

Abstract
Androgen receptor (AR) pathway inhibitors are the mainstay treatment for advanced prostate cancer, but resistance to therapy is common. Here, we used a CRISPR activation screen in metastatic castration-sensitive prostate cancer cells to identify genes that promote resistance to AR inhibitors. Activation of the TGF-β target gene PRRX2 promoted enzalutamide resistance. PRRX2 expression was highest in double-negative prostate cancer (DNPC), which lack AR signaling and neuroendocrine differentiation, and a PRRX2-related gene signature identified a subset of DNPC patients with reduced overall survival. PRRX2-expressing cells showed alterations in the CDK4/6/Rb/E2F and BCL2 pathways. Accordingly, treatment with CDK4/6 and BCL2 inhibitors sensitized PRRX2-expressing, castration-resistant tumors to enzalutamide. Overall, PRRX2 was identified as a driver of enzalutamide resistance. The PRRX2 signature merits investigation as a biomarker of enzalutamide resistance in prostate cancer that could be reversed with CDK4/6 and BCL2 inhibitors.
Keywords: Enzalutamide, PRRX2, CRISPRa screen, DNPC, TGF-β signaling, CDK4/6, BCL2
Introduction
The androgen receptor (AR) is the single most important therapeutic target in prostate cancer (PC). Hence, androgen-deprivation therapy (ADT) and potent AR pathway inhibitors (ARPI) such as enzalutamide (ENZ) are widely used to treat this disease (1). However, the majority of patients eventually develop resistance, at which point therapeutic options are scarce.
Most of the mechanisms underlying ARPI resistance rely on re-activation of the AR pathway. Therefore, the development of new and more potent anti-AR inhibitors is an active field of research (2). However, a growing number of patients develop alterations in pathways that bypass the need for AR activation, giving rise to androgen-independent (AI) ARPI-resistant neuroendocrine (NEPC) or double negative PC (DNPC) (3, 4). Furthermore, recent evidence suggests that ENZ treatment itself can select AR low/indifferent pre-existing clones, promote a lineage switch from luminal to basal phenotype or activate certain oncogenic pathways that contribute to disease progression (5, 6).
To identify genes that confer enzalutamide resistance, which may inform new therapeutic strategies, we used an in vitro genome-wide CRISPR activation (CRISPRa) screen in the androgen-sensitive LNCaP cell line. In addition, our cell line had an AR activity reporter (ARE-GFP) which allowed for classification of the population of enriched sgRNAs between those with androgen-dependent (AD) versus androgen indenendent (AI) mechanisms.
Our screen identified the Paired Related Homeobox-2 (PRRX2) transcription factor as one of the top hits. PRRX2 is a target of the TGF-β pathway and helps mediate the epithelial to mesenchymal transition (EMT) in breast cancer (7). In this study, we show that PRRX2 is an oncogene in PC and that PRRX2 over-expression mediates ENZ resistance, which can be overcome via BCL2 and CDK4/6 inhibition.
Materials and Methods
Cell lines
LNCaP, 22RV1 and DU145 cells were obtained from the ATCC. LNRII cells were a generous gift from Dr. Jindan Yu at Northwestern University. All cells were grown in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic solution. For assays in charcoal stripped medium (CSS), cells were first hormone starved in RPMI 1640 media without Phenol red supplemented with 10% charcoal stripped FBS and 1% penicillin/streptomycin for 72h before start of assay. LAPC9-AI cells were a generous gift from Dr. Dean Tang group at Roswell Park. These cells were maintained in castrated mice. All cells were verified as mycoplasma free and genetically authenticated by ATCC.
CRISPRa screen
For the CRISPRa screen we used the Calabrese library Set A which contains 3 different sgRNAs to activate each gene (8). Briefly, LNCaP cells were transduced with a GFP reporter under the control of an androgen responsive element (ARE) (9). Then they were transduced with the dCas9-VP64-Blast and with the PXPR_502 vector for sgRNA expression at low MOI. Cells were then treated with DMSO (Veh) or ENZ (25μM) during 7.5 or 9 weeks. Cells at 7.5 weeks were sorted into GFP(+) or GFP (−) using FACS. DNA was extracted, sequenced and analyzed. For details see supplementary materials and methods.
sgRNA validation
To generate stable LNCaP-sgPRRX2 cells, we cloned the individual sgPRRX2 (Supplementary Table 1) into the PXPR_502 (Addgene #61425) backbone as described (8). For details, see supplementary materials and methods.
Organoid assays
To generate organoids, fresh tumors were shredded mechanically and then digested with collagenase (Gibco, 17018) in complete RPMI 1640 media for 2 h at 37 °C. Then, they were incubated with Trypsin for 5 min at 37 °C. Digested tissues were then treated with DNase I (Sigma,), and filtered with a 40 μm filter to obtain single cells. Cells were then frozen or used for organoid culture. For details, see supplementary materials and methods.
Mice experiments
NSG mice (Jackson Laboratory) used in this study were housed in a pathogen-free animal barrier facility. When mice were 6–8 weeks old, they were injected subcutaneously with different cell lines according to experiment. All experiments and procedures were performed in compliance with ethical regulations and the approval of the Northwestern University Institutional Animal Care and Use Committee. For details, see supplementary materials and methods.
In vivo drug treatments
ENZ (Axon Chemistry, 1613) 10 mg/kg or its vehicle (corn oil) were injected via intraperitoneal injection (IP) 3 times a week. ENZ was dissolved in DMSO (2.5%) and corn oil.
Veneto (MedChem Express, HY-15531) was prepared in DMSO (5%), Tween 80 (5%), PEG300 (40%) and PBS (50%). 1M NaOH was added to adjust the pH to 7 and help solubilize the compound. Veneto 100 mg/kg or its vehicle were given via oral gavage (P.O) 5 days on and 2 days off.
Palbo (MedChem Express, HY-50767A) was dissolved in 50 mmol/L sodium lactate pH 4, and was given via oral gavage 5 days on 2 days off. To avoid an interaction between Veneto and Palbo were administered with at least a 4 hour difference.
RNA-seq
LNCaP cells were treated with DMSO control or ENZ 5 μM for 7 days. Three biological replicates per condition were used for this experiment. For details, see supplementary materials and methods.
ATAC-seq
For ATAC-seq experiment cells were treated with ENZ (5μM) or DMSO for 4 days. Two replicates of each conditions were used for this experiment. For details, see supplementary materials and methods.
Human prostate cancer datasets
For information regarding human prostate cancer datasets see supplementary materials and methods.
Gene enrichment analysis
Gene enrichment analysis was performed using the Enrichr program (10–12). See supplementary materials and methods for detail information.
Hierarchical clustering and PRRX2 signature
For hierarchical clustering and similarity matrix construction an input matrix with gene expression values of AR, NEPC and PRRX2 signature genes was inputted into Clustergrammer (13).
Similarity matrix row distance was calculated using 1 - cosine-distance. For patient hierarchical clustering, the rows and columns were clustered using the cosine distance and average linkage parameters.
For more information, see supplementary materials and methods.
PRRX2, AR and NEPC scores
To calculate the different scores we downloaded the expression of the AR, PRRX2 and NEPC signature genes from cBioPortal. We then calculated the absolute value of the expression of each gene. Subsequently, we ranked the gene expression from 1 to n, where n = Number of patients. The highest rank was given to the patient with the highest expression of the gene. To obtain the final score we added the ranks of the genes with a z-score > 0 and subtracted the ones with z-score <0.
To classify ARPC, NEPC, DNPC and ARPC/NEPC populations from the SU2C 2019 in dataset from Fig. 5A, we normalized the scores from 0–1 and arbitrarily set up a threshold of 0.5. Patients with AR score and NEPC scores < 0.5 were classified as ARPC (n=85). Patients with NEPC score > 0.5 and AR score < 0.5 were classified within the NEPC group (n=22). DNPC patients had both AR score and NEPC scores <0.5 (n=100). Finally, NEPC/AR patients had AR and NEPC scores >0.5 (n=5).
Fig. 5. PRRX2 expression and a PRRX2 signature are elevated in DNPC.
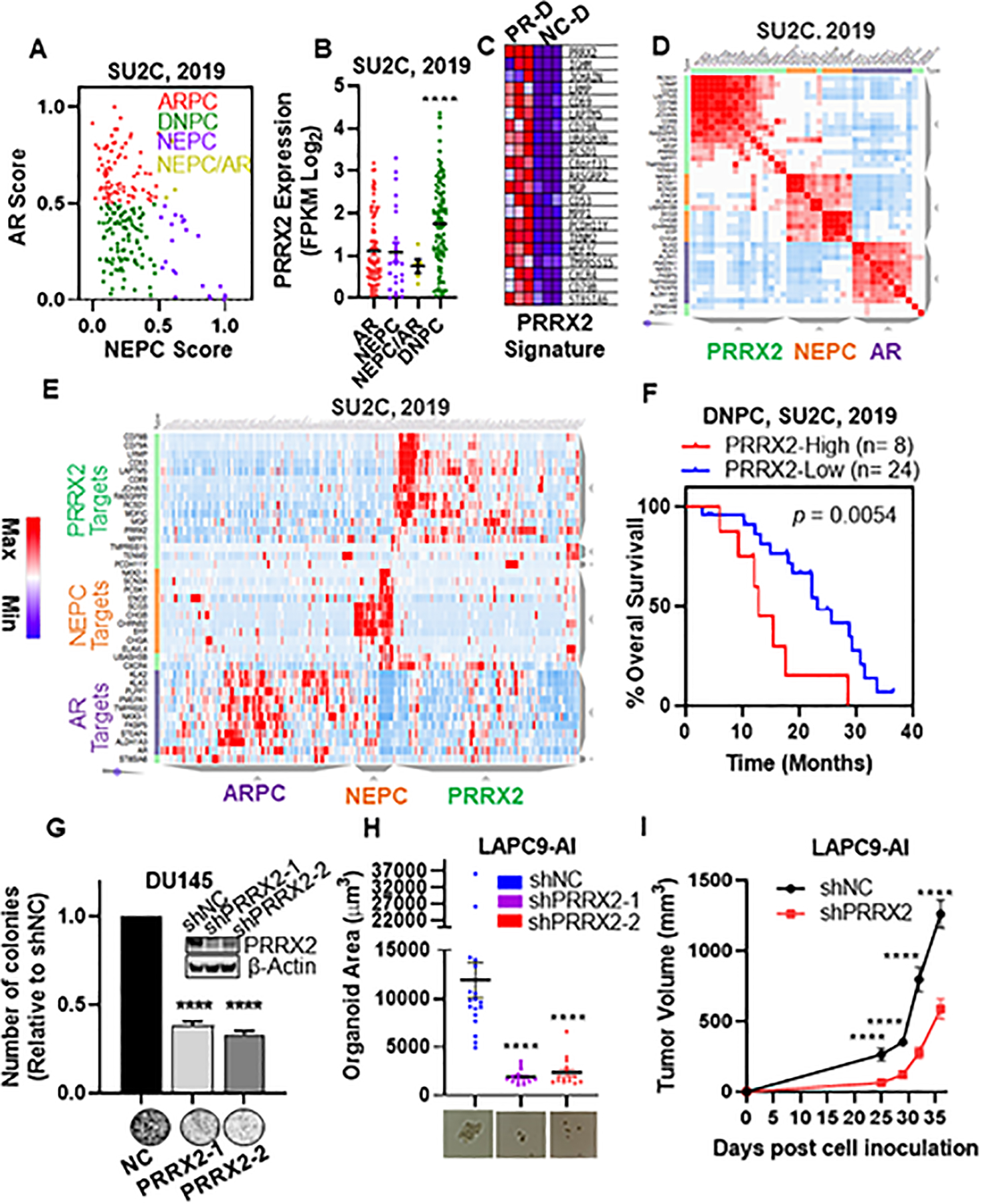
A) Scatter plot of AR and NEPC scores within the SU2C patient dataset. ARPC defined as AR Score < 0.5 and NEPC Score <0.5 (n=85). NEPC defined as NEPC Score > 0.5, AR Score < 0.5 (n=22). DNPC defined as AR score < 0.5 and NEPC score < 0.5 (n=71). ARPC/NEPC defined as both AR Score and NEPC score >0.5 (n= 5). B) PRRX2 expression levels in ARPC, NEPC, NEPC/ARPC and DNPC subgroups. C) Heatmap of the top 21 differentially expressed genes between LNCaP-sgNC-DMSO (NC-D) and LNCaP-sgPRRX2-DMSO (PR-D) used for the PRRX2 signature. D) Similarity matrix of patients in the SU2C cohort using AR, NEPC and PRRX2 signature genes. E) Hierarchical clustering of the SU2C, 2019 patients according to the expression of AR, NEPC and PRRX2 signature genes. F) Overall survival plot of DNPC patients classified into PRRX2 high (25th percentile of PRRX2 score) and PRRX2 low (rest of the patients). G) Colony formation assay of DU145 after PRRX2 knockdown using two different shRNAs. Experiment performed in 3 independent biological replicates H) LAPC9-AI organoid size after PRRX2 knockdown with two different shRNAs. Experiment performed in 3 independent biological replicates I) Tumor growth of LAPC9-AI-shNC and LAPC9-AI-shPRRX2–1 inoculated into castrated mice. Clustering: D, E Clustergrammer was used to build the matrix using 1-cosine distance and average linkage. Statistical analysis: Log-rank analysis used for survival analysis. Unpaired two tail t-test for G, H, I. Error bars represent S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Statistical analyses
Statistical analyses were performed using unpaired two-tailed Student’s t test or one way ANOVA, unless otherwise indicated. Survival studies were analyzed by Log-rank (Mantel–Cox) test. Correlations were analyzed by Spearman’s correlation coefficient (r). Data are presented as mean ± standard error of the mean (S.E.M.), unless otherwise indicated. For all analyses, results were considered statistically significant with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Cell proliferation, IHC, Immunofluorescence, Western Blot, qPCR, siRNA transfection, lentivirus production and plasmids
Detailed protocols for these techniques are described in supplementary materials and methods.
Data Availability Statement
The data analyzed in this study were obtained from Gene Expression Omnibus (GEO). The RNA-seq and ATAC-seq generated in this study are available in GEO with the accession number: GSE199539. For analysis using the Stelloo RNA-seq and ChIP-seq data were obtained from the gene ontology database with the accession numbers: GSE120741 and GSE120738, respectively (14). The Pomerantz ChIP-seq data was obtained from GSE70079 (15). ChIP-seq data from the PC3 and LNCaP cell lines were obtained from (GSM3145503) (4) and (GSE14092) (16) respectively. Patient data of paired patients before and after ADT treatment was obtained from GSE48403 (17). All the other data supporting the findings of this study are available within the article and its Supplementary Information files and from the corresponding author upon reasonable request.
Results
CRISPRa genome-wide screen identifies PRRX2 as a driver of enzalutamide resistance
To idenfity new targets of enzalutamide resistance, we conducted a CRISPR activation (CRISPRa) screen using the hormone-sensitive prostate cancer cell line LNCaP. The positive selection screen allows for enrichment of cells that have upregulated the expression of resistance-mediating genes (Figure 1A). In addition, to discern between androgen dependent (AD) and androgen independent (AI) resistance, our cells expressed GFP under the control of the androgen responsive element (ARE) (Figure S1A). We validated the reporter system by stimulating and inhibiting the AR using dihydrotestosterone (DHT) or ENZ, respectively (Figure S1B).
Fig. 1. CRISPRa genome-wide screen identifies PRRX2 as a driver of enzalutamide resistance.
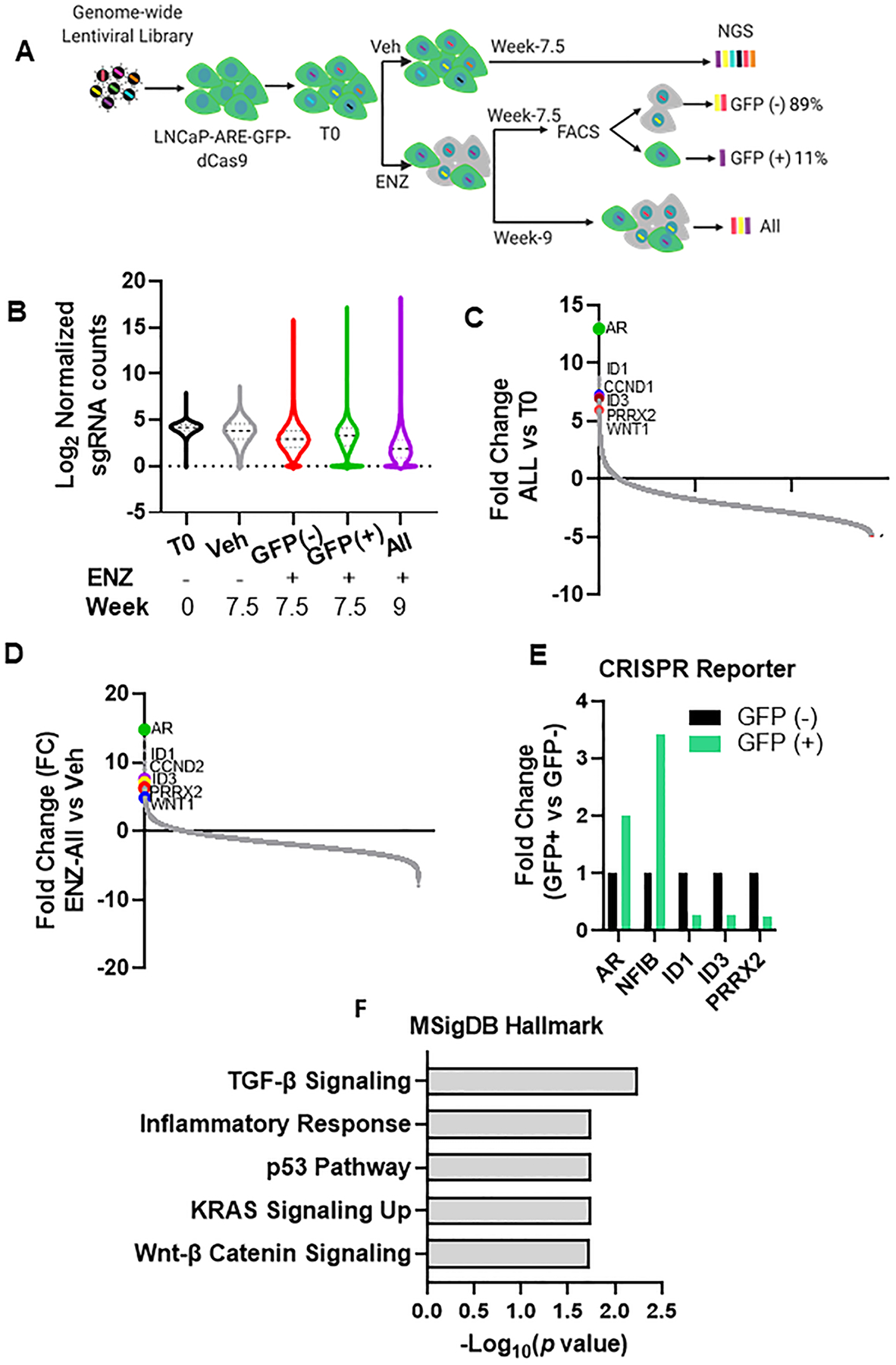
A) CRISPRa Screen design. Cells were transduced with the Calabrese library at a low MOI (0.3–0.5) and 4 days after transfection, cells were treated with vehicle (Veh) or 25 μM enzalutamide (ENZ) for 7.5 or 9 weeks before sequencing. ENZ treated cells at 7.5 weeks were sorted into GFP (+) and GFP (–) cells using FACS. We sequenced the sgRNAs from week 0 (T0), Vehicle (Veh), ENZ-GFP (+), ENZ-GFP(−) and ENZ unsorted cells at week 9 (ENZ-All). B) Violin plot of log2 sgRNA counts representation within the different populations. Lines represent the median and the interquartile range. C,D) Ranked-ordered plot of normalized counts for the fold change (FC) of the ENZ-All population vs T0 (C) and the ENZ-All vs Veh (D). E) Fold change of the sgRNA counts comparison between GFP (+) vs GFP (−) for genes shown in graph. F) Significantly over-represented pathways (q<0.15) for genes with >3.5 FC in the ENZ-All vs T0.
We then transduced the LNCaP-ARE-GFP cells with the dCas9-VP64 vector and subsequently with the sgRNA library at a low MOI to obtain only one sgRNA per cell (Figure S1C) (see Materials and Methods). Four days post-infection, we divided the cells into 2 groups: Vehicle (Veh) and ENZ and treated them for 7.5 and 9 weeks to allow for sgRNA enrichment. To separate AR-dependent (AD) from AR-independent (AI) mechanisms of resistance, we sorted the ENZ-treated population at 7.5 weeks into GFP(+) and GFP(−) fractions (Figure 1A).
Analysis of the distribution of the different sgRNA populations showed that all the ENZ treated groups had wider distributions with enriched and depleted sgRNAs indicating the selection pressure exerted by ENZ (Figure 1B). Comparison between the unsorted ENZ-All population to untreated (T0) or Veh groups revealed AR as the most enriched sgRNA after ENZ treatment (Figure 1C,D). This result validates our experimental approach, as AR overexpression is one of the main mechanisms of ENZ resistance (18, 19). Additionally, our screen identified other genes previously linked to ENZ resistance such as WNT1, ID1, ID3 and CCND2 (Figure 1C, D)(5, 20, 21). Overall, 422 guides were enriched (FC> 3.5) and 1040 (FC<−3.5) guides depleted post-ENZ compared to Veh (Supplementary Table 2).
To identify genes specifically enriched in AD or AI populations, we compared the sgRNA distribution in the ENZ-GFP(+) and ENZ-GFP(−) groups. FACS sorting revealed that 89% of the cells were GFP(−), suggesting that only a small population of cells activated the reporter in the presence of ENZ (Figure S1D). Surprisingly, AR was also enriched in the GFP (−) population (Figure S1E,F). This could be due to an incomplete separation of GFP (+) and GFP (−) cells during FACS. Alternatively, the ARE reporter may not have been a faithful indicator of functional AR activity in some cells. Nevertheless, direct comparison between the ENZ-GFP(+) vs ENZ-GFP(−) sgRNA reads revealed that AR was 2 fold more enriched in the ENZ-GFP(+) versus the ENZ-GFP(−) population (Figure 1E, 1SG). To further validate our reporter, we analyzed the distribution of NFIB, a transcription factor (TF) that co-regulates the expression of AR target genes (22). We found that NFIB was 3.14 times more enriched in the ENZ-GFP(+) population. In contrast, ID1 and ID3 were more enriched in the GFP(−) compared to the GFP(+) population. These genes mediate ENZ- resistance via an AI mechanism (5).
To identify the main pathways related to ENZ resistance we performed a gene enrichment analysis using enriched sgRNAs in the ENZ-All vs Veh groups as input. We found an over-representation of the TGF-β, TP53 and WNT-β catenin signaling pathways (Figure 1F). Of note, all these pathways have been linked to ENZ resistance and PC aggressiveness in different studies (23–26).
PRRX2 mediates enzalutamide resistance in vitro and in vivo
Among the top hits, we identified PRRX2 a homeobox TF that mediates epithelial to mesenchymal transition (EMT) and metastasis in breast and colon cancer (7, 27). PRRX2 was enriched in our ENZ treated population and in the GFP (−) vs GFP (+) population (Figure 1C–E). Of note, the role of PRRX2 in PC is unknown. To explore the novel resistance mechanisms potentially mediated by PRRX2, we focused on this gene for the rest of the study.
To validate the CRISPRa screen results, we generated a stable LNCaP over-expressing PRRX2 cell line using our CRISPRa system (LNCaP-sgPRRX2) and a control cell line (LNCaP-sgNC). PRRX2 over-expression enhanced growth in the presence of ENZ and in androgen-deprived charcoal stripped (CSS media) conditions (Figure 2A,B). In addition, PRRX2 over-expression increased colony formation in DMSO and in ENZ-treated conditions compared to control cells (Figure 2C). Furthermore, LNCaP-sgPRRX2 cells grew better as spheroids in 3D culture conditions in CSS media and in the presence of ENZ compared to the control cells (Figure 2D).
Fig. 2. PRRX2 mediates enzalutamide resistance in vitro and in vivo.
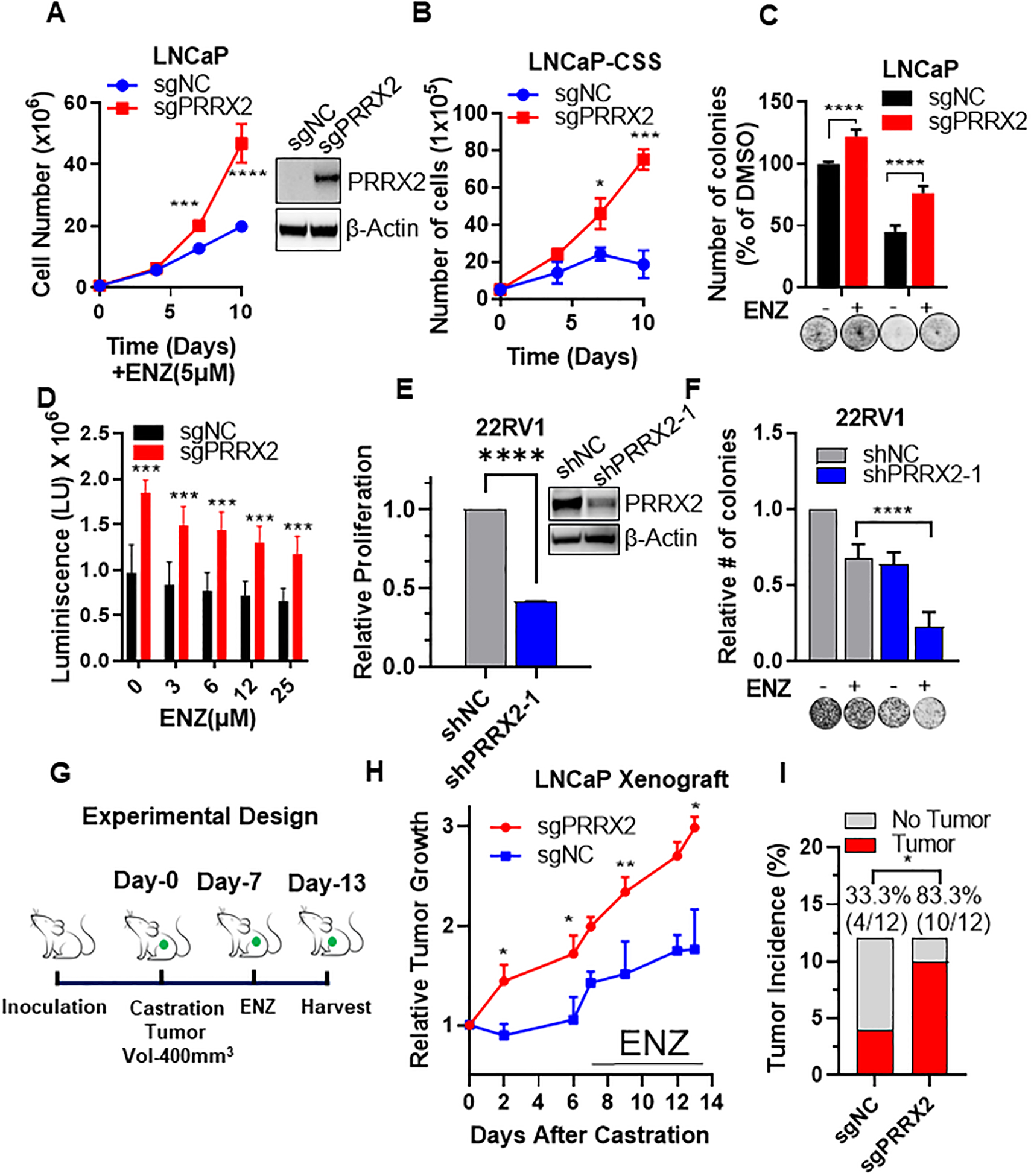
A) Cell counts of LNCaP-sgNC and LNCaP-sgPRRX2 cells after treatment with ENZ (5μM) at different time-points. Insert: Western blot comparing PRRX2 levels in LNCaP-sgNC vs LNCaP-sgPRRX2 cells. B) Cell counts of LNCaP-sgNC and LNCaP-sgPRRX2 cells in charcoal stripped (CSS) media at different time-points. C) Colony formation assay after 14 days of treatment with ENZ (5μM) or DMSO. D) Growth in low attachment plates (GILA) assay for 3D spheroids of LNCaP-sgNC vs LNCaP-sgPRRX2 cells grown in CSS and different concentrations of ENZ for 7 days. E) Cell proliferation assay of 22RV1 cells after PRRX2 knockdown using shRNA. F) Colony formation assay of 22RV1-shNC and 22RV1-shPRRX2–1 cells after treatment with ENZ (40μM) for 14 days. G) In vivo experimental design. H) Relative tumor growth of LNCaP-sgNC and LNCaP-sgPRRX2 xenografts in castrated mice treated with ENZ (10mg/kg) IP 3 times/week. I) Tumor incidence (volume ≥ 100mm3) after 1 month. All viability experiments were performed in at least 3 independent biological replicates. Statistical tests: p-value determined by two-sided t-test (A-H). Chi-squared test for I. Error bars represent S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Next, we knocked down PRRX2 by shRNA in the ENZ resistant 22RV1 PC cells, which express the AR variant ARV7. Down-regulation of PRRX2 significantly impaired cell proliferation and colony formation of 22RV1 cells (Figure 2E,F, S2A), and sensitized these cells to ENZ (Figure 2F). Interestingly, PRRX2 knockdown decreased the levels of AR and ARV7 in 22RV1 cells, which might partially explain why they became sensitive to ENZ (Figure S2B). These results indicate that PRRX2 mediates resistance to androgen deprivation and to ENZ in vitro.
To extend these results in vivo, we injected LNCaP-sgPRRX2 or LNCaP-sgNC cells subcutaneously into intact non-castrated mice and allowed for tumors to be established. To model the effects of ADT and ENZ treatment, we castrated the mice after the tumors reached approximately 400mm3 and then treated them with ENZ (Figure 2G,S2C). LNCaP-sgPRRX2 xenografts were able to re-grow after castration and in the presence of ENZ in contrast to LNCaP-sgNC cells (Figure 2H). Of note, 83.3% (10/12) of the injected sites developed a tumor (> 100mm3 after 1 month of injection) in the LNCaP-sgPRRX2 group, compared to only 33.3% (4/12) in the control group, indicating that PRRX2 is oncogenic in PC (Figure 2I). Of note, LNCaP-sgNC tumors did not express PRRX2, suggesting that tumor growth on these cells was mediated by a PRRX2-independent mechanism (Figure S2D). Overall, these results show that over-expression of PRRX2 is able to drive resistance to ENZ in vitro and in vivo.
PRRX2 modulates the expression of enzalutamide-regulated genes
To gain further insight into the role of PRRX2 in ENZ resistance, we performed RNA-seq comparing LNCaP-sgPRRX2 vs LNCap-sgNC cells under DMSO (NC-D vs PR-D) and ENZ treated (NC-E vs PR-E) conditions (Figure 3A, Supplementary Table 3). We clustered all the genes into four different clusters (Figure 3B). Cluster 1 (C1) contains genes that are repressed by ENZ and induced by PRRX2. In this cluster, we found that PRRX2 attenuates the repressive effect of ENZ. Of note, MYC target genes, mTORC1 signaling and androgen response genes are enriched in this cluster (Figure 3C). Cluster 2 (C2) contains genes induced by PRRX2 regardless of ENZ treatment. Interestingly, genes involved in regulating cell cycle pathways such as the E2F target genes and G2-M checkpoint are enriched in C1 and C2, suggesting that PRRX2 plays an important role in the regulation of this process (Figure 3C,D). Cluster 3 contains genes repressed by PRRX2. The low number of genes in this cluster indicates that PRRX2 is mainly a transcriptional activator. Interestingly, P16 (CDKN2A), a tumor suppressor gene that impairs cell cycle progression, belongs to this cluster. Finally, in Cluster 4 we observed that PRRX2 cooperates with ENZ to further induce the expression of certain genes. Genes and pathways implicated in cancer progression and ENZ resistance, e.g. CXCR4, CD44, WNT, TGF-β and EMT are enriched in this cluster (Figure 3E).
Fig. 3. PRRX2 modulates the expression of enzalutamide-regulated genes.
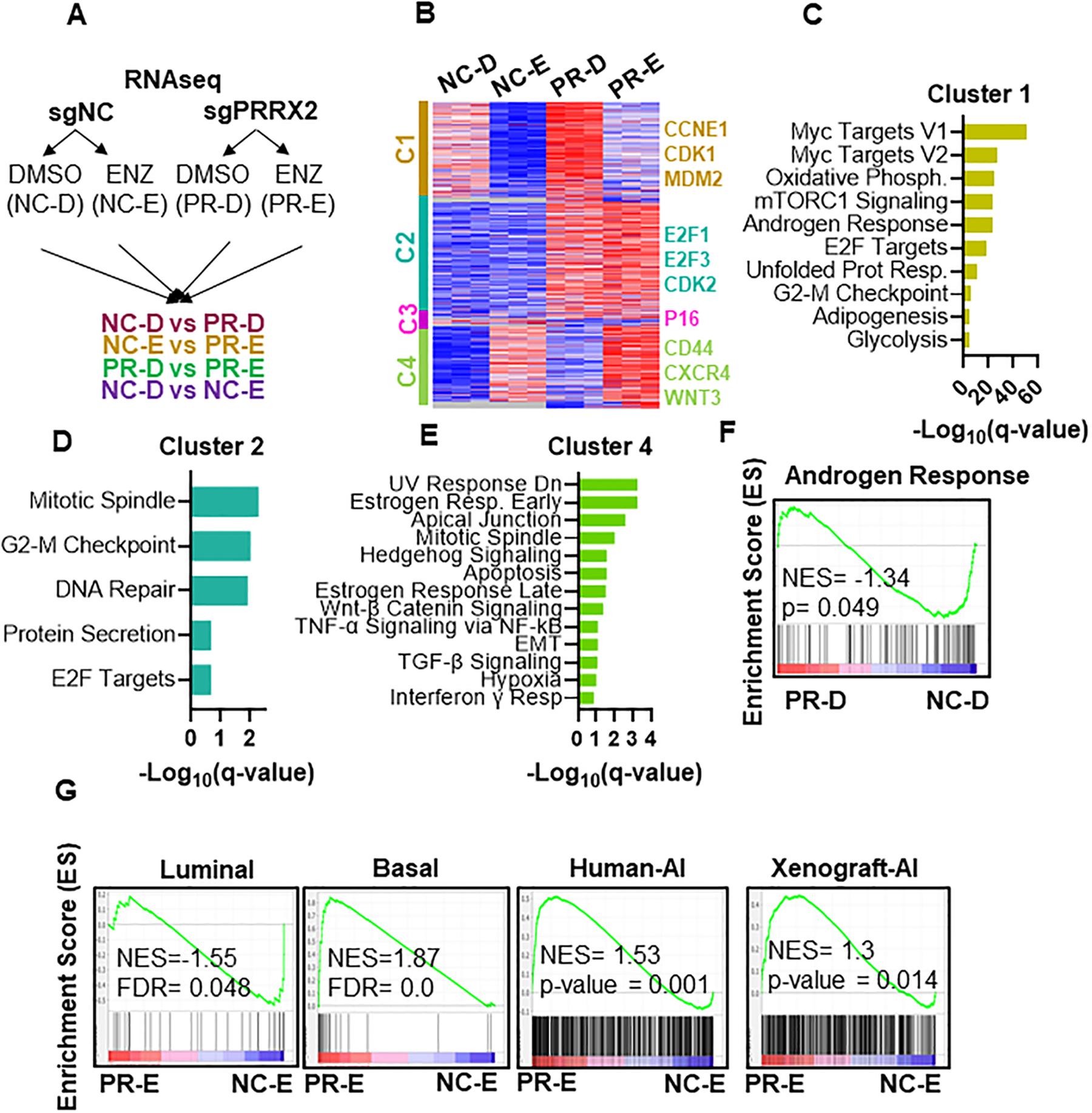
A) Experimental design of RNA-seq experiment. LNCaP-sgNC and LNCaP-sgPRRX2 cells were treated with 5μM ENZ (E) or DMSO (D) for 7 days. B) Heatmap of RNA-seq data in LNCaP-sgNC-DMSO (NC-D), LNCap-sgNC-ENZ (NC-E), LNCaP-sgPRRX2-DMSO (PR-D) and LNCaP-sgPRRX2-Enz (PR-E). Clusters were generated using K-means clustering function in Morpheus software. C, D, E) Top significant pathways enriched in Cluster 1, 2 and 4 respectively. F) GSEA plot of Androgen Response geneset (Hallmark dataset) enriched in LNCaP-sgPRRX2 cells in DMSO conditions. G) GSEA analysis of different signatures in PR-E vs NC-E cells.
To study the RNA-seq data in more detail, we compared the PR-D vs NC-D and PR-E vs NC-E cells using gene set enrichment analysis (GSEA). We found that EMT was the most significantly enriched pathway in both DMSO and ENZ conditions with PRRX2 overexpression confirming the role of PRRX2 in EMT. Interestingly, E2F target genes and G2-M pathways were enriched only when comparing PR-E vs NC-E cells. In addition, pathways related to the immune system were also enriched in PRRX2 over-expressing cells (Figure S3A,B).
Of note, we found that the Androgen Response pathway was negatively enriched (NES= −1.34) in the PR-D vs NC-D cells (Figure 3F), raising the possibility that PRRX2 promotes cell transition from an AR-dependent luminal to an AR-independent basal phenotype. Supporting this hypothesis, PR-E cells had a negative enrichment score for the luminal phenotype (NES = −1.55) and a positive NES score for the basal phenotype (NES = 1.87) when compared to NC-E cells (Figure 3G). To further explore the concept of an AR-independent phenotype triggered by PRRX2, we used two gene signatures. One consists of up-regulated genes in AI (ADT resistant) vs AD (therapy naïve), and the other one consists of genes up-regulated in AI (castrated) vs AD (intact) xenografts (28). We found that in PR-E cells were enriched for both of these signatures compared to NC-E cells (Figure 3G).
Together, these results show that in the presence of ENZ, PRRX2 helps maintain the activation of cell cycle genes and further enhances the expression of pro-tumorigenic ENZ-induced genes/pathways.
The AR and TGF-β pathways converge to cross-regulate PRRX2
Due to the importance of AR in PC and ENZ resistance, we next investigated the relationship between AR and PRRX2. We first analyzed the expression correlation between AR and PRRX2 across four different datasets and found a significant negative correlation between these genes (Figure S4A). Furthermore, AR activity (measured by an AR score (29, 30)) and PRRX2 expression were also negatively correlated in the TCGA (Spearman r = −0.46) and the SU2C datasets (Spearman r = −0.34) (Figure 4A).
Fig. 4. The AR and TGF-β pathways converge to cross-regulate PRRX2.
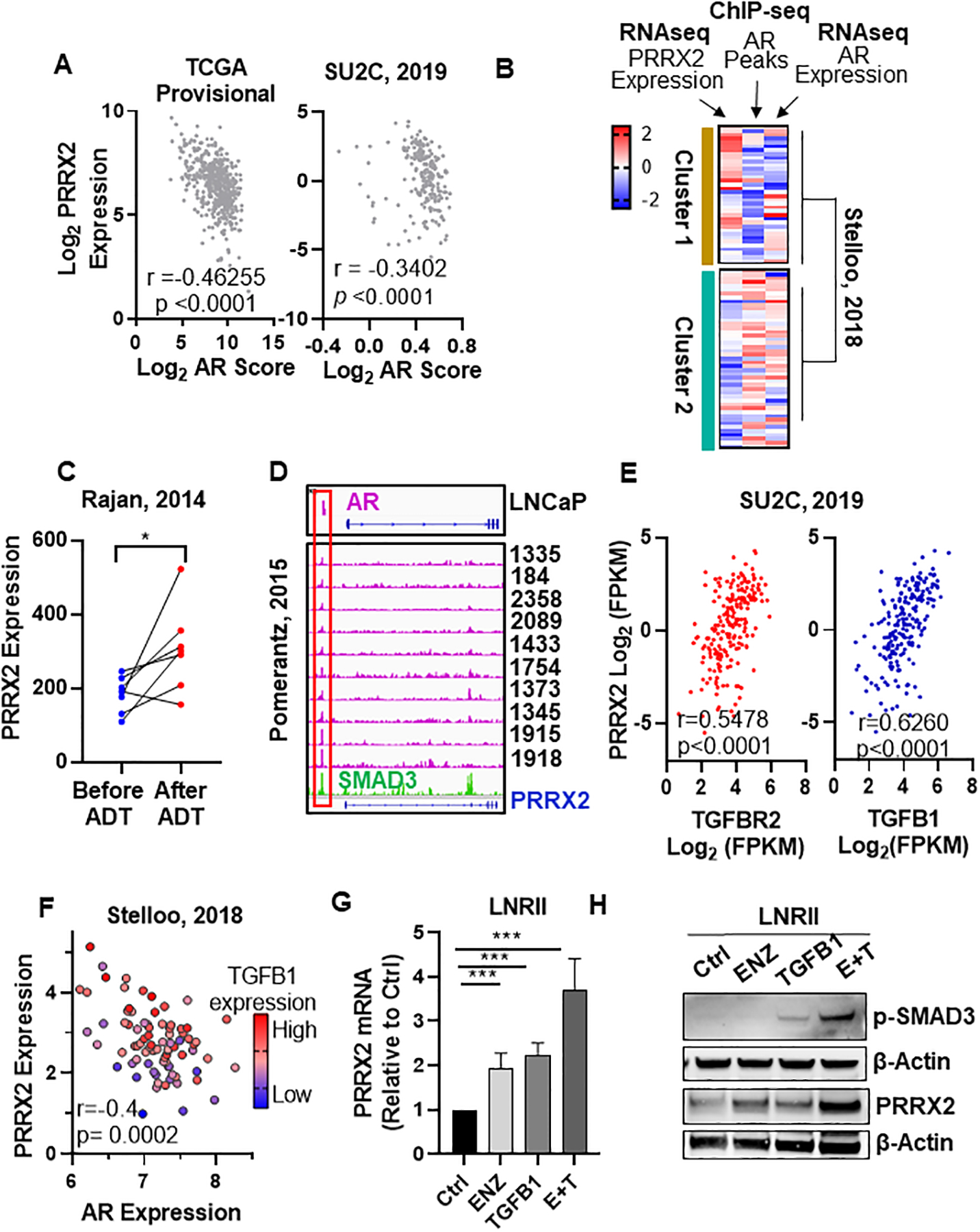
A) Spearman r correlation between AR score and PRRX2 log2 expression levels. AR score and expression data were obtained from cBioPortal TCGA provisional database (n = 499) and SU2C (n = 209). B) PC patients from Stelloo et al (n = 83) were clustered using K-means clustering according to their PRRX2 and AR expression (z-values) and number of AR ChIP peaks (z-values). C) PRRX2 expression in paired patient samples (Rajan et al) before and after 22 weeks of ADT treatment. * *p<0.05 by paired two tail t-test. D) ChIP-seq data of AR and SMAD3 binding peaks near the PRRX2 promoter in different PC patients and cell lines. IGV was used for visualization E) Spearman r correlation between PRRX2, TGFB1 and TGFBR2 expression. Data obtained from cBioPortal (SU2C, 2019) n=208. F) Scatter plot of AR and PRRX2 expression. Circle color represents TGFB1 expression. Spearman r coefficient shown for AR and PRRX2 expression. Data obtained from Stelloo et al. (n = 82) G, H) PRRX2 expression measured by RT-qPCR (G) or Western blot (H) in LNRII (LNCaP cells expressing TGFBR2) after treatment with ENZ (5μM), TGFB1 (5ng/ml) or the combination during 48h. β-Actin served as loading control. Experiments performed in 3 independent biological replicates. Statistical analysis in G done with unpaired two-tail t-test. Error bars represent S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To analyze the link between AR activity and PRRX2 further, we studied AR chromatin occupancy using an AR ChIP-seq patient dataset from Stelloo et al (14). We clustered these patients according to AR occupancy, measured by the number of AR peaks (ChIP-seq), and PRRX2 expression (RNA-seq) in each patient. As noted earlier (14), AR expression did not always correlate with AR chromatin occupancy (Figure 4B), suggesting, that AR expression may not be an accurate measure of AR activity. Our clustering revealed two subgroups, Cluster 1 contains higher numbers of AR chromatin binding peaks and lower PRRX2 expression. In contrast, Cluster 2 has patients with lower number of AR peaks and higher PRRX2 expression (Figure 4B), suggesting that patients with higher AR activity have lower levels of PRRX2 expression and vice versa.
Based on these results, we hypothesized that AR negatively regulates PRRX2. To address this hypothesis, we first examined changes in PRRX2 expression using an RNA-seq data from paired patient samples before and after ADT (17). We found an increase in PRRX2 expression after ADT treatment suggesting that PRRX2 expression is a potential AR target gene (Figure 4C). Next, we analyzed AR binding into the PRRX2 promoter region using ChIP-seq patient and cell lines datasets. Of note, we found an AR peak near the PRRX2 promoter present in LNCaP cells. Importantly, the same AR peak was present in 11 out of 14 patient samples from the Pomerantz dataset (Figure 4D) (15). We confirmed the presence of this peak using ChIP-qPCR in LNCaP cells (Figure S4B). To analyze the functional relevance of this peak, we knocked-down AR/ARV7 in 22RV1 cells using siRNA and found an increase in PRRX2 mRNA and protein levels (Figure S4C,D). We also treated LNCaP cells with ENZ. However, we found no effect in PRRX2 expression levels suggesting that AR alone is not sufficient to regulate PRRX2 in this model (Figure S4E).
The TGF-β pathway is known to induce PRRX2 expression. SMAD3 is one of the main signal transducers of the TGF-β pathway (31). To study the relationship between TGF-β and PRRX2 in PC, we analyzed a ChIP-seq dataset of SMAD3 binding in PC3 cells (4).Of note, we found a SMAD3 peak near the PRRX2 promoter coinciding with the location of the AR peak, suggesting that they could potentially co-regulate PRRX2 expression (Figure 4D). This peak was confirmed in DU145 cells stimulated with TGF-β1 using ChIP-qPCR in DU145 cells (Figure S4B). Further analysis of PC datasets showed a strong correlation between the expression of PRRX2, the TGFB1 receptor 2 (TGFBR2) (r=0.5478) and of its ligand (TGFB1) (r=0.6260) (Figure 4E). This result was also validated in the Stelloo dataset where we observed that patients with the highest expression of PRRX2 had lower AR expression and higher TGFB1 levels (Figure 4F). In addition, stimulation of DU145 cells with TGF-β1 lead to an increase in PRRX2 transcription (Figure S4F).
LNCaP cells are insensitive to TGF-β1 stimulation due to the lack of TGFBR2 expression, which may explain the lack of PRRX2 induction upon AR inhibition in LNCaP cells. To test if AR and the TGF-β pathway cross-regulate PRRX2, we used LNRII cells, a cell line derived from LNCaP that exogenously expresses the TGFBR2 gene, making it competent for TGF-β1 stimulation (32). We found that stimulation of LNRII cells with TGF-β1 or treatment with ENZ alone increased the levels of PRRX2 expression at mRNA and protein levels in LNRII cells. However, treatment with both ENZ and TGF-β1 led to the highest increase in PRRX2 mRNA and protein levels (Figure 4G–H). This increase was accompanied by an increase in p-SMAD3 levels. Of note, triple immunofluorescence staining of AR, PRRX2 and p-SMAD2/3 proteins in LNRII cells revealed that cells with the lowest expression of AR expressed higher levels of both PRRX2 and p-SMAD2/3 (Figure S4G). Taken together, these data suggest that full PRRX2 induction in tumors requires AR inhibition and activation of the TGF-β pathway.
A PRRX2 signature stratifies DNPC and correlates with poor survival
Given that AR negatively regulates PRRX2, we hypothesized that PRRX2 is mainly expressed in low AR settings. NEPC and DNPC are two subtypes of PC characterized by low AR expression or activity. To investigate if PRRX2 was over-expressed in these groups, we classified patients from the SU2C dataset into four categories (ARPC, NEPC, AR/NEPC, DNPC) according to their AR and NEPC scores (Figure 5A). Analysis of PRRX2 expression in each of the clusters revealed that DNPC had the highest expression of PRRX2 (Figure 5B). In addition, the frequency of patients with PRRX2 upregulation was highest in DNPC (24.4%) compared to NEPC (13.6%) and ARPC (9.7%) (Figure S5A). Of note, LNCaP-sgPRRX2 cells treated with ENZ had a positive enrichment of a DNPC patient gene signature (Figure S5B).
To further study the clinical role of PRRX2 in PC, we used the 21 significantly up-regulated genes (top 15th percentile of the up-regulated genes) in the PR-D vs NC-D cells to create a PRRX2 signature (Figure 5C). We then analyzed if the PRRX2 signature genes were co-expressed in PC patients by performed a similarity matrix with the PRRX2, AR, and NEPC-signature genes in four different PC datasets. The similarity matrix revealed three unique clusters corresponding to PRRX2, NEPC and AR in all datasets (Figure 5D, S5C, D).
We then classified the patients according to the expression of the PRRX2 signature genes using unsupervised hierarchical clustering. Analysis of the TCGA and the MSKCC, datasets revealed only two groups denominated PRRX2-high and PRRX2-low clusters (Figure S5E). Of note, the PRRX2-high clusters had lower expression of AR target genes. The SU2C, Beltran and DKFZ datasets formed three clusters denominated ARPC, NEPC and PRRX2 (Figure 5E, S5F). Similar to our previous results, patients within the PRRX2 cluster had lower expression of AR and NEPC target genes, further supporting our hypothesis about higher PRRX2 expression in DNPC patients. In addition, we found a 71.7% overlap between the patients in the PRRX2 cluster and the patients within the DNPC cluster in the SU2C dataset (Figure S5G).
Further characterization of the PRRX2 cluster revealed an over-representation of bone tumor metastases (Figure S5H). Furthermore, PRRX2 expression itself was higher in bone samples compared to other tissues (Figure S5I,J). To further characterize the different clusters, we analyzed the expression of genes significantly over-expressed in the PRRX2 cluster compared to the ARPC and NEPC clusters. We found an enrichment of immune related pathway such as the interferon gamma and alpha signaling in the PRRX2 cluster (Figure S5K). These results correlate with previous observations, which show that DNPC is associated with immune-related pathways (33). Overall, these results show that a PRRX2 signature is able to divide PC patients within a specific subgroup that shares certain characteristics with the DNPC patients, such as bone metastasis and enrichment of immune-related pathways.
PRRX2 is an oncogene in DNPC
To study the significance of PRRX2 score in PC prognosis, we calculated a PRRX2 score using the genes within the PRRX2 signature (Figure 5C). Overall, our score was distinct from the AR and NEPC scores and matched with the patient stratification previously made by hierarchical clustering (Figure S5L–N). We then calculated the disease-free survival (DFS) between patients with the highest PRRX2 score (25th percentile) compared with the rest of the patients in the TCGA and the MSKCC datasets, but found no difference in DFS in any of these datasets (Figure S5O,P). Of note, patients with high clinical Gleason scores were mainly located within the PRRX2-high score group. This result was consistent in three different datasets (Figure S5Q–S). Furthermore, the gene expression of PRRX2 was higher in samples with Gleason score higher than 8 (Figure S5T).
Analysis of the SU2C dataset, which contains advanced metastatic PC patients, showed no difference in overall survival (OS) between high (top 25th percentile) and low (rest of the patients) PRRX2 score patients (Figure S5U). However, stratification of the DNPC patients using our PRRX2 signature score revealed that the OS of the patients with the highest PRRX2 score (25th percentile) was significantly lower compared to the rest of the patients (12.88 vs 25.66 months, p=0.0054) (Figure 5F).
To further study the role of PRRX2 in AR-negative cells, we knocked-down PRRX2 in DU145 cells, considered a model of DNPC (34). We found a significant decrease in colony formation in DU145-shPRRX2 compared to control cells using two different shRNAs (Figure 5G). To gain more insight into the role of PRRX2 in DNPC, we used the LAPC9 androgen independent (LAPC9-AI), an ENZ-resistant DNPC patient-derived-xenograft model (35). LAPC9-AI was derived from the androgen-dependent parental cells, LAPC9-AD. LAPC9-AI xenografts grow in castrated mice and express low levels of AR target genes KLK3, FKBP5 and of the NEPC gene, SYP. Of note, we found that LAPC9-AI xenografts express significantly higher levels of PRRX2 compared to LAPC9-AD cells (Figure S5V).
Similar to our observations in DU145 cells, PRRX2 downregulation in LAPC9-AI cells led to a significant decrease in the organoid size compared to control cells (Figure 5H, S5W). We validated these results in vivo after injection of LAPC9-AI-shPRRX2–1 cells into castrated NSG mice. We observed that PRRX2 downregulation delayed tumor growth and decreased proliferation, as evidenced by lower Ki67 expression (Figure S5X), suggesting that PRRX2 is necessary for cell survival in AR negative/DNPC cells (Figure 5I). Together, these results show that PRRX2 is an oncogene and might play a role in the aggressiveness of PC within the DNPC population.
PRRX2 regulates the Rb1/E2F and BCL2 pathways
We sought to study the transcriptional function of PRRX2 in more detail by ChIP-seq experiments but were limited by the lack of suitable ChIP antibodies. Instead, we conducted an ATAC-Seq experiment using the LNCaP-sgNC vs LNCaP-sgPRRX2 cells under ENZ treatment. We then annotated the genes that had ATAC-seq peaks and found 2069 (p<0.05) differentially opened chromatin regions (Supplementary Table 4). We found that open chromatin regions in PRRX2 overexpressing cells corresponded to genes such as the WNT receptor, FZD1, and the TF TRSP1, which plays a role in cell proliferation. Closed chromatin regions corresponded to the pro-apoptotic cadherin (CDH11) (36) (Figure 6A).
Fig. 6. PRRX2 regulates the Rb1/E2F and BCL2 pathways.
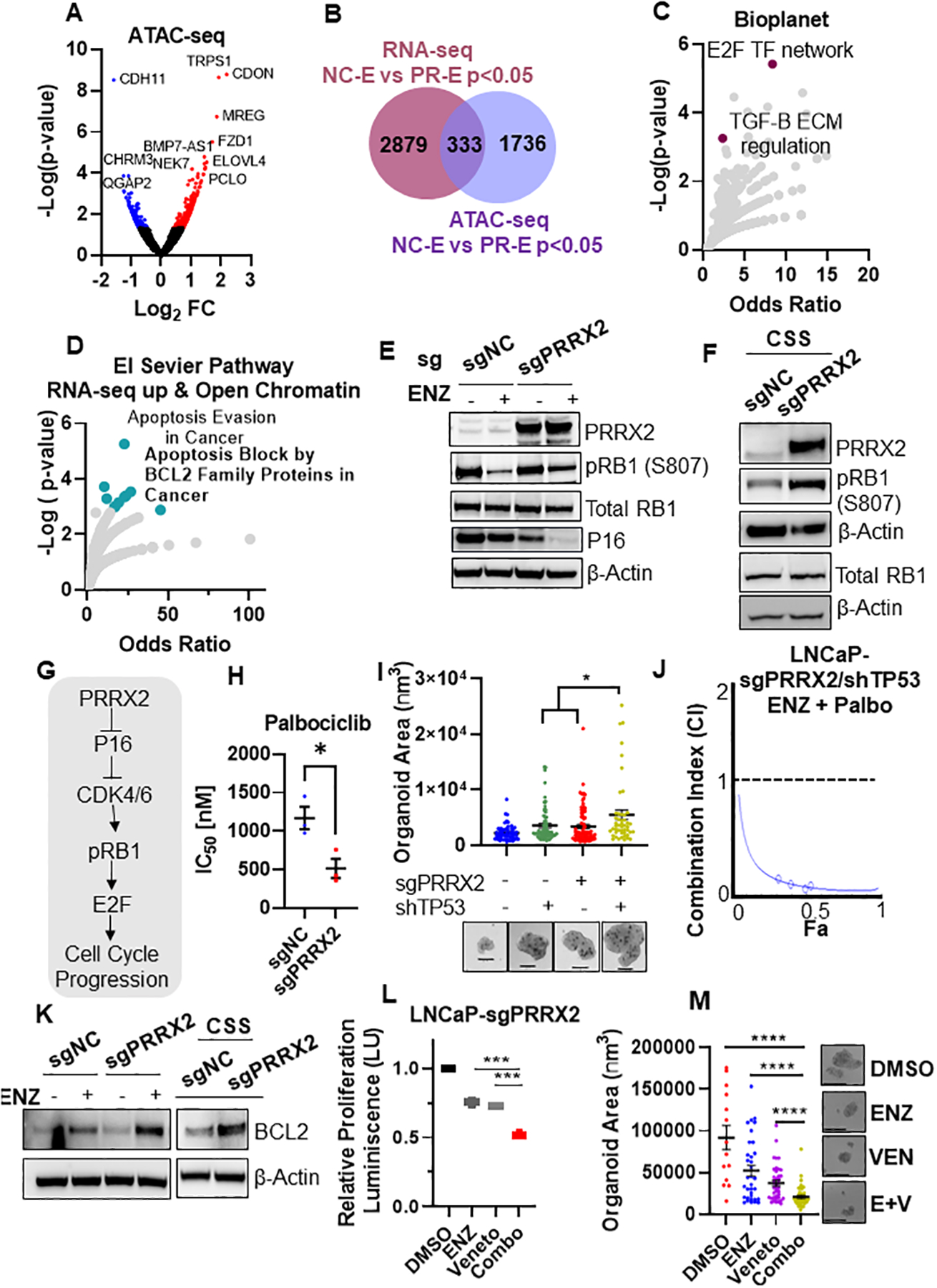
A) Volcano plot of ATAC-seq genes. Colored genes represent p-value < 0.05 B) Venn diagram showing the overlap between RNAseq (p<0.05) and ATAC-seq (p<0.05) genes. C) Enrichment analysis of different pathways using the list of genes that overlap (n=333) between RNAseq and ATAC-seq experiments in B. D) Enrichment analysis using the genes up-regulated in PR-D vs NC-D RNAseq (p<0.05) and the open chromatin regions in the PR-D vs NC-D (p<0.05) (n=100). E) Western blot analysis of LNCaP-sgNC and LNCaP-sgPRRX2 cells treated with ENZ (5μM) for 10 days. F) Western blot analysis of LNCaP-sgNC and LNCaP-sgPRRX2 cells grown in CSS for 7 days. G) Proposed link of PRRX2 to the Rb/E2F pathway. H) Palbociclib (Palbo) IC50 of LNCaP-sgNC and LNCaP-sgPRRX2 cells after treatment for 4 days. I) Area quantification of GILA assay spheroids. Cells were grown in CSS media and treated with ENZ (5μM) for 7 days. Representative images shown. Scale bar = 50μm. J) Compusyn was used to calculate the combination index (CI) for cells were treated with Palbo and ENZ for 4 days. K) Western blot analysis of LNCaP-sgNC and LNCaP-sgPRRX2 cells treated with ENZ (5μM) or grown in CSS media for 7 days. L) GILA assay for LNCaP-sgPRRX2 cells grown in androgen free CSS media and treated with ENZ (5μM), 12μM venetoclax (Veneto) or the combination for 4 days. M) Sphere size quantification of experiment in C. Scale bar = 200μm. All experiments were performed at least in 3 independent biological replicates. Statistical tests: Unpaired two tail t-test (H, I, L, M). Error bars represent S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To gain more insight into the molecular mechanisms driven by PRRX2, we integrated the RNA-seq and the ATAC-seq data. We found an overlap of 333 differentially expressed genes between ATAC-seq (p<0.05) and RNA-seq (p<0.05) (Figure 6B). Pathway analysis using all of the 333 overlap genes revealed that the E2F transcription factor network was within the top pathways, consistent with our RNA-seq analysis (Figure 6C). In addition, analysis of the pathways enriched using just the up-regulated genes (RNA-seq) and the open chromatin regions (ATAC-seq) (n = 100 genes) revealed an enrichment of apoptosis block by BCL2 family proteins and apoptosis evasion in cancer pathways (Figure 6D).
To validate these results, we analyzed the expression of RB1, which is upstream E2F, in LNCaP-sgPRRX2 and sgNC cells after ENZ treatment. RB1 loss via genetic mechanisms or via RB1 protein inactivation through hyper-phosphorylation (hp-RB1) leads to the activation of E2F TFs and subsequent cell cycle progression (37). We found that ENZ treatment decreased p-RB1 (inactive) levels in both cell lines. However, p-RB1 reduction was more pronounced in the control cells compared to the LNCaP-sgPRRX2 cells (Figure 6E). In addition, LNCaP-sgPRRX2 cells had higher levels of hp-RB1 in androgen deprived CSS media compared to control cells (Figure 6F). Of note, P16 (CDKN2A), a tumor suppressor that inhibits CDK4/6-mediated RB1 phosphorylation, was significantly lower in the PRRX2 over-expressing cells compared to control (Figure 6E) confirming that the LNCaP-sgPRRX2 cells had a more active cell cycle than control cells (Figure 6G). Importantly, LNCaP-sgPRRX2 cells were more sensitive to the CDK4/6 inhibitor Palbociclib (Palbo) than control cells (Figure 6H), validating the functional significance of this pathway in PRRX2 expressing cells (Figure 6G).
Genetic loss of RB1 and TP53 co-occur in a subset of PC patients and is associated with resistance to ENZ (6). We hypothesizes that PRRX2 expression leading to RB1 inactivation via hyper-phosphorylation will phenocopy RB1 loss and cooperate with TP53 loss to promote ENZ resistance. To test this hypothesis, we knocked-down TP53 in LNCaP-sgPRRX2 cells (Figure S6A). Importantly, we found that LNCaP-sgPRRX2-shTP53 cells were the most resistant to ENZ compared to control cells and to each alteration alone (Figure 6I, S6B). Of note, the combination of ENZ and Palbo, synergized to inhibit LNCaP-sgPRRX2/shTP53 cells (Figure 6J).
In addition to the E2F pathway, we noted that anti-apoptotic pathways were enriched in ENZ-treated PRRX2 over-expressing cells. Treatment with ENZ increased BCL2 levels in both cell lines. However, the increase was higher in the LNCaP-sgPRRX2 cells compared to control. Similarly, we found higher levels of BCL2 in LNCaP-sgPRRX2 cells grown in CSS conditions (Figure 6K). Notably, ENZ and BCL2 inhibition using Venetoclax (Veneto) cooperated in inhibiting LNCaP-sgPRRX2 organoids (Figure 6L,M). In addition, BCL2 knock-down using shRNAs decreased cell proliferation of LNCaP-sgPRRX2 cells under ENZ-treatment (Figure S6C,D).
To extend the translational relevance of these findings, we then studied the relationship between PRRX2 and the expression of E2F downstream genes (CCND2, CCND3) and of BCL2 in the SU2C dataset. We found a positive correlation between the expression of PRRX2 with CCND2 and with BCL2 (Figure S6E,F). Of note, CCND2, CCND3 and BCL2 were also overexpressed in the PRRX2 patient cluster (Figure S6G,H). Importantly, BCL2 protein levels were increased in the PRRX2-high cluster within the TCGA dataset (Figure S6I). Altogether, these results suggest that PRRX2 plays a role in the regulation of the cell cycle, via the RB1/E2F pathway and of cell survival via the BCL2 pathway.
Pharmacological inhibition of CDK4/6 and BCL2 sensitizes PRRX2 over-expressing CRPC to ENZ.
To test whether CDK4/6 and BCL2 inhibition will reverse PRRX2-driven ENZ resistance, we established LNCaP-sgPRRX2 xenografts in NSG mice and castrated the mice when the tumor reached approximately 300mm3. Following recurrence of tumors to ~400mm3 after castration, we started treatment with ENZ alone or combinations of ENZ with Palbo, Veneto or both. We did not include a Veneto or a Palbo only group based on our in vitro results, which show that treatment with ENZ is necessary to induce E2F pathway activation and BCL2 expression.
In contrast to parental LNCaP cells that are sensitive to castration and ENZ treatment, we found that castration and ENZ were not able to slow down the growth of LNCaP-sgPRRX2 tumors (Figure 7A). The addition of Veneto and of Palbo to ENZ improved the response by significantly delaying tumor growth. However, the triple combination had the highest effect in suppressing tumor growth (Figure 7A). Importantly, the drug regimen administered was well-tolerated by mice (Figure S7A).
Fig. 7. Pharmacological inhibition of CDK4/6 and BCL2 sensitizes PRRX2 over-expressing CRPC to enzalutamide.
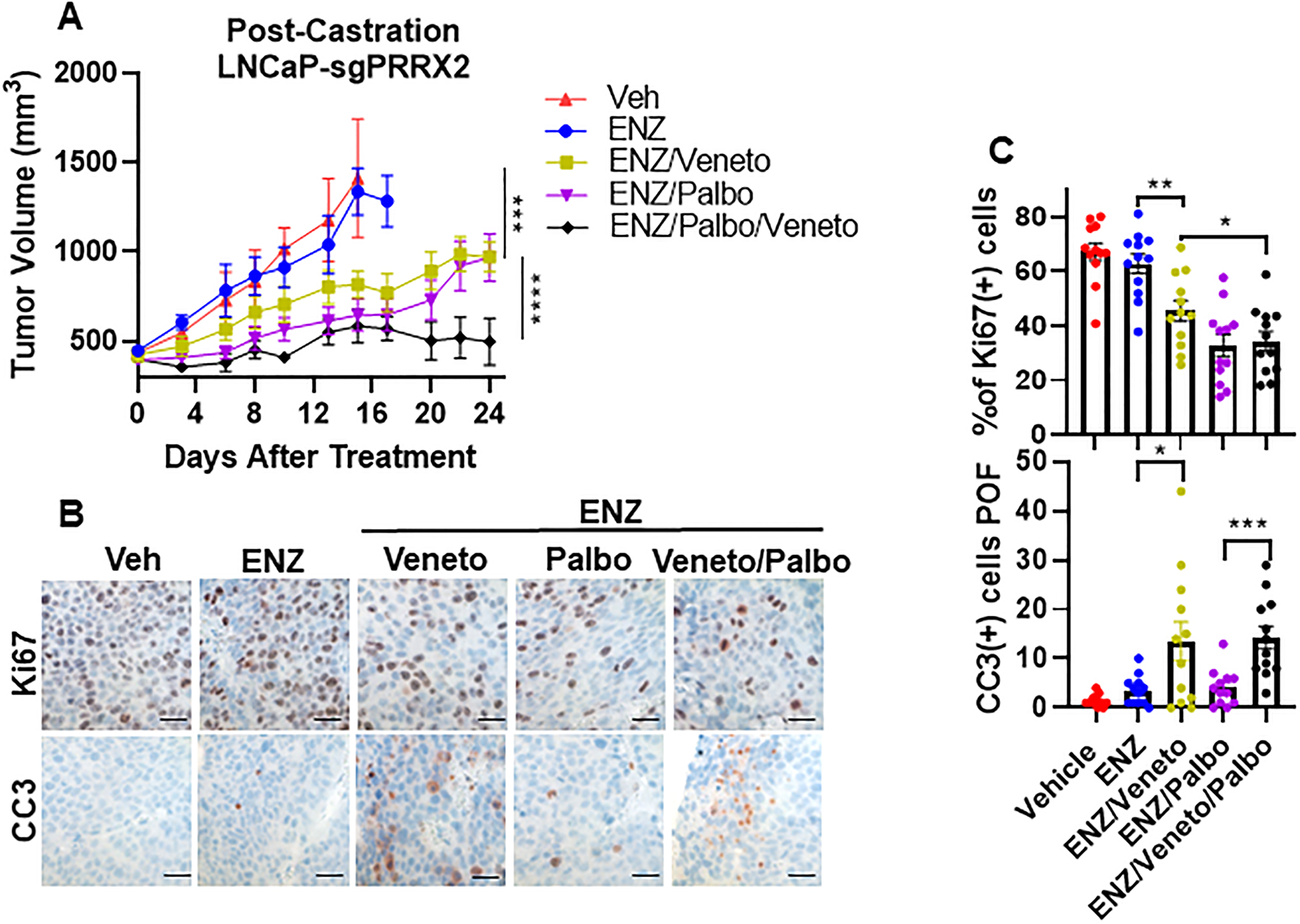
A) Established LNCaP-sgPRRX2 xenograft bearing NSG mice were castrated. When tumor re-grew, they were treated with vehicle (Veh), 10mg/kg enzalutamide thrice weekly (ENZ), 100 mg/kg/d venetoclax (Veneto), 100 mg/kg palbociclib, 5 days on, 2 days off (Palbo) alone or in combinations as indicated. Veh (n=4), ENZ (n=6), ENZ + Veneto (n=6), ENZ + Palbo (n=6), ENZ + Veneto + Palbo (n=6). Statistics: ANOVA comparison between (Veh, ENZ, ENZ+Veneto) and between (ENZ + Veneto, ENZ + Palbo, ENZ + Veneto + Palbo) B) Immunostaining (IHC) images of Ki67 and cleaved caspase 3 (CC3) (quantified as number of cells per optical field POF). Scale bar = 50 μm. C) Quantification of data from B. Four tumors were stained per condition and 3 images/slide were analyzed. Statistical tests: Unpaired two tail t-test. Error bars represent S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Cell proliferation measured by Ki67 staining confirmed that ENZ treatment did not have any effect in tumor cell proliferation compared to the Veh group, consistent with the results in tumor growth (Figure 7B,C). In contrast, ENZ + Palbo and the triple combination had the highest effect decreasing cell proliferation. In addition, groups that received Veneto treatment showed increased apoptosis (Figure 7B,C). Overall, these results show that inhibition of CDK4/6 and BCL2 can overcome ENZ resistance in a PC model with high PRRX2 expression.
Discussion
Despite good initial response to ARPI such as ENZ, resistance remains a major challenge for the treatment of CRPC. With the advent of new and more potent ARPI, resistance mediated via AR-independent mechanisms is becoming more common, in particular through the development of AR negative/low PC such as NEPC and DNPC. Understanding the molecular mechanisms driving ENZ resistance is important for the development of new therapeutic strategies to treat these patients.
This study sought to identify mediators of ENZ resistance via a genome-wide CRISPRa screen. Of note, the top hit of our screen was AR, reiterating the importance of AR as a therapeutic target even in late stages of PC. Other interesting hits were WNT1, ID1 and ID3. The WNT pathway has been previously linked to AR-independent PC and to ENZ resistance, ID1 and ID3 proteins are TFs that are up-regulated in ENZ-resistant PC models and confer resistance in vitro and in vivo (5, 20, 21).
In this study, we focused on the homeobox transcription factor PRRX2. This gene is up-regulated in human PC compared to healthy glands (38). We found that PRRX2 is mainly expressed in DNPC and a PRRX2 signature was able to stratify DNPC patients with worse OS, suggesting that this gene is particularly important for AR-low tumors. PRRX2 can mediate EMT, suggesting that it could contribute to maintain the cells in an undifferentiated stem-like phenotype. Interestingly, PRRX2 is over-expressed in a cluster of prostate basal stem cells (39). In our study, PRRX2 over-expression decreased the expression of an AR target gene/luminal signatures. Furthermore, ENZ treatment accentuated the decrease in expression of luminal signature genes while increasing expression of basal signature genes. However, it does not appear that PRRX2 is sufficient to drive a lineage switch from AR positive to AR low phenotype.
The higher expression of PRRX2 in AR low/DNPC tumors could be explained by co-regulation of PRRX2 by AR and the TGF-β pathway. Of note, PRRX2 is induced by TGF-β in several models (7). Interestingly, the TGF-β pathway is up-regulated after ENZ treatment and has also been shown to mediate ENZ resistance (24, 26, 32). Of note, there is an ongoing clinical trial with the combination of the TGF-β receptor inhibitor, Galunisertib, with ENZ for mCRPC patients (NCT02452008).
Previous studies have linked the oncogenic effects of PRRX2 to activation of the WNT β-catenin and EMT pathways (27, 40). In this study, we expanded the oncogenic role of PRRX2 to the regulation of the cell cycle and anti-apoptotic BCL2-mediated pathways. Interestingly, other groups have shown that BCL2 is upregulated after ENZ treatment and is over-expressed in AR-independent PC (35, 41). Here, we show that PRRX2 enhances the effect of ENZ-mediated BCL2 up-regulation. Of note, the combination of Venetoclax and ENZ is currently under clinical trial for patients with mCRPC (NCT03751436). Our study suggests that treatment with Palbociclib and Venetoclax will reverse enzalutamide resistance particularly in DNPC with high PRRX2 expression. Notably, the combination of Palbociclib, Venetoclax and the anti-hormonal agent fulvestrant is currently undergoing a clinical trial for ER+ breast cancer (NCT03900884) (42).
Supplementary Material
Statement of significance:
PRRX2 mediates enzalutamide resistance via activation of the E2F and BCL2 pathways, which can be targeted with CDK4/6 and BCL2 inhibitors to reverse resistance.
Acknowledgements:
We thank Dr. Matt Clutter from the Northwestern High-throughput Core facility for helping with the CRISPRa screen design and execution. Dr. Ching Man Wai, Dr. Matthew J. Schipma and Priyam Patel at the NUSeq Facility for ATAC-seq execution and data analysis. Carolina Ostiguin from the The Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core for help in flow cytometry analysis. The American Association of University Women (AAUW) - International Fellowship for their support. All the Abdulkadir lab members for their valuable discussion.
Funding:
National Cancer Institute P50CA180995, F30CA50248 and F30CA50196. Prostate Cancer Foundation Challenge Award.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest
References
- 1.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97. [DOI] [PubMed] [Google Scholar]
- 2.Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N Engl J Med. 2018;378(15):1408–18. [DOI] [PubMed] [Google Scholar]
- 3.Bhagirath D, Yang TL, Tabatabai ZL, Majid S, Dahiya R, Tanaka Y, et al. BRN4 Is a Novel Driver of Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer and Is Selectively Released in Extracellular Vesicles with BRN2. Clin Cancer Res. 2019;25(21):6532–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo H, Ci X, Ahmed M, Hua JT, Soares F, Lin D, et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat Commun. 2019;10(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32(4):474–89 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355(6320):84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juang YL, Jeng YM, Chen CL, Lien HC. PRRX2 as a novel TGF-beta-induced factor enhances invasion and migration in mammary epithelial cell and correlates with poor prognosis in breast cancer. Mol Carcinog. 2016;55(12):2247–59. [DOI] [PubMed] [Google Scholar]
- 8.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun. 2018;9(1):5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vatapalli R, Sagar V, Rodriguez Y, Zhao JC, Unno K, Pamarthy S, et al. Histone methyltransferase DOT1L coordinates AR and MYC stability in prostate cancer. Nat Commun. 2020;11(1):4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc. 2021;1(3):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez NF, Gundersen GW, Rahman A, Grimes ML, Rikova K, Hornbeck P, et al. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci Data. 2017;4:170151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stelloo S, Nevedomskaya E, Kim Y, Schuurman K, Valle-Encinas E, Lobo J, et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun. 2018;9(1):4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47(11):1346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajan P, Sudbery IM, Villasevil ME, Mui E, Fleming J, Davis M, et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol. 2014;66(1):32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3(9):1020–9. [DOI] [PubMed] [Google Scholar]
- 19.Takeda DY, Spisak S, Seo JH, Bell C, O’Connor E, Korthauer K, et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell. 2018;174(2):422–32 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Liu J, Cheng L, Li C, Zhang Z, Bai Y, et al. Inhibition of noncanonical Wnt pathway overcomes enzalutamide resistance in castration-resistant prostate cancer. Prostate. 2020;80(3):256–66. [DOI] [PubMed] [Google Scholar]
- 21.He Y, Wei T, Ye Z, Orme JJ, Lin D, Sheng H, et al. A noncanonical AR addiction drives enzalutamide resistance in prostate cancer. Nat Commun. 2021;12(1):1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanda JS, Awadallah WN, Kohrt SE, Popovics P, Cates JMM, Mirosevich J, et al. Increased nuclear factor I/B expression in prostate cancer correlates with AR expression. Prostate. 2020;80(13):1058–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Laere B, Oeyen S, Mayrhofer M, Whitington T, van Dam PJ, Van Oyen P, et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res. 2019;25(6):1766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He MX, Cuoco MS, Crowdis J, Bosma-Moody A, Zhang Z, Bi K, et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat Med. 2021;27(3):426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paller C, Pu H, Begemann DE, Wade CA, Hensley PJ, Kyprianou N. TGF-beta receptor I inhibitor enhances response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate. 2019;79(1):31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tewari AK, Cheung ATM, Crowdis J, Conway JR, Camp SY, Wankowicz SA, et al. Molecular features of exceptional response to neoadjuvant anti-androgen therapy in high-risk localized prostate cancer. Cell Rep. 2021;36(10):109665. [DOI] [PubMed] [Google Scholar]
- 27.Chai WX, Sun LG, Dai FH, Shao HS, Zheng NG, Cai HY. Inhibition of PRRX2 suppressed colon cancer liver metastasis via inactivation of Wnt/beta-catenin signaling pathway. Pathol Res Pract. 2019;215(10):152593. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Liu Y, Chen W, Fang Y, Xu H, Zhu HH, et al. TOP2Ahigh is the phenotype of recurrence and metastasis whereas TOP2Aneg cells represent cancer stem cells in prostate cancer. Oncotarget. 2014;5(19):9498–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–82. [DOI] [PubMed] [Google Scholar]
- 32.Song B, Park SH, Zhao JC, Fong KW, Li S, Lee Y, et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J Clin Invest. 2019;129(2):569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest. 2019;129(10):4492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su W, Han HH, Wang Y, Zhang B, Zhou B, Cheng Y, et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell. 2019;36(2):139–55 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun. 2018;9(1):3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Ying J, Li H, Zhang Y, Shu X, Fan Y, et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/beta-catenin signaling and silenced in common carcinomas. Oncogene. 2012;31(34):3901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaikhibrahim Z, Lindstrot A, Langer B, Buettner R, Wernert N. Comprehensive gene expression microarray analysis of Ets-1 blockade in PC3 prostate cancer cells and correlations with prostate cancer tissues: Insights into genes involved in the metastatic cascade. Int J Mol Med. 2011;27(6):811–9. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Xu H, Cheng C, Ji Z, Zhao H, Sheng Y, et al. Identification of a Zeb1 expressing basal stem cell subpopulation in the prostate. Nat Commun. 2020;11(1):706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lv ZD, Wang HB, Liu XP, Jin LY, Shen RW, Wang XG, et al. Silencing of Prrx2 Inhibits the Invasion and Metastasis of Breast Cancer both In Vitro and In Vivo by Reversing Epithelial-Mesenchymal Transition. Cell Physiol Biochem. 2017;42(5):1847–56. [DOI] [PubMed] [Google Scholar]
- 41.Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM, Xiang J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007;17(6):531–6. [DOI] [PubMed] [Google Scholar]
- 42.Whittle JR, Vaillant F, Surgenor E, Policheni AN, Giner G, Capaldo BD, et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res. 2020;26(15):4120–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data analyzed in this study were obtained from Gene Expression Omnibus (GEO). The RNA-seq and ATAC-seq generated in this study are available in GEO with the accession number: GSE199539. For analysis using the Stelloo RNA-seq and ChIP-seq data were obtained from the gene ontology database with the accession numbers: GSE120741 and GSE120738, respectively (14). The Pomerantz ChIP-seq data was obtained from GSE70079 (15). ChIP-seq data from the PC3 and LNCaP cell lines were obtained from (GSM3145503) (4) and (GSE14092) (16) respectively. Patient data of paired patients before and after ADT treatment was obtained from GSE48403 (17). All the other data supporting the findings of this study are available within the article and its Supplementary Information files and from the corresponding author upon reasonable request.