

Abstract
To avoid acquired variants found in the blood, cultured skin fibroblasts are a recommended DNA source for germline genetic testing in patients with hematologic disorders, but data are lacking regarding practicality and limitations. We conducted a retrospective cohort study of 350 subjects with hematologic disorders who underwent skin fibroblast culture for germline genetic testing. We analyzed next‐generation sequencing data from the targeted capture of 144 inherited cancer and bonemarrow failure genes to identify variants at heterozygous and subclonal variant allele frequencies. Sixteen (5%) biopsies failed to culture. Culture failure was more likely in samples with delays in culture initiation (OR = 4.3; p < 0.01) or a pathogenic variant in a telomere gene (OR = 42.6; p < 0.01). Median culture time was 28 days (IQR 22−29 days). Culture time was longer for subjects with prior allogeneic stem cell transplantation (+10.7%; p = 0.02) and shorter in subjects with a heterozygous pathogenic variant (−11.9%; p < 0.01), larger biopsy size (−10.6%; p < 0.01), or lymphoid malignancy (−8.4%; p < 0.01). Subclonal variants were identified in 10 (4%) and confirmed in five (56%) of eight with alternate samples available. Subclonal and discordant variants illustrate that germline testing from cultured skin fibroblasts requires phenotypic correlation and, in rare cases, follow‐up studies for optimal interpretation.
Keywords: germline genetics, inherited, leukemia, lymphoma, myelodysplastic syndrome, skin fibroblasts
1. INTRODUCTION
Peripheral blood is the standard tissue collected for germline genetic testing in most clinical scenarios. In patients with hematologic malignancies (HMs), however, peripheral blood frequently contains tumor‐ or clonal hematopoiesis‐related acquired genetic variants, often occurring in genes that can also cause inherited cancer susceptibility disorders if present in the germline (Tawana et al., 2018). Thus, an alternative source of DNA is needed for accurate germline testing.
Cultured skin fibroblasts have been promoted as the best DNA source for germline testing for susceptibility to HMs for several reasons. First, the culturing process removes contaminating blood cells. Thus, unlike many other sources of potential germline material, such as saliva, buccal swabs, and nail clippings which all contain contaminating blood cells to varying degrees (Padron et al., 2018; Theda et al., 2018), leukemia‐ and myelodysplastic syndrome (MDS)‐related acquired genetic events that can even persist in blood and bone marrow during clinical remission should not be detected in DNA derived from cultured skin fibroblasts. Second, skin fibroblasts have demonstrated efficacy in reliably detecting germline variants in challenging scenarios encountered in bone marrow failure disorders that make detection in the peripheral blood difficult. For example, hematopoietic stem cells in multiple bone marrow failure disorders can lose the germline pathogenic variant(s) by multiple mechanisms, such as a somatic reversion event that corrects the germline variant as seen in Fanconi anemia (Gregory et al., 2001) or acquired monosomy 7 that favors loss of the germline variant carrying chromosome 7 in ataxia pancytopenia syndrome (Chen et al., 2016; Tesi et al., 2017). Lastly, cultured skin fibroblasts provide ample DNA quantity in contrast to alternate sources such as hair follicles or nail clippings, which provide limited quantities often insufficient for the majority of clinical genetic testing laboratories.
With these potential advantages and recent clinical guidelines recommending germline genetic testing for a growing proportion of patients with hematologic disorders (Arber et al., 2016; DeZern & Churpek, 2021; National Comprehensive Cancer Network, 2021, 2022), there is a growing incentive for clinical genetic testing laboratories to grow and/or test DNA from cultured skin fibroblasts. However, because prior experience with cultured skin fibroblasts for germline genetic testing in this context has been limited, there is a knowledge gap in terms of practical considerations as well as potential limitations of their use. For example, expected culture failure rates, the turnaround time from culture initiation to sufficient growth for DNA extraction, the impact of sample shipment on growth, and clinical predictors of failure or extended times in culture are unknown. Additionally, normal‐appearing human skin has recently been shown to harbor a high burden of acquired variants, including in genes relevant to inherited cancer risk such as TP53 (Martincorena et al., 2015; Yizhak et al., 2019). If these acquired variants are present in enough cells in the piece of biopsied skin put into culture or if the culturing process itself induces or selects for acquired variants that provide a growth advantage, there is the possibility that this acquired mosaicism could contribute to false‐positive genetic testing results.
To address these knowledge gaps, we conducted a retrospective cohort study of 350 subjects with hematologic disorders who had skin fibroblasts cultured for germline genetic testing. Here we report the observed failure rates and turnaround time from biopsy to the growth of a sufficient number of cells for DNA extraction for genetic testing using a standardized culture protocol. We identify clinical, genetic, and technical factors associated with these outcomes which can be utilized to optimize the use of skin fibroblasts for genetic testing. Finally, we report expected times in culture for subjects found to carry heterozygous and subclonal pathogenic/likely pathogenic (P/LP) variants in genes relevant to inherited blood disorders and cancer risk, and whether these variants are present or absent in blood or bone marrow samples.
2. MATERIALS AND METHODS
2.1. Study population
All subjects with hematologic disorders who had skin specimens cultured for germline genetic testing using a standard protocol (see Supporting Information) in the University of Chicago Constitutional Cytogenetics Laboratory between April 1, 2013 and June 1, 2018 were eligible (Figure S1). Those lacking clinical data or consent were excluded. The University of Chicago and the University of Wisconsin‐Madison's Institutional Review Boards approved this study. This study was conducted in accordance with the Helsinki Declaration.
2.2. Skin biopsy and culture protocol
Skin samples were obtained by a 3‐mm punch biopsy from any site or removal of a skin ellipse from the site of a bone marrow biopsy or other surgical incision and placed in sterile RPMI media at room temperature for transport. Samples were processed and cultured using a standardized protocol (see Supporting Information Methods). DNA extraction was performed when a T75 flask reached 80%−100% confluency.
2.3. Data abstraction
Skin biopsy specimen and processing information, including time from biopsy to culture initiation and to DNA extraction, shipment from an outside institution, culture failure (defined as insufficient growth to reach 80% confluency in a T75 flask), and biopsy size, was abstracted from tissue culture logs. Clinical data were abstracted from medical records.
2.4. Genetic testing and variant interpretation
Next‐generation sequencing (NGS) data from the targeted capture of 144 inherited cancer, clonal hematopoiesis, and bone marrow failure disorder genes (Table S1) obtained for clinical genetic testing purposes in the University of Chicago Genetic Services Laboratory per established methods (Guidugli et al., 2017) were analyzed to identify variants at both heterozygous (40%−60%) and subclonal (10%−40%) variant allele frequencies (VAFs) using a custom pipeline (Figure S2). Variants were defined as P or LP according to The American College of Medical Genetics and Genomics/The Association for Molecular Pathology guidelines and the Clinical Genome Sequence Variant Interpretation Working Group recommendations (Biesecker et al., 2018; Richards et al., 2015; Riggs et al., 2020; Tavtigian et al., 2018). Subjects were considered to be carriers if they carried a heterozygous P/LP variant in a gene with an autosomal recessive inheritance pattern. Carrier variants were excluded from analyses unless otherwise stated.
2.5. Variant validation and tissue source comparisons
All P/LP variants were validated using residual DNA from the original fibroblast culture specimen, either by Sanger sequencing for single nucleotide and small insertion−deletion variants, or quantitative real‐time polymerase chain reaction or custom high‐density gene‐targeted array comparative genomic hybridization for copy number alterations. VAFs from NGS testing of blood or bone marrow (OncoPlus; Kadri et al., 2017) of P/LP variants were assessed in all available samples. For subclonal variants or variants not fitting expected clinical presentations, a second skin biopsy with repeat fibroblast culture was also performed to confirm variant presence or absence when possible.
2.6. Statistical analysis
Fisher's exact tests and logistic regression models were used to assess significant associations with culture failure. t tests, Mann Whitney U tests, and linear regression models were used to assess factors associated with log time from culture initiation to sufficient growth for DNA extraction. Statistical analysis was performed in Stata v.16 (StataCorp).
3. RESULTS
3.1. Study population
Among 458 skin samples processed during the study period, 350 samples from unique, consented subjects with clinical data available constituted the study population (Figure S1 and Table 1). Except for three pediatric subjects, subjects were adults with a median age at the time of skin biopsy of 60 years (range: 1−88). The majority had an active HM (n = 211; 60%) at the time of biopsy with acute myeloid leukemia (AML) (n = 110; 32%) and MDS (n = 48; 14%) as the most frequent diagnoses. Seventy‐eight subjects (22%) had hematopoietic stem cell transplantation (HSCT) before biopsy, among whom 45 subjects had an autologous transplant, 41 allogeneic, and eight both.
Table 1.
Subject and skin biopsy characteristics, overall and by culture status.
| Total n (%) | Successful culture, n (%) | Culture failure, n (%) | p valued | |
|---|---|---|---|---|
| Total | 350 (100) | 334 (95) | 16 (5) | |
| Age | 0.68 | |||
| Median in years (range) | 60 (1−88) | 59 (1−88) | 60 (17−82) | |
| Sex | 0.12 | |||
| Female | 182 (52) | 177 (53) | 5 (31) | |
| Race | 0.22 | |||
| White, non‐Hispanic | 235 (67) | 225 (67) | 10 (63) | |
| White, Hispanic | 14 (4) | 12 (4) | 2 (13) | |
| Black | 49 (14) | 46 (14) | 3 (19) | |
| Other | 21 (6) | 21 (6) | 0 (0) | |
| Missing | 31 (9) | 30 (9) | 1 (6) | |
| Smoking status | 0.34 | |||
| Current | 20 (6) | 18 (5) | 2 (13) | |
| Former | 137 (39) | 132 (40) | 5 (31) | |
| Never | 163 (47) | 155 (46) | 8 (50) | |
| Missing | 30 (9) | 29 (9) | 1 (6) | |
| Body mass index (n = 311) | 0.40 | |||
| Mean in kilograms/m2 (SD) | 28.4 (6.7) | 28.5 (6.8) | 27.2 (5.4) | |
| Hematologic diagnosis | 0.49 | |||
| Acute myeloid leukemia | 110 (32) | 101 (30) | 9 (56) | |
| Myelodysplastic syndrome | 48 (14) | 47 (14) | 1 (6) | |
| Myeloproliferative neoplasm | 26 (7) | 26 (8) | 0 (0) | |
| Therapy‐related myeloid neoplasm | 16 (5) | 16 (5) | 0 (0) | |
| Plasma cell neoplasm | 42 (12) | 40 (12) | 2 (13) | |
| Acute lymphoblastic and mixed lineage leukemia | 30 (9) | 30 (9) | 0 (0) | |
| Mature T‐ and B‐cell neoplasms | 43 (12) | 41 (12) | 2 (13) | |
| Aplastic anemia and other unexplained cytopenias | 21 (6) | 19 (6) | 2 (13) | |
| Other benign hematologic disease | 12 (3) | 12 (4) | 0 (0) | |
| Missing | 2 (1) | 2 (1) | 0 (0) | |
| Prior chemotherapy | 1.00 | |||
| Never | 74 (21) | 71 (21) | 3 (19) | |
| <6 months before skin biopsy | 192 (55) | 183 (55) | 9 (56) | |
| ≥6 months before skin biopsy | 51 (15) | 49 (15) | 2 (13) | |
| Missing | 33 (9) | 31 (9) | 2 (13) | |
| Prior radiation | 1.00 | |||
| Yes | 64 (18) | 61 (18) | 3 (19) | |
| Missing | 33 (9) | 32 (10) | 1 (6) | |
| Prior transplanta | ||||
| Allogeneic | 41 (12) | 40 (12) | 1 (6) | 0.70 |
| Autologous | 45 (13) | 43 (13) | 2 (13) | 1.00 |
| Missing | 28 (8) | 27 (8) | 1 (6) | |
| Skin biopsy size (n = 343) | 0.51 | |||
| Median in cm2 (range) | 0.07 (0.01−0.56) | 0.07 (0.01−0.56) | 0.07 (0.03−0.15) | |
| Skin biopsy site | 0.04 | |||
| Buttock at site of bone marrow biopsy | 221 (63) | 211 (63) | 10 (63) | |
| Upper inner arm | 50 (14) | 48 (14) | 2 (13) | |
| Scapula | 28 (8) | 28 (8) | 0 (0) | |
| Other | 13 (4) | 10 (3) | 3 (19) | |
| Missing | 38 (11) | 37 (11) | 1 (6) | |
| Timing of skin culture initiation | <0.01 | |||
| Same day | 292 (83) | 283 (85) | 9 (56) | |
| Next day | 48 (14) | 45 (13) | 3 (19) | |
| Two or more days | 10 (3) | 6 (2) | 4 (25) | |
| Heterozygous pathogenic/likely pathogenic variant(s)b | 0.40 | |||
| None | 253 (72) | 246 (74) | 7 (44) | |
| One or more | 58 (17) | 55 (16) | 3 (19) | |
| Missing | 39 (11) | 33 (10) | 6 (38) | |
| Heterozygous pathogenic/likely pathogenic variant(s) in a telomere‐associated geneb,c | <0.01 | |||
| No | 305 (87) | 298 (89) | 7 (44) | |
| Yes | 6 (2) | 3 (1) | 3 (19) | |
| Missing | 39 (11) | 33 (10) | 6 (38) |
Note: Bold values are those that are statistically significant (p à 0.05).
Eight subjects had both an autologous and allogeneic stem cell transplant.
Identified on initial fibroblast culture or via alternative tissue if culture failed.
Pathogenic or likely pathogenic variant in PARN, RTEL1, TERC, or TERT.
Comparison between successful culture and culture failure populations.
3.2. Skin biopsy characteristics
The median biopsy size was 0.07 cm2 (range: 0.01–0.56 cm2). The most common body sites for skin biopsy were at the site of a bone marrow biopsy incision (n = 221; 63%) and the upper inner arm (n = 50; 14%). Twenty‐eight samples (8%) were shipped from outside institutions for culture.
3.3. Culture failure rate and predictors
Using a standardized protocol, 16 of 350 skin biopsies (5%) failed to culture (Table 1). Technical factors significantly associated with culture failure included site of biopsy and timing of culture initiation post‐biopsy (Tables 1 and S2). Compared with obtaining the skin biopsy from the buttock at the site of a bone marrow biopsy, the odds of culture failure were similar for those obtained from the upper inner arm or scapula (OR: 0.6; 95% CI: 0.1−2.7) but increased for those obtained from other body sites (e.g., the thigh) (OR: 6.8; 95% CI: 1.6−28.1). Compared with culture initiation on the day of biopsy, culture initiation the following day or two or more days later was associated with a two‐fold (OR: 2.1; 95% CI: 0.6−8.0) and 21‐fold (OR: 21.0; 95% CI: 5.0−87.5) increased odds of culture failure, respectively. Nine of 292 samples (3%) with culture initiation the same day as the biopsy failed, compared to three of 48 samples (6%) initiated 1 day later, and four of 10 samples (40%) initiated 2 or more days later. The only clinical factor associated with culture failure was possession of a heterozygous P/LP variant in a telomere‐associated gene (OR: 42.6; 95% CI: 7.3−249.2) (Table S2). Three of six (50%) samples with a heterozygous P/LP variant in either PARN, RTEL1, TERC, or TERT failed, versus only seven of 305 (2%) samples without such variants (p < 0.01) (Tables 1, S3 and S4). One of these individuals had telomere lengths measured by Flow FISH (Johns Hopkins) and had critically short (<1st percentile) lymphocyte telomere lengths for age in the setting of a hypocellular bone marrow and skin features consistent with dyskeratosis congenita (Table S4). Shipment of the biopsy from an outside institution via overnight courier was not associated with culture failure—only one out of 28 shipped samples (4%) failed. A second biopsy and culture were performed for five of the 16 initial failures (Table S5). Of these, all five, including one with an RTEL1 and one with a TERT P/LP variant, cultured successfully. For three of these samples, the initial skin biopsy and culture attempt had technical factors that may have contributed to the original culture failure, including delays in culture initiation by 2 or more days in two and by 1 day in one subject with a heterozygous TERT variant. These adverse technical factors were not present on the second biopsy attempt, potentially contributing to their success.
3.4. Time from culture initiation to sufficient growth for DNA extraction and predictors
Samples took a median of 27.6 days (range: 14−80) to reach the growth threshold for DNA extraction (Figures 1 and S3). Clinical factors associated with an increased time from culture initiation to DNA extraction included prior allogeneic HSCT (16.1% longer; p < 0.01), increasing age (0.2% longer per 1 year increase in age; p = 0.01), and carrying a heterozygous P/LP variant in a telomere‐associated gene (31.9% longer; p = 0.05; Table 2). In contrast, a sample from a subject with a lymphoid malignancy (vs. a myeloid malignancy) (7.1% shorter; p = 0.01), of increasing size (9.7% shorter for every one log cm2 increase in skin biopsy size; p < 0.01), those shipped from an outside institution (11.8% shorter vs. those not shipped; p = 0.01), and those from a subject with any heterozygous P/LP variant (8.1% shorter vs. those without a P/LP variant; p = 0.02) were factors associated with a decreased time to DNA extraction (Table 2). All of these retained significance in a multivariable model except shipped from an outside institution and presence of a heterozygous P/LP variant in a telomere‐associated gene (Table 2).
Figure 1.
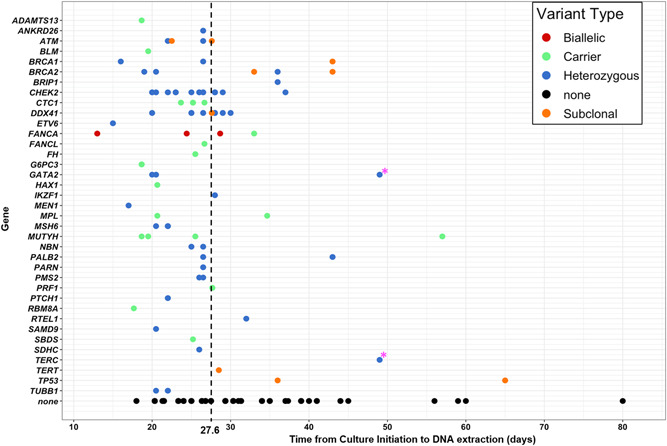
Time from culture initiation to sufficient growth for DNA extraction by gene and heterozygous, carrier, and subclonal variant status. Black dashed line indicates median time from culture initiation to DNA extraction for all n = 334 skin samples that were successfully cultured. Pink star (*) indicates two variants (both genes included in a heterozygous large deletion event) in a single subject that was not confirmed in a repeat skin biopsy sample.
Table 2.
ccc initiation to sufficient growth for DNA extraction. a
| Simple linear regression | Multiple linear regression | ||||||
|---|---|---|---|---|---|---|---|
| % change | 95% CI | p value | % change | 95% CI | p value | ||
| Clinical factors | |||||||
| Patient age (years) | 0.2% | 0.04% to 0.4% | 0.01 | 0.3% | 0.1% to 0.5% | <0.01 | |
| Female | −2.5% | −7.8% to 2.9% | 0.36 | −1.4% | −6.5% to 4.1% | 0.61 | |
| Race (vs. White, non‐Hispanic) | |||||||
| Black | −2.2% | −9.5% to 5.8% | 0.58 | 1.1% | −6.3% to 9.1% | 0.78 | |
| Other | −2.5% | −10.9% to 6.7% | 0.59 | 1.7% | −7.4% to 11.7% | 0.73 | |
| Smoking history (vs. never) | |||||||
| Former | 2.1% | −3.6% to 8.1% | 0.48 | −1.4% | −7.0% to 4.6% | 0.65 | |
| Current | 3.4% | −8.4% to 16.6% | 0.60 | 1.3% | −9.4% to 13.2% | 0.82 | |
| BMI (kg/m2) | −0.1% | −0.5% to 0.3% | 0.66 | −0.2% | −0.6% to 0.2% | 0.31 | |
| Hematologic malignancy diagnosis (vs. myeloid) | |||||||
| Lymphoid | −5.9% | −11.2% to −0.3% | 0.04 | −8.4% | −13.9% to −2.5% | <0.01 | |
| Benign | −2.7% | −11.4% to 6.8% | 0.56 | 3.4% | −7.6% to 15.7% | 0.56 | |
| Prior chemotherapy (vs. never) | |||||||
| <6 mo before skin biopsy | 5.0% | −1.8% to 12.3% | 0.15 | −1.0% | −8.0% to 6.5% | 0.78 | |
| ≥6 mo before skin biopsy | −0.3% | −8.8% to 9.1% | 0.95 | −7.2% | −15.6% to 2.0% | 0.12 | |
| Prior radiation | 5.3% | −1.7% to 12.9% | 0.14 | 2.0% | −4.9% to 9.4% | 0.59 | |
| Prior allogeneic stem cell transplant | 16.1% | 7.0% to 25.7% | <0.01 | 10.7% | 1.5% to 20.7% | 0.02 | |
| Prior autologous stem cell transplant | 2.3% | −5.5% to 10.8% | 0.57 | 5.2% | −3.3% to 14.4% | 0.24 | |
| Technical factors | |||||||
| Size of biopsy (log cm2) | −9.7% | −13.4% to −5.8% | <0.01 | −10.6% | −15.0% to −6.0% | <0.01 | |
| Body site of biopsy (vs. buttock at site of bone marrow biopsy incision) | |||||||
| Upper inner arm or scapula | 1.9% | −4.4% to 8.7% | 0.56 | 2.8% | −4.1% to 10.1% | 0.43 | |
| Other site | −5.9% | −19.5% to 10.1% | 0.45 | −4.0% | −16.9% to 10.8% | 0.58 | |
| Skin culture initiation timing (vs. same day) | |||||||
| Next day | 0.4% | −7.1% to 8.5% | 0.92 | 11.9% | 2.7% to 21.9% | 0.01 | |
| 2 or more days | 17.2% | −4.1% to 43.2% | 0.12 | 16.7% | −4.0% to 41.9% | 0.12 | |
| Sample shipped from outside institution | −11.8% | −19.9% to −2.9% | 0.01 | — | — | — | |
| Genetic factors | |||||||
| Heterozygous pathogenic/likely pathogenic variant(s) | −8.1% | −14.4% to −1.2% | 0.02 | −11.9% | −17.9% to −5.5% | <0.01 | |
| Heterozygous pathogenic/likely pathogenic variant in a telomere‐associated geneb | 31.9% | −0.2% to 74.4% | 0.05 | 33.5% | −2.4% to 82.5% | 0.07 | |
Note: Bold values are those that are statistically significant (p à 0.05).
Modeling was performed with log transformation of time from culture initiation to DNA extraction in days. The % change column is derived from model β‐coefficients by e^(β). For example, time in culture (in days) for a skin biopsy from a subject with a prior allogeneic stem cell transplant is (e^0.149 = 1.16) is 16% longer than one from a subject who has not had a prior allogeneic stem cell transplant.
Pathogenic or likely pathogenic variant in PARN, RTEL1, TERC, or TERT.=variable dropped due to collinearity.
3.5. Heterozygous P/LP variant detection, impact on skin fibroblast culture, and comparison across tissue types
Among the 350‐subject initial study population, 311 underwent genetic testing (Figure S1). Of these, 58 subjects (19%) had one or more heterozygous P/LP variant(s) associated with a known hereditary cancer or bone marrow failure disorder (Table S3). CHEK2 (n = 16; 25%), followed by DDX41 (n = 9; 11%), BRCA2 (n = 3; 1%), FANCA (n = 3, compound heterozygotes; 1%), GATA2 (n = 3; 1%), and MSH6 (n = 3; 1%) were the most frequent. Our analysis confirmed 52 of 52 P/LP variants reported on the original clinical test report and identified six additional P/LP variants, all of which were in genes not included among the original genetic testing request. One additional variant previously reported as P/LP was downgraded to a variant of uncertain significance based on updated ACMG/ClinGen criteria. A P/LP carrier status variant was identified in 24 subjects (8%), of whom five (21%) also carried a heterozygous P/LP causative variant (Table S3).
Despite the increased time from culture initiation to DNA extraction for samples from subjects with a heterozygous P/LP variant in a telomere‐associated gene, among the entire study population, those carrying a heterozygous P/LP variant in any gene exhibited decreased time in culture (25.7 vs. 28.1 days in those without a P/LP variant; p = 0.02) (Figure S3). Although numbers of samples from carriers of P/LP variants in individual genes are limited (Figure 1), times in culture for genetic variants known to associate with proliferation in hematopoietic tissues, such as CHEK2 (Bao et al., 2020), were also observed to have skin fibroblast culture growth times shorter than average.
Of 74 subjects with either one or more heterozygous P/LP causative or carrier variants, 35 had NGS covering the P/LP variant(s) in a repeat skin biopsy and culture (n = 2) and/or blood/bone marrow specimens (n = 34) (Figure 2 and Table S3). Thirty‐five of 36 (97%) total variants were confirmed, although a single large deletion event including both GATA2 and TERC found in a sample that took 48 days to culture (Figure 1, pink stars) was not present on a second skin biopsy and culture specimen (Figures 2, pink star and S4). Overall, VAFs were similar between skin fibroblast and blood/bone marrow specimens across two different NGS assays, with the exception of two cases (Figure 3). One of subject 268's two P/LP compound heterozygous FANCA variants was detected in blood at 11% VAF versus 54% VAF in cultured skin fibroblasts. She had a prior history of severe cytopenias in her mid‐teens and was being considered for an allogeneic HSCT but had spontaneous improvement in her blood counts maintained until her germline testing at age 35. Subject 283's SAMD9 de novo missense LP variant was detected at 50% VAF in skin versus 30% in a bone marrow sample diagnosed as refractory cytopenia of childhood with monosomy 7 in 80% of cells. The bone marrow sample had two additional acquired SAMD9 variants (p.Arg2639His at 15% VAF; and p.Val853Ile at 3% VAF).
Figure 2.
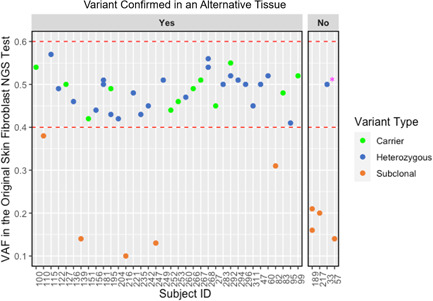
Variants identified in cultured skin fibroblasts at heterozygous variant allele frequencies are confirmed in an alternative tissue more often than variants at subclonal variant allele frequencies (VAF). Alternative tissue specimens included blood or bone marrow or a second skin biopsy and culture sample. In total, 36 of 37 heterozygous (blue dots) or carrier status (green dots) variants at VAF 40%−60% (designated by dashed red lines) identified on the original skin fibroblast next‐generation sequencing test were confirmed in an alternative tissue specimen. One variant (pink star), a large deletion variant encompassing both GATA2 and TERC, was not confirmed on a second skin biopsy and culture sample despite a VAF of 50% from the original culture and confirmation on a microarray (Figure S4). This subject did not have clinical features of either GATA2 deficiency syndrome or a telomere biology disorder. Five of nine subclonal variants (orange dots) were confirmed in alternative tissues.
Figure 3.
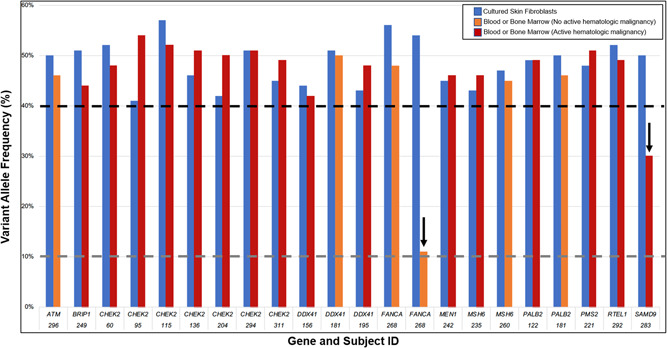
Pathogenic/likely pathogenic heterozygous and carrier status variants at variant allele frequencies (VAFs) 40%−60% in cultured skin fibroblasts have a similar VAF in blood or bone marrow samples on a somatic NGS assay (OncoPlus) across subjects, genes, and disease status with exceptions. Black dashed line indicates heterozygous VAF cut‐off. Dark gray dashed line indicates VAF limit of detection for germline pipeline. Arrows indicate two samples with blood or bone marrow VAF < 40%.
3.6. Subclonal P/LP variant detection, association with skin fibroblast culture time, and comparison across tissue types
Eleven P/LP subclonal variants were identified in 10 subjects (4%) out of the 252 with sequence data available (Figures S1, 4a, and Table S6). The median VAF was 16% (IQR: 14%−26%). Median time in culture of these samples was 28.5 days (vs. 27 in those without a subclonal variant; p = 0.21) (Figure 1). Clinical and technical factors associated with the presence of a subclonal P/LP variant included: prior autologous HSCT (OR: 4.34; 95% CI: 1.11−16.98), prior allogeneic HSCT (OR: 7.35; 95% CI: 1.84−29.39); and time in culture (OR: 7.93; 95% CI: 1.00−63.24) (Table S7).
Figure 4.
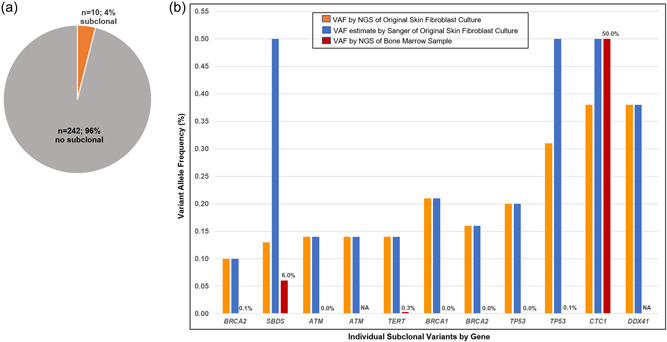
Subclonal pathogenic/likely pathogenic variants (VAF 10%‐40%) are (a) Identified in 4% of samples and (b) have varying VAF when estimated by Sanger of the original skin fibroblast culture versus by NGS of a bone marrow sample. NA, sample not available.
Attempts were made to confirm the presence of these subclonal variants. First, Sanger sequencing of residual DNA from the original skin fibroblast culture confirmed the presence of all of these subclonal variants. Of the 11 variants, Sanger sequencing data for eight were consistent with the initial NGS assay VAFs (Figures 4b, S5 and Table S6). However, Sanger sequencing peak heights for the remaining three were consistent with a heterozygous state, including one CTC1 (38% VAF in original skin NGS and heterozygous by Sanger of a bone marrow sample as well), one SBDS (13% VAF in original skin NGS and present in 22 of 335 reads in NGS of a bone marrow sample), and one TP53 (31% VAF in original skin NGS, present in 2 of 2528 reads by NGS of a bone marrow sample, and not present by Sanger of a new skin biopsy and culture) variant. The SBDS variant lies in a region with a pseudogene, creating challenges for targeted capture. The etiology of the TP53 variant, whether due to mosaicism, selection in culture, or technical factors was not clear. In total, NGS data from bone marrow samples were available for nine of the 11, of which five (56%) detected the variant (Figure 2, orange circles). The majority of these were present at a very low VAF (Figure 4b and Table S6). Three of these subjects had a repeat skin biopsy cultured for confirmation studies. The variant was not confirmed in the second culture in any of these. In contrast, thawing and further culture of a frozen aliquot of skin fibroblasts from the original culture for Subject 189, who had subclonal LP variants in BRCA1 and BRCA2 at 21% and 16% VAFs, respectively, detected in the initial culture that took 42 days to grow, yielded VAFs of 52% and 48% with retesting. Neither of these variants were identified in this subject's NGS assay (OncoPlus; Kadri et al., 2017) performed using a bone marrow sample. In total, three of these 11 subclonal variants (27%) are likely heterozygous, including the CTC1 at 38% VAF, DDX41 at 38% VAF, and SBDS at 13% VAF in the original skin NGS assay. Subject 216's BRCA2 variant is likely a sequencing error at a homopolymer site. The others could represent sequencing error or bias, somatic mosaicism in skin alone or in multiple body tissues, or culture artifacts.
3.7. Donor cell contamination of cultured skin fibroblasts
Among 23 subjects who had allogeneic HSCT before skin biopsy with cultured skin fibroblast DNA available, there was no evidence of donor genotype on variable number tandem repeat (VNTR) analysis (Supporting Information Methods).
4. DISCUSSION
Our findings demonstrate that culturing of skin fibroblasts is successful for the majority of samples, including those requiring shipment to another facility for culturing, and adds about 28 days to genetic testing turnaround time. Culture failure, a clinically challenging scenario leading to further delays, can be minimized by setting up the initial culture on the same day or at most, the day following the skin biopsy. Importantly, this standardized protocol was successful for culturing and genetic testing from patients with multiple hereditary hematologic disorders, whose samples interestingly had a shorter than average time in culture compared to those without a P/LP heterozygous germline variant even after adjusting for age, size of biopsy, and other potential confounders. In contrast, in keeping with prior studies demonstrating the relationship between replicative capacity and telomere length (Allsopp et al., 1992; Harley et al., 1990) and showing abnormal fibroblast growth in association with known telomere disorders (Dokal et al., 1992), our samples from subjects with a telomere biology disorder often failed to culture (50% failure rate) or took a significantly longer time to reach sufficient cell numbers. Finally, comparisons of VAFs found in cultured skin fibroblasts of heterozygous and subclonal variants, suggest a benefit of using cultured skin fibroblasts in cases with the possibility of germline allelic loss in blood or bone marrow, as well as the possibility of rare false positive, false negative, or subclonal variants of unclear etiology with use of this sample type.
Our data suggest that cultured skin fibroblasts represent a feasible sample type for germline genetic testing for adults with multiple benign and malignant hematologic disorders, including those with recent chemotherapy, radiation, or autologous or allogeneic HSCT and those ultimately diagnosed with hereditary cancer or inherited bone marrow failure disorder. The exception may be subjects expected to have a telomere biology disorder, in whom culture failure and/or extended times in culture can be expected with this protocol, suggesting that protocol modifications and/or considering additional back‐up sample types, such as an additional skin biopsy for immediate DNA extraction without culture, may be necessary. Subjects requiring an urgent HSCT or other germline genetic testing‐directed therapeutic decision may also not be able to wait the additional 28 days. For these cases, genetic testing from peripheral blood or DNA extracted from a skin biopsy sample without culture may be necessary, but a skin biopsy for fibroblast culture could be taken at the same time for confirmation of findings. If a specific genetic variant is already known (e.g., a genetic panel performed on malignant cells for prognostic/therapeutic purposes identified a P/LP variant in a gene causative of a hereditary hematologic disorder but the acquired vs. germline status is unknown), targeted testing of family members may also be helpful in some cases.
Overall, culture failure is a rare event, even among samples shipped for culturing, providing reassurance for healthcare systems expected to utilize external commercial or academic laboratories. Importantly, though, our findings suggest that samples should be shipped on the same day as the biopsy with the goal of initiating the culture as soon as possible to avoid delays of greater than 1 day post‐biopsy to minimize failure events. Also of note, among the 16 samples that failed to culture, repeat biopsy and culture were successful for all five samples in which this was attempted, suggesting that this is a reasonable option for cases in which skin fibroblasts are the ideal sample type (e.g., post allogeneic HSCT). Success of a second culture is especially likely if there were correctable technical factors, such as a delay in initiating the original culture set‐up or use of a nonideal body site, that impacted the first biopsy.
Time from culture initiation to having sufficient cell numbers for DNA extraction was impacted by several clinical and technical factors, such as older age and prior allogeneic HSCT being associated with longer time in culture, and larger size of biopsy being associated with a shorter time in culture. Other factors, such as those with a lymphoid malignancy and those with a heterozygous P/LP variant, both being associated with a shorter time in culture even after adjustment for skin biopsy characteristics, age, and other clinical factors, merit consideration. Differences in types or cumulative doses of chemotherapy or other cytotoxic exposures and/or underlying germline genetic differences could account for the lymphoid malignancy finding.
The shorter time in culture for those with a heterozygous P/LP variant, most of whom had variants in DNA repair‐associated genes (34 of 58, including ATM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, MSH6, PALB2, and PMS2) is of interest. Although 25.7‐ versus 28‐day average times in culture may not significantly impact genetic testing turnaround times, our data add to the literature on success rates and expected time in culture for patients with multiple different inherited disorders. Our data are concordant with prior observations of a longer time in culture for individuals with a telomere biology disorder (Allsopp et al., 1992; Dokal et al., 1992; Harley et al., 1990), but did not replicate longer times in culture previously observed for individuals with P/LP heterozygous SAMD9 (21 days) or compound heterozygous FANCA (median 26 days) variants (Narumi et al., 2016; Pinto et al., 2009). Individual patient factors or culture protocols may contribute to these differences.
From a biological perspective, our data also add to accumulating literature that germline variants in diverse hereditary cancer and bone marrow failure genes impact tissue proliferation rates. We observed the shortest culture times in individuals with P/LP compound heterozygous FANCA (14 days) or heterozygous BRCA1 (16 days), MEN1 (17 days), or BRCA2 (19 days) variants. This observation fits well with recent data showing that germline and acquired loss of function of genes in the Fanconi anemia pathway stimulate proliferation of the epidermis (Ruiz‐Torres et al., 2021). Bao et al. (2020) also demonstrate that germline CHEK2 variants increase the proliferation of hematopoietic cells. Additional research into gene by tissue effects is warranted.
We also found rare but intriguing instances of discordant results between samples from the same subject, both in variants at the subclonal level and at the putative germline level. In select cases, there was an apparent explanation for the discrepancy, such as difficult regions to capture and/or sequence due to pseudogenes or homopolymers. However, in others, the possibilities of new variants arising as an artifact of culturing, as has been seen at the chromosomal level in cancer cytogenetic cultures, or an acquired variant present at a low level in normal skin (Martincorena et al., 2015; Yizhak et al., 2019) being selected for during the culturing process, remain and could lead to a false‐positive germline test result. Repeat skin biopsy and culture from a different skin site were negative for the P/LP variant in three such challenging cases in our series, suggesting that a repeat skin biopsy and/or alternative tissue sample should be tested if the clinical picture does not fit the diagnosis. False‐negative genetic testing results may occur if variants well below the usual 40% are not analyzed and/or confirmed using Sanger or other alternatives in cultured skin fibroblast DNA and/or if blood samples are used as the primary tissue source in subjects with the possibility of somatic reversion or other mechanisms that could favor the loss of the germline allele below the 10% VAF threshold of detection at many germline genetic testing centers.
Our study has limitations. First, all of these samples were grown using a standardized protocol including cytokines. It is possible that alternative protocols may yield different findings. Additionally, we were not able to detect subclonal variants reliably at allelic frequencies below 10%; thus, we cannot rule out the possibility that subclonal variants are present in a larger proportion of skin fibroblast cultures. However, we have evaluated their presence at the VAFs relevant to most germline genetic testing platforms.
In conclusion, cultured skin fibroblasts are a feasible and reliable tissue sample for germline genetic testing in adults with many HMs, cytopenias, and inherited bone marrow failure disorders. Knowledge of predictors of culture failure and time in culture can help inform clinical expectations for individual patients and laboratories/practices beginning to utilize this tissue source. Differences in times in culture by genotype are intriguing and warrant future studies to understand the biological underpinnings. Finally, careful assessment of the clinical phenotype is always warranted when interpreting germline genetic results and confirmatory testing in alternative tissues, skin to blood and vice versa should be considered before making a formal germline diagnosis when the clinical phenotype is discordant with genetic testing results.
AUTHOR CONTRIBUTIONS
Lia DeRoin and Jane E. Churpek designed and coordinated the study, and collected, analyzed, and interpreted the data; Marcela Cavalcante de Andrade Silva contributed to data collection and analysis and figure/table preparation; Kristin Petras, James McElherne, Megan Theissen, Renee Briese, and Carrie Fitzpatrick established the skin fibroblast culture protocol, cultured all skin samples, and maintained culture logs; Lucy A. Godley and Jane E. Churpek contributed to subject recruitment; Lia DeRoin, Marcela Cavalcante de Andrade Silva, Kelly Arndt, Nathaniel Phillips, Pankhuri Wanjari, Hari Prasanna Subramanian, David Montes, Soma Das, Lucy A. Godley, Jeremy Segal, Daniela del Gaudio, Carrie Fitzpatrick, and Jane E. Churpek contributed to genomic screening, analysis, and interpretation of sequencing and bioinformatics data; Lia DeRoin and Jane E. Churpek wrote the manuscript; and all authors edited and approved the final version. Jane E. Churpek is the guarantor of this study and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
CONFLICTS OF INTEREST
The authors report the following conflicts of interest: Honoraria for an educational article, UpToDate, Inc. (J. E. C. and L. A. G.).
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank all of the patients who participated in this study and James P. Zacny, Ph.D. for editorial assistance. Funding sources that supported this study were the American Society of Hematology (Minority Medical Student Award Program [L. D.]), the National Heart, Lung, and Blood Institute (R25HL096383‐09 [L. D.]) and R03 HL145253 [J. E. C.), and CAPES/PDSE/8888.1/188484/2018‐01 (M. C. A. S.).
DeRoin, L. , Silva, M. C. d. A. , Petras, K. , Arndt, K. , Phillips, N. , Wanjari, P. , Subramanian, H. P. , Montes, D. , McElherne, J. , Theissen, M. , Briese, R. , Das, S. , Godley, L. A. , Segal, J. , del Gaudio, D. , Fitzpatrick, C. , & Churpek, J. E. (2022). Feasibility and limitations of cultured skin fibroblasts for germline genetic testing in hematologic disorders. Human Mutation, 43, 950–962. 10.1002/humu.24374
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the supplementary material of this article. The curation of all P/LP variants is detailed in Tables S3 and S6. These variants are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Allsopp, R. C. , Vaziri, H. , Patterson, C. , Goldstein, S. , Younglai, E. V. , Futcher, A. B. , Greider, C. W. , & Harley, C. B. (1992). telomere length predicts replicative capacity of human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America, 89(21), 10114–10118. 10.1073/pnas.89.21.10114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber, D. A. , Orazi, A. , Hasserjian, R. , Thiele, J. , Borowitz, M. J. , Le Beau, M. M. , Bloomfield, C. D. , Cazzola, M. , & Vardiman, J. W. (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 127(20), 2391–2405. 10.1182/blood-2016-03-643544 [DOI] [PubMed] [Google Scholar]
- Bao, E. L. , Nandakumar, S. K. , Liao, X. , Bick, A. G. , Karjalainen, J. , Tabaka, M. , Gan, O. I. , Havulinna, A. S. , Kiiskinen, T. T. J. , Lareau, C. A. , de Lapuente Portilla, A. L. , Li, B. , Emdin, C. , Codd, V. , Nelson, C. P. , Walker, C. J. , Churchhouse, C. , de la Chapelle, A. , Klein, D. E. , … Sankaran, V. G. (2020). Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature, 586(7831), 769–775. 10.1038/s41586-020-2786-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker, L. G. , Harrison, S. M. , & ClinGen Sequence Variant Interpretation Working, G. (2018). The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genetics in Medicine, 20(12), 1687–1688. 10.1038/gim.2018.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D. H. , Below, J. E. , Shimamura, A. , Keel, S. B. , Matsushita, M. , Wolff, J. , Sul, Y. , Bonkowski, E. , Castella, M. , Taniguchi, T. , Nickerson, D. , Papayannopoulou, T. , Bird, T. D. , & Raskind, W. H. (2016). Ataxia‐pancytopenia syndrome is caused by missense mutations in SAMD9L. American Journal of Human Genetics, 98(6), 1146–1158. 10.1016/j.ajhg.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeZern, A. E. , & Churpek, J. E. (2021). Approach to the diagnosis of aplastic anemia. Blood Advances, 5(12), 2660–2671. 10.1182/bloodadvances.2021004345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokal, I. , Bungey, J. , Williamson, P. , Oscier, D. , Hows, J. , & Luzzatto, L. (1992). Dyskeratosis congenita fibroblasts are abnormal and have unbalanced chromosomal rearrangements. Blood, 80(12), 3090–3096. https://www.ncbi.nlm.nih.gov/pubmed/1361371 [PubMed] [Google Scholar]
