

Abstract
Machine learning (ML) is revolutionizing our ability to understand and predict the complex relationships between protein sequence, structure, and function. Predictive sequence-function models are enabling protein engineers to efficiently search sequence space for useful proteins with broad applications in biotechnology. In this review, we highlight recent advances applying machine learning to protein engineering. We discuss supervised learning methods that infer the sequence-function mapping from experimental data and new sequence representation strategies for data-efficient modeling. We then describe the various ways ML can be incorporated into protein engineering workflows, including purely in silico searches, machine learning-assisted directed evolution, and generative models that learn the underlying distribution of protein function in sequence space. ML-driven protein engineering will become increasingly powerful with continued advances in high-throughput data generation, data science, and deep learning.
Keywords: Machine learning, protein engineering, deep neural networks, protein fitness, landscapes
Introduction
Evolution has shaped proteins to perform complex chemical and biological functions with exceptional proficiency, accuracy, and specificity. These naturally occurring proteins represent a huge potential to solve some of the world’s most pressing problems. Protein engineering aims to realize this potential by altering protein functions for applications in biotechnology, industry, and medicine [1–3]. Amino acid sequences encode the overall function and properties of proteins. The relationship between protein sequence and function can be imagined by the sequence-function ‘fitness landscape’ wherein protein sequences are mapped to a corresponding fitness value that represents a measurable protein property like catalytic activity or thermostability [4]. Protein engineering aims to search this landscape for high fitness sequences.
Directed evolution navigates this landscape through iterative rounds of mutation, high-throughput functional assays, and selection to ‘walk’ uphill toward more fit sequences. Despite its widespread utility and success, directed evolution is resource- and time-intensive, and the search process is largely ignorant of the underlying sequence-function landscape. This local search is inefficient and can ultimately limit fitness achieved in a protein engineering experiment. Machine learning (ML) has recently emerged as a highly effective method for inferring the sequence-function landscape [5–7]. ML-guided protein engineering can discover highly fit sequences more efficiently and with less experimental screening than traditional directed evolution [8]. In this review, we highlight recent approaches that leverage ML to explore, exploit, and expand functional sequence space to engineer proteins for improved function.
Supervised learning for modeling the protein-fitness landscape
Rational protein engineering requires a detailed, quantitative understanding of the complex biophysical interactions underlying protein sequence and function. Machine learning can infer these interactions from experimental data to generate predictive models for protein design. While a diverse range of ML models are suitable for this task, here we focus our discussion on recent advances in deep neural networks due to their state-of-the-art performance and flexibility. We also discuss the importance of model evaluation and how this influences our ability to compare different modeling approaches.
Supervised deep neural networks combine multiple layers of nonlinear functions to learn highly complex input-output relationships. There are several commonly used architectures capable of modeling the sequence-function landscape, and each one carries inductive biases that make different assumptions about the relationship. Common architectures include multi-layer perceptrons (MLPs) that learn nonlinear interactions between inputs, convolutional neural networks (CNNs) that consider information within a local “receptive field” in the input space, recurrent neural networks (RNNs) that process information in a sequential manner, and transformers that can learn specific long-range interactions between inputs using attention mechanisms. Both CNNs and RNNs outperform baseline linear models for protein function prediction [9–12]. CNNs also show promise for predicting the effects of mutations that were not experimentally tested. This capacity may be useful when characterizing mutations is costly or expanding the set of beneficial mutations that can be used to design a protein [9,13].
Supervised deep learning relies on large sequence-function datasets for model training and evaluation, but many proteins have limited (10s–100s) functional data due to the lack of high-throughput functional assays. One effort to reduce the data requirement for training is to apply more informative protein representations to supervised learning tasks [14]. Rather than simply representing a protein as a sequence of amino acids, which is very high dimensional, it’s possible to devise other protein representations that may capture key physiochemical properties such as total charge, evolutionary features such as site conservation, and structural characteristics such as solvent accessible surface areas. A carefully designed set of features can distill essential aspects of protein function and enable learning with less data [15,16]. A more recent and data-driven approach uses unsupervised machine learning to learn informative protein representations from the large amounts of unlabeled sequence data available in databases such as UniProt [17–22]. These models effectively learn condensed representations of protein sequences by identifying correlations and patterns within protein families. These low-dimensional protein representations can be used as inputs for supervised models to improve predictive accuracy and enable learning from smaller sequence-function data sets [8,23]. Variational autoencoders (VAEs), long short-term memory (LSTM) RNNs, and transformers trained on natural sequence information consistently show top performance in learning effective protein representations for supervised ML [19,23–26].
The potential for machine learning in protein engineering has resulted in rapid development of new ML models and strategies, which demand rigorous performance benchmarks and metrics for model comparison. Differences in datasets, train-test splits, and evaluation metrics make it non-trivial to compare ML modeling approaches as these factors all impact model performance. Recent efforts to standardize evaluation of learned representations aim to address these issues by outlining standard datasets, splits, and tasks for assessing representation learning models [19], but evaluation of supervised models has yet to be standardized. An important factor in standardizing evaluation of supervised models is assessing model performance. Models are often evaluated by their average prediction error, but there are more relevant metrics for evaluating models for protein engineering applications. To address this, Mater et al. [13] devised five metrics to evaluate a model’s ability to handle extrapolation, epistasis, sparse data, and variability in sequence length. Incorporating these kinds of tasks into a standard benchmark for supervised ML models will allow protein engineers to better evaluate ML models and find a model that best suits their needs. Finally, a common goal of recent supervised learning papers has been to create models and training protocols that are more accessible for comparing ML approaches and for use by non-ML experts [9,11,27–29]. As the field of ML-based protein engineering expands, it is important to make these tools readily accessible to all.
In silico optimization of the protein fitness landscape
An accurate supervised sequence-function model can be used to guide the search through sequence space for new and improved proteins (Figure 1). These models can extrapolate the learned relationship beyond the training set and identify highly fit sequences. We refer to this approach as “in silico optimization” because candidate proteins are identified by optimizing the sequence-function model.
Figure 1.
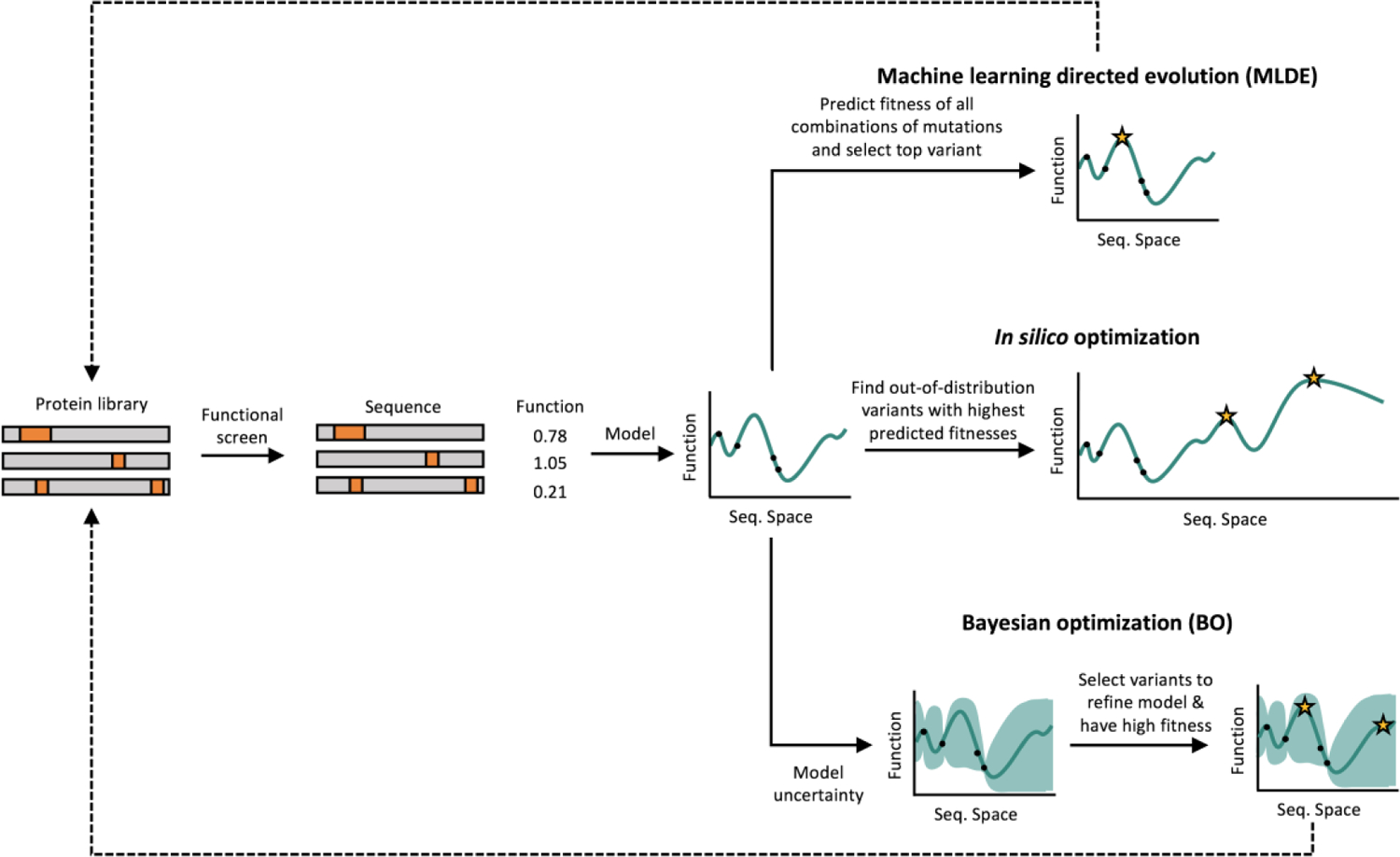
Machine learning-driven protein optimization strategies typically require an initial protein sequence library, typically created using error-prone PCR, site-saturated mutagenesis, or chimeragenesis. This protein sequence library is screened experimentally to determine a fitness value for each protein variant. Protein sequence-function data is modeled using supervised machine learning. The model can then be used to design improved variants in multiple ways. In machine learning directed evolution (MLDE), the fitness for other combinations of mutations in a combinatorial library are predicted and the best variant is selected for further rounds of mutagenesis, screening, and modeling. In silico optimization, however, uses optimization strategies to find highly fit protein variants far from the initial training library in the larger protein-sequence space. Bayesian optimization involves iterative rounds of mutagenesis, screening, and learning to maximize protein fitness over fewer rounds of experimental characterization by proposing protein variants that will help refine the model and have high fitness.
In silico optimization strategies often involve landscape search heuristics such as hill climbing, simulated annealing, and genetic algorithms to identify sequences that are predicted to have high fitness values [9,12,30]. We recently applied a hill climbing approach to design GB1 sequences based on neural network models trained on deep mutational scanning data. We found the top designed variant with 10 mutations from wildtype GB1 was stable when expressed in E. coli and exhibited substantially increased binding affinity for IgG. It is also important to consider the diversity of candidate sequences in order to increase the independence of designs. Diversification strategies attempt to maximize the predicted fitness of designs while ensuring that they occupy distinct regions of the landscape [30,31].
In silico optimization can also be applied in small data settings using learned low-dimensional protein sequence representations [23]. The learned representations distill information pertinent to function beyond just sequence information alone and enable ML-based protein engineering in remarkably small data regimes. eUniRep is a representation model trained on 24 million unlabeled protein sequences and was used in a supervised setting to design improved GFP variants with fewer than 100 labeled sequence-function examples. The eUniRep representation appears to guide design proposals away from non-functional sequence space. Embeddings may therefore be useful in scenarios where ML algorithms are attempting significant extrapolation beyond the training set. Despite these recent successes, there are still many open questions in the field related to the best sequence representations, supervised models, and limits of extrapolation.
Active learning to iteratively search protein sequence space
The in silico optimization approach described above learns from a sequence-function dataset to design improved proteins in a one-step process. Active machine learning takes a different approach, where an iterative design-test-learn cycle is implemented to make the search through sequence space more efficient. While this approach requires multiple rounds of sequence proposal and refining the model with experimental data, the total screening burden is drastically reduced in comparison to both traditional directed evolution and most in silico optimization methods.
Machine learning-assisted directed evolution (MLDE) is a new approach that supplements traditional directed evolution with an underlying sequence-function model to effectively screen larger regions of sequence space [8,32]. MLDE begins with a combinatorial site-saturation mutagenesis library from which a small number of variants are screened for a function of interest. The resulting sequence-function data is used to train an ML model that predicts the function for all remaining variants in the combinatorial space. The top variant’s mutations are identified and fixed to provide the parent sequence for the next round of MLDE. By repeating this process, it’s possible to efficiently traverse large swathes of sequence space to find the optimal protein. Recent work has found that using biophysical, structural, and evolutionary information to filter the training data for diverse or highly fit variants helped achieve the maximum fitness more often [8,33]. MLDE augments the power of traditional directed evolution by providing a guided-walk through sequence space while reducing overall screening burden and accounting for epistasis.
Bayesian optimization (BO) is another approach that enables the engineering of proteins with relatively few experimental measurements. BO employs an iterative design-test-learn cycle where a model is trained on sequence-function data and used to propose new protein sequences to test that are simultaneously informative for the model and are predicted to have high fitness. Experimentally testing these sequences refines the model and also identifies new fitness peaks [34]. Gaussian process models have long been the standard for BO of the protein fitness landscape due to their ability to explicitly model uncertainty [35,36]. We recently applied BO to engineer acyl-ACP reductases for improved in vivo fatty alcohol production [37]. Over the course of ten design-test-learn cycles and less than 100 experimental measurements, we were able to engineer enzymes that produce over two-fold more fatty alcohols than the starting natural sequences. However, gaussian processes are relatively simple models and are limited in their ability to learn highly complex relationships compared to deep learning models. Ensembles of deep learning models, such as CNNs and RNNs, can be used to estimate model uncertainty for BO and improve the maximum fitness achieved relative to Gaussian processes-based optimization [38]. Thus, combining active learning techniques with state-of-the-art supervised ML models is a promising approach for engineering proteins that cannot be screened in high-throughput.
Targeted exploration of protein sequence space with generative modeling
Supervised methods are powerful for inferring the protein landscape from labeled sequence-function data; however, there are millions of sequences in databases such as UniProt that lack functional data [22]. These data are referred to as “unlabeled” because they have unknown functional characteristics, but they still provide valuable clues regarding the underlying landscape. Generative models learn the distribution of unlabeled protein sequence data with the assumption that any sequence falling within this distribution will exhibit similar properties to the training data. By proposing sequences from this distribution, generative models can sample novel sequences that “look” and function like natural sequences [39,40]. However, it’s difficult for these generative models to propose designs with improved properties without explicit fitness or function information. Here we discuss modeling strategies to direct generative models toward proteins with improved function (Figure 2). For a comprehensive review on the broad applications of generative modeling in protein engineering, we refer readers to the recent review by Wu et al. [5].
Figure 2.
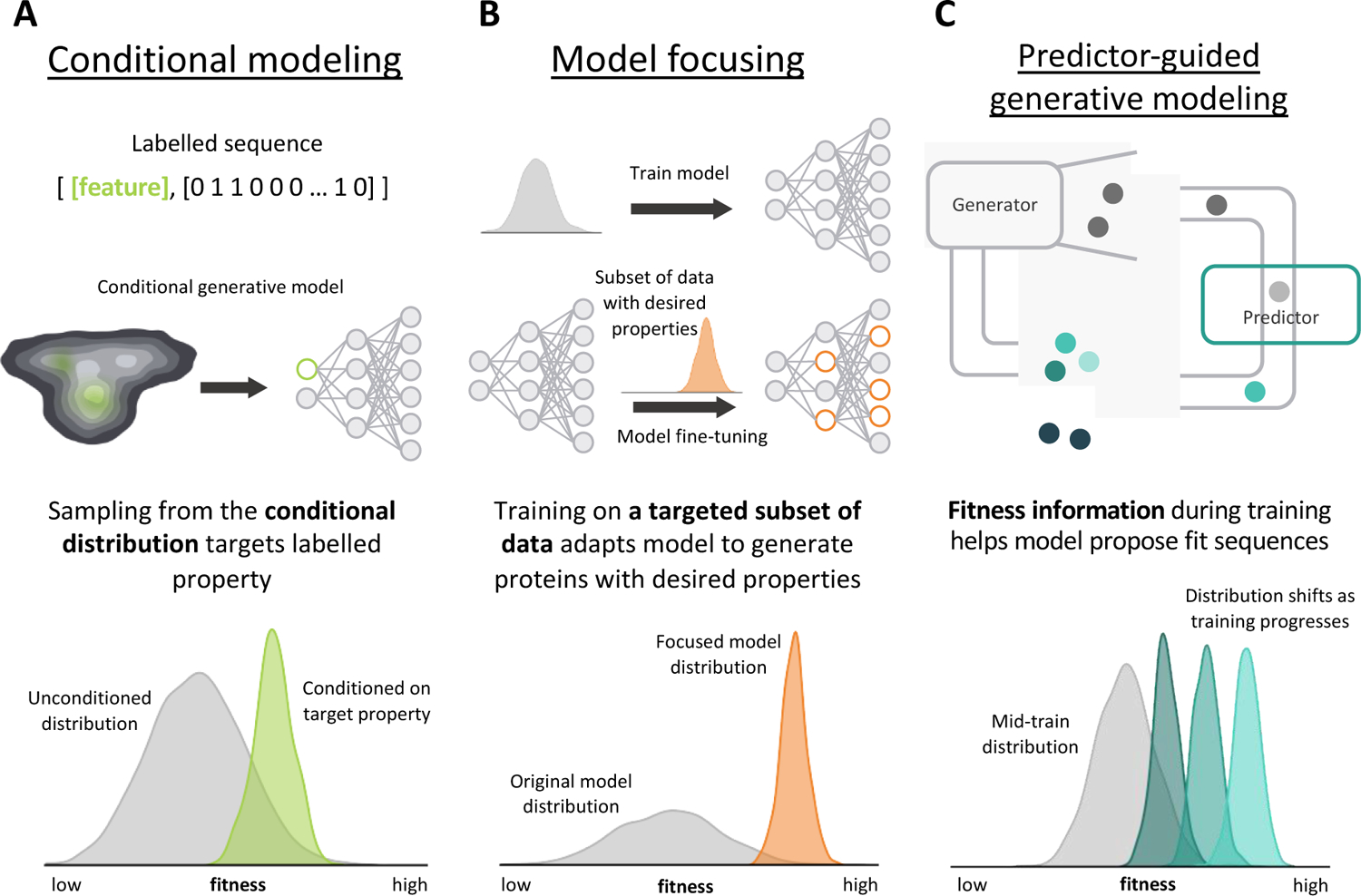
Strategies for protein engineering with generative models. A) Conditional modeling labels training examples with a feature vector to identify sequences with a target attribute. Conditioning on a specific attribute generates a posterior distribution that preferentially samples proteins with the target attribute. B) Model focusing adapts a general model to a specialized task with additional training on a targeted subset of data. Model focusing biases the model to generate proteins with properties that resemble the targeted training set. C) Predictor-guided generative modeling incorporates fitness information into generator training, which can be provided by any model that predicts a property. As generator training progresses, the model proposes more highly fit examples.
Conditional generative models can be used to bias the sampling process to engineer proteins with specific properties. These models learn a probability distribution over sequences and a set of defined protein attributes; this distribution can be conditioned on a desired attribute to enable controlled generation of proteins with the target attribute (Figure 2a) [41–44]. This strategy has been used on a diverse set of protein engineering tasks, including engineering luciferases with specified solubility levels [42]. Hawkins-Hooker et al. trained a conditional VAE on luciferase sequences labelled with a low, medium, or high solubility level. Conditioning on a particular solubility resulted in novel luciferases that displayed the conditioned solubility while retaining native luciferase activity. While this study targets one attribute, multiple attributes can be conditioned on simultaneously to generate proteins with many desired attributes, and provides a flexible modeling framework suited to diverse applications [45,46].
Another strategy to bias generative models toward improved sequences is model focusing, where a general model’s parameters are fine-tuned on a subset of data with a desired property (Figure 2b). The resulting model is biased to sample proteins that closely match the characteristics of the tuning data. For example, Amimeur et al. [47] trained a generative adversarial network (GAN) to produce human-like antibodies. They then fine-tuned this model’s parameters by focusing the training on a subset of sequences with desirable properties, such as decreased immunogenicity, that biased the model to design sequences with the target property.
Other generative protein engineering strategies explicitly incorporate predicted fitness into a generator’s training process such that over time, the model is taught to propose more highly fit sequences (Figure 2c). These predictions can be from supervised learning models or any other biophysical or structure-based model. Feedback GAN (fbGAN) couples a GAN architecture with a predictive model that predicts the fitness of generated sequences during the training process [48]. Fit sequences are identified and fed back into the model’s training data. While fbGAN modifies the training data, the Deep Exploration Network (DEN) developed by Linder et al. [30] ties the predicted function of generated sequences directly into the generator’s cost function. The DEN performed better than other state-of-the-art methods, including fbGAN, across a variety of applications, including the design of highly fit and diverse avGFP variants. The DEN also incorporates an explicit diversity and likelihood metric to efficiently search multiple regions of sequence space for fit variants with high confidence for success. Generative models can find functional protein sequences far removed from the training data – coupling this explorative power with functional information can guide this exploration toward fit proteins for effective protein engineering.
Conclusion
Deep learning is revolutionizing our ability to decipher the complex relationships between protein sequence, structure, and function [49–51]. Predictive models of the sequence-function landscape are enabling protein engineers to efficiently and reliably search sequence space for new and useful proteins. Approaches that minimize experimental data requirements, including information-rich protein representations and integrated design-test-learn cycles, will push the boundaries of protein engineering to functions that rival the performance and complexity of natural systems. This new frontier of biomolecular engineering will address global grand challenges in human health, agriculture, the environment, and energy.
Acknowledgements
This work was funded by the National Institutes of Health (R35GM119854).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV: Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol 2006, 24:198–204. [DOI] [PubMed] [Google Scholar]
- 2.Wu Z, Yang KK, Liszka MJ, Lee A, Batzilla A, Wernick D, Weiner DP, Arnold FH: Signal Peptides Generated by Attention-Based Neural Networks. ACS Synth Biol 2020, 9:2154–2161. [DOI] [PubMed] [Google Scholar]
- 3.Chevalier A, Silva D-A, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam K-H, Yao G, et al. : Massively parallel de novo protein design for targeted therapeutics. Nature 2017, 550:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romero PA, Arnold FH: Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol 2009, 10:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z, Johnston KE, Arnold FH, Yang KK: Protein sequence design with deep generative models. Current Opinion in Chemical Biology 2021, 65:18–27. [DOI] [PubMed] [Google Scholar]
- 6.Wittmann BJ, Johnston KE, Wu Z, Arnold FH: Advances in machine learning for directed evolution. Current Opinion in Structural Biology 2021, 69:11–18. [DOI] [PubMed] [Google Scholar]
- 7.Yang KK, Wu Z, Arnold FH: Machine-learning-guided directed evolution for protein engineering. Nat Methods 2019, 16:687–694. [DOI] [PubMed] [Google Scholar]
- 8.Wittmann BJ, Yue Y, Arnold FH: Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Systems 2021, 12:1026–1045.e7. [DOI] [PubMed] [Google Scholar]
- 9.Gelman S, Fahlberg SA, Heinzelman P, Romero PA, Gitter A: Neural networks to learn protein sequence–function relationships from deep mutational scanning data. Proc Natl Acad Sci USA 2021, 118:e2104878118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Verma D, Sheridan RP, Liaw A, Ma J, Marshall NM, McIntosh J, Sherer EC, Svetnik V, Johnston JM: Deep Dive into Machine Learning Models for Protein Engineering. J Chem Inf Model 2020, 60:2773–2790. [DOI] [PubMed] [Google Scholar]
- 11.Griffith D, Holehouse AS: PARROT is a flexible recurrent neural network framework for analysis of large protein datasets. eLife 2021, 10:e70576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryant DH, Bashir A, Sinai S, Jain NK, Ogden PJ, Riley PF, Church GM, Colwell LJ, Kelsic ED: Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 2021, 39:691–696. [DOI] [PubMed] [Google Scholar]
- 13.Mater AC, Sandhu M, Jackson C: The NK Landscape as a Versatile Benchmark for Machine Learning Driven Protein Engineering. bioRxiv 2020, doi: 10.1101/2020.09.30.319780. [DOI] [Google Scholar]
- 14.Bepler T, Berger B: Learning the protein language: Evolution, structure, and function. Cell Systems 2021, 12:654–669.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlin DA, Caster RW, Wang X, Betzenderfer SA, Chen CX, Duong VM, Ryklansky CV, Alpekin A, Beaumont N, Kapoor H, et al. : Kinetic Characterization of 100 Glycoside Hydrolase Mutants Enables the Discovery of Structural Features Correlated with Kinetic Constants. PLOS ONE 2016, 11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A, et al. : Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 2017, 357:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alley EC, Khimulya G, Biswas S, AlQuraishi M, Church GM: Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 2019, 16:1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinzinger M, Elnaggar A, Wang Y, Dallago C, Nechaev D, Matthes F, Rost B: Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics 2019, 20:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao R, Bhattacharya N, Thomas N, Duan Y, Chen X, Canny J, Abbeel P, Song YS: Evaluating Protein Transfer Learning with TAPE. Adv Neural Inf Process Syst 2019, 32:9689–9701. [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Y, Jiang G, Yu T, Liu Y, Vo L, Ding H, Su Y, Qian WW, Zhao H, Peng J: ECNet is an evolutionary context-integrated deep learning framework for protein engineering. Nat Commun 2021, 12:5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang KK, Wu Z, Bedbrook CN, Arnold FH: Learned protein embeddings for machine learning. Bioinformatics 2018, 34:2642–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The UniProt Consortium: The Universal Protein Resource (UniProt). Nucleic Acids Research 2007, 36:D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM: Low-N protein engineering with data-efficient deep learning. Nat Methods 2021, 18:389–396. [DOI] [PubMed] [Google Scholar]
- 24.Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, Guo D, Ott M, Zitnick CL, Ma J, et al. : Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci USA 2021, 118:e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu C, Nisonoff H, Fannjiang C, Listgarten J: Combining evolutionary and assay-labelled data for protein fitness prediction. bioRxiv 2021, doi: 10.1101/2021.03.28.437402. [DOI] [PubMed] [Google Scholar]
- 26.Ding X, Zou Z, Brooks CL III: Deciphering protein evolution and fitness landscapes with latent space models. Nat Commun 2019, 10:5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siedhoff NE, Illig A-M, Schwaneberg U, Davari MD: PyPEF—An Integrated Framework for Data-Driven Protein Engineering. J Chem Inf Model 2021, 61:3463–3476. [DOI] [PubMed] [Google Scholar]
- 28.Chen KM, Cofer EM, Zhou J, Troyanskaya OG: Selene: a PyTorch-based deep learning library for sequence data. Nat Methods 2019, 16:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Favor A, Jayapurna I: Evaluating eUniRep and other protein feature representations for in silico directed evolution. Authorea 2020, doi: 10.22541/au.159683529.96283070. [DOI] [Google Scholar]
- 30.Linder J, Bogard N, Rosenberg AB, Seelig G: A Generative Neural Network for Maximizing Fitness and Diversity of Synthetic DNA and Protein Sequences. Cell Systems 2020, 11:49–62.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu D, Brookes DH, Busia A, Carneiro A, Fannjiang C, Popova G, Shin D, Chang EF, Nowakowski TJ, Listgarten J, et al. : Machine learning-based library design improves packaging and diversity of adeno-associated virus (AAV) libraries. bioRxiv 2021, doi: 10.1101/2021.11.02.467003. [DOI] [Google Scholar]
- 32.Wu Z, Kan SBJ, Lewis RD, Wittmann BJ, Arnold FH: Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc Natl Acad Sci USA 2019, 116:8852–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu Y, Hu J, Wei G-W: Cluster learning-assisted directed evolution. Nat Comput Sci 2021, 1:809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hie BL, Yang KK: Adaptive machine learning for protein engineering. Current Opinion in Structural Biology 2022, 72:145–152. [DOI] [PubMed] [Google Scholar]
- 35.Romero PA, Krause A, Arnold FH: Navigating the protein fitness landscape with Gaussian processes. Proceedings of the National Academy of Sciences 2013, 110:E193–E201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedbrook CN, Yang KK, Rice AJ, Gradinaru V, Arnold FH: Machine learning to design integral membrane channelrhodopsins for efficient eukaryotic expression and plasma membrane localization. PLoS Comput Biol 2017, 13:e1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenhalgh JC, Fahlberg SA, Pfleger BF, Romero PA: Machine learning-guided acyl-ACP reductase engineering for improved in vivo fatty alcohol production. Nat Commun 2021, 12:5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruver N, Stanton S, Kirichenko P, Finzi M, Maffettone P, Myers V, Delaney E, Greenside P, Wilson AG: Effective Surrogate Models for Protein Design with Bayesian Optimization. In ICML 2021 Workshop on Computational Biology; 2021. [Google Scholar]
- 39.Repecka D, Jauniskis V, Karpus L, Rembeza E, Rokaitis I, Zrimec J, Poviloniene S, Laurynenas A, Viknander S, Abuajwa W, et al. : Expanding functional protein sequence spaces using generative adversarial networks. Nat Mach Intell 2021, 3:324–333. [Google Scholar]
- 40.Madani A, Krause B, Greene ER, Subramanian S, Mohr BP, Holton JM, Olmos JL, Xiong C, Sun ZZ, Socher R, et al. : Deep neural language modeling enables functional protein generation across families. bioRxiv 2021, doi: 10.1101/2021.07.18.452833. [DOI] [Google Scholar]
- 41.Sohn K, Lee H, Yan X: Learning Structured Output Representation using Deep Conditional Generative Models. In Advances in Neural Information Processing Systems. Edited by Cortes C, Lawrence N, Lee D, Sugiyama M, Garnett R. Curran Associates, Inc.; 2015. [Google Scholar]
- 42.Hawkins-Hooker A, Depardieu F, Baur S, Couairon G, Chen A, Bikard D: Generating functional protein variants with variational autoencoders. PLoS Comput Biol 2021, 17:e1008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brookes DH, Park H, Listgarten J: Conditioning by adaptive sampling for robust design. arXiv 2021. arXiv:1901.10060. [Google Scholar]
- 44.Chan A, Madani A, Krause B, Naik N: Deep Extrapolation for Attribute-Enhanced Generation. arXiv 2021. arXiv:2107.02968. [Google Scholar]
- 45.Karimi M, Zhu S, Cao Y, Shen Y: De Novo Protein Design for Novel Folds Using Guided Conditional Wasserstein Generative Adversarial Networks. J Chem Inf Model 2020, 60:5667–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das P, Sercu T, Wadhawan K, Padhi I, Gehrmann S, Cipcigan F, Chenthamarakshan V, Strobelt H, dos Santos C, Chen P-Y, et al. : Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat Biomed Eng 2021, 5:613–623. [DOI] [PubMed] [Google Scholar]
- 47.Amimeur T, Shaver JM, Ketchem RR, Taylor JA, Clark RH, Smith J, Van Citters D, Siska CC, Smidt P, Sprague M, et al. : Designing Feature-Controlled Humanoid Antibody Discovery Libraries Using Generative Adversarial Networks. bioRxiv 2020, doi: 10.1101/2020.04.12.024844. [DOI] [Google Scholar]
- 48.Gupta A, Zou J: Feedback GAN for DNA optimizes protein functions. Nat Mach Intell 2019, 1:105–111. [Google Scholar]
- 49.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. : Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eguchi RR, Anand N, Choe CA, Huang P-S: IG-VAE: Generative Modeling of Immunoglobulin Proteins by Direct 3D Coordinate Generation. bioRxiv 2020, doi: 10.1101/2020.08.07.242347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norn C, Wicky BIM, Juergens D, Liu S, Kim D, Tischer D, Koepnick B, Anishchenko I, Foldit Players, Baker D, et al. : Protein sequence design by conformational landscape optimization. Proc Natl Acad Sci USA 2021, 118:e2017228118. [DOI] [PMC free article] [PubMed] [Google Scholar]