

Summary
Unlike most kinases, phosphatidylinositol 5-phosphate 4-kinase β (PI5P4Kβ) utilizes GTP as a physiological phosphate donor and regulates cell growth under stress (i.e., GTP-dependent stress resilience). However, the genesis and evolution of its GTP-responsiveness remain unknown. Here, we reveal that PI5P4Kβ has acquired GTP-preference via generating a short dual nucleotide-recognizing motif, GEA (Guanine Efficient-Association). Comparison of nucleobase recognition with 660 kinases and 128 G-proteins has uncovered that most kinases and PI5P4Kβ use their mainchain atoms for adenine recognition, while the sidechain atoms are required for guanine recognition. Mutational analysis of the GEA motif revealed that the acquisition of the GTP-reactivity is accompanied by an extended activity to ITP and XTP. Along with the evolutionary analysis data that points strong negative selection of the GEA motif, these results suggest that the GTP-responsiveness of PI5P4Kβ is evolved by a compromised trade-off between activity and specificity, underpinning the development of the GTP-dependent stress resilience.
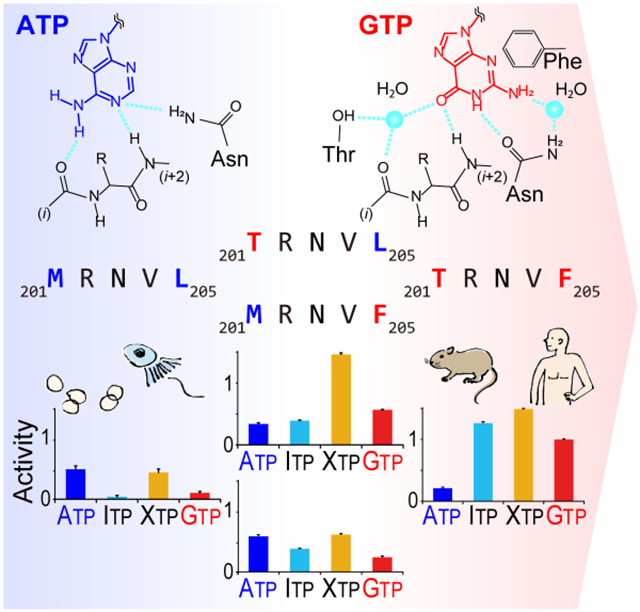
Graphical Abstract
eTOC blurb
PI5P4Kβ is a unique GTP-utilizing kinase that functions as an intracellular GTP sensor. Takeuchi et al. show that GTP binding requires the GTP/ATP dual recognition motif, named GEA, which emerged in a vertebrate. Further analyses reveal that limited substitutions in the GEA motif turned PI5P4Kβ into an unusual GTP-utilizing kinase.
Introduction
Kinases are essential for various cellular processes, including signal transduction, transcription, and metabolism. Protein kinases, which represent the largest superfamily consisting of over 500 genes in the human genome, share a conserved catalytic domain and structural motif that serves for adenosine triphosphate (ATP) recognition, followed by hydrolysis and phosphotransfer to their substrates (Bossemeyer, 1995; Endicott et al., 2012; Huse and Kuriyan, 2002; Shen et al., 2005; Shi et al., 2006; Taylor et al., 1992; Wang and Cole, 2014). On the other hand, phosphoinositide kinases (PI-kinases) and inositol phosphate kinases (IP-kinases, including inositol kinases) form distinct families that target the hydroxy groups of inositol moieties (Burke, 2018; Hammond and Burke, 2020). Although there is extraordinary diversity in their structure, substrate specificity, and participating pathways, all these kinases use ATP as the physiological phosphate donor (Clarke and Irvine, 2011; Loijens et al., 1996; Shears and Wang, 2019; Vadas et al., 2011). However, how and why kinases possess the ATP preference among other nucleoside triphosphates remain largely unknown.
Similar to this widely known ATP-preference of kinases, small G-proteins and heterotrimeric G-protein complexes are representative enzymes that exclusively use guanine nucleotides (i.e., guanosine triphosphate (GTP) and guanosine diphosphate (GDP)) to exert their biological functions. For G-proteins, evolutionally conserved G-domain functions to recognize GTP and GDP (Wennerberg et al., 2005; Wittinghofer and Vetter, 2011). The major determinant of the guanine base-binding specificity is the NKxD motif in the G-domain, where the conserved Asp side chain typically forms a bifurcated hydrogen bond with the guanine moiety (Vetter and Wittinghofer, 2001; Wittinghofer and Vetter, 2011).
Given that the overwhelmingly greater frequency of ATP-preferring kinases has raised the consensus that kinases function with ATP, the strong GTP-preference of phosphatidylinositol 5-phosphate 4-kinase β (PI5P4Kβ) was a surprising discovery (Sumita et al., 2016). PI5P4K, also called Type II PIPK, is a member of the phosphoinositide kinase superfamily. PI5P4K converts the second lipid messenger phosphatidylinositol 5-phosphate, PI(5)P, to phosphatidylinositol 4,5-diphosphate, PI(4,5)P2 (Rameh et al., 1997). Despite the higher intracellular concentration of ATP (1–5 mM) relative to GTP (0.1–0.5 mM), PI5P4Kβ exhibits a strong preference for GTP even in the presence of ATP and acts as an intracellular GTP sensor, which plays a critical role in metabolic adaptation and tumorigenesis (Sumita et al., 2016; Takeuchi et al., 2016). For the GTP recognition by enzymes, the guanine base cannot interact in the same way as the adenine base in the nucleotide-binding pocket due to the distinct arrangement of hydrogen donors and acceptors at the 1st and 6th positions of guanine (i.e., N(1) and O(6)) and adenine (i.e., NH(1) and NH2(6)) (Fig. 1). PI5P4Kβ achieved the dual interaction to ATP and GTP through two distinctive binding modes, at least in part by utilizing the short stretch of sequence containing the 201TRNVF205 sequence in its catalytic center, as shown in Fig. 2. Interestingly, the other isotypes of PI5P4K, PI5P4Kα and PI5P4Kγ, have unqualified features as a GTP sensor, even though they share high overall sequence similarity and can use GTP as phosphodonor (Bazenet et al., 1990; Sumita et al., 2016). Likewise, phosphatidylinositol 4-phosphate 5-kinase (PI4P5K), the closest subfamily of PI5P4K, uses ATP as the preferred phosphate donor (Jenkins et al., 1994; Sumita et al., 2016). Importantly, a recent report suggests that PI5P4K has diverged from the PI4P5K family at the ancestral lineage of Choanoflagellates and Filasterea (Khadka and Gupta, 2019). In the ancestorial linage, however, there was only one isotype of PI5P4K, until three isotypes (α, β, and γ) of PI5P4K appeared in vertebrates. These data argue that PI5P4K is originally an ATP-type kinase and becomes a GTP-sensor at some point in the evolution. However, how PI5P4Kβ evolutionarily acquired GTP preference to function as an intracellular GTP-sensor remains unclear.
Figure 1. Chemical structures of PNTs.

Chemical structures of ATP and GTP, along with other PNTs used in this study.
Figure 2. Distinct ATP- and GTP-binding modes of PI5P4Kβ.

(A) ATP- and (B) GTP-binding modes of PI5P4Kβ determined in our previous study (Sumita et al., 2016). The hydrogen bonds between PNTs and PI5P4Kβ are shown in red dotted lines. PDB ID for the ATP- and GTP-bound structures are 6K4H and 6K4G, respectively. See also Table S6.
In the present study, we performed a rigorous comparison of nucleotide-bound structures of 660 kinases and 128 G-proteins available in the protein data bank (PDB) (Berman et al., 2003), which uncovers a shared ATP recognition mode among kinases and PI5P4Kβ, and the uniqueness of GTP recognition by PI5P4Kβ. A comprehensive biochemical and structural analysis of PI5P4Kβ, using the structurally deduced rational mutants and the purine nucleoside triphosphate (PNT) analogs (Fig. 1), enabled us to delineate further a few critical substitutions of amino acid residues in a short stretch of dual nucleotide recognizing motif, GEA (Guanine Efficient-Association). Along with the evolutional conservation of the GEA sequence among the vertebrates, our results uncover a compromised trade-off relationship between the GTP-dependent activity and nucleotide specificity of PI5P4Kβ, as an evolutionary basis for PI5P4Kβ to function as an intracellular GTP sensor.
Results
The ATP-base recognition mode of PI5P4Kβ is shared among other kinases.
The catalytic domain of the kinases consists of two lobes harboring an ATP-binding site at the hinge region (Figs. 3A and S1A). To grasp the consensus ATP-base recognition mode of the human kinome and compare them with that of PI5P4Kβ (Fig. 2), we analyzed the amino acid residue(s) responsible for the adenine base recognition in nucleotide-bound structures for 600 protein kinase and 10 PI-kinase structures in the PDB. Besides, 50 structures of IP-kinases are included. Note that we focused on how the proteins recognize the nucleotide-base; thus, other parts of ATP and the overall folding of the ATP-recognizing kinases are not considered. Since some PDB deposited structures contain multiple ATP molecules in the coordinates, the analysis identified interactions to 968 ATP and its analogs in the 660 kinase structures (Fig. 3B, Table S1, and Data S1). This analysis revealed hydrogen bond distances between the adenine base and protein atoms with the median values of 3.0 Å and 2.9 Å for the N(1)-(i+2)th and NH2(6)-ith distances, respectively (Fig. 3B, Table S1, and Data S1). There are 10 cases that the distances exceed those for typical hydrogen bonds; these are mostly associated with low resolution and/or high local-temperature factors. Also, an exceptional case is observed in PIM1 kinase, which possesses Pro residue in the (i+2)th position and cannot form a hydrogen bond to N(1) (Fig. S1A). Excluding the above exceptions, the N(1) and NH2(6) positions of ATP adenine base form two hydrogen bonds between the mainchain amide and carbonyl groups from the (i+2)th and ith residues, respectively.
Figure 3. Nucleotide-base binding by kinases and G-proteins.

(A and C) Schematic representations of typical hydrogen-bond interactions between nucleotide bases and proteins are shown for (A) kinases and (C) G-proteins. Representative nucleotide-complex structures from major protein families are also shown with the PDB IDs. The hydrogen bonds are shown in red dotted lines. (B and D) Histogram of hydrogen bond distances between (B) kinases and adenine bases, and between (D) G-proteins and guanine bases. The interactions are listed in supplemental materials for kinases (Data S1) and G-proteins (Data S2). See also Fig. S1 for structural figures of other kinases and G-proteins, and Table S1 and S2 for the list of PDB ID, respectively.
Next, we assessed the structure of the ATP-binding mode of PI5P4Kβ (Fig. 2A). Consistent with the ATP-mode of many other kinases (Figs. 3A and S1A), PI5P4Kβ uses mainchains—the amide group of Val-204 and the carbonyl oxygen of Arg-202—to recognize the N(1) and NH2(6) of ATP, respectively. The hydrogen-bond distances between N(1) and NH2(6) to the respective sites are 3.3 Å and 3.4 Å, both of which are 0.3 Å and 0.5 Å longer than the aforementioned median values. The N(1) of ATP also forms a hydrogen bond with the Nδ of Asn-203 (2.9 Å). The related ATP kinase PI4P5Kα also interacts with ATP in the same binding mode (Fig. 3A) (Muftuoglu et al., 2016). These results reveal that the ATP-binding mode of currently available kinase structures and PI5P4Kβ share the common mechanism in which they utilize the mainchain amide and carbonyl groups to recognize N(1) and NH2(6) positions of an adenine base, respectively.
The GTP-binding mode of PI5P4Kβ by the GEA motif is distinctive from G-protein.
Next, we exploited the same strategy and analyzed the amino acid residue(s) that are responsible for the guanine base recognition and their distance to the guanine base, using 128 G-proteins, such as small-GTPases and α-subunit of heterotrimeric G proteins, in the PDB (Figs. 3C, 3D, and S1B, Table S2, and Data S2). In G-proteins, the N(1) and NH2(2) of the guanine base are simultaneously recognized by the sidechain carboxylate of Asp in the NKXD motif (Vetter and Wittinghofer, 2001; Wittinghofer and Vetter, 2011), the medians of the hydrogen bond distances are 2.8 Å and 2.9 Å, respectively (Fig. 3D). In most cases, the N(7) of the guanine base forms a hydrogen bond with the Nδ of Asn in the NKXD motif (distance median, 3.1 Å, Fig. 3D). In addition, O(6) forms a hydrogen bond(s) with a neighboring i+1th Lys and/or a remote mainchain amide group(s). Intriguingly, the number of interactions between O(6) and neighboring atoms is significantly larger than other GTP atoms; O(6) has 4 interactions on average.
Like G-proteins, PI5P4Kβ also uses sidechain amino acids to form hydrogen bonds to N(1), NH2(2), and O(6) of the guanine ring (Asn-203 for N(1) and NH2(2), and Thr-201 for O(6)). However, the interacting residues are distinct from those of the G-protein; PI5P4Kβ utilizes the TRNVF sequence (residues 201-205 in humans) to recognize GTP (Fig. 4). Asn-203 in the 201TRNVF205 sequence is structurally located at the corresponding position of the conserved Asp residue of the G-protein, as its Oδ and Nδ atoms form direct and indirect hydrogen bonds with N(1) and NH2(2), respectively (Fig. 2B). The distance of the direct hydrogen bond between Asn-203 Oδ and N(1) was 2.7 Å, comparable to that of the NKXD motif and N(1). Another characteristic feature of PI5P4Kβ is a hydrogen-bond network around O(6) of the guanine base involving Thr-201, Arg-202, and Val-204 in the TRNVF sequence and a water molecule (Fig. 2B). The GTP interactions are enabled by a 1.5 Å shift of the base moiety relative to the ATP and the presence of a water molecule that mediates hydrogen bonds to the O(6) from Thr-201 Oγ and Arg-202 carbonyl. As mentioned above, the O(6) atom of the guanine base is extensively recognized in G-proteins; however, water mediates hydrogen bonds are rather rare (16 cases relative to 1167 direct interactions).
Figure 4. Sequence alignment of PI5P4K and PI4P5K family proteins.

The GTP-recognizing TRNVF sequences and the ATP-recognizing MNNψL sequences are colored in red and blue, respectively. Asterisks indicate the positions Thr-201, Asn-203, and Phe-205 in human PI5P4Kβ.
Together, the results of structural comparisons of human kinome and G-protein with PI5P4Kβ reveal three critical features of PI5P4Kβ: 1) Because the arrangement of a hydrogen donor and acceptor in the guanine base differs from those of the adenine, PI5P4Kβ has a specific GTP-binding mode using both sidechains and mainchains of the 201TRNVF205 sequence (Fig. 2B). We designate this dual nucleotide recognizing motif as GEA (Guanine Efficient-Association). 2) The GTP-binding mode of PI5P4Kβ is, thus, different from that of the G-proteins, which utilize the conserved N/TKXD motif for guanine base recognition (Figs. 2B and 3C). 3) While G-proteins typically have pico to nanomolar affinity to GTP, the affinity of PI5P4Kβ to GTP seems to be much weaker, as its KM value is only ~100 μM (Sumita et al., 2016). The indirect hydrogen bond between the Asn-203 Nδ and NH2(2), mediated by a water molecule, could account for the weaker affinity of PI5P4Kβ compared to that of the G-protein.
The strong contribution of the hydrogen-bond network around O(6) for the GTP recognition by PI5P4Kβ.
To further delineate the relative contribution of each amino acid in the GEA motif, we took advantage of the fragment molecular orbital (FMO) calculation, which enables deducing the ab initio total energies of molecules in superior accuracy by dividing molecules into fragments (Fedorov and Kitaura, 2009; Fedorov et al., 2012; Nakano et al., 2002; Tanaka et al., 2014). For the FMO calculation, the crystal structure of the PI5P4Kβ-GTP complex (PDB: 6K4G) was fragmented into amino acid units at bonds between the C and Cα atoms of the main chain; GTP was fragmented into the base, sugar, and phosphate groups. As shown in Fig. 5, the results of FMO analysis indicate that both Val-204 and the water molecule held by Thr-201 possess an energetically favored interaction with O(6) of the guanine base. It is worth noting that the deduced interaction is even stronger than the aforementioned Asn-203 plus water interactions with the NH2(2) position. The shift of the base position promotes a formation of aromatic-aromatic interactions with Phe-205 in the GEA motif (Fig. 5A), which is unique to guanine base recognition. Interestingly, the guanine and adenine base recognition of PI5P4Kβ and casein kinase II (CKII)—a known bimodal kinase that equally uses GTP and ATP (Niefind et al., 1999)— has similarity in the hydrogen-bond networks around N(1) and O(6) as well as the 1.5 Å shift of the guanine base compared to that of the adenine ring (Fig. 2B and S2) (Niefind et al., 1999); however, CKII uses only mainchain atoms for the base recognitions. The results of the FMO analysis further verified the significant contribution of the hydrogen-bond network around O(6) for guanine base recognition by PI5P4Kβ.
Figure 5. FMO calculation of the PI5P4Kβ-GTP complex.

(A) The energetic contributions of each residue to PI5P4Kβ-guanine base interaction are indicated. The energetic contributions of two water molecules bound to the NH2(2) and O(6) positions of the guanine base moieties are also shown. (B) The energetic contributions of each residue of PI5P4Kβ and GTP to the interaction with water molecules that are bound to the (top) NH2(2) and (bottom) O(6) positions, respectively, of guanine base moieties are indicated. (C) Interacting residues are shown (stick model in magenta) in the PI5P4Kβ-GMPPNP complex structure determined in our previous study (15) (PDB ID 6K4G). The water molecules that interact with NH2(2) and O(6) positions are shown in orange and cyan, respectively.
NH(1) and O(6) are the minimal requirements for the GTP-binding mode.
The results of structural analyses of the PI5P4Kβ–GTP interactions suggest the critical importance of sidechains of Thr-201, Asn-203, and Phe-205 to recognize the guanine base (Figs. 2B and 5). To further investigate the structural requirements of nucleotide bases in the ATP- and GTP-type interactions, we exploited 10 PNTs with different functional groups or protonation states at the 1st, 2nd, and 6th positions of the purine base (Fig. 1). The effects of the differences in functional groups to recognition and activity were analyzed using X-ray crystallography and the nuclear magnetic resonance (NMR)-based nucleotide hydrolysis assay, which faithfully recapitulates the characteristic GTP-preference of the kinase (Sumita et al., 2016). In the NMR analysis, recombinant PI5P4Kβ was incubated with the PNTs, and their conversion to diphosphate form was quantified (Fig. 6). The concentration of PNTs was set to 250 μM for the sake of comparison, which falls within the physiological concentrations of GTP (Traut, 1994). While the GTP concentrations were not optimal for the GTP-hydrolysis as the nucleotide concentration is significantly lower than its KM value (Table S3), PI5P4Kβ still hydrolyzed GTP 13 times more efficiently than ATP as previously reported (Fig. 6A)(Sumita et al., 2016).
Figure 6. PNT hydrolysis activity of PI5P4Kβ.

(A) PNT hydrolysis activity of PI5P4Kβ. The PNT hydrolysis activity (nmol/hr) of PI5P4Kβ was calculated from the signal intensities of diphosphorylated and triphosphorylated nucleotides NMR signals after the reaction (see Star Methods and panel B). In the PNT-hydrolysis assay, 250 μM PNTs were mixed with 2 μM PI5P4Kβ. The average values from three experiments are shown with error bars (standard deviations; S.D.). The values from each experiment are shown as gray dots. Highly hydrolyzed nucleotides (> 0.1 of the ratios) are indicated by red. (B) Representative spectra in the NMR-based PNT hydrolysis assay. The intensities of H8 protons of diphosphorylated (D)/triphosphorylated (T) nucleotide signals are quantified from the spectra. See also Fig. S3.
Furthermore, the NMR results were verified by the competition profile of the in vitro kinase assay using radiolabeled 32P-GTP and a series of non-radiolabeled PNTs as competitors (Fig. S3). The NMR results revealed that PI5P4Kβ possesses substantial hydrolysis activity with inosine triphosphate (ITP), xanthosine triphosphate (XTP), 6-thio-GTP, and 2-amino-ATP (2a-ATP) (Fig. 6A). Consistently, the results of the competitive in vitro kinase assay showed that the GTP-dependent PI(5)P phosphorylation activity was strongly inhibited by ITP, XTP, and 6-thio-GTP (Fig. S3). The results indicate that NH2(2) of GTP is dispensable for the binding to PI5P4Kβ, since both ITP and XTP lack the NH2(2) moiety. On the other hand, O(6) appeared to be required for the binding. ITP and XTP, both of which have the O(6) moiety, showed substantial hydrolysis by PI5P4Kβ (Fig. 6). In contrast, O6-methyl-GTP (O6-me-GTP) and 2-amino-6Cl-purine triphosphate (2a-6Cl-PTP), which lack the O(6) moiety, showed only low hydrolysis activity. In line with this notion, 6-thio-GTP, which possesses a structurally and electrostatically similar sulfate instead of oxygen in the 6th position, can also be utilized by PI5P4Kβ, but significantly low level, indicating a strict requirement of an oxygen atom at the position (Fig. 6). Given the large energetic contribution of the O(6) in the GTP-PI5P4Kβ interactions (Fig. 5), the activity of PI5P4Kβ extends beyond GTP to the purine nucleoside triphosphate with the O(6) moiety probably due to the strong dependence on the O(6) interaction in the nucleotide recognition.
Crystal structures of PI5P4Kβ-ITP, PI5P4Kβ-XTP, and PI5P4Kβ-2a-ATP complexes support the notions deduced from the NMR analysis (Fig. 7). The 2a-ATP binds to PI5P4Kβ with a binding mode similar to that of ATP (Figs. 7A and S4A), suggesting that N(1) and NH2(6) are essential for the ATP-mode binding. The NH2(2), an additional functional group unique to 2a-ATP, forms an additional van der Waals interaction with Phe-205, probably causing the stronger binding of 2a-ATP than ATP. Both ITP and XTP utilize NH(1) and O(6) groups to interact with PI5P4Kβ, and their binding modes are essentially the same as that of the GTP (Figs. 7B and C, and S4). Interestingly, XTP has an alternative binding mode (Figs. 7D and S4D). In the alternative binding mode, the base of the XTP is flipped by 180° respective to the first one (Fig. 7C and D). Even after the base flip, XTP forms a hydrogen-bond network similar to the first one; the N(1) occupies an almost identical position within 1 Å difference, and the positions of O(2) and O(6) are merely swapped. As a result, XTP can bind to PI5P4Kβ in two different ways. In either case, the NH group interacts with Asn-203 Oδ, and O(6) or O(2) forms bifurcated hydrogen bonds to Val-204 and a water molecule, which in turn forms a hydrogen bond with Oγ of Thr-201 (Fig. 7C and D). These alternate interactions of XTP clearly show that NH(1) and O(6) are the minimal requirements for the GTP-binding mode.
Figure 7. 2a-ATP, ITP, and XTP binding modes of PI5P4Kβ.

(A) Interaction of 2a-ATP to PI5P4Kβ (PDB ID 7EM3). (B) Interactions of the ITP to PI5P4Kβ(PDB ID 7EM1). (C and D) Interactions of XTP to PI5P4Kβ (C) in the mode similar to GTP (GTP-binding mode) and (D) in the mode unique to XTP (XTP-binding mode) (PDB ID 7EM2). Red dotted lines represent hydrogen bonds between the PNTs and PI5P4Kβ. As for the ITP complex, the water position visible with a lower σ value is also indicated with a gray circle. See also Fig. S4 for overlay of the 2a-ATP, ITP, and XTP binding modes with the ATP or GTP binding modes. See also Table S6 for structural statistics.
Functional dissection of the GEA-motif residues for ATP and GTP recognition.
Next, functional contributions of critical side chains of PI5P4Kβ for ATP- and GTP-dependent kinase and hydrolysis activity were examined using amino acid substitutions. We compared the effect of mutations of Thr-201, Asn-203, and Phe-205 in the GEA motif (Fig. 8 and Table S3-4). Arg-202 and Val-204 are not mutated as these sidechains are pointing to the opposite side of the nucleotide-binding pocket. To assess the evolutionary significance of amino acid substitutions, Thr-201 and Phe-205 were substituted to Met and Leu in PI4P5K (or Type I PIPK) (Fig. 4), respectively. We have already reported that the T201M (PI5P4KβT201M) and F205L (PI5P4KβF205L) mutations decreased GTP-dependent kinase activity without losing ATP-dependent kinase activity (Sumita et al., 2016). The decreased GTP-dependent kinase activity of PI5P4KβT201M has been explained by the loss of the hydrogen-bond network around O(6) by the mutation (Fig. 2B), which is reflected in the larger KM value of the GTP hydrolysis activity (Table S3). In the case of PI5P4KβF205L, the loss of the π-π interaction between the guanine base and the aromatic ring of Phe-205 caused the reduction of the GTP-dependent kinase activity (Sumita et al., 2016). As a reflection, the reduction of the hydrolysis activity is mainly due to the reduction of the catalytic rate (kcat) (Table S4), which might indicate that PI5P4KβF205L cannot hold the nucleotide to productive conformation as efficient as WT. In contrast, the ATP-dependent kinase and hydrolysis activity was unaffected by the F205L mutation (Sumita et al., 2016).
Figure 8. PNT hydrolysis activities of PI5P4Kβ mutants.

(A) PNT-hydrolysis activities of the WT PI5P4Kβ and T201M, N203D, N203A, F205L, and T201M_F205L double mutants. The PNT hydrolysis activity (nmol/hr) of PI5P4Kβ was calculated from the signal intensity of diphosphorylated nucleotides after the reaction (see Materials and Methods section for details). For the PNT-hydrolysis assay, 250 μM PNTs were mixed with 2 μM PI5P4Kβ. The average values from the three experiments are shown with error bars (S.D.). The values from each experiment are shown as gray dots. (B) The kcat/KM values of PNT hydrolysis activities of the WT PI5P4Kβ and T201M, N203D, F205L, and T201M_F205L double mutants. The values are calculated from KM and kcat values shown in Table S3 and S4, respectively. N.D.: not determined. For the determination of KM and kcat values, the reactions were carried out for different concentrations of PNTs from 63.5 μM, 125 μM, 250 μM, 500 μM, 1 mM, and 2 mM.
To further probe the functional role of the GEA motif, we mutated Asn-203, which is required for ATP and GTP-binding and the only invariant residue in the base-recognition loop of PI4P5K and PI5P4K. The mutation of Asn-203 to alanine, N203A (PI5P4KβN203A), markedly reduced the hydrolysis activity for both ATP and GTP (Fig. 8). Since Asn-203 plays a role of a hydrogen donor for N(1) of the ATP (Fig. 2A), the reduced ATP-hydrolysis activity of N203A can be explained by the loss of the hydrogen bond with N(1) of the adenine ring. PI5P4KβN203D showed a substantial decrease in the GTP-hydrolysis activity. The loss of the hydrogen bond between Nδ of Asn-203 and the carbonyl oxygen of Arg-145 seems to destabilize the χ1 rotameric state of Asn-203, which may, in turn, induce dissociation of the water molecule bound to NH2(2) (Fig. S5). Consistent with these results, the FMO calculation indicates the importance of the hydrogen bond between guanine base and Asn-203; approximately 1/4 of the interaction energy between the nucleotide base and PI5P4Kβ is contributed by this interaction (Fig. 5). Since Asn residue can play a role for both hydrogen donor and acceptor, Asn-203 is a critical residue for interacting both ATP and GTP to recognize the atom at the 1st position to hold these nucleotides to the proper position to be catalyzed. The increased activity to ITP in PI5P4KβN203D cannot be fully deduced from the structure (Fig. S6) as it is mainly due to the elevation of the kcat value (Table S4), and the some of the catalytically important loops are missing in our structure. The KM value of the XTP is increased in PI5P4KβN203D (Table S3). Since XTP is more anionic in neutral pH than GTP and ITP, the N203D mutation might cause an electrostatic repulsion that prevents the binding of XTP to the nucleotide-binding pocket. In the line of this notion, the rotameric state of Asp-203 of PI5P4KβN203D in the XTP bound state is different from those of wild type PI5P4Kβ in the XTP bound state as well as those of PI5P4KβN203D in the ITP bound state (Fig. S7).
A trade-off between activity and specificity in evolutionary steps toward GTP recognition.
Thr-201 and Phe-205 in PI5P4Kβ are substituted to Met and Leu, respectively, in PI4P5K, while Asn-203 is conserved in both PI5P4Kβ and PI4P5K. To further analyze the way that PI5P4Kβ evolutionarily acquired the GTP-dependent activity, we prepared the PI5P4KβT201M_F205L double mutant, which has the same amino acids as ATP-dependent ancestral kinase, PI4P5K (i.e., Met-201 and Leu-205). GTP, XTP, ITP, and ATP hydrolysis activities of the PI5P4KβT201M_F205L double mutant were compared with PI5P4KβT201M and PI5P4KβF205L mutants, which reproduces the evolutionary steps toward the WT PI5P4Kβ. It should be noted that the crystal structures of the mutants with PNTs are similar to those of the WT PI5P4Kβ (Fig. 8 and 9).
Figure 9. A trade-off between activity and specificity, and evolution of PI5P4Kβ.

(A) Normalized GTP, XTP, ITP, and ATP hydrolysis activity of each mutant compared to WT (for GTP, XTP, and ITP) and PI5P4KβT201M_F205L. The average values taken from Figure 8A were used. (B) Nucleotide fraction of each site that encodes a GEA motif of PI5P4Kβ. The species used are shown in Figure 4 and are listed in Star Methods.
The hydrolysis activities of each mutant at 250 μM nucleotide concentration are shown in Fig. 8. These are parallel to the kcat/KM values of the hydrolysis activities determined from the PNT concentration dependence (Fig. S5). In Fig. 9A, the hydrolysis activities to GTP, XTP, and ITP are normalized against the WT PI5P4Kβ, while that of ATP was normalized against the PI5P4KβT201M_F205L double mutant. While the double mutant largely abolished the activity to GTP, ITP, and XTP, it showed strong ATP-hydrolysis activity; consequently, the PI5P4KβT201M_F205L mutant is reverted from the GTP-dependent kinase to its ancestral ATP-dependent kinase at least for the nucleotide-binding and hydrolysis activity.
Intriguingly, acquisition of the Thr residue at 201st position (see PI5P4KβF205L) makes the kinase more active to GTP, while largely retaining the activity to ATP. However, the gain in the GTP-hydrolysis activity comes with a much stronger activity to XTP. The acquisition of Phe at the 205th position (see PI5P4KβT201M) did not reduce its hydrolysis activity to ATP and slightly enhanced the activity to GTP. Again, the hydrolysis activity to XTP and ITP was also increased. The strongest GTP-dependent kinase activity and GTP-hydrolysis activity were achieved when both Thr-201 and Phe-205 were acquired; however, the resultant WT PI5P4Kβ still retains substantial activity to XTP and ITP. Nevertheless, as shown in the inhibition profile of the GTP-dependent kinase activity by low (<16 μM) concentration of ITP and XTP (Fig. S3), negligible cellular concentrations of these nucleotide triphosphates might not affect the GTP-dependent kinase activity of the WT PI5P4Kβ in living cells.
Evolutional selection for the emergence of the GEA motif.
A recent study has proposed that PI5P4K has diverged from the PI4P5K family at the ancestral lineage of Choanoflagellates and Filasterea (Khadka and Gupta, 2019). Given that human PI4P5Kα is ATP-preferential kinase (Loijens and Anderson, 1996; Sumita et al., 2016), it is conceivable that PI5P4K was originally an ATP-type kinase and became a GTP-sensor at some point in the evolution. Thus, next, we studied the genesis and evolution of the GEA motif. We estimated evolutional selection and retention of the amino acid sequence of PI5P4Kβ from genomic sequences and codon usage of 12 diversified vertebrate species shown in Fig. 4 using the Single-Likelihood Ancestor Counting (SLAC) program in Datamonkey (Pond and Frost, 2005). We found that codons corresponding to Thr-201 through Phe-205 amino acid positions have two to six synonymous mutations, while no mutations change the amino-acid sequences of proteins (Fig. 9B and Table S5). Those results suggest that the GTP-recognizing sequence, 201TRNVF205, is under negative selection.
In contrast, among the ancestral ATP-dependent kinase, PI4P5Ks, the MNNψL sequence is conserved, where "ψ" is donated for branched-chain amino acids (Fig. 4). These analyses indicate that PI5P4Kβ has evolved to acquire the atypical GTP-binding mode by introducing a few substitutions in the MNNψL sequence, without largely changing the canonical ATP-binding mode (Figs. 2A and 3A). Especially, the introduction of Thr-201 and Phe-205 and the retention of Asn-203, whose sidechains establish the interactions with the guanine base, would be of importance due to their unique contributions to the guanine-base recognition in a way that is distinct from G-proteins (Figs. 2B and 3C). Considering the compromised trade-off between activity and specificity (Fig. 9), these results reveal how evolutional selection of the GEA motif occurred to gain a new activity for the GTP-responsive PI5P4Kβ signaling.
Discussion
Unlike most kinases, PI5P4Kβ utilizes GTP as a physiological phosphate donor, and this unique GTP preference makes PI5P4Kβ an intra-cellular GTP sensor that regulates GTP-dependent stress resilience. Here we analyzed how PI5P4Kβ evolutionarily acquired the GTP preference contrary to the prevailing rule that kinase utilizes ATP. Based on the inductive rule of the nucleotide recognition derived from a comprehensive analysis of nucleotide bound tertiary structures, we have concluded that the ATP binding mode of PI5P4Kβ is similar to other kinases, while the GTP binding mode is unique and not the canonical one found in the G-proteins (Figs. 2 and 3).
Role of two evolutionarily acquired sidechains in the GEA motif: a trade-off between GTP-dependent activity and nucleotide specificity.
Evolutionarily, PI5P4Kβ is considered to have diverged from the ATP-type kinase PI4P5K (Khadka and Gupta, 2019). Our analyses provide a novel insight into the molecular evolution by which PI5P4Kβ acquired a GTP preference. Human PI5P4Kβ has the GEA (201TRNVF205) motif as a dual base-recognition fragment that is conserved among PI5P4Kβ from very ancient vertebrates, such as the elephant sharks and coelacanths, to humans (Fig. 4) (Sumita et al., 2016). While all residues in the GEA motif are involved in the specific interaction with the guanine base in PI5P4Kβ (Fig. 2B), PI5P4Kβ has evolved functional roles for the sidechains of the 1st and 5th residues of the GEA motif (201TRNVF205) to achieve the interaction with the guanine base necessary to function as a GTP sensor (Figs. 2B and 5). Our biochemical analysis revealed an unexpected activity of WT PI5P4Kβ extending to ITP and XTP (Fig. 6); these PNTs structurally share O(6) as a proton acceptor (Fig. 1). The FMO analysis revealed that the hydrogen-bonding interactions to the O(6) position by Val-204 HN and a water molecule stabilized by Thr-201 play critical roles in the base recognition (Fig. 5). Indeed, PI5P4KβT201M shows a substantially reduced hydrolysis activity for GTP and its analogs (Figs. 8). The O(6) position of the nucleotide bases is the site that chemically differentiates ATP from GTP; thus, the strong interaction with PI5P4Kβ is reasonable for its acquisition of the GTP preference. Note that the structures of the nucleotide-base binding pocket are mostly invariant among PNT bound states, except some rotameric changes in sidechains are observed (Thr201 and Asn203; Figs. 2, 7, S4, S6 and S7).
However, the benefit only comes with extended activity to XTP and ITP (Fig. 6). Phe-205 also contributes to the extended activity via π-π interaction. These results suggest a trade-off between the GTP-dependent activity and nucleotide specificity in the WT PI5P4Kβ. The kinase seems to benefit from the GTP-dependent function without much sacrifice, as the intracellular concentrations of ITP and XTP are negligibly small in vivo. Thus, the extended biochemical activity on XTP and ITP is not detrimental to the biological function of PI5P4Kβ.
Due to higher cellular concentrations of ATP than GTP, half of the PI5P4Kβ activity still originates from ATP at physiological nucleotide concentrations (Sumita et al., 2016). In addition, the T201M_F205L double mutant of PI5P4Kβ, which has the nucleotide interaction sequence closer to its ancestral kinase PI4P5K, showed much stronger ATP-hydrolysis activity, while almost completely abolishing GTP- hydrolysis activity (Fig. 8 and 9). Although the role of the “basal” ATP-dependent activity of PI5P4Kβ is still unclear, it might contribute as a buffer that attenuates small changes in nucleotide concentrations, including ITP and XTP, and provides stability to the energy homeostasis system.
Non-detrimental mutations provide a smooth transition in a functional evolution.
PI5P4Kβ shares the kinase fold with other PI-kinases that utilize ATP as the phosphodonor. In the course of evolution, PI5P4Kβ made a compromise to acquire the cellular GTP-sensing function under the given environment and the restraint of the phosphoinositide kinase fold. Considering that the GEA motif of the PI5P4Kβ isotype is conserved among vertebrates under strong negative selection (Figs. 4 and 9B), this compromise appears to be a favored evolutionary solution for the GTP recognition within the premised structure and sequence of the phosphoinositide kinases. It should be noted that neither PI5P4KβT201M nor PI5P4KβF205L loses its original ATP-dependent activity (Fig. 8). Therefore, having a semi-optimal sequence between the GTP-recognizing 201TRNVF205 sequence and the original MNNψL sequence might not be detrimental. This notion would be supported by the fact that we have successfully established a cell line that only has PI5P4KβF205L in our previous paper (Sumita et al., 2016). Such non-detrimental cryptic mutations might allow a smooth evolutional transition of an ATP-dependent kinase to a GTP-sensing kinase. It should also be noted that the highest GTP-dependent activity was observed for the WT PI5P4Kβ. Given the importance of the GTP concentration in controlling energy metabolism and proliferation under stress conditions (Sumita et al., 2016), establishing a new mode of signaling that utilizes GTP would be crucial for the resilience of energy homeostasis in higher animals. We showed that the evolution of PI5P4Kβ or, more specifically, two critical amino acid substitutions in the GEA motif were vital to achieve this evolutionarily important step. Thus, the results also illuminate that a new form of biological resilience could be accomplished by acquiring a few cryptic mutations that are already implemented in genome sequences but have not yet manifested their functions.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Koh Takeuchi (koh-takeuchi@mol.f.u-tokyo.ac.jp).
Materials availability
This study did not generate new unique reagents. Plasmids produced in this study will be available upon request.
Data and code availability
Atomic coordinates and structure factors have been deposited in the Protein Data Bank. Accession codes are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21 (DE3) | Nippon Gene | 312-06534 |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth with agar (Miller) | Sigma-Aldrich | L3147 |
| Kanamycin sulfate | Fujifilm Wako | 113-00343 |
| LB Broth (Miller) | Sigma-Aldrich | L3522 |
| Isopropyl-β-D-thiogalactoside | Sigma-Aldrich | I5502 |
| d-myo-phosphatidylinositol 5-phosphate | Echelon Biosciences | P-0016d |
| 1,2-dipalmitoyl-phosphatidylserine | Echelon Biosciences | L-3116 |
| GTP | TriLink BioTechnologies | N-1512 |
| ATP | TriLink BioTechnologies | N-1510 |
| ITP | TriLink BioTechnologies | N-4017 |
| XTP | TriLink BioTechnologies | N-1023 |
| 6-thio-GTP | TriLink BioTechnologies | N-8007 |
| 6-OMe-GTP | TriLink BioTechnologies | N-1031 |
| 2a-6Cl-PNP | TriLink BioTechnologies | N-1002 |
| 2a-ATP | TriLink BioTechnologies | N-1001 |
| 6Cl-PNP | TriLink BioTechnologies | N-2009 |
| 2-oxo-ATP | TriLink BioTechnologies | N-1099 |
| Deposited data | ||
| Crystal structure of WT-GMPPNP complex | This Work | 6K4G |
| Crystal structure of WT-AMPPNP complex | This Work | 6K4H |
| Crystal structure of WT-ITP complex | This Work | 7EM1 |
| Crystal structure of WT-XTP complex | This Work | 7EM2 |
| Crystal structure of WT-2a-ATP complex | This Work | 7EM3 |
| Crystal structure of F205L-ITP complex | This Work | 7EM4 |
| Crystal structure of F205L-XTP complex | This Work | 7EM5 |
| Crystal structure of N203D-ITP complex | This Work | 7EM6 |
| Crystal structure of N203D-XTP complex | This Work | 7EM7 |
| Crystal structure of T201M-2a-ATP complex | This Work | 7EM8 |
| Oligonucleotides | ||
| T201M-f 5'-ATGGTGGTTATGAGGAACGTGTTC-3’ | Eurofins | N/A |
| T201M-r 5’-CACGTTCCTCATAACCACCATGTA-3’ | Eurofins | N/A |
| N203D-f 5’-GGTGGTTACCAGGGACGTGTTCAGCCATC-3’ | Eurofins | N/A |
| N203D-r 5’-GATGGCTGAACACGTCCCTGGTAACCACC-3’ | Eurofins | N/A |
| N203A-f 5’-CATGGTGGTTACCAGGGCGGTGTTCAGCCATCGG-3’ | Eurofins | N/A |
| N203A-r 5’-CCGATGGCTGAACACCGCCCTGGTAACCACCATG-3’ | Eurofins | N/A |
| F205L-f 5'-CCAGGAACGTGTTGAGCCATCGG-3’ | Eurofins | N/A |
| F205L-r 5’-ATGGCTCAACACGTTCCTGGTAACC-3’ | Eurofins | N/A |
| Software and algorithms | ||
| Pymol (Version 2.4.0) | Schrödinger | https://pymol.org/ |
| Topspin | Bruker | https://www.bruker.com/en/products-and-solutions/mr/nmr-software.html |
| Origin 2020 | Origin Lab Corporation | https://www.originlab.com/ |
| XDS | (Kabsch, 2010) | https://xds.mr.mpg.de/ |
| XSCALE | (Kabsch, 2010) | https://xds.mr.mpg.de/ |
| PHENIX | (Liebschner et al., 2019) | https://phenix-online.org/ |
| phenix.refine | (Liebschner et al., 2019) | https://phenix-online.org/ |
| PyMOL | Schrödinger, LLC. | https://pymol.org/2/ |
| Molecular Operating Environment | Chemical Computing Group, | https://www.chemcomp.com/Products.htm |
| ABINIT-MP | (Mochizuki et al., 2019; Nakano et al., 2006) | https://www.hpci-office.jp/pages/e_appli_abinit-mp |
| L-INS-i | MAFFT version 7.205 | https://mafft.cbrc.jp/alignment/software/ |
| TranslatorX | (Abascal et al., 2010) | https://translatorx.org/ |
| Single-Likelihood Ancestor Counting (SLAC) | Datamonkey | http://www.datamonkey.org/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For expression of PI5P4Kβ proteins, E. coli BL21 (DE3) strain (Nippon Gene) was used.
METHOD DETAILS
Cloning
Expression vectors
Expression vectors of human PI5P4Kβ were generated by the standard polymerase chain reaction (PCR) method, as described by Sumita et al. (2016). The full-length PI5P4Kβ was used in biochemical and NMR assays, while the first 31 amino-acid residues of PI5P4K were further deleted by QuikChange mutagenesis for X-ray crystallography as described by Sumita et al. (Sumita et al., 2016). The N-terminal truncated PI5P4Kβ (a.a. 32-416) showed the same enzymatic activity as the full-length PI5P4Kβ; however, it gave crystals with better diffraction. The T201M-, N203D-, N203A-, F205L, and T201M_F205L-PI5P4Kβ mutants were generated using primer pairs 1, 2, 3, and 4, respectively.
Materials
Unless otherwise noted, all materials were purchased from Sigma-Aldrich or Fujifilm Wako Pure Chemical Corporation. Phospholipids were purchased from Echelon Biosciences. PNTs were purchased from TriLink BioTechnologies.
Buffers
Buffer A: 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 5 mM β-mercaptoethanol (βMe), and 10 mM imidazole
Buffer B: 50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5 mM βMe, and 300 mM imidazole
Buffer C: 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, and 2 mM DTT
Crystallization buffer (reservoir solution): 100 mM sodium-citrate (pH 6.0), 10 mM magnesium-acetate, 100 mM lithium-acetate, and 12% (v/v) polyethylene glycol-4000 (PEG-4000)
NMR buffer: 10 mM sodium phosphate (pH 6.8), 100 mM NaCl, 10 mM MgCl2, 2 mM DTT, and 99.6% D2O
Expression and purification of PI5P4Kβ
The human PI5P4Kβ was expressed and purified, as described previously (Sumita et al., 2016). Briefly, the BL21(DE3) harboring the PI5P4Kβ expression vector was inoculated in 5 ml of LB medium containing 50 μg/ml of kanamycin at 37 °C overnight. Cells were collected by centrifugation and further inoculated into 1 liter of Luria-Bertani medium. When the bacterial culture was grown to OD600 = ~0.6, 0.6 mM of isopropyl-β-D-thiogalactopyranoside was added to induce the protein overexpression. The induced culture was further incubated at 25 °C for 16 h and harvested by centrifugation.
The cells were lysed by sonication in Buffer A. The lysate was centrifuged to remove cell debris, and the supernatant was subjected to a Ni-affinity chromatography using 2 ml of Ni-NTA agarose (QIAGEN). The column was washed with Buffer A, and the PI5P4Kβ protein was eluted from the column with buffer B. The elution was concentrated to about 10 ml by ultrafiltration using Amicon Ultra with MWCO of 10K (Millipore), and 80 μl of PreScission protease was added per 10 mg of PI5P4Kβ. The solution was dialyzed against Buffer C overnight at 4 °C and applied on a Resource Q column (GE Healthcare) that was equilibrated with Buffer C. PI5P4Kβ was eluted with a linear gradient of 30 column volumes and an increasing ionic strength up to 400 mM NaCl. The fractions corresponding to the PI5P4Kβ dimer, which would be eluted around 250 mM NaCl concentration, were collected and concentrated to 20 mg/ml and stored at −80 °C. The expression and purification procedures of PI5P4Kβ mutants were essentially identical to those for the WT PI5P4Kβ.
NMR spectroscopy
All experiments were performed on Bruker Avance 700 MHz spectrometer equipped with a triple resonance probe. All spectra were collected in the NMR buffer (see buffer list above) at 25 °C. Spectra were processed using TOPSPIN (Bruker).
For the PNT-hydrolysis assay, 250 μM PNTs were mixed with 2 μM PI5P4Kβ, and hydrolysis reactions were carried out at 25 °C for 20 h. Sample volume was 500 μL. The reaction was monitored by the ratio of the intensity of the H8 position signal from the dinucleotide- over trinucleotide-forms in the 1D 1H experiments (Fig. 6B) and the amount of produced dinucleotide-forms of each nucleotide was plotted as the PNT hydrolysis activity. For determination of KM and kcat values, the same reaction is carried out for different concentrations of PNTs from 63.5 μM, 125 μM, 250 μM, 500 μM, 1 mM, and 2 mM. The activity (a) at each PNT concentration ([PNT]) were fitted against the Michaelis–Menten equation:
| (1) |
where, Vmax is the maximal velocity of the enzymatic reaction, and KM is the Michaelis–Menten constant. The catalytic constant kcat is calculated using the concentration of PI5P4Kβ ([PI5P4Kβ]) as follows:
| (2) |
Least-square fitting was conducted using Origin 2020 software (Origin Lab Corporation).
Crystallization, data collection, and structure determination
PI5P4Kβ was crystallized with the sitting-drop vapor diffusion method by mixing 1.5 μl of protein solutions (20 mg/ml), and 1.5 μl of crystallization buffer (reservoir solution) (see buffers in the supplemental materials) at 4°C; these conditions were optimized based on the previously reported crystallization conditions (Sumita et al., 2016). Under the optimized conditions, plate-shaped crystals with approximate dimensions of 0.4 × 0.4 × 0.1 mm3 appeared in 1–2 days.
To prepare PI5P4Kβ-PNT complex crystals, we applied the multi-step soaking method (Senda et al., 2016). In the first step, apo-PI5P4Kβ crystals were soaked in a reservoir solution containing 12.5–25.0% (w/v) PVP-K15 and 0.5–10 mM of PNTs for 1–2 days at 20 °C. The crystals were then transferred to another reservoir solution supplemented with 12.5% (w/v) PVP-K15, 10–15% (v/v) ethylene glycol (EG), and 0.5–10 mM of the PNTs for 30 sec. Diffraction data were collected at beamline BL17A and NE3A of the PF in KEK (Tsukuba, Japan) and beamline X06SA of SLS in PSI (Villigen, Switzerland). The wavelength and temperature were set to 0.9800 Å or 1.0000 Å and −178 °C, respectively. The diffraction data were processed and scaled using the programs XDS and XSCALE, respectively (Kabsch, 2010). The apo-PI5P4Kβ crystal before soaking belonged to space group C2221, with unit-cell parameters a = 107.1 Å, b = 182.4 Å, c = 105.4 Å.
The structures of the PI5P4Kβ-PNT complexes were determined by the molecular replacement method using the PHENIX program (Liebschner et al., 2019). The structure of human PI5P4Kβ (PDB ID: 3WZZ) in the apo form was used as a search model (Sumita et al., 2016). A simulated annealing mFo-DFc omit map corresponding to the individual nucleotide, and the TRVNF sequences were used to confirm the conformation of the bound nucleotides (Fig. S8). Water molecules were modeled only for those nucleotides clearly visible (> 3σ) in mFo-DFc maps. Crystallographic refinement was performed by the phenix.refine program (Liebschner et al., 2019).
Data collection and structure refinement statistics are listed in Table S6. For WT-PI5P4Kβ, the favored Ramachandran values were, 98%, 96%, 97%, 98%, and 97% for WT-GMPPNP, WT-AMPPNP, WT-ITP, WT-XTP, and WT-2A-a-ATP complexes, respectively. For the PI5P4Kβ mutants, the favored Ramachandran values were, 97%, 95%, 94%, 96% and 97% for the T201M-2a-ATP, N203D-ITP, N203D-XTP, F205L-ITP, and F205L-XTP complexes, respectively. Molecular graphics were prepared by PyMOL (Schrödinger, LLC.).
All atomic coordinates obtained in the present study have been deposited in the PDB under the accession codes, as shown in the statistics table (Table S6).
Structural analysis of kinases and G-proteins
660 protein kinase, PI-kinase, and IP-kinase (including inositol kinase) structures in complex with any types of ATP and ATP analogs, including ATPγS, AMPPNP, AMPPCP, and ADP, were collected from PDB and the hydrogen-bonding interaction of the kinases that are described in the repository were analyzed. As for G-proteins, 122 small G-proteins, and 6 α-subunits of heterotrimeric G-proteins in complex with any types of GTP and GTP analogs, including GTPγS, GMPPNP, GMPPCP, and GDP, were collected from the PDB and the hydrogen-bonding interaction of the G-proteins that are described in the repository were analyzed. The structural data used in this study are listed in the Supplementary Materials.
Fragment molecular orbital (FMO) calculations
Ab initio FMO calculation (Fedorov and Kitaura, 2009; Fedorov et al., 2012; Tanaka et al., 2014) was performed on the crystal structure of the PI5P4Kβ-GTP complex. While PI5P4Kβ is a homodimer, only the monomeric structure was utilized for the FMO calculation, and inter-subunit interactions were neglected. The crystal structure was modified before performing the FMO calculation. First, all water molecules except those interacting with the nucleotide base were deleted from the crystal structure. Second, hydrogen atoms were added based on the assignment of the protonation state that was calculated using the Protonate 3D function of the Molecular Operating Environment program package (Chemical Computing Group, Montreal, Canada). Third, the energy of hydrogen atoms was minimized with the Amber10: EHT force field. Then, FMO calculations for the PI5P4Kβ structure were performed using ABINIT-MP software ((Mochizuki et al., 2019; Nakano et al., 2006). The second-order Møller-Plesset perturbation (MP2) (Mochizuki et al., 2004a; Mochizuki et al., 2004b)method was used with the 6-31G* basis function as a theoretical calculation level; namely, the FMO-MP2/6-31G* level of theory was used. For the FMO calculation, PI5P4Kβ was fragmented into amino acid units at bonds between the C and Cα atoms of the mainchain. GTP was fragmented into the base, sugar, and phosphate groups. Each water molecule was treated as a single fragment. This fragmentation treatment made it possible to easily calculate the electronic structure of the whole complex and the inter-fragment interaction energies (IFIEs).
Evolutionary analysis of PI5P4Kβ
We obtained nucleotide sequences of PI5P4Kβ from the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) as follows: NM_003559.5 for Homo sapiens, XP_003786482.1 for Otolemur garnettii, NM_054051.1 for Mus musculus, NM_001192196.1 for Bos taurus, XP_010592684.1 for Loxodonta Africana, XM_015299518.2 for Gallus gallus, XP_003222566.1 for Anolis carolinensis, XP_002940195.1 for Xenopus tropicalis, XM_014497621.1 for Latimeria chalumnae, XP_011610877.1 for Takifugu rubripes, XP_004084068.1 for Oryzias latipes, and XM_007910909.1 for Callorhinchus milii. For each sequence, we extracted coding regions and translated them. We then make multiple alignments of the amino acid sequences using L-INS-i program in the MAFFT version 7.205 (Katoh and Standley, 2013), and based on the amino acid alignment, we generated nucleotide alignment using TranslatorX (Abascal et al., 2010). We then calculated synonymous and nonsynonymous substations for each codon and statistically evaluated their frequencies using the Single-Likelihood Ancestor Counting (SLAC) program in Datamonkey (Pond and Frost, 2005).
QUANTIFICATION AND STATISTICAL ANALYSIS
For NMR hydrolysis activity assays, we provide the averages of three independent measurements and standard deviations. Fitting errors for the KM and kcat values were calculated by Origin 2020 software (OriginLab Corporation). Data collection and refinement statistics presented in Table S6 were calculated using the XSCALE (Kabsch, 2010) and phenix.refine (Liebschner et al., 2019) programs, respectively.
Supplementary Material
Data S2 List of distances between G-proteins and GTP and its analogs
Related to Figure 2. The table retains the PDB IDs, chain ID, residue number of interacting residues, type of nucleotide, and respective distances. The table contains six different tabs; each contains the distances to N(1) and NH2(2), distances between O(6)-N(i+1), distances between O(6)-N(not i+1), distance to N(7), and distances to N(7) with OD1 swap in PDB.
Data S1 List of distances between kinases and ATP and its analogs
Related to Figure 2. The table retains the PDB IDs, chain ID, residue number of interacting residues, type of nucleotide, and respective distances.
Highlights.
GTP sensing of PI5P4Kβ is enabled by the dual ATP/GTP recognizing GEA motif.
As in kinases, the mainchain atoms of the GEA motif contribute to adenine binding
Similar to G-proteins, the sidechain atoms are required for guanine recognition.
Proposal for the evolutional path of how PI5P4Kβ became a GTP sensing kinase.
Acknowledgment
We thank Ms. Tomomi Sato for her contribution in the early stage of this work. We thank Ms. Emily Dobbs and Dr. Eric P. Smith for their excellent proofreading.
This work was supported in part by a UC College of Medicine Research Innovation grant, a Marlene Harris Ride Cincinnati grant, JSPS KAKENHI (20H03165), and NIH grants (R21NS100077, R01NS089815, and R01CA255331) to A.T.S. Support was also provided by Project for Cancer Research and Therapeutic Evolution (P-CREATE; JP20cm0106173 to A.T.S., T.S., and K.T.) from Japan Agency for Medical Research and Development (AMED), Generating research infrastructure and novel technologies for anti-infective drug and vaccine discovery from AMED (21gm1610003h0201 to KT), and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Numbers JP19am0101071 (TS) and JP19am0101113 (KF) (support numbers 0586 and 2194), JST, CREST Grant Number JP20356709, Japan, and Swiss Light Source (proposal number 20191094 and 20191134). The work was also supported in part by KAKENHI (grant number 20H03378 and 20H04722 to KT) from Japan Society for the Promotion of Science (JSPS). his work was supported by the public accession beam time of Photon Factory, Institute of Materials Structure Science, High Energy Accelerator Research Organization (Accession numbers: 2017G147, 2019G063, and 2021G054).
Footnotes
Competing interests
The authors have declared that no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abascal F, Zardoya R, and Telford MJ (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res 38, W7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazenet CE, Ruano AR, Brockman JL, and Anderson RA (1990). The human erythrocyte contains two forms of phosphatidylinositol-4-phosphate 5-kinase which are differentially active toward membranes. Journal of Biological Chemistry 265, 18012–18022. [PubMed] [Google Scholar]
- Berman H, Henrick K, and Nakamura H (2003). Announcing the worldwide Protein Data Bank. Nature Structural & Molecular Biology 10, 980–980. [DOI] [PubMed] [Google Scholar]
- Bossemeyer D (1995). Protein kinases--structure and function. FEBS Lett 369, 57–61. [DOI] [PubMed] [Google Scholar]
- Burke JE (2018). Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol Cell 71, 653–673. [DOI] [PubMed] [Google Scholar]
- Clarke JH, and Irvine RF (2011). The activity, evolution and association of phosphatidylinositol 5-phosphate 4-kinases. Adv Enzyme Regul. S0065-2571(11)00050-1 [pii] [DOI] [PubMed] [Google Scholar]
- Endicott JA, Noble MEM, and Johnson LN (2012). The Structural Basis for Control of Eukaryotic Protein Kinases. Annual Review of Biochemistry 81, 587–613. [DOI] [PubMed] [Google Scholar]
- Fedorov D, and Kitaura K (2009). The Fragment Molecular Orbital Method: Practical Applications to Large Molecular Systems (CRC Press; ). [Google Scholar]
- Fedorov DG, Nagata T, and Kitaura K (2012). Exploring chemistry with the fragment molecular orbital method. Physical Chemistry Chemical Physics 14, 7562–7577. [DOI] [PubMed] [Google Scholar]
- Hammond GRV, and Burke JE (2020). Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr Opin Cell Biol 63, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, and Kuriyan J (2002). The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Jenkins GH, Fisette PL, and Anderson RA (1994). Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J Biol Chem 269, 11547–11554. [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallographica Section D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadka B, and Gupta RS (2019). Novel Molecular Signatures in the PIP4K/PIP5K Family of Proteins Specific for Different Isozymes and Subfamilies Provide Important Insights into the Evolutionary Divergence of this Protein Family. Genes (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung L-W, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loijens JC, and Anderson RA (1996). Type I Phosphatidylinositol-4-phosphate 5-Kinases Are Distinct Members of This Novel Lipid Kinase Family *. Journal of Biological Chemistry 271, 32937–32943. [DOI] [PubMed] [Google Scholar]
- Loijens JC, Boronenkov IV, Parker GJ, and Anderson RA (1996). The phosphatidylinositol 4-phosphate 5-kinase family. Adv Enzyme Regul 36, 115–140. [DOI] [PubMed] [Google Scholar]
- Mochizuki Y, Koikegami S, Nakano T, Amari S, and Kitaura K (2004a). Large scale MP2 calculations with fragment molecular orbital scheme. Chemical Physics Letters 396, 473–479. [Google Scholar]
- Mochizuki Y, Nakano T, Koikegami S, Tanimori S, Abe Y, Nagashima U, and Kitaura K (2004b). A parallelized integral-direct second-order Møller–Plesset perturbation theory method with a fragment molecular orbital scheme. Theoretical Chemistry Accounts 112, 442–452. [Google Scholar]
- Mochizuki Y, Nakano T, Okiyama Y, Sakakura K, Akinaga Y, Watanabe H, Kato K, Sato S, Yamamoto J, Yamashita Y, et al. (2019). ABINIT-MP - Open Ver.1. [Google Scholar]
- Muftuoglu Y, Xue Y, Gao X, Wu D, and Ha Y (2016). Mechanism of substrate specificity of phosphatidylinositol phosphate kinases. Proceedings of the National Academy of Sciences 113, 8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Kaminuma T, Sato T, Fukuzawa K, Akiyama Y, Uebayasi M, and Kitaura K (2002). Fragment molecular orbital method: use of approximate electrostatic potential. Chemical Physics Letters 351, 475–480. [Google Scholar]
- Nakano T, Mochizuki Y, Fukuzawa K, Amari S, and Tanaka S (2006). CHAPTER 2 - Developments and applications of ABINIT-MP software based on the fragment molecular orbital method. In Modern Methods for Theoretical Physical Chemistry of Biopolymers, Starikov EB, Lewis JP, and Tanaka S, eds. (Elsevier Science; ), pp. 39–52. [Google Scholar]
- Niefind K, Pütter M, Guerra B, Issinger OG, and Schomburg D (1999). GTP plus water mimic ATP in the active site of protein kinase CK2. Nat Struct Biol 6, 1100–1103. [DOI] [PubMed] [Google Scholar]
- Pond SL, and Frost SD (2005). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Tolias KF, Duckworth BC, and Cantley LC (1997). A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 390, 192–196. [DOI] [PubMed] [Google Scholar]
- Shears SB, and Wang H (2019). Inositol phosphate kinases: Expanding the biological significance of the universal core of the protein kinase fold. Adv Biol Regul 71, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Hines AC, Schwarzer D, Pickin KA, and Cole PA (2005). Protein kinase structure and function analysis with chemical tools. Biochim Biophys Acta 1754, 65–78. [DOI] [PubMed] [Google Scholar]
- Shi Z, Resing KA, and Ahn NG (2006). Networks for the allosteric control of protein kinases. Current Opinion in Structural Biology 16, 686–692. [DOI] [PubMed] [Google Scholar]
- Sumita K, Lo Y-H, Takeuchi K, Senda M, Kofuji S, Ikeda Y, Terakawa J, Sasaki M, Yoshino H, Majd N, et al. (2016). The Lipid Kinase PI5P4K beta Is an Intracellular GTP Sensor for Metabolism and Tumorigenesis. Molecular Cell 61, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Senda M, Lo YH, Kofuji S, Ikeda Y, Sasaki AT, and Senda T (2016). Structural reverse genetics study of the PI5P4Kβ-nucleotide complexes reveals the presence of the GTP bioenergetic system in mammalian cells. FEBS J 283, 3556–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Mochizuki Y, Komeiji Y, Okiyama Y, and Fukuzawa K (2014). Electron-correlated fragment-molecular-orbital calculations for biomolecular and nano systems. Physical Chemistry Chemical Physics 16, 10310–10344. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, and Sowadski JM (1992). Structural framework for the protein kinase family. Annu Rev Cell Biol 8, 429–462. [DOI] [PubMed] [Google Scholar]
- Traut T (1994). Physiological concentrations of purines and pyrimidines. Molecular and Cellular Biochemistry 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Vadas O, Burke JE, Zhang X, Berndt A, and Williams RL (2011). Structural Basis for Activation and Inhibition of Class I Phosphoinositide 3-Kinases. Science Signaling 4, re2. [DOI] [PubMed] [Google Scholar]
- Vetter IR, and Wittinghofer A (2001). The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 294, 1299. [DOI] [PubMed] [Google Scholar]
- Wang Z, and Cole PA (2014). Catalytic mechanisms and regulation of protein kinases. Methods Enzymol 548, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Rossman KL, and Der CJ (2005). The Ras superfamily at a glance. Journal of Cell Science 118, 843. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, and Vetter IR (2011). Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80, 943–971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S2 List of distances between G-proteins and GTP and its analogs
Related to Figure 2. The table retains the PDB IDs, chain ID, residue number of interacting residues, type of nucleotide, and respective distances. The table contains six different tabs; each contains the distances to N(1) and NH2(2), distances between O(6)-N(i+1), distances between O(6)-N(not i+1), distance to N(7), and distances to N(7) with OD1 swap in PDB.
Data S1 List of distances between kinases and ATP and its analogs
Related to Figure 2. The table retains the PDB IDs, chain ID, residue number of interacting residues, type of nucleotide, and respective distances.
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the Protein Data Bank. Accession codes are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21 (DE3) | Nippon Gene | 312-06534 |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth with agar (Miller) | Sigma-Aldrich | L3147 |
| Kanamycin sulfate | Fujifilm Wako | 113-00343 |
| LB Broth (Miller) | Sigma-Aldrich | L3522 |
| Isopropyl-β-D-thiogalactoside | Sigma-Aldrich | I5502 |
| d-myo-phosphatidylinositol 5-phosphate | Echelon Biosciences | P-0016d |
| 1,2-dipalmitoyl-phosphatidylserine | Echelon Biosciences | L-3116 |
| GTP | TriLink BioTechnologies | N-1512 |
| ATP | TriLink BioTechnologies | N-1510 |
| ITP | TriLink BioTechnologies | N-4017 |
| XTP | TriLink BioTechnologies | N-1023 |
| 6-thio-GTP | TriLink BioTechnologies | N-8007 |
| 6-OMe-GTP | TriLink BioTechnologies | N-1031 |
| 2a-6Cl-PNP | TriLink BioTechnologies | N-1002 |
| 2a-ATP | TriLink BioTechnologies | N-1001 |
| 6Cl-PNP | TriLink BioTechnologies | N-2009 |
| 2-oxo-ATP | TriLink BioTechnologies | N-1099 |
| Deposited data | ||
| Crystal structure of WT-GMPPNP complex | This Work | 6K4G |
| Crystal structure of WT-AMPPNP complex | This Work | 6K4H |
| Crystal structure of WT-ITP complex | This Work | 7EM1 |
| Crystal structure of WT-XTP complex | This Work | 7EM2 |
| Crystal structure of WT-2a-ATP complex | This Work | 7EM3 |
| Crystal structure of F205L-ITP complex | This Work | 7EM4 |
| Crystal structure of F205L-XTP complex | This Work | 7EM5 |
| Crystal structure of N203D-ITP complex | This Work | 7EM6 |
| Crystal structure of N203D-XTP complex | This Work | 7EM7 |
| Crystal structure of T201M-2a-ATP complex | This Work | 7EM8 |
| Oligonucleotides | ||
| T201M-f 5'-ATGGTGGTTATGAGGAACGTGTTC-3’ | Eurofins | N/A |
| T201M-r 5’-CACGTTCCTCATAACCACCATGTA-3’ | Eurofins | N/A |
| N203D-f 5’-GGTGGTTACCAGGGACGTGTTCAGCCATC-3’ | Eurofins | N/A |
| N203D-r 5’-GATGGCTGAACACGTCCCTGGTAACCACC-3’ | Eurofins | N/A |
| N203A-f 5’-CATGGTGGTTACCAGGGCGGTGTTCAGCCATCGG-3’ | Eurofins | N/A |
| N203A-r 5’-CCGATGGCTGAACACCGCCCTGGTAACCACCATG-3’ | Eurofins | N/A |
| F205L-f 5'-CCAGGAACGTGTTGAGCCATCGG-3’ | Eurofins | N/A |
| F205L-r 5’-ATGGCTCAACACGTTCCTGGTAACC-3’ | Eurofins | N/A |
| Software and algorithms | ||
| Pymol (Version 2.4.0) | Schrödinger | https://pymol.org/ |
| Topspin | Bruker | https://www.bruker.com/en/products-and-solutions/mr/nmr-software.html |
| Origin 2020 | Origin Lab Corporation | https://www.originlab.com/ |
| XDS | (Kabsch, 2010) | https://xds.mr.mpg.de/ |
| XSCALE | (Kabsch, 2010) | https://xds.mr.mpg.de/ |
| PHENIX | (Liebschner et al., 2019) | https://phenix-online.org/ |
| phenix.refine | (Liebschner et al., 2019) | https://phenix-online.org/ |
| PyMOL | Schrödinger, LLC. | https://pymol.org/2/ |
| Molecular Operating Environment | Chemical Computing Group, | https://www.chemcomp.com/Products.htm |
| ABINIT-MP | (Mochizuki et al., 2019; Nakano et al., 2006) | https://www.hpci-office.jp/pages/e_appli_abinit-mp |
| L-INS-i | MAFFT version 7.205 | https://mafft.cbrc.jp/alignment/software/ |
| TranslatorX | (Abascal et al., 2010) | https://translatorx.org/ |
| Single-Likelihood Ancestor Counting (SLAC) | Datamonkey | http://www.datamonkey.org/ |