- Gregory, J. J., Jr. , Wagner, J. E. , Verlander, P. C. , Levran, O. , Batish, S. D. , Eide, C. R. , Steffenhagen, A. , Hirsch, B. , & Auerbach, A. D. (2001). Somatic mosaicism in Fanconi anemia: Evidence of genotypic reversion in lymphohematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2532–2537. 10.1073/pnas.051609898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidugli, L. , Johnson, A. K. , Alkorta‐Aranburu, G. , Nelakuditi, V. , Arndt, K. , Churpek, J. E. , Godley, L. A. , Townsley, D. , Young, N. S. , Fitzpatrick, C. , Del Gaudio, D. , Das, S. , & Li, Z. (2017).Clinical utility of gene panel‐based testing for hereditary myelodysplastic syndrome/acute leukemia predisposition syndromes. Leukemia, 31(5), 1226–1229. 10.1038/leu.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley, C. B. , Futcher, A. B. , & Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345(6274), 458–460. 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- Kadri, S. , Long, B. C. , Mujacic, I. , Zhen, C. J. , Wurst, M. N. , Sharma, S. , McDonald, N. , Niu, N. , Benhamed, S. , Tuteja, J. H. , Seiwert, T. Y. , White, K. P. , McNerney, M. E. , Fitzpatrick, C. , Wang, Y. L. , Furtado, L. V. , & Segal, J. P. (2017). Clinical validation of a next‐generation sequencing genomic oncology panel via cross‐platform benchmarking against established amplicon sequencing assays. Journal of Molecular Diagnostics, 19(1), 43–56. 10.1016/j.jmoldx.2016.07.012 [DOI] [PubMed] [Google Scholar]
- Martincorena, I. , Roshan, A. , Gerstung, M. , Ellis, P. , Van Loo, P. , McLaren, S. , Wedge, D. C. , Fullam, A. , Alexandrov, L. B. , Tubio, J. M. , Stebbings, L. , Menzies, A. , Widaa, S. , Stratton, M. R. , Jones, P. H. , & Campbell, P. J. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science, 348(6237), 880–886. 10.1126/science.aaa6806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumi, S. , Amano, N. , Ishii, T. , Katsumata, N. , Muroya, K. , Adachi, M. , Toyoshima, K. , Tanaka, Y. , Fukuzawa, R. , Miyako, K. , Kinjo, S. , Ohga, S. , Ihara, K. , Inoue, H. , Kinjo, T. , Hara, T. , Kohno, M. , Yamada, S. , Urano, H. , … Hasegawa, T. (2016). SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nature Genetics, 48(7), 792–797. 10.1038/ng.3569 [DOI] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network . (2021). Myelodysplastic Syndromes (version 2.2021). https://www.nccn.org/patients/guidelines/content/PDF/mds-patient.pdf [DOI] [PMC free article] [PubMed]
- National Comprehensive Cancer Network . (2022). Acute Myeloid Leukemia (version 1.2022). https://www.nccn.org/patients/guidelines/content/PDF/aml-patient.pdf
- Padron, E. , Ball, M. C. , Teer, J. K. , Painter, J. S. , Yoder, S. J. , Zhang, C. , Zhang, L. , Moscinski, L. C. , Rollison, D. E. , Gore, S. D. , Bejar, R. , Walter, M. J. , Sekeres, M. A. , Komrokji, R. S. , & Epling‐Burnette, P. K. (2018). Germ line tissues for optimal detection of somatic variants in myelodysplastic syndromes. Blood, 131(21), 2402–2405. 10.1182/blood-2018-01-827881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, F. O. , Leblanc, T. , Chamousset, D. , Le Roux, G. , Brethon, B. , Cassinat, B. , Larghero, J. , de Villartay, J. P. , Stoppa‐Lyonnet, D. , Baruchel, A. , Socie, G. , Gluckman, E. , & Soulier, J. (2009). Diagnosis of Fanconi anemia in patients with bone marrow failure. Haematologica, 94(4), 487–495. 10.3324/haematol.13592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs, E. R. , Andersen, E. F. , Cherry, A. M. , Kantarci, S. , Kearney, H. , Patel, A. , Raca, G. , Ritter, D. I. , South, S. T. , Thorland, E. C. , Pineda‐Alvarez, D. , Aradhya, S. , & Martin, C. L. (2020). Technical standards for the interpretation and reporting of constitutional copy‐number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genetics in Medicine, 22(2), 245–257. 10.1038/s41436-019-0686-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Torres, S. , Brusadelli, M. G. , Witte, D. P. , Wikenheiser‐Brokamp, K. A. , Sauter, S. , Nelson, A. S. , Sertorio, M. , Chlon, T. M. , Lane, A. , Mehta, P. A. , Myers, K. C. , Bedard, M. C. , Pal, B. , Supp, D. M. , Lambert, P. F. , Komurov, K. , Kovacic, M. B. , Davies, S. M. , & Wells, S. I. (2021). Inherited DNA repair defects disrupt the structure and function of human skin. Cell Stem Cell, 28(3), 424–435.e426. 10.1016/j.stem.2020.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Greenblatt, M. S. , Harrison, S. M. , Nussbaum, R. L. , Prabhu, S. A. , Boucher, K. M. , Biesecker, L. G. , & ClinGen Sequence Variant Interpretation Working, G. (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genetics in Medicine, 20(9), 1054–1060. 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawana, K. , Drazer, M. W. , & Churpek, J. E. (2018). Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: Are we there yet? Leukemia, 32(7), 1482–1492. 10.1038/s41375-018-0051-y [DOI] [PubMed] [Google Scholar]
- Tesi, B. , Davidsson, J. , Voss, M. , Rahikkala, E. , Holmes, T. D. , Chiang, S. C. C. , Komulainen‐Ebrahim, J. , Gorcenco, S. , Rundberg Nilsson, A. , Ripperger, T. , Kokkonen, H. , Bryder, D. , Fioretos, T. , Henter, J. I. , Mottonen, M. , Niinimaki, R. , Nilsson, L. , Pronk, C. J. , Puschmann, A. , … Bryceson, Y. T. (2017). Gain‐of‐function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood, 129(16), 2266–2279. 10.1182/blood-2016-10-743302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theda, C. , Hwang, S. H. , Czajko, A. , Loke, Y. J. , Leong, P. , & Craig, J. M. (2018). Quantitation of the cellular content of saliva and buccal swab samples. Scientific Reports, 8(1), 6944. 10.1038/s41598-018-25311-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak, K. , Aguet, F. , Kim, J. , Hess, J. M. , Kubler, K. , Grimsby, J. , Frazer, R. , Zhang, H. , Haradhvala, N. J. , Rosebrock, D. , Livitz, D. , Li, X. , Arich‐Landkof, E. , Shoresh, N. , Stewart, C. , Segre, A. V. , Branton, P. A. , Polak, P. , Ardlie, K. G. , & Getz, G. (2019). RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science, 364(6444). 10.1126/science.aaw0726 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article. The curation of all P/LP variants is detailed in Tables S3 and S6. These variants are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).