

Abstract
Transcriptome sequencing (RNA-seq) is widely used to detect gene rearrangements and quantitate gene expression in acute lymphoblastic leukemia (ALL), but its utility and accuracy in identifying CNVs has not been well described. CNV information inferred from RNA-seq can be highly informative to guide disease classification and risk stratification in ALL due to the high incidence of aneuploid subtypes within this disease. Here we describe RNAseqCNV, a method to detect large scale copy number variations from RNA-seq data. We used models based on normalized gene expression and minor allele frequency to classify arm level CNVs with high accuracy in ALL (99.1% overall and 98.3% for non-diploid chromosome arms, respectively), and the model was further validated with excellent performance in acute myeloid leukemia (accuracy 99.8% overall and 99.4% for non-diploid chromosome arms). RNAseqCNV outperforms alternative RNA-seq based algorithms in calling CNVs in the ALL dataset, especially in samples with a high proportion of CNVs. The CNV calls were highly concordant with DNA-based CNV results and more reliable than conventional cytogenetic-based karyotypes. RNAseqCNV provides a method to robustly identify copy number alterations in the absence of DNA-based analyses, further enhancing the ability of RNA-seq to classify ALL subtype.
INTRODUCTION
The majority of cases of acute lymphoblastic leukemia (ALL) are characterized by chromosomal aneuploidy, gene rearrangements, and sequence mutations with associated distinct gene expression profiles supporting the importance of these alterations as initiating and subtype-defining events in leukemogenesis 1–3. With the comprehensive, genome-wide representation of RNA expression and high sensitivity and accuracy in calling gene rearrangements, transcriptome sequencing (RNA-seq) has been proven highly effective in detecting the driver genetic lesions and classifying ALL subtypes. In contrast, detection of copy number variations (CNVs) from bulk RNA-seq data is rarely addressed.
Recently, several methods have been described to accomplish this. CNVkit4 is a CNV caller that uses second-generation sequencing data that provides an option to deploy the segmentation algorithms on the RNA-seq data to identify CNVs. CaSpER5 attempts to detect both gene level and large-scale CNVs while applying median filtering to the expression signal and allelic frequency shift signal. Both tools provide a wide range of functionalities but lack a robust approach to identify large-scale CNVs. As a result, there is a lack of reliable methods for calling chromosomal and arm-level CNVs, in particular for highly aneuploid samples. Large-scale CNVs are pivotal for subtyping up to 30% of ALL cases harboring large number of whole chromosome gains or losses 6, such as the high hyperdiploid (modal chromosome number ≥51), low hypodiploid (chromosome number 31–39) and near haploid (chromosome number 24–30). As RNA-seq is being increasingly utilized clinically7, and offers the potential to profile all key modalities of leukemia-classifying alterations, it would be highly valuable to extract large-scale CNV information from RNA-seq, especially when the DNA level data are unavailable for determining the aneuploid subtypes.
The relationship between genomic copy number and RNA expression level is influenced by multiple factors in addition to the copy number per se, including regulation by transcription factors, DNA methylation, chromatin modification, GC bias, as well as technical factors such as methods of library preparation and sequencing. In addition, the relatively sparse coverage of the genome by RNA-seq makes the detection of precise CNV breakpoints and small CNV segments highly challenging or even impossible. Additionally, for human samples with extreme aneuploidy, e.g. greater than 60 or less than 30 chromosomes, gene expression normalization can be significantly biased, complicating interpretation of the baseline expression level of diploid regions and subsequent CNV inference.
Many CNV calling methods of SNP array or genome/exome sequencing data use both signal intensity (similar to total sequencing depth) combined with B-allele frequency (BAF; similar to minor allele frequency (MAF) by sequencing) data8–11. The signal intensity correlates with the copy number, while the BAF guides discerning the exact copy number. On diploid chromosomes, the theoretical BAF value is 0, 1 or 0.5. However, BAF can be influenced by CNV (e.g., BAF of 0.33 and 0.66 after 1 copy gain). Based on this principle, we developed a user-friendly software package implemented in R to call CNVs from RNA-seq - RNAseqCNV. In addition to fast analysis and intuitive visualization, the algorithm has been incorporated into a Shiny R package to enable interactive curation of the reported CNVs.
METHODS
Data processing
RNAseqCNV requires two types of information for each sample: the read count per gene, and the minor allele fraction (MAF) of single nucleotide variations (Figure 1). Samples can be analyzed in either batch or per sample mode. In batch analysis, all the input samples are used for normalization and gene expression centering (log2 fold change calculation). In this mode, the majority of samples should not contain multiple large-scale CNVs. For optimal results, the samples should be of the same cancer type and library preparation protocol to reduce the effect of differential expression due to these factors. In per sample analysis, each sample will be analyzed against the in-built or user-provided diploid reference samples. The in-built diploid reference contains gene expression data from 40 ALL samples (subset of EGAS00001003266) 2 without large-scale CNVs. The diploid nature of these samples was confirmed with Affymetrix SNP Array 6.0 analyzed by RawCopy12.
Figure 1. Workflow of RNAseqCNV.
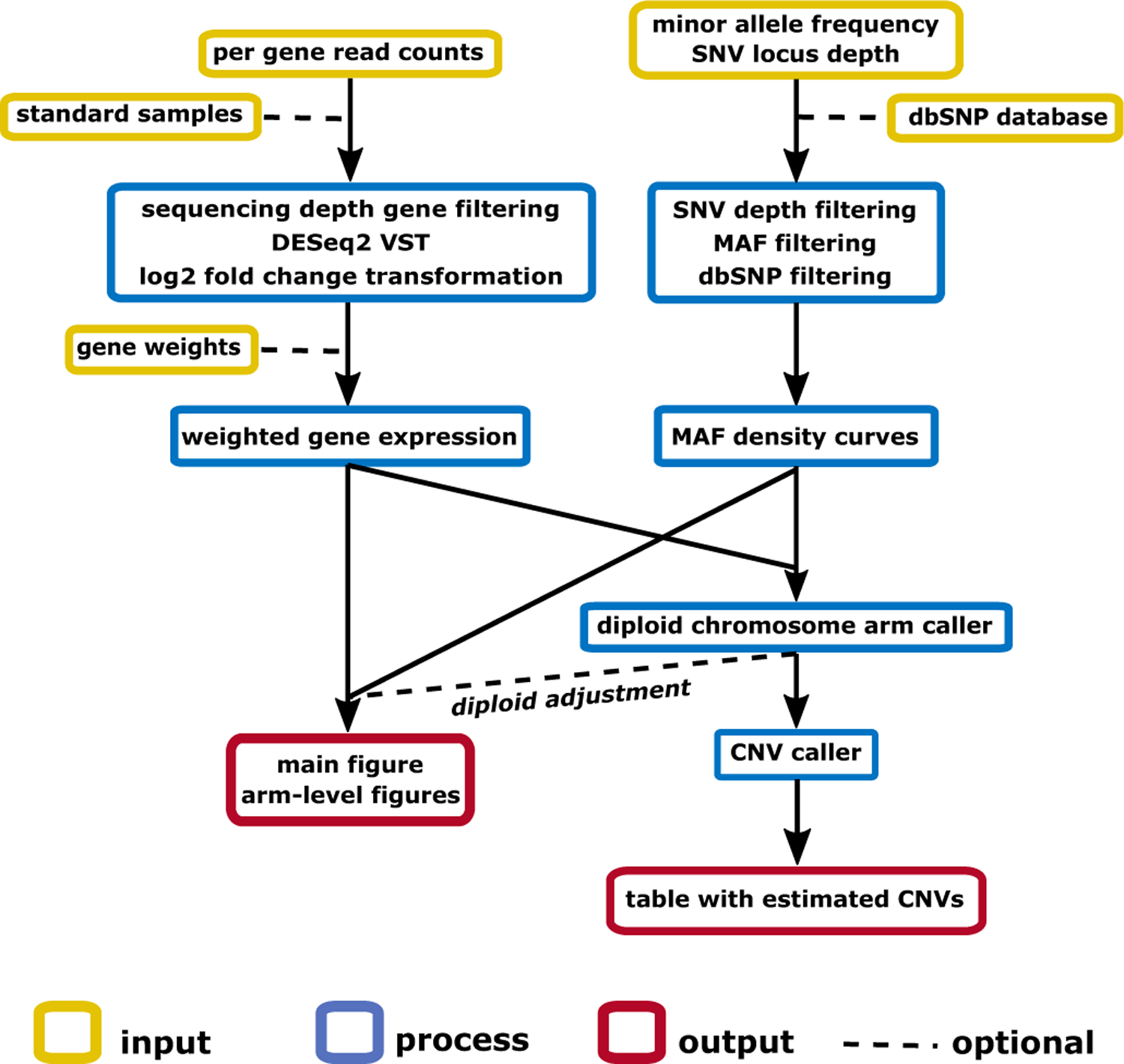
Per-gene read counts and SNVs information are input for the analysis. Each sample is analyzed against standard samples (in-built or user provided). The DESeq2 variance stabilizing transformation (VST) is used for gene expression level normalization. Then log2 fold change per gene is calculated against the median expression of the gene across the cohort. Each gene is assigned a weight according to the (in-built or user provided) weight matrix. Subsequently, weighted quantiles per-chromosome are used to generate boxplots. High quality SNVs are kept based sequencