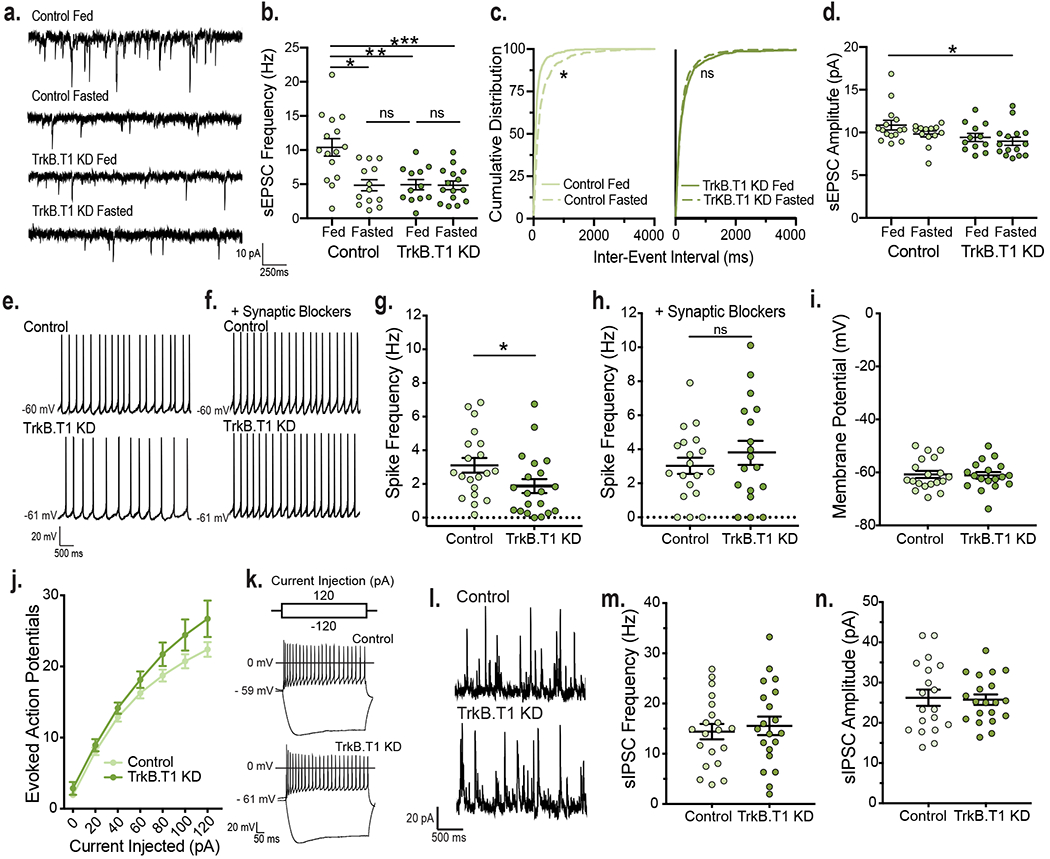
a, Representative traces and b, sEPSC frequency from fed (n = 12 cells) and fasted (n = 15 cells) TrkB.T1 KD and fed (n = 15 cells) and fasted (n = 13 cells) control animals (from 4 – 5 mice per group). Ordinary Two-way ANOVA; Genotype, p = 0.004; Energy Status, p = 0.004; Interaction of Genotype and Energy Status, p = 0.005. Tukey multiple comparisons, *, p = 0.0005; **; p = 0.0008; ***, p = 0.0003 . c, Cumulative distributions of inter-event interval generated from 50 random events per recorded cell for Control fed and fasted (*, p < 0.0001, KS = 0.30, Kolmogorov-Smirnov, two-sided) and TrkB.T1 KD fed and fasted mice (*, p = 0.25, KS = 0.05, Kolmogorov-Smirnov, two-sided). d, sEPSC amplitude (Control Fed n = 15 cells; Control Fasted n = 13 cells; TrkB.T1 KD Fed n = 12 cells; TrkB.T1 KD Fasted n = 15 cells, from 4 – 5 mice per group). Ordinary Two-way ANOVA; *, Genotype, p = 0.0216; Energy Status, p = 0.129; Interaction of Genotype and Energy Status, p = 0.55. e, Representative traces of spike frequency in the absence f, and presence of synaptic blockers. g, Spike frequency (Hz) of VMH neurons from fed control and TrkB.T1 KD males (n = 20 cells, from 5 - 6 mice per group). Students unpaired two-sided t-test, *, p = 0.04. h, Spike frequency of VMH neurons from fed TrkB.T1 KD (n = 18) and control (n = 19) (4 mice per group) males in the presence of synaptic blockers 10 uM SR 95531, 50 uM NBQX and 10 uM CPP. Student’s unpaired two-sided t-test, NS. i, Membrane potential of VMH neurons from fed TrkB.T1 KD (n = 18) and control (n = 19) (4 mice per group) mice in the presence of synaptic blockers. Student’s unpaired two-sided t-test, NS. j, Input-output curves of VMH neurons from fed TrkB.T1 KD (n = 14) and control (n = 15) mice (4 – 6 mice per group). Two-way RM ANOVA: Interaction, p = 0.2; Genotype, p = 0.09. k, Representative traces. l, Representative traces and m, Frequency (Hz) and n, amplitude of sIPSC from fed control and TrkB.T1 KD VMH neurons (n = 20 cells, 4 – 5 mice per group). Student’s unpaired two-sided t-test, NS. Data represented as mean +/− SEM.
The diminished excitatory drive onto VMH neurons in fed TrkB.T1 KD mice resulted in a significantly lower firing rate compared to fed controls (Fig. 5e and g). To assess whether alterations in intrinsic excitability were also responsible, neuronal firing was measured in the presence of synaptic blockers for GABAA, AMPA, and NMDA receptors. Under these conditions, firing rate was no longer different across groups, indicating that changes in the synaptic physiology but not in the intrinsic excitability of VMH neurons were at play (Fig. 5f and h). In support, resting membrane potential was unchanged in mutant cells (Fig. 5i). Moreover, the number of evoked action potentials following depolarizing current injections was comparable in fed TrkB.T1 KD and control mice (Fig. 5j and k).
We observed decreases in VMH excitatory synapse number and neuronal activity in BDNF2L/2L:CK-Cre mice (Fig. 1). Moreover, astrocytes secrete factors regulating excitatory synapse assembly38–40. Thus, we asked whether diminished BDNF/TrkB.T1 signaling in VMH astrocytes impedes excitatory synaptogenesis, diminishing the excitatory drive onto neurons in this region. No significant differences were observed in TrkB.T1 KD mutants compared to control (Extended Data Fig. 8a and b), indicating that other mechanisms drive the reduced activity of neurons in mice lacking TrkB.T1 in VMH astrocytes.
Astrocytes have been shown to regulate inhibitory synapse formation and GABAergic signaling in other regions of the brain41–44. To determine whether inhibitory transmission was altered by knockdown of astrocytic TrkB.T1, spontaneous inhibitory post-synaptic currents (sIPSCs) were recorded in VMH neurons. Frequency and amplitude of sIPSCs were normal in TrkB.T1 KD mice (Fig. 5l, m and n). The findings suggest that alterations in GABAergic transmission do not contribute to the hypoactivity of VMH neurons in TrkB.T1 KD mutants.
The collective data show that astrocytic TrkB.T1 signaling critically regulates the excitatory drive onto and activity of VMH neurons. Moreover, it is essential for effects of caloric status promoting excitatory synaptic plasticity in the VMH.
TrkB.T1 signaling in VMH astrocytes inhibits glutamate clearance at VMH synapses
GLT-1 expression and glutamate uptake at VMH synapses were increased in fasted WT and fed BDNF2L/2L:CK-Cre mice, leading to decreases in neuronal activity (Fig. 2). We hypothesized that diminished BDNF signaling via astrocytic TrkB.T1 contributed significantly to these effects. We found that similar to BDNF2L/2L:CK-Cre mice, TrkB.T1 KD mice exhibited a significant increase in GLT-1 and normal levels of GLAST protein in the VMH at 5 weeks post-surgery (Fig. 6a, b and c). Furthermore, the glutamate transporter inhibitor, DL-TBOA, had a significantly greater effect on the decay time of NMDAR currents in fasted compared to fed controls (Fig. 6d and e). Fed TrkB.T1 KD mice also exhibited enhanced glutamate clearance at VMH synapses compared to fed controls. Notably, the effect of fasting increasing synaptic glutamate clearance observed in control mice was absent in TrkB.T1 KD mice (Fig. 6d and e). Baseline kinetics of NMDAR receptors were similar across groups (Extended Data Fig. 9).
Figure 6: Specific depletion of TrkB.T1 from VMH astrocytes in adult mice leads to increased glutamate uptake at excitatory synapses.
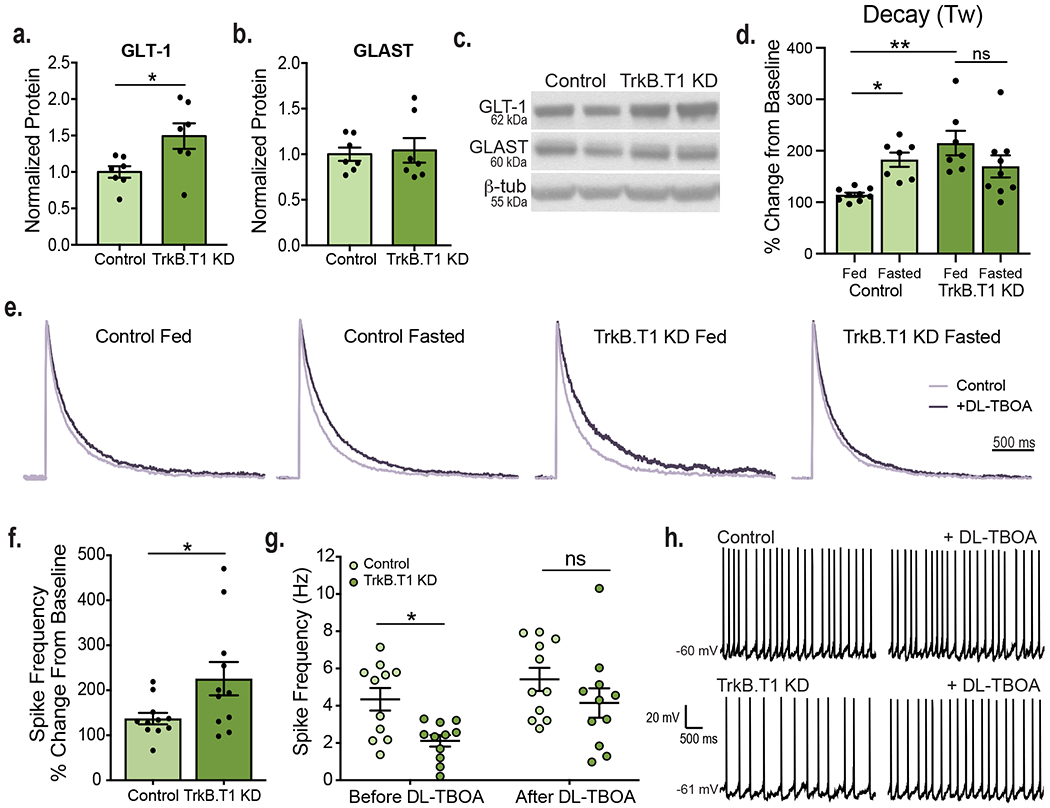
VMH GLT-1 a, and GLAST b, protein expression in TrkB.T1 KD and control mice 5 weeks post-surgery (n = 7). Students unpaired two-sided t-test, *, p = 0.02. c, Representative western blots of GLT-1 and GLAST expression in the VMH (Data collected from two experiments). d, Effect of DL-TBOA on the NMDAR current decay (Tau weighted, ms) in fed and fasted TrkB.T1 KD and control animals (Control Fed n = 9, Control Fasted n = 7, TrkB.T1 KD Fed n = 7, TrkB.T1 KD Fasted n = 9, 5 – 7 mice per group). Ordinary Two-way ANOVA; Genotype, p = 0.0175; Energy Status p = 0.51; Interaction of Genotype and Energy Status, p = 0.003. Tukey multiple comparisons, *, p = 0.044; **, p = 0.0017. e, Average amplitude-normalized NMDAR responses at baseline (light purple) and following application of 100 uM DL-TBOA (dark purple). f, Percent change in neuronal firing rate (Hz) in response to bath DL-TBOA (100 uM) application (n = 11 cells, 4 - 5 mice, per group). Student’s unpaired two-sided t-test, *, p = 0.03. g, Spike frequency of VMH neurons of fed control and TrkB.T1 KD mice before and after bath application of 100 uM DL-TBOA (n = 11 cells, 4 - 5 mice per group). Two-way ANOVA; Treatment, p = 0.014; Genotype, p = 0.006; Treatment x Genotype Interaction, p = 0.43. Tukey multiple comparisons, *, p = 0.059. h, Representative traces in current clamp showing VMH neuronal firing. Data are represented as mean +/− SEM.
To determine whether increased synaptic glutamate uptake drove the reduced VMH neuronal activity in TrkB.T1 KD mice, firing rate was measured before and after bath application of DL-TBOA. Whereas firing rate following DL-TBOA application was 137% that of baseline in control mice, it was 225% that of baseline in TrkB.T1 KD mice (Fig. 6f, g and h). Importantly, firing rates of VMH neurons in control and mutant mice were no longer significantly different following glutamate transporter blockade.
The findings indicate that BDNF/TrkB.T1 signaling in VMH astrocytes is the primary mechanism driving decreases in synaptic glutamate uptake in the fed state.
TrkB.T1 in VMH astrocytes regulates the organization of the tripartite synapse
The rate of glial glutamate uptake at synapses is determined by both the number of astrocytic glutamate transporters and the proximity of PAPs to the synapse, with increased process protrusion leading to greater glutamate clearance12. We hypothesized that BDNF/TrkB.T1 signaling in VMH astrocytes diminishes glutamate reuptake by reducing perisynaptic astrocyte invasion of excitatory synapses in addition to inhibiting GLT-1 expression. We performed ultrastructural analysis of synapses within the dm/c VMH in control and TrkB.T1 KD mice 5 weeks post-surgery (Fig. 7a). Asymmetric excitatory synapses were identified based on the presence of presynaptic vesicles and a thick prominent opposed post-synaptic density. Astrocytes were identified base on their irregular contouring borders, pale cytoplasm void of vesicles, and rounded processes. The percentage of synapses contacted by astrocytes was similar in controls and mutants. However, there was a trend towards a significant increase in bilateral coverage of synapses by PAPs in TrkB.T1 KD mice compared to controls (Fig. 7b and c). Moreover, synapse invasion by astrocytic processes was significantly increased in TrkB.T1 KD mice compared to control animals, with an average protrusion of 58 nm compared to 26 nm, respectively (Fig. 7d and e). There were no differences in synapse structure based on the length of the post-synaptic density and active site, and in the distance of astrocyte processes to the post-synaptic density (Fig. 7f – i).
Figure 7: Knockdown of TrkB.T1 in VMH astrocytes increases astrocyte invasion of VMH synapses.
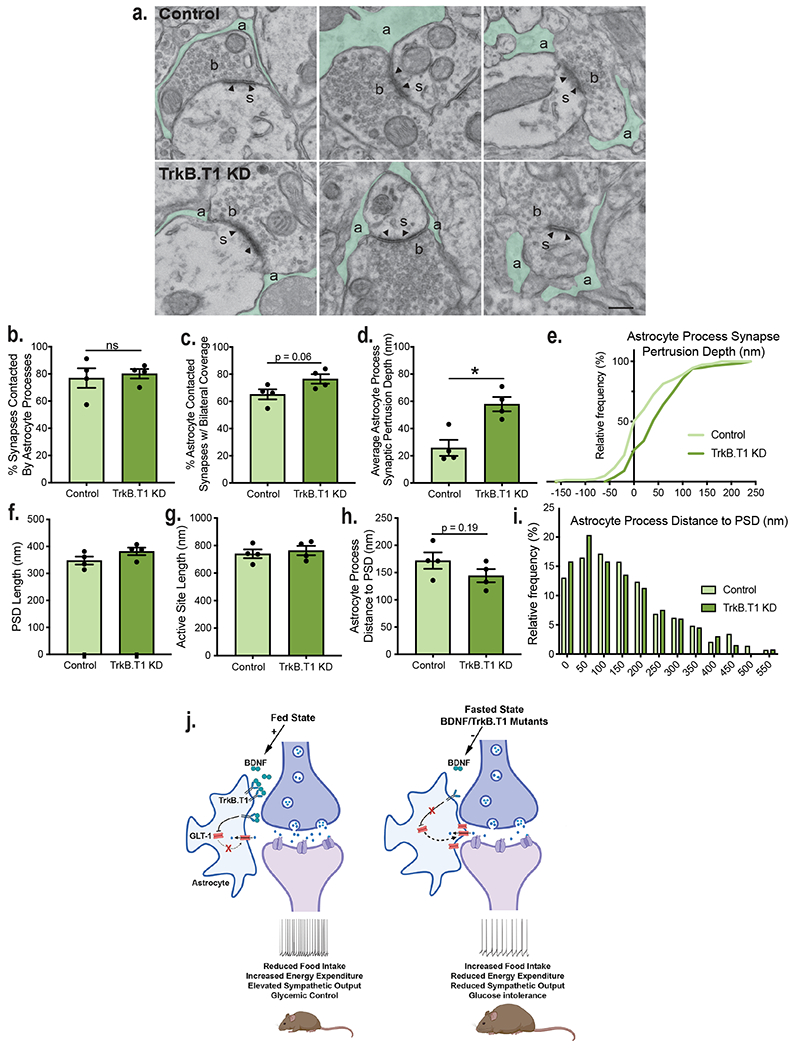
a, Representative electron microscopy images showing astrocyte processes (a, green) surrounding VMH synapses in control and TrkB.T1 KD mice 5 weeks post-surgery. Axonal bouton (b), dendritic spine (s), post-synaptic density (arrowhead). Scale bar 250 nm. For all of following panels, analysis was performed in control (n = 198 synapses) and TrkB.T1 KD (n = 166 synapses) animals (n = 4). b, Percentage of synapses contacted by astrocyte processes. Student’s unpaired two-sided t-test, NS. c, Percentage of astrocytes contacting synapses bilaterally. Students unpaired two-sided t-test, p = 0.06. d, Astrocyte process protrusion depth (nm) into synapses. Students unpaired two-sided t-test, *, p = 0.006. e, Cumulative distribution of astrocyte process synaptic protrusion depth (p < 0.0001, KS = 0.2912, Kolmogorov-Smirnov, two-sided). f, Post-synaptic density length (nm). g, Active site length (nm). h, Astrocyte process distance to the post-synaptic density. Students unpaired two-sided t-test, p = 0.19. i, Histogram of astrocyte process distance to post-synaptic density (p = 0.3, KS = 0.1165. Kolmogorov-Smirnov, two-sided). j, Model of how caloric status and BDNF signaling regulate the structural and functional plasticity of VMH astrocytes to regulate neuronal activity and energy and glucose balance. Created with Biorender.com. Data are represented as mean +/− SEM.
The data show that astrocyte coverage of VMH excitatory synapses is regulated by TrkB.T1 signaling. This is likely a contributing factor to the increased glutamate uptake observed in TrkB.T1 KD mice and provides a presynaptic mechanism by which VMH neuronal activity can be regulated by BDNF signaling to ensure that neuronal output matches the metabolic demands of the animal.
Discussion
In this study we identify VMH astrocytes as key regulators of excitatory transmission in neural circuits mediating energy and glucose homeostasis and as essential substrates for actions of BDNF promoting metabolic health. We show that these cells exhibit functional plasticity in response to changes in caloric status in a BDNF-TrkB.T1 signaling-dependent manner. In support, selective depletion of TrkB.T1 in VMH astrocytes prevented energy status-induced changes in excitatory transmission and elicited metabolic dysfunction and leptin resistance. These multifaceted effects of TrkB.T1 are driven by structural and molecular changes in astrocytes that inhibit synaptic glutamate clearance and increase synaptic glutamate availability, thereby augmenting neuronal activity and sympathetic output in the periphery (Fig. 7j).
We found that fasting and brain-wide deletion of Bdnf, conditions that lead to reduced BDNF content in the VMH, result in diminished activity of VMH neurons. This is consistent with the reported satiety effects of BDNF in this region, containing primarily glutamatergic anorexigenic neurons1. This BDNF-driven plasticity involves changes in the excitatory synaptic tone onto VMH neurons but not in alterations in intrinsic excitability. Accordingly, the frequency of EPSCs and the density of excitatory synapses in the VMH were reduced in both fasted WT and fed BDNF2L/2L:CK-Cre mice. Interestingly, these synaptic remodeling events were not observed in the vlVMH but only in dm/cVMH, which has a higher content of receptors for nutrients and metabolic hormones and receives input from feeding-regulating centers of the hypothalamus34,45–47. Our studies indicate that BDNF signals directly onto VMH astrocytes to control neuronal activity and metabolic function. Evidentiary is the increased feeding and body weight gain, hypoactivity and reduced thermogenesis observed in TrkB.T1 KD animals fed a chow diet. Moreover, these mutants also exhibit body weight-independent deficits in glucose tolerance, lipid metabolism and leptin sensitivity. The VMH is a highly heterogenous nucleus, with both distinct and overlapping neuronal populations regulating different behavioral and physiological processes48,49. Previous work has centered on genetic manipulations in discrete cell populations, including SF1+ neurons, which clearly play pivotal roles in energy and glucose balance control32,33,50. Because processes from a single astrocyte can contact up to 100,000 synapses in rodents and one million in humans51, these cells are in a privileged position to serve as master regulators of neuronal subpopulations that are functionally diverse and that control different aspects of whole animal physiology in a concerted manner. Additionally, astrocytes communicate via gap junctions and form networks spanning long distances52.
Whereas VMH GABAergic neurotransmission was normal in TrkB.T1 KD mutants, neuronal excitatory drive and activity was reduced, similar to our observations in fasted WT and fed BDNF2L/2LCK:Cre mice. Unlike BDNF2L/2LCK:Cre mice, excitatory synapse density was not altered in TrkB.T1 KD mutants, suggesting that the effects of BDNF on excitatory synapse assembly do not involve VMH astrocytes but likely rely on neuronal BDNF-TrkB.FL signaling mechanisms53–55. This is in contrast with cortical astrocytes, which are unable to mediate excitatory synaptogenesis in culture when lacking TrkB.T120, indicating context-dependent effects of astrocytic BDNF signaling and the functional heterogeneity of astrocytes.
While we cannot completely rule out post synaptic mechanisms, our studies indicate that astrocytic glutamate reuptake at VMH synapses is a chief presynaptic mechanism mediating changes in neuronal activity induced by caloric status and BDNF. Regulation of synaptic glutamate levels is mediated by content of astrocytic GLT-1 as well as proximity of PAPs to the synapse12,56. Both of these parameters were elevated in TrkB.T1 KD mutants, and GLT-1 expression was increased in fasted WT and fed BDNF2L/2LCK:Cre mice. Models show that increasing astrocyte protrusion depth of a synapse from 0 to 150 nm causes a stepwise increase in astrocyte GLT-1 currents, while decreasing neuronal excitatory currents12. Studies in other brain regions indicate that astrocytic processes can rapidly extend and retract to change their proximity to post synaptic dendritic spines9,57. Within the ARC, disruptions in astrocyte nutrient sensing alters PAP coverage of and synaptic input onto POMC and AgRP neurons58–60. Additionally, astrocytes are able to respond at the timescale of meals to rapidly fine-tune neuronal activity in a manner that meets the energy demands of the animal61. Further attesting to a presynaptic mechanism is the finding that whereas baseline NMDAR currents were not affected by fasting or depletion of central BDNF or TrkB.T1 in VMH astrocytes, synaptic glutamate clearance was enhanced in all of those conditions. Importantly, glutamate transporter blockade with DL-TBOA, which eliminates observed differences in glutamate reuptake and presynaptic glutamate availability between groups, was sufficient to abolish reductions in the firing rates of VMH neurons in fasted WT and TrkB.T1 and BDNF mutants compared to controls. It is important to note that BDNF signaling in the VMH is both necessary and sufficient for effects of feeding status on astrocytic function and excitatory neurotransmission. Accordingly, ICV delivery of BDNF in WT mice was sufficient to fully reverse the increase in astrocytic glutamate reuptake and decreased excitatory tone induced by fasting. Moreover, the effects of caloric status on glutamate clearance and excitatory drive onto VMH neurons were absent in TrkB.T1 KD mutants, indicating the necessity of astrocytic BDNF signaling.
Viral knockdown of TrkB.T1 in VMH astrocytes also elicited a decrease in VMH levels of TrkB.FL, which is expressed by neurons but not by astrocytes. As the viral-mediated TrkB.T1 KD in the VMH is astrocyte-specific, decreases in TrkB.FL are likely a homeostatic response to the reductions in neuronal activity in TrkB.T1 KD animals. Indeed, neuronal TrkB.FL expression is known to be activity-dependent24,26. Reduced neuronal TrkB signaling is unlikely to contribute significantly to the weight gain observed in TrkB.T1 KD mutants as TrkB depletion in VMH neurons did not produce significant effects on body weight, feeding, thermogenesis or locomotor activity. In far contrast, TrkB.T1 KD mutant mice display many of the metabolic phenotypes present in BDNF2L/2L:CK-Cre mutants5. Unlike astrocytic TrkB.T1 KD mutants, neuronal TrkB.FL mice do not exhibit significant reductions in NE in inguinal and gonadal white adipose tissues. While they exhibit a significant reduction in NE in BAT and increases in serum leptin, these effects are milder than those observed in astrocytic TrkB.T1 mutants. Interestingly, neuronal TrkB knockdown led to impaired glucose homeostasis. Indeed, studies from our laboratory and others have shown that genetic manipulation signaling pathways downstream of BDNF in VMH neuronal populations can elicit impairments in glucose homeostasis independent of body weight62,63. Thus, neuronal TrkB signaling in the VMH contributes to glycemic control, whereas signaling in astrocytes is a chief mechanism underlying the effects of BDNF on feeding, energy expenditure and body weight.
How does reduced neuronal activity lead to the metabolic dysfunction in TrkB.T1 KD mutants? VMH neurons project to several energy balance-regulating brain regions64. For example, cells in the dm/cVMH send excitatory inputs to anorexigenic POMC neurons in the arcuate nucleus and the strength of this synaptic input is dynamically regulated by caloric status14. Reduced excitatory drive onto those cells in TrkB.T1 KD mutants could contribute to energy balance dysregulation. VMH fibers also reach autonomic centers in the hindbrain controlling sympathetic output to metabolic organs influencing glycemic control and thermogenesis. We showed that glucose intolerance and reduced thermogenesis in TrkB.T1 KD mutants is accompanied by reduced sympathetic tone in BAT and WAT. Notably, TrkB.T1 KD mice exhibited reduced neuronal activity in the VMH in response to a bolus of glucose.
In summary, our results identify VMH astrocytes as an essential cellular substrate for actions of BDNF facilitating energy and glucose homeostasis. We propose that BDNF acts as an energy-relaying signal to these cells, allowing them to provide contextual guidance to VMH neurons via alterations in synaptic glutamate so that the activity of relevant neuronal populations reflects and meets the metabolic demands of the animal.
Methods
Animals
All procedures were approved by the Institutional Animal Care and Use Committee at Tufts University and conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals guidelines. Animals were housed in a 14-dark 10-dark light cycle with an ambient temperature of 68-74° F and 30-70% humidity and had free access to water and chow (18.6% protein, 6.2 % fat and a caloric content of 3.1 kcal/g, Envigo, 2918) unless indicated. BDNF2L/2LCK:Cre mice were used for the experiments shown in Figures 1d–m, 2a–h and Extended Data Figures 1a, c–d and 2, with littermates used as WT controls5. These mice are in a C57Bl6J x 129 hybrid background and were generated by crossing floxed BDNF mice with Cam kinase-cre mice. BDNF depletion in these animals begins at post-natal day 0 as is completed by the end of the third postnatal week, when BDNF stores are almost entirely depleted everywhere in the brain, except the cerebellum, as BDNF is primarily expressed by excitatory neurons. Mice used for experiments shown in Figure 1b–c, Figure 1n – q, Figure 2i–j, Figure 3a–c, Extended Data Figure 1b, Extended Data Figure 3, and Extended Data Figure 5c were male C57Bl6J mice obtained from Jackson laboratories. TrkB.T1 KD and neuronal TrkB KD mice were used for experiments analyzing the role of astrocytic and neuronal BDNF signaling in the VMH, respectively, (Figures 3–7 and Extended Data Figures 4–9). These animals are in a C57Bl6 background and were generated as described in the stereotaxic surgeries section below. Different cohorts of TrkB.T1 KD and control mice were used for results shown in Figure 3h–I versus those in Figure 3k–o and Figure 4a–l because experiments in 3h-l required a 25-week post-surgery examination period whereas the other experiments needed to be performed 3-6 weeks post-surgery. This was also the case for Neuronal TrkB mutants and controls used to generate data shown in Supplemental Figure 7a-b versus 7c-k. All animals were male except those used in Figure 3i and Extended Figure 5b. Littermates and age-matched animals were used as controls to reduce variability. For experiments requiring tissue harvesting for molecular and electrophysiological analysis, animals were euthanized between 8 AM and 10 AM to control for circadian rhythms. For experiments involving comparisons of fed versus fasted mice, fed mice had ad libitum access to chow while fasted animals were food restricted for 24 hours with free access to water. The number of animals used for each experiment is described in the corresponding figure legend.
Stereotaxic Surgeries
Stereotaxic surgeries were performed in floxed TrkB animals (a gift from Dr. Baoji Xu23) and C57Bl6 mice at 8 to 9 weeks of age. Animals were anesthetized using the Kent Scientific SomnoSuite Low-Flow Isoflurane Anesthesia System. Stereotaxic coordinates A/P −1.33, M −0.55, L 0.45 and D/V −5.85 from Bregma were used to bilaterally target the VMH. Post-hoc targeting was confirmed using immunofluorescent labeling of GFP or western blot analysis of TrkB expression in every animal. Mistargeted animals were excluded from all analyses. Viruses used were obtained from the University of North Carolina Viral Vector Core. To generate TrkB.T1 KD mutants and control mice and to examine the effect of viral delivery to the VMH in WT animals (Supplemental Fig. 5c), 150 nL containing 36 x 107 viral particles of AAV5-GFAP-GFP-Cre (#R41277) or AAV5-GFAP-eGFP (#R41276), respectively, were delivered bilaterally to the VMH of floxed TrkB mice and wild type C57Bl6 mice. To generate neuronal TrkB mutants and controls, 100 nL containing 17.6 x 107 viral particles of AAV2-CMV-Cre-GFP, (#R41573) or AAV2-CMV-GFP (#R42072) were delivered bilaterally to the VMH of floxed TrkB mice, respectively.
Food Intake and Body Weight Measurements
After stereotaxic surgery, mice were individually housed and had unrestricted access to water and chow. Body weights were measured weekly post-surgery at the same time of day. Food intake was measured during weeks 4 – 6 post-surgery by weighing weekly food administered and food remaining.
Locomotor Activity Analysis
Locomotor activity measurements were performed in TrkB.T1 KD and control mice at 5 weeks post-surgery. Animals were individually housed in the testing room (12-hr light/dark cycle) for 6 days for acclimation prior to recording. Home cages were placed in a Smart Frame Activity System photobeam frame, which records locomotor activity based on continuous recording of beam breaks by the MotorMonitor Software (Hamilton/Kinder). Day one of data collection was discarded. The remaining 6 days were binned in 12 increments and separated based on light cycle and averaged across test days.
Body Temperature Measurements
Core body temperature was measured in TrkB.T1 KD and control mice at 6 weeks post-surgery using the MicroThermal 2T Hand-Held Thermometer (Braintree Scientific) fitted with a mouse rectal probe (part #RET3, ¾” L, 0.28 dia, 0.065 tip). Mice were handled and conditioned to the procedure on day one, then experimental measures were recorded on day two and three and averaged. All measurements were made between the hours of 10 – 11 AM.
Glucose and Insulin Tolerance Testing
Glucose and insulin tolerance tests were performed in in TrkB.T1 KD and control mice at 6-7 weeks post-surgery. Mice were fasted for 16 or 6 hours prior to glucose tolerance testing or insulin tolerance testing, respectively. Blood glucose levels (mg/dL) were obtained by slicing a lateral nick in the tail of the mouse that allowed for analysis of blood droplets using the Freestyle Blood Glucose Monitoring System (Abbot Diabetes Care Inc.; Alameda, CA). A baseline measurement (time 0) was recorded. Following this measurement, 2.5 g/kg of D-Glucose (Sigma, G8270) was administered intraperitoneally for glucose tolerance testing, and 0.85U/kg Inulin (Humulin R U-100) was administered intraperitoneally for insulin tolerance testing. Blood glucose was then measured at 15, 30, 60 and 120 minutes post-injection.
c-Fos Expression Analysis after Glucose Administration
TrkB.T1 KD and control mice were fasted for 16-hours and then injected intraperitoneally with 2.5 g/kg of D-Glucose 7 weeks post-surgery. Sixty minutes following glucose delivery, they were perfused with 4% paraformaldehyde in 1x PBS (pH 7.4) and post-fixed for 24 hours prior to OCT embedding. Thirty-micron thick coronal sections were obtained using a Leica CM1900 cryostat. Free floating brain sections were blocked in 1% BSA, 5% NDS, and 0.4% Triton X-100 in PBS 1x. Sections were immunolabeled using goat anti-c-fos (1:200, Santa Cruz SC-52-G) and chicken anti-GFP (1:800, Abcam ab13970), followed by secondary antibodies donkey anti chicken 488 (1:500, JaxIR) and donkey ant goat Cy3 (1:500, JaxIR). A Nikon A1R microscope was used to capture images. 10x images capturing the VMH were acquired and the total number of c-fos+ cells in two independent VMH images per animal were counted using ImageJ software.
Norepinephrine Measurements
Catecholamine analysis was performed in serum, muscle, liver, BAT, iWAT and gWAT from TrkB.T1 KD and Control males, and neuronal TrkB KD and Control males 5-weeks post-surgery. Serum was treated with EGTA-Glutathione upon extraction to prevent catecholamine degradation. Tissue was weighed, lysed and treated with 5mM Glutathione/0.4N Perchloric acid (1 mL per 100 mg tissue). Norepinephrine content was analyzed by the Vanderbilt Hormone Assay & Analytical Services Core (Vanderbilt University School of Medicine, Nashville, TN) using HPLC methods.
Lipid Measurements
Lipid analysis was performed in liver, iWAT, gWAT and BAT from TrkB.T1 KD and Control males, and neuronal TrkB KD and Control males 5-weeks post-surgery by the Vanderbilt Hormone Assay & Analytical Services Lipid Core Vanderbilt University School of Medicine, Nashville, TN).
Serum Insulin and Leptin Measurements
Serum was isolated from collected blood from using BD Microtainer tubes (Fisher, 365967). Serum insulin content was quantified from 24-hour fasted animals using the Rat/Mouse Insulin ELISA kit (Millipore Sigma, EZRMI-13K). Serum leptin content was quantified from ad libitum fed animals using the Mouse Leptin ELISA kit (Sigma, RAB0334). Measurements were performed at 5 weeks post-viral delivery to the VMH.
Leptin Administration Studies
The effect of leptin administration on body weight and food intake in TrkB.T1 KD and control animals was examined 4-weeks post-surgery. Daily food intake was measured both during the habituation phase (3 days) and drug injection phase (3 days) and averaged based on condition. To habituate mice to handling and injections, mice were injected intraperitoneally with 150 uL of 20mM Tris-HCL twice daily for 3 days (8AM and 7PM). Following this, mice were injected intraperitoneally with a dosage of 3 ug/g of leptin (0.5 ug/uL in 20 nM Tris HCL; Recombinant Mouse Leptin Protein, CF, RND Systems, 498-0B-05M) twice daily for 3 days (8AM and 7PM). Final reported body weights were taken on the morning following the final leptin injection.
pSTAT3 Expression Analysis in Leptin Treated Animals
in TrkB.T1 KD and control mice were fasted for 16-hours and then injected intraperitoneally with a dosage of 5 ug/g of leptin (0.5 ug/uL in 20 nM Tris HCL; Recombinant Mouse Leptin Protein, CF, RND Systems, 498-0B-05M). Forty-five minutes after leptin delivery, animals were perfused with 4% paraformaldehyde in 1x PBS (pH 7.4) and post-fixed for 24 hours prior to OCT embedding. Thirty-micron thick floating coronal sections were obtained using a Leica CM1900 cryostat. Sections were permeabilized in 1% NaOH for 2 minutes prior to blocking in 5% NGS and 0.3% triton X-100 in 1X PBS and immunolabeled using rabbit anti phospho-Stat3 (1:250, Cell Signaling 9145S) and chicken anti-GFP (1:800, Abcam ab13970), followed by secondary antibodies goat anti chicken 488 (1:500, JaxIR) and goat ant rabbit Cy3 (1:500, JaxIR). A Nikon A1R microscope was used to capture images. 40x images capturing the dm/cVMH of animals were acquired and total pSTAT3 was counted using ImageJ software. Two images of bilateral VMH were analyzed and averaged per animal. Studies were performed at 4-weeks post-surgery
Synapse Density Quantitation
BDNF2L/2LCK:Cre mice and their corresponding controls (10-12 weeks of age) and TrkB.T1 KD mutants and controls (five weeks post surgeries) were perfused using 4% paraformaldehyde in 1x PBS (pH 7.4) and post-fixed for 24 hours prior to OCT embedding. Thirty-micron thick floating coronal sections were obtained using a Leica CM1900 cryostat. Antibodies were used for the following proteins: rabbit anti-PSD95 (1:500, ThermoFisher 51-6900), guinea pig anti-vGlut2 (1:500, Millipore AB2251-I), chicken anti-GFP (1:800, Abcam ab13970), rabbit anti Iba1 (1:500, WAKO 019-19741), mouse anti NeuN (1:250, Millipore MAB377), rabbit anti Sox9 (1:1000, Millipore MAB5535) and rabbit anti TrkB (1:250, Santa Cruz C-13 SC119). For PSD95 and vGlut2 immunolabeling, sections were post fixed in 4% paraformaldehyde for 10 minutes, washed in PBS 1x, then blocked for 30 minutes at room temperature in PBS with 10% NGS, 1% milk, and 0.5% triton x-100. For all other antibodies, a blocking solution containing 1% BSA, 5% NGS and 0.3% triton x-100 in PBS was used. A Nikon A1R microscope configured for confocal microscopy was used to capture images of all brain sections. Excitatory synapse density was quantified from 64x confocal images using the Synapse Counter plugin (SynPuCo/SynapseCounter) on NIH ImageJ software. Two images were quantified and averaged per animal per indicated VMH region (dm/cVMH and vlVMH).
Electrophysiological Analysis
Electrophysiological recordings were performed in BDNF2L/2LCK:Cre mice and their corresponding controls at 8-12 weeks of age and in TrkB.T1 KD mutants and controls at five weeks post surgeries. For this, 300 uM-coronal sections were prepared using a Leica Vibrating Blade Microtome (Leica VT1000S). Sections were prepared and maintained in oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF) containing 26 mM NaHCO3, 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4, 2 mM CaCl2 and 10 mM D-Glucose and kept at 32 degrees Celsius for 1 hour prior to recording. All recordings were obtained within 5 hours of slicing and performed at 33-34 degrees Celsius. Whole-cell recordings were made using the Nikon Eclipse FN1 microscope, a MultiClamp 700B amplifier (Axon instruments), Digidata 1440A Digitizer (Axon Instruments), pClampex 10.2 program software (Axon Instruments) and sampled at 10 kHz. Recording electrodes with a resistance between 3 – 6 megaohms were used. For voltage clamp recordings a cesium-based internal solution was used containing 120 mM D-gluconic acid, 10 mM Hepes, 0.5 mM CaCl2, 20 mM TEA-Cl, 120 mM CsOH, 10 mM EGTA, 2mM ATP and 0.3 mM GTP, with a final osmolality between 295 – 300 mOsm. For current clamp recordings a potassium based internal solution was used containing 135 mM KMeSO3, 3 mM KCl, 10 mM Hepes, 1 mM EGTA, 0.1 mM CaCl2, 8 mM Na2-phosphocreatine, 4 mM ATP and 0.3 mM GTP with a final osmolality of 295 mOsm.
For functional analysis of glutamate transporter activity whole cell recordings were performed from dm/cVMH neurons. NMDAR sensitive currents were isolated by application of 10 uM DNQX (Tocris, 0189) and 10 uM SR 95531 (Tocris, 1262), and by holding the neurons at +40 mV to remove the magnesium block from NMDARs. A concentric bipolar stimulator electrode (CBARC75, FHC) was placed in the inferior lateral portion of the VMH. Stimulation was controlled using a Grass S88 Stimulator (Grass Instruments) and a Grass SIU5 Stimulus Isolation Unit (Grass Instruments). NMDAR currents were recorded at 2x threshold in response to a 1 stim 0.1 ms pulse. Responses were recorded before and after bath application of 100 uM DL-TBOA (Tocris, 1223). Final recordings were made in the presence of NMDAR inhibitor 50 uM APV (Tocris, 0106) to ensure that recorded traces are specific NMDAR responses. Trace amplitude (pA), total charge transfer (pA*ms) and decay (weighted tau) were calculated using ClampFit 10.2 (Axon Instruments).
For sEPSC measurements, whole cell recordings were performed in dm/cVMH neurons in slices submerged in aCSF containing 10 uM SR 95531 (Tocris, 1262). Neurons were held at −60 mV and recorded for 3 minutes. For sIPSC measurements, whole cell recordings were performed in dm/cVMH neurons in slices submerged in aCSF. Neurons were held at 0 mV to isolate inhibitory currents and recorded for 3 minutes. sEPSC and sIPSC recordings were analyzed using Mini Analysis Program 6.03 (Synaptosoft).
Current clamp experiments were performed in slices submerged in aCSF. Neurons were current clamped with 0 pA current injected for spike frequency recordings. Membrane potentials were corrected for an ~8-mV liquid junction potential. For recordings of changes in spike frequency firing activity was recorded before and after 100 uM DL-TBOA (Tocris, 1223) was washed on. For intrinsic excitability analysis, synaptic activity was blocked using 50 uM NBQX (Tocris, 1044), 10 uM CPP (Abcam, ab120160) and 10 uM SR 95531 (Tocris, 1262). Spike frequency recordings were performed. The number of action potentials generated in response to a series of 500-ms current injections from −200 to 200 pA in 20-pA steps were measured in the current-clamp configuration.
For all experiments, access resistance was monitored before and after experimental recordings and neurons with a greater than 25% change in access were excluded. Unless otherwise indicated all analysis was performed using Matlab and Clampfit (Axon Instruments). For electrophysiological experiments performed on TrkB.T1 KD and Control animals, proper surgical targeting was confirmed via slice fixation and subsequent immunofluorescent analysis of GFP expression. Slices were fixed for 24-hours, permeabilized in 0.5% Triton X-100 for one hour, followed by blocking in 1% BSA, 5% NGS, 0.3% Triton X-100 in PBS 1x for 2 hours. Slices were incubated with chicken anti-GFP (1:800, Abcam ab13970) overnight in blocking solution, then probed with goat anti-chicken Alexa 488 (1:500, JaxIR) for one hour. Slices were washed in progressing concentrations of glycerol and mounted in 100% glycerol. GFP fluorescence was analyzed using a Zeiss Axioplan 2 microscope. Only cells samples from sections containing GFP stain were used in analysis.
BDNF Rescue Experiments
To examine the effect of BDNF administration on astrocytic glutamate uptake and excitatory drive onto VMH neurons, 8-week old C57Bl6 males were single housed and either fasted or allowed ad libitum access to food for 24 hours. Mice were given a single i.c.v. injection into their lateral ventricle of either 4 uL aCSF or 2.5 ug BDNF (R&D Systems, 248-BDB-050/CF) diluted in aCSF. Stereotaxic coordinates used to target the lateral ventricle were A/P −0.22, M/L + 1.00 and D/V −2.50. Animals then recovered for 2 hours, after which brain sections were prepared for electrophysiological analysis as previously described.
Western Blot Analysis
VMH tissue was extracted by coronal sectioning of fresh brain tissue using the Leica Vibratome 1000S. A 800 um coronal section was extracted, from which the VMH was microdissected using a dissection scope. Tissue was then frozen and stored in −80 degrees Celsius until use. Tissue was lysed in either a sodium phosphate-based lysis buffer (20 mM sodium phosphate, 1% SDS, plus protease and phosphatase inhibitors – buffer used for glutamate transporter analysis) or a lysis buffer containing 140 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 10% Glycerol in 20 mM Tris-HCL, containing protease and phosphatase inhibitors. Lysates were sonicated and centrifuged at 14000G for 7 minutes. Supernatant was then collected, and protein concentration was quantified using the Pierce BCA Protein Assay Kit (ThermoFisher, 23225). For glutamate transporter analysis, samples were reduced for 30 minutes at 32 degrees Celsius using a DTT based reducing reagent (NuPAGE Sample Reducing Agent and LDS Sample buffer, ThermoFisher NP0009 and NP0007), and ran using the xCELL sureLock Minigel electrophoresis kit (LifeTechnologies, EI0001). Samples were run in NuPAGE MOPS SDS Running Buffer (ThermoFisher, NP0001) with NuPAGE Antioxidant (ThermoFisher, NP0005) on NuPAGE 4 – 12% Bis-Tris Gels (ThermoFisher, NP0336BOX). Transfer was performed using the iBlot 2 Dry Blotting System (ThermoFisher, IB2001) onto nitrocellulose membranes (ThermoFisher, IB23001). For all other proteins, samples were reduced in a 5x beta-mercaptoethanol based loading buffer (1 mL glycerol, 1g SDS, 3.1 mL 1M Tris pH 6.8, 100 uL 5% bromophenol blue, 2.5 mL beta-mercaptoethanol and dH20) for 30 minutes at 32 degrees Celsius and ran using the xCELL sureLock Minigel electrophoresis kit (LifeTechnologies, EI0001). Samples were run in Tris-Glycine SDS Running Buffer (ThermoFischer, LC2675) on 4-12% Tris-Glycine gels (ThermoFisher, XP04122BOX). Wet transfer was performed in Tris-Glycine Transfer Buffer (ThermoFisher, LC3675) onto PVDF membranes at 300 mA for 1 hour. Blots were blocked in 5% non-fat dry milk 1x TBS-T for 1 hr at room temperature. Primary antibodies were incubated in blocking buffer overnight at +4 degrees Celsius. Secondary antibodies were incubated for 1 hour in blocking buffer at room temperature. Blots were developed using ECL Prime (SigmaAldrich, RPN2232) onto HyBlot ES Autoradiography film (Denville, 1156P37). Densitometry analysis was performed in ImageJ.
Primary antibodies used are as follows: rabbit anti ß-Tubulin (1:10000, Abcam ab 6046), mouse anti GLT-1 (1:3000, Millipore MAB 2262), rabbit anti GLAST (1:500, Danbolt Lab AB314), rabbit anti TrkB (80E3) (1:1000, Cell Signaling 46032).
Quantitative RT-PCR analysis
Tissue collected for RNA analysis was flash frozen in liquid nitrogen immediately after extraction and stored in −80 degrees Celsius until use. RNA was extracted using TRIzol reagent (ThermoFisher, 15596026) followed by DNAse treatment using the RNase-Free DNase Set (Qiagen, 79254). RNA quality and quantity were assessed using a Nanodrop instrument. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher, 4368814) and quantified using a Nanodrop instrument.
Real-time qPCR analysis was performed using the Stratagene Mx3000p Real-Time aPCR machine, with SYBR green PCR master mix provided by the Tufts Genomics Core. Brain tissue was normalized using GAPDH. qPCR primers used are listed in table 1 the Supplementary Materials.
Isolation of Astrocytes from Acute VMH Tissue
Astrocytes were isolated from VMH freshly extracted from C57Bl6 mice (8-10 weeks of age) using the MiltenyilBiotec Anti-ACSA-2 MicroBead kit (Mouse, 130-097-679) and related Anti-ACSA-2 antibodies (Mouse, 130-099-138) according to the protocol. Magnetic isolation of astrocytes was performed two times to ensure purity. Due to the small size of the VMH, two animals per sample were pooled. Following astrocyte (will only contain antibody labeled astrocytes) and flowthrough (will contain neurons, microglia, oligodendrocytes, unlabeled astrocytes and any other cell types) isolation, RNA was collected using the PicoPure RNA Isolation Kit (Arcturus, KIT0202) with DNAse treatment performed using the RNase-Free DNase Set (Qiagen, 79254). cDNA synthesis and real-time PCR analysis was performed as previously described. Astrocyte isolation was considered successful if samples were enriched for GLT-1 and did not contain neuronal ß3-tubulin.
Electron Microscopy Analysis
At 5 weeks post-surgery, TrkB.T1 KD and controls were perfused in cold 0.1M PB, followed by fresh 4% PFA 2.5% glutaraldehyde in 0.1M PB (pH 7.2). Extracted brains were post fixed in fresh 4% PFA 2.5% glutaraldehyde in 0.1M PB (pH 7.2) overnight. 150 uM-thick coronal sections were made in cold 0.1M PB using a Leica VT100S Vibratome. Floating sections were further processed by the Electron Microscopy Facility at Harvard Medical School. Sections were washed in 0.1 M Sodium Cacodylate buffer (pH 7.4) and postfixed with 1% Osmiumtetroxide (OsO4)/1.5% Potassium ferrocyanide (KFeCN6) for 30 minutes, washed twice in water and one time in 50mM Maleate buffer pH 5.15 (MB) and incubated in 1% uranyl acetate in MB for 30 minutes followed by 2 washes in water and subsequent dehydration in grades of alcohol (10 min each; 50%, 70%, 90%, 2x 10 min 100%). The samples were then incubated overnight at 4C in a 1:1 mixture of propylene oxide and TAAB Epon (TAAB Laboratories Equipment Ltd, https://taab.co.uk). The following day samples were embedded flat in TAAB Epon between two sheets of aclar plastic (Electron Microscopy Sciences) and polymerized at 60C for 48 hours. Ultrathin sections (about 80 nm) were cut on a Reichert Ultracut-S microtome, placed onto copper grids, stained with uranyl acetate and lead citrate and examined in a JEOL 1200EX transmission electron microscope. Images were recorded with an AMT 2k CCD camera.
Analysis of images was performed in ImageJ. The analysis was performed after blinding of image conditions. Excitatory asymmetric synapses were identified based on presence of thick opposed post-synaptic densities and presynaptic prominent rounded vesicles exceeding the post synaptic density length. Astrocyte processes surrounding synapses were identified based on pale cytoplasm, irregular contouring borders, rounded processes and the absence of vesicles.
STATISTICAL ANALYSIS
GraphPad Prism (GraphPad, 7.0a & 8.4.3) software was used to perform all statistical analysis. Tests used to analyze data were: Student’s t-test, One-Way ANOVA with Tukey multiple comparisons, and Two-Way ANOVA with Tukey or Bonferroni multiple comparisons. Comparisons were determined to be statistically significant when p < 0.05. Statistics used for each dataset are indicated in the figure legends. N for each dataset is indicated in figure legends. For electrophysiology experiments, n is given as both the number of cells analyzed, and the number of animals used. All values are depicted as mean ± SEM.
Extended Data
Extended Data Fig. 1. Intrinsic excitability of VMH neurons is not regulated by energy status or BDNF signaling.
a. Amplitude of sEPSCs in VMH neurons of fed (n = 21) and fasted WT (n = 21) and fed BDNF2L/2LCK:Cre mice (n = 19) (4 – 6 mice per group), Ordinary One-way ANOVA, p = 0.3
b. Amplitude of sEPSCs in VMH neurons of in WT Fed + aCSF (n = 13), Fasted + aCSF (n = 13) and Fasted + BDNF conditions (n = 12) (4 mice per group), Ordinary One-way ANOVA, p = 0.136
c. Input-output curves from VMH neurons in fed (n = 19) and fasted WT (n = 18) and fed BDNF2L/2LCK-Cre mice (n = 17) (3 - 4 mice per group). Two-way repeated measures ANOVA: Interaction, p = 0.7; Genotype, p = 0.3.
d. Representative Traces showing resulting hyperpolarization or evoked action potentials in response to current injection steps of −120 mV and 120 mV.
Data represented as mean +/− SEM
Extended Data Fig. 2. Energy status and BDNF regulate astrocytic glutamate uptake at VMH synapses.
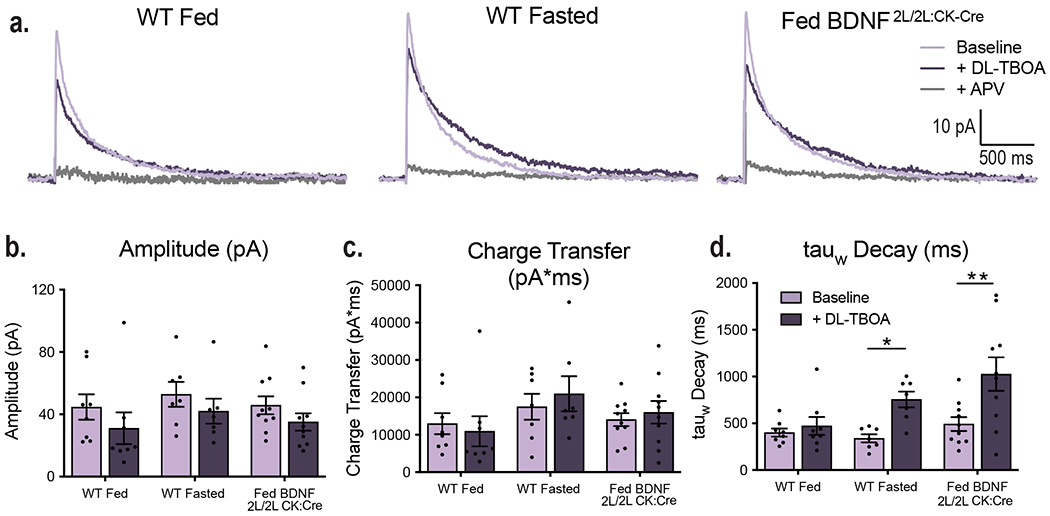
a. Representative traces of raw NMDAR responses (light purple), responses +100 uM DL-TBOA (dark purple), and responses + 50 uM APV (light gray). For following panels, WT Fed (n = 8), WT Fasted (n = 7), and Fed BDNF 2L/2L CK:Cre (n = 10)
b. Amplitude (pA) of NMDAR responses before and after DL-TBOA application.
c. Charge transfer (pA*ms) of NMDAR responses before and after DL-TBOA application.
d. Decay (weighted tau) of NMDAR responses before and after DL-TBOA application. Two-way ANOVA: Genotype, p = 0.01; p = 0.0005; Interaction of genotype and DL-TBOA, p = 0.09. Bonferroni multiple comparisons, *, p = 0.001.
Data represented as mean +/− SEM
Extended Data Fig. 3. Energy status regulates astrocytic glutamate uptake at VMH synapses via BDNF signaling.
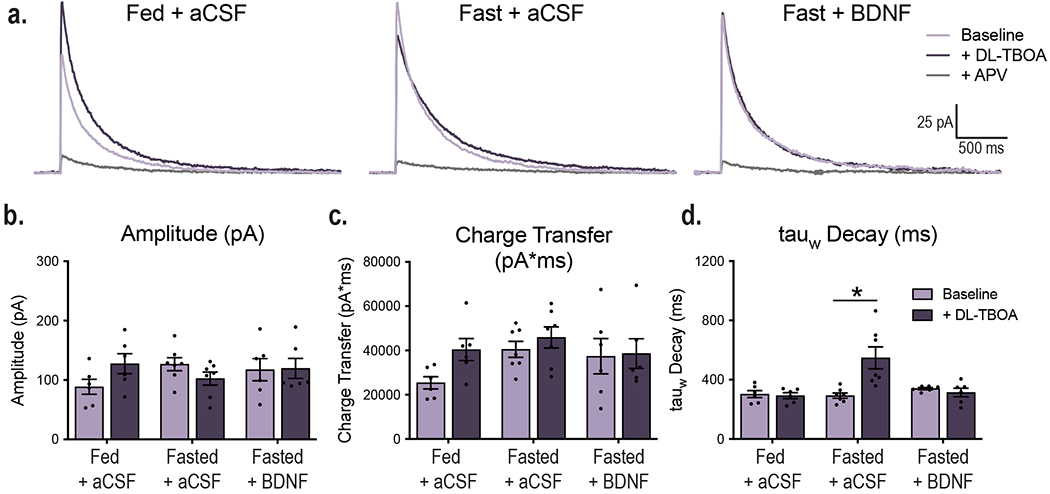
a. Representative traces of raw NMDAR responses (light purple), responses +100 uM DL-TBOA (dark purple), and responses + 50 uM APV (light gray). For following panels, Fed + aCSF (n = 6), Fasted + aCSF (n = 7), Fasted + BDNF (n = 6)
b. Amplitude (pA) of NMDAR responses before and after DL-TBOA application.
c. Charge transfer (pA*ms) of NMDAR responses before and after DL-TBOA application.
d. Decay (weighted tau) of NMDAR responses before and after DL-TBOA application. Two-way ANOVA: Condition, p = 0.007; DL-TBOA, p = 0.02; Interaction of condition and DL-TBOA, p = 0.0009. Bonferroni multiple comparisons, *, p < 0.0001.
Data represented as mean +/− SEM
Extended Data Fig. 4. TrkB.T1 in VMH astrocytes is an essential regulator of body weight under standard chow conditions.
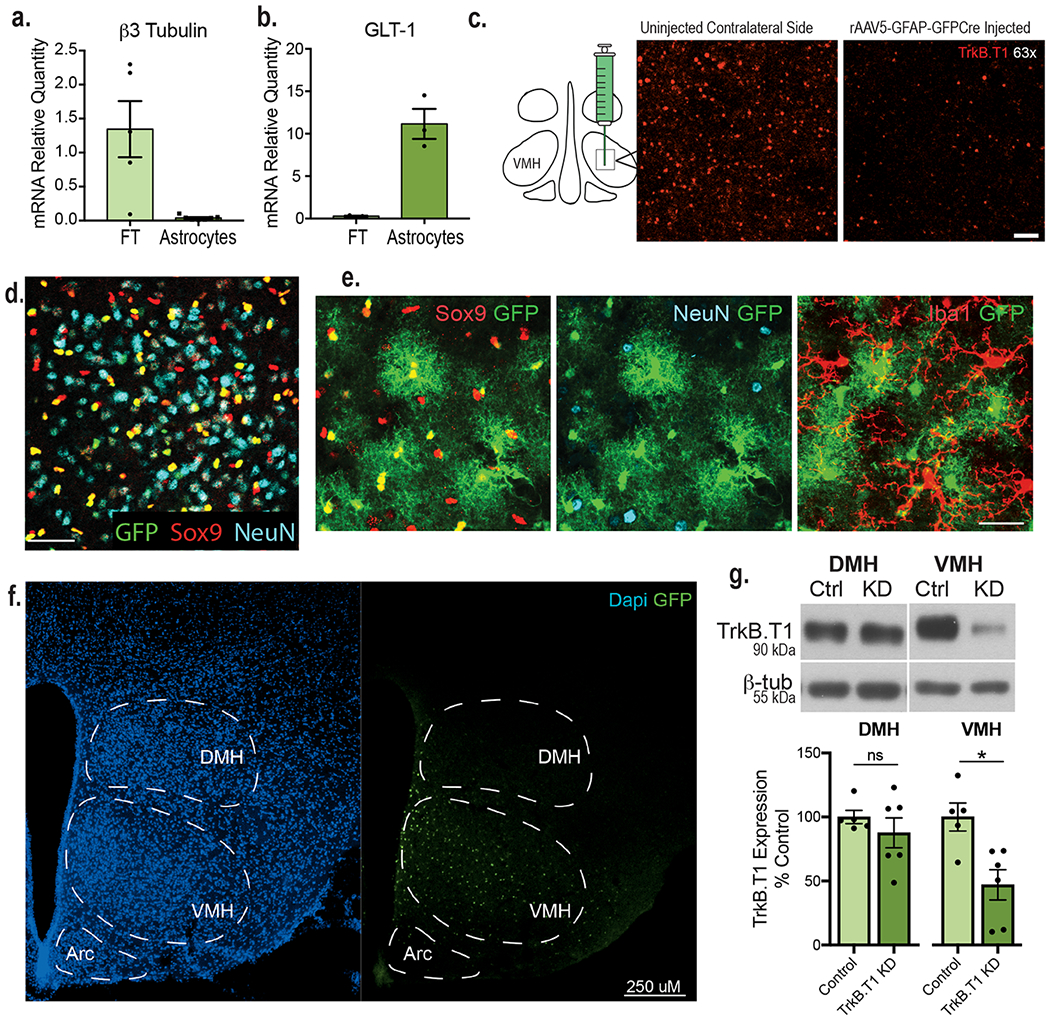
a. Expression of the neuronal marker b3 tubulin (FT n = 5, Astrocyte n = 7) and
b. GLT-1 in mRNA isolated from VMH astrocytes and flowthrough (FT n = 3, Astrocyte n = 3).
c. Immunolabeling of TrkB.T1 in the VMH of a TrkB F/F animal injected unilaterally with AAV5-GFAP-GFP-Cre 4 weeks post-surgery. Scale bar 15 uM.
d. Co-localization of AAV5-GFAP-GFP-Cre with GFP signal with astrocytes (Sox9 + cells) and exclusion from neurons (NeuN + cells). Scale bar 50 uM.
e. Co-localization of AAV5-GFAP-driven GFP signal with astrocytic (Sox 9), neuronal (NeuN) and microglial (Iba)-specific markers. Scale bar 150 nM.
f. Image showing that viral spread is limited to the VMH.
g. Western blot and analysis showing TrkB.T1 expression in Control and TrkB.T1 KD mice within the VMH (n = 6) and the DMH of mice (n = 5). Data collected from one experiment. Student’s two-sided t-test, * , p = 0.01. Data represented as mean +/− SEM.
Extended Data Fig. 5. Depletion of TrkB.T1 from VMH astrocytes leads to increased body weight and alterations in locomotor activity, sympathetic tone and leptin insensitivity.
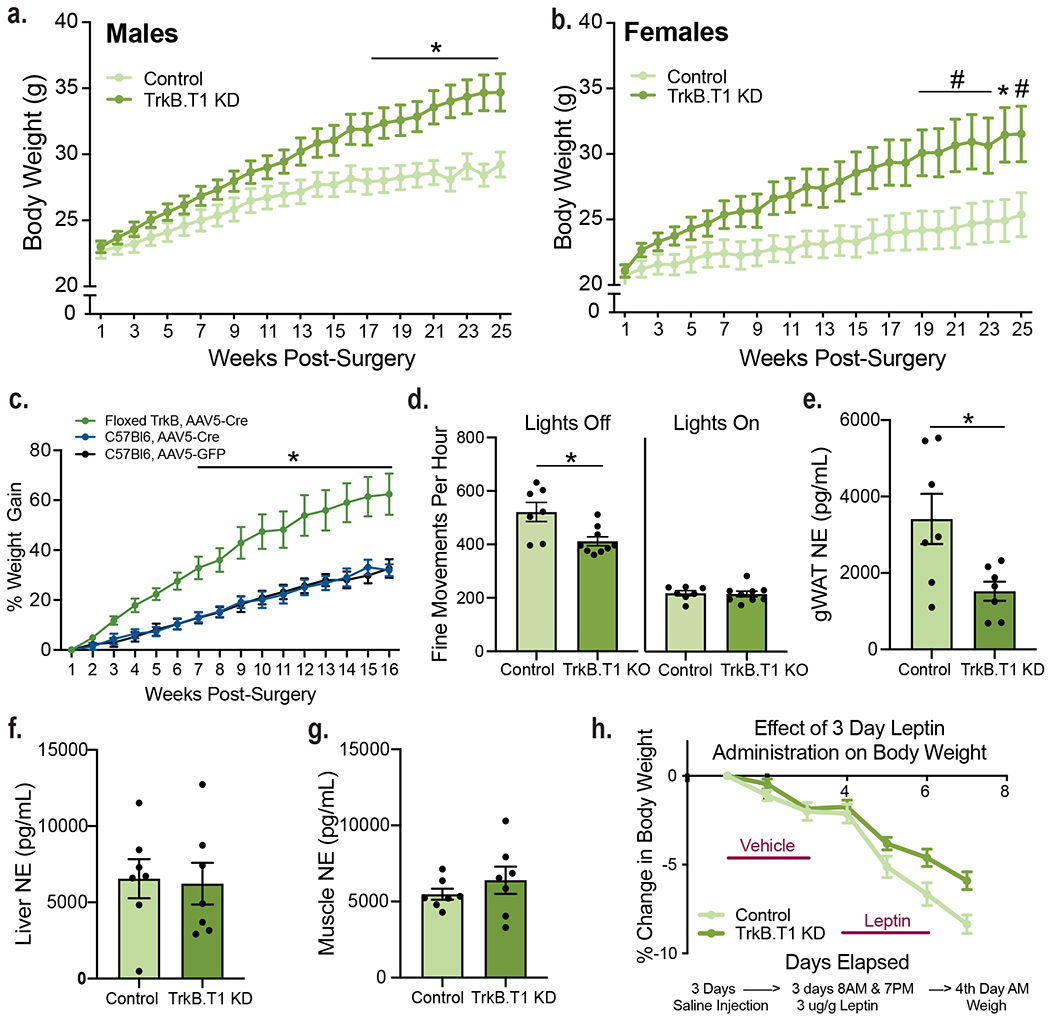
a. Body weights of TrkB.T1 KD (n = 10) and control males (n = 13). Two-way RM ANOVA: Genotype, p = 0.02; Time, p < 0.0001; Time x Genotype Interaction, p <0.0001; Bonferroni multiple comparisons, *, p < 0.05.
b. Body weights of TrkB.T1 KD (n = 11) and control females (n= 8). Two-way RM ANOVA: Genotype, p = 0.04; Time, p < 0.0001; Time x Genotype Interaction, p < 0.0001; Bonferroni multiple comparisons, *, p < 0.05, #, p = 0.09.
c. Percentage body weight gain in TrkB.T1 KD (n = 7) and wild type C57Bl6 males (n = 4) delivered AAV5-GFAP-GFP or AAV5-GFAP-GFP-Cre to the VMH. Two-way RM ANOVA: Genotype, p = 0.01; Bonferroni multiple comparisons, *, p < 0.5.
d.. Fine movements per hour of TrkB.T1 KD and control animals over 6 days (n = 7 - 9). Students two-sided t-test, *, p = 0.009.
e – g. Norepinephrine levels in tissues from TrkB.T1 KD and control animals (n = 7). Student’s two-sided t-test, *, p = 0.01.
h. Experimental design and daily weight change in TrkB.T1 KD and control male mice 4 weeks post-surgery in response to IP administration of vehicle for 3 days followed by administration of leptin (3 ug/g) for 3 days.
Data are represented as mean +/− SEM.
Extended Data Fig. 6. The use of serotype AAV2 and a CMV promoter to knockdown TrkB is specific to neurons.
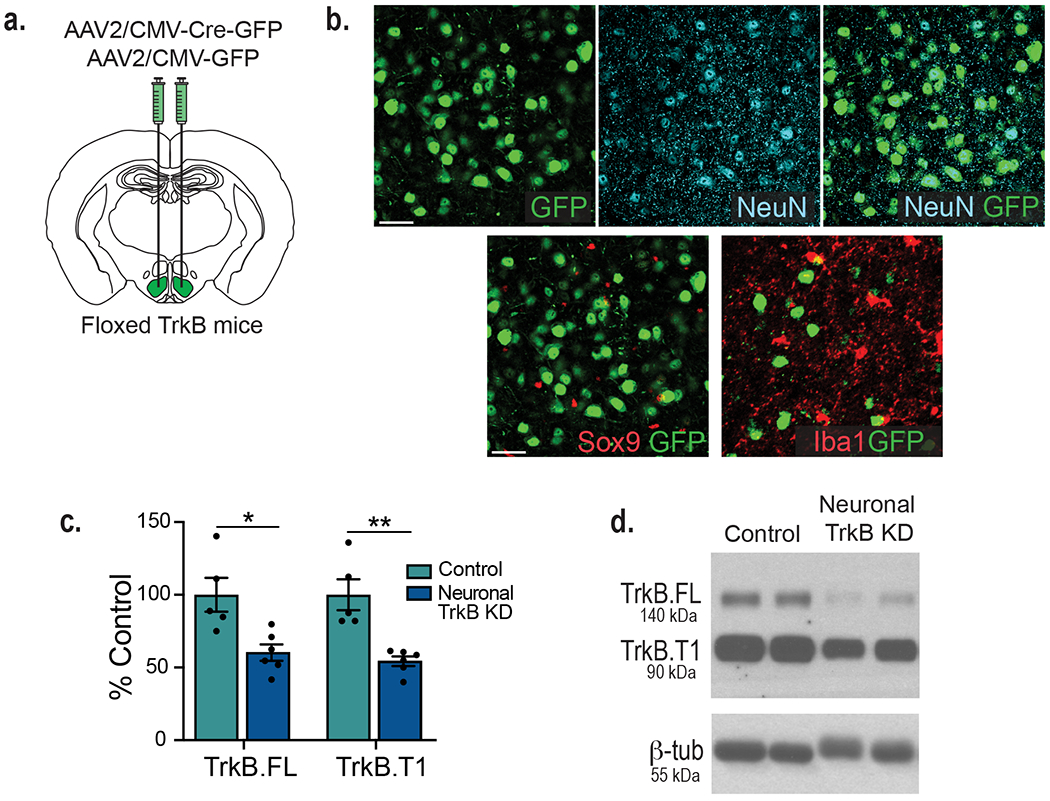
a. Diagram showing experimental approach for depleting TrkB from neurons bilaterally in the VMH of adult mice.
b. Co-immunolabeling of VMH showing co-localization of AAV2-CMV-driven GFP signal with the neuronal marker NeuN but not with the astrocytic marker Sox9 or the microglial marker Iba1. Scale bar 50 nM.
c. TrkB.T1 and TrkB.FL expression in floxed TrkB mice delivered AAV2-CMV-GFP (control) or AAV2-CMV-GFPCre (TrkB KD) to the VMH (n = 6). Student’s two-sided t-test, *, p = 0.01, **, p = 0.0016. Data represented as mean +/− SEM.
d. Representative western blot showing viral knockdown of TrkB in VMH. Data collected from one experiment.
Extended Data Fig. 7. TrkB in VMH neurons is not required for the regulation of energy balance under chow conditions but is essential for glycemic control.
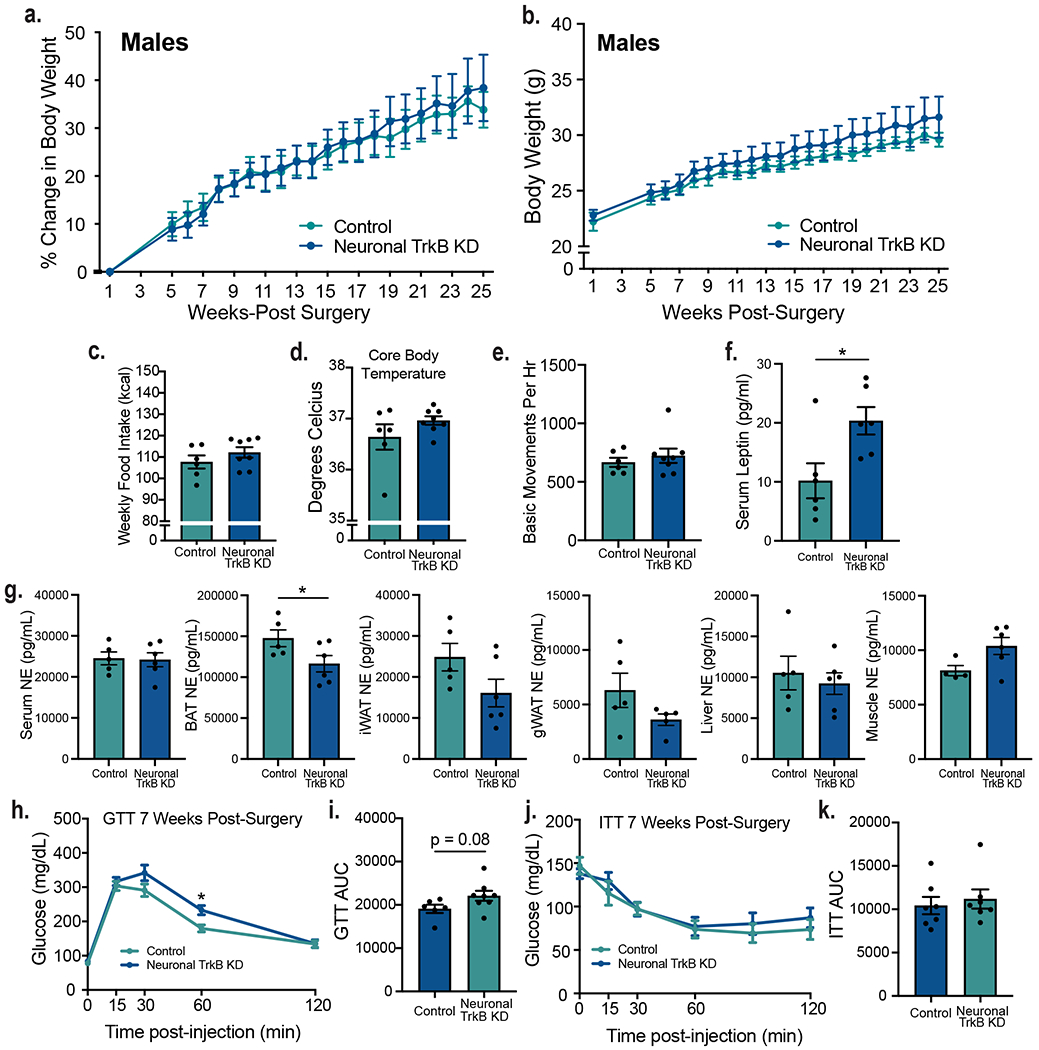
A . Percent body weight gain in Neuronal TrkB KD and control mice (n = 6). Two-way RM ANOVA: Genotype, p = 0.88; Time, p <0.0001; Interaction, p = 0.85; Subjects (matching), p <0.0001.
B. Body weights of Neuronal TrkB KD and control mice (n = 7). Two-way RM ANOVA: Genotype, p = 0.44; Time, p <0.0001; Interaction, p 0.76; Subjects (matching), p <0.0001.
C. Average weekly food intake weeks 3 - 6 post-surgery in neuronal TrkB KD (n = 8) and control (n = 6) mice. Student’s two-sided t-test, NS.
D. Core body temperatures in neuronal TrkB KD (n = 8) and control (n = 6) mice. Student’s two-sided t-test, NS.
E. Basic movements per hour of neuronal TrkB KD (n = 8) and control (n = 6) mice recorded over 6 days.
F. Serum levels of leptin (pg/mL) in fed animals (n = 6). Students two-sided t-test, *, p = 0.02.
G. Norepinephrine levels in indicated tissues in neuronal TrkB KD (n = 6) and control (n = 5) mice. Student’s two-sided t-test, *, p = 0.05.
H. Glucose tolerance test of neuronal TrkB KD (n = 8) and control (n = 6) mice. Two-way RM ANOVA; Time, p < 0.0001; Genotype, p = 0.1; Interaction, p = 0.01. Bonferroni multiple comparisons, *, p = 0.04.
I. GTT area under the curve. Students two-sided t-test, p = 0.08.
J. Insulin tolerance test (n = 7). Two-way RM ANOVA; Time, p < 0.0001; Genotype, p = 0.65; Interaction, p = 0.48. Bonferroni multiple comparisons.
K. ITT area under the curve. Students t-test, NS. Data represented as mean +/− SEM.
Extended Data Fig. 8. Selective depletion of TrkB.T1 from VMH astrocytes in adult mice does not alter VMH neuronal excitatory synapse density.
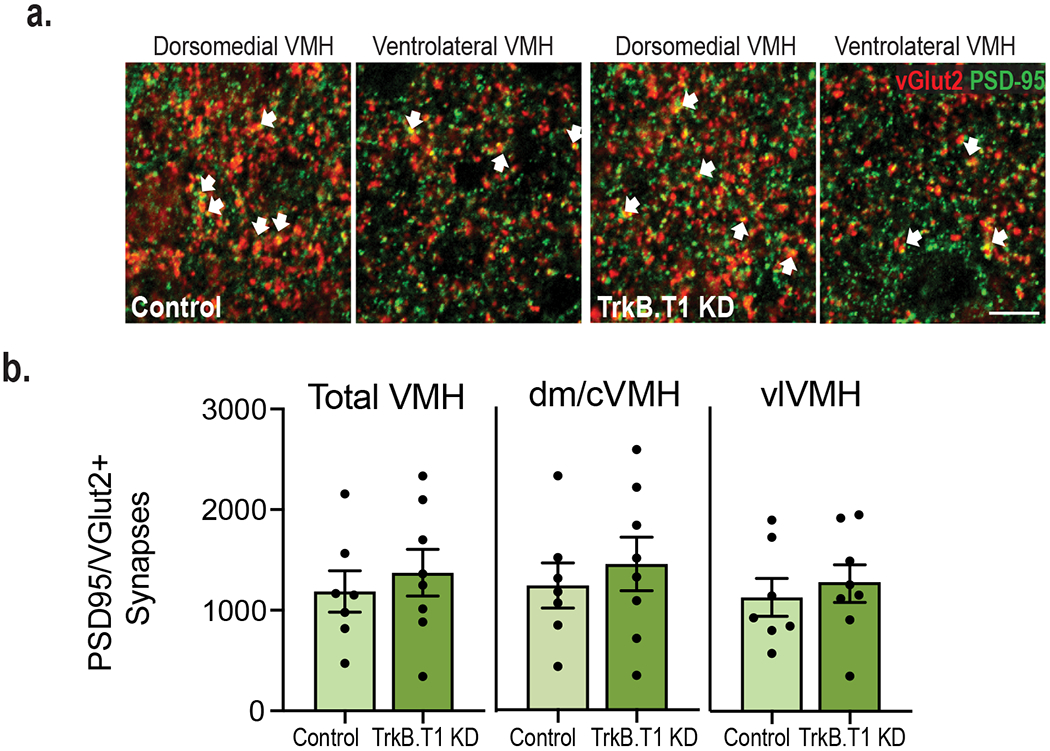
a. Representative images of VMH co-immunolabeled with anti-PDS95 and anti-vGlut2. Arrows indicate PSD95 and vGlut2 co-localization (scale bar 15 uM).
b. Density of excitatory synapses (co-localization of vGlut2 and PSD95) in the VMH of TrkB.T1 KD (n = 8) and control mice (n = 7). Students two-tailed t-test, NS
Data are represented as mean +/− SEM.
Extended Data Fig. 9. Selective depletion of TrkB.T1 from VMH astrocytes in adult mice leads to increased glutamate uptake at synapses.
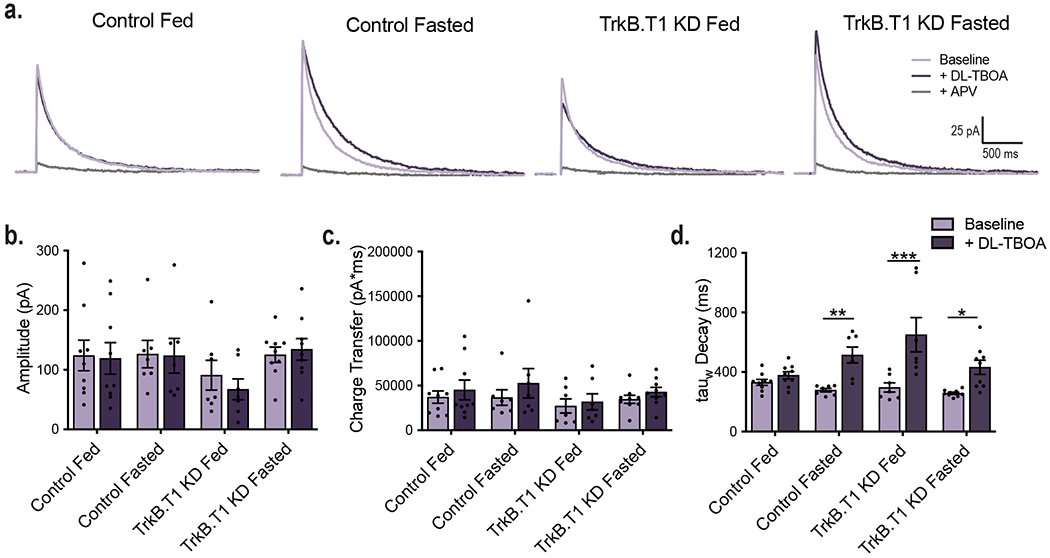
a. Representative traces of raw NMDAR responses (light purple), responses + 100 uM DL-TBOA (dark purple), and responses + 50 uM APV (light gray). For following panels, Control Fed (n = 9), Control Fasted (n = 7), TrkB.T1 Fed (n = 7), TrkB.T1 Fasted (n = 9).
b. Amplitude (pA) of NMDAR responses before and after DL-TBOA application.
c. Charge transfer (pA*ms) of NMDAR responses before and after DL-TBOA application.
d. Decay (weighted tau) of NMDAR responses before and after DL-TBOA application. Two-way ANOVA: Genotype, p = 0.03; Treatment, p < 0.0001; Genotype x Treatment Interaction, p = 0.01. Bonferroni multiple comparisons, *, p = 0.02. **, p = 0.005, ***, p < 0.0001. Data represented as +/− SEM.
Supplementary Material
Acknowledgments
We thank Dr. Baoji Xu for the floxed TrkB animals. We thank the HMS Electron Microscopy Facility and Maria Ericsson for electron microscopy imaging, consultation, and services. We thank the Vanderbilt Hormone Assay and Analytical Services Core and Lipid Core supported by NIH grants DK059637 and DK020593, specifically Dale Edgerton, Eric Allen and Carla Harris for assisting in tissue and serum catecholamine and lipid analysis. We thank the Imaging, Genomics, and Circuits Behavior Cores at the Tufts Center for Neuroscience Research. This work was supported by grants by the National Institute of Diabetes and Digestive and Kidney Diseases awarded to Maribel Rios (1R21NS091871 and 1R01DK117935-01) and Dominique Ameroso (1F31DK118789-01A1), the Synapse Neurobiology Training Program awarded to Dominique Ameroso (5T32NS061764-09), and the Training Program in Nutrition, Obesity and Metabolic Disorders awarded to Alice Meng (T32DK124170).
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data Availability Statement
All data associated with this study are presented in the paper or the extended data figures. Source data for every main or extended figure are provided with this manuscript.
References
- 1.Fu LY & van den Pol AN Agouti-related peptide and MC3/4 receptor agonists both inhibit excitatory hypothalamic ventromedial nucleus neurons. J Neurosci 28, 5433–5449, doi: 10.1523/JNEUROSCI.0749-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang EJ & Reichardt LF Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72, 609–642, doi: 10.1146/annurev.biochem.72.121801.161629 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Tran PV et al. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol 498, 637–648, doi: 10.1002/cne.21070 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M & Rios M Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci 27, 14265–14274, doi: 10.1523/JNEUROSCI.3308-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rios M et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15, 1748–1757, doi: 10.1210/mend.15.10.0706 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Chen ZY et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143, doi: 10.1126/science.1129663 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han JC et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 359, 918–927, doi: 10.1056/NEJMoa0801119 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speliotes EK et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42, 937–948, doi: 10.1038/ng.686 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haber M, Zhou L & Murai KK Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci 26, 8881–8891, doi: 10.1523/JNEUROSCI.1302-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirrlinger J, Hulsmann S & Kirchhoff F Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci 20, 2235–2239, doi: 10.1111/j.1460-9568.2004.03689.x (2004). [DOI] [PubMed] [Google Scholar]
- 11.Lippman JJ, Lordkipanidze T, Buell ME, Yoon SO & Dunaevsky A Morphogenesis and regulation of Bergmann glial processes during Purkinje cell dendritic spine ensheathment and synaptogenesis. Glia 56, 1463–1477, doi: 10.1002/glia.20712 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pannasch U et al. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat Neurosci 17, 549–558, doi: 10.1038/nn.3662 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Dietrich MO & Horvath TL Feeding signals and brain circuitry. Eur J Neurosci 30, 1688–1696, doi: 10.1111/j.1460-9568.2009.06963.x (2009). [DOI] [PubMed] [Google Scholar]
- 14.Sternson SM, Shepherd GM & Friedman JM Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci 8, 1356–1363, doi: 10.1038/nn1550 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Valenstein ES, Cox VC & Kakolewski JW Modification of motivated behavior elicited by electrical stimulation of the hypothalamus. Science 159, 1119–1121, doi: 10.1126/science.159.3819.1119 (1968). [DOI] [PubMed] [Google Scholar]
- 16.Danbolt NC Glutamate uptake. Prog Neurobiol 65, 1–105, doi: 10.1016/s0301-0082(00)00067-8 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Lehre KP & Danbolt NC The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18, 8751–8757 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanson E et al. Astrocytic glutamate uptake is slow and does not limit neuronal NMDA receptor activation in the neonatal neocortex. Glia 63, 1784–1796, doi: 10.1002/glia.22844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baxter GT et al. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J Neurosci 17, 2683–2690 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holt LM et al. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife 8, doi: 10.7554/eLife.44667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohira K et al. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J Neurosci 25, 1343–1353, doi: 10.1523/JNEUROSCI.4436-04.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose CR et al. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 426, 74–78, doi: 10.1038/nature01983 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Baydyuk M et al. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc Natl Acad Sci U S A 108, 1669–1674, doi: 10.1073/pnas.1004744108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du J, Feng L, Yang F & Lu B Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150, 1423–1434, doi: 10.1083/jcb.150.6.1423 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haapasalo A et al. Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 277, 43160–43167, doi: 10.1074/jbc.M205202200 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Meyer-Franke A et al. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21, 681–693, doi: 10.1016/s0896-6273(00)80586-3 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carim-Todd L et al. Endogenous truncated TrkB.T1 receptor regulates neuronal complexity and TrkB kinase receptor function in vivo. J Neurosci 29, 678–685, doi: 10.1523/JNEUROSCI.5060-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yacoubian TA & Lo DC Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci 3, 342–349, doi: 10.1038/73911 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Lindberg D, Chen P & Li C Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J Comp Neurol 521, 3167–3190, doi: 10.1002/cne.23338 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Nguyen NL et al. Separate and shared sympathetic outflow to white and brown fat coordinately regulates thermoregulation and beige adipocyte recruitment. Am J Physiol Regul Integr Comp Physiol 312, R132–R145, doi: 10.1152/ajpregu.00344.2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruffin M & Nicolaidis S Electrical stimulation of the ventromedial hypothalamus enhances both fat utilization and metabolic rate that precede and parallel the inhibition of feeding behavior. Brain Res 846, 23–29, doi: 10.1016/s0006-8993(99)01922-8 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Bingham NC, Anderson KK, Reuter AL, Stallings NR & Parker KL Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology 149, 2138–2148, doi: 10.1210/en.2007-1200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhillon H et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203, doi: 10.1016/j.neuron.2005.12.021 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Elmquist JK, Ahima RS, Elias CF, Flier JS & Saper CB Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A 95, 741–746, doi: 10.1073/pnas.95.2.741 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satoh N et al. Sympathetic activation of leptin via the ventromedial hypothalamus: leptin-induced increase in catecholamine secretion. Diabetes 48, 1787–1793, doi: 10.2337/diabetes.48.9.1787 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Scott MM et al. Leptin targets in the mouse brain. J Comp Neurol 514, 518–532, doi: 10.1002/cne.22025 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komori T, Morikawa Y, Nanjo K & Senba E Induction of brain-derived neurotrophic factor by leptin in the ventromedial hypothalamus. Neuroscience 139, 1107–1115, doi: 10.1016/j.neuroscience.2005.12.066 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Allen NJ et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414, doi: 10.1038/nature11059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldwin KT & Eroglu C Molecular mechanisms of astrocyte-induced synaptogenesis. Curr Opin Neurobiol 45, 113–120, doi: 10.1016/j.conb.2017.05.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung WS, Allen NJ & Eroglu C Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol 7, a020370, doi: 10.1101/cshperspect.a020370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elmariah SB, Oh EJ, Hughes EG & Balice-Gordon RJ Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci 25, 3638–3650, doi: 10.1523/JNEUROSCI.3980-04.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon BE & Lee CJ GABA as a rising gliotransmitter. Front Neural Circuits 8, 141, doi: 10.3389/fncir.2014.00141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mederos S & Perea G GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 67, 1842–1851, doi: 10.1002/glia.23644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takano T et al. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature 588, 296–302, doi: 10.1038/s41586-020-2926-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chee MJ, Myers MG Jr., Price CJ & Colmers WF Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J Neurosci 30, 3380–3390, doi: 10.1523/JNEUROSCI.4031-09.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klockener T et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci 14, 911–918, doi: 10.1038/nn.2847 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ono T et al. Glucoresponsive neurons in rat ventromedial hypothalamic tissue slices in vitro. Brain Res 232, 494–499, doi: 10.1016/0006-8993(82)90295-5 (1982). [DOI] [PubMed] [Google Scholar]
- 48.Choi YH, Fujikawa T, Lee J, Reuter A & Kim KW Revisiting the Ventral Medial Nucleus of the Hypothalamus: The Roles of SF-1 Neurons in Energy Homeostasis. Front Neurosci 7, 71, doi: 10.3389/fnins.2013.00071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams KW & Elmquist JK From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci 15, 1350–1355, doi: 10.1038/nn.3217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi YH et al. Melanocortin receptors mediate leptin effects on feeding and body weight but not adipose apoptosis. Physiol Behav 79, 795–801, doi: 10.1016/s0031-9384(03)00205-1 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Halassa MM, Fellin T, Takano H, Dong JH & Haydon PG Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27, 6473–6477, doi: 10.1523/JNEUROSCI.1419-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagy JI & Rash JE Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev 32, 29–44, doi: 10.1016/s0165-0173(99)00066-1 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Genoud C, Knott GW, Sakata K, Lu B & Welker E Altered synapse formation in the adult somatosensory cortex of brain-derived neurotrophic factor heterozygote mice. J Neurosci 24, 2394–2400, doi: 10.1523/JNEUROSCI.4040-03.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez A et al. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci 18, 7336–7350 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otal R, Martinez A & Soriano E Lack of TrkB and TrkC signaling alters the synaptogenesis and maturation of mossy fiber terminals in the hippocampus. Cell Tissue Res 319, 349–358, doi: 10.1007/s00441-004-1020-5 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Genoud C et al. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol 4, e343, doi: 10.1371/journal.pbio.0040343 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murai KK, Nguyen LN, Irie F, Yamaguchi Y & Pasquale EB Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci 6, 153–160, doi: 10.1038/nn994 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Garcia-Caceres C et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166, 867–880, doi: 10.1016/j.cell.2016.07.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horvath TL et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A 107, 14875–14880, doi: 10.1073/pnas.1004282107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim JG et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17, 908–910, doi: 10.1038/nn.3725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nuzzaci D et al. Postprandial Hyperglycemia Stimulates Neuroglial Plasticity in Hypothalamic POMC Neurons after a Balanced Meal. Cell Rep 30, 3067–3078 e3065, doi: 10.1016/j.celrep.2020.02.029 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Fagan MP et al. Essential and sex-specific effects of mGluR5 in ventromedial hypothalamus regulating estrogen signaling and glucose balance. Proc Natl Acad Sci U S A 117, 19566–19577, doi: 10.1073/pnas.2011228117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Felsted JA et al. Alpha2delta-1 in SF1(+) Neurons of the Ventromedial Hypothalamus Is an Essential Regulator of Glucose and Lipid Homeostasis. Cell Rep 21, 2737–2747, doi: 10.1016/j.celrep.2017.11.048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McClellan KM, Parker KL & Tobet S Development of the ventromedial nucleus of the hypothalamus. Front Neuroendocrinol 27, 193–209, doi: 10.1016/j.yfrne.2006.02.002 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are presented in the paper or the extended data figures. Source data for every main or extended figure are provided with this manuscript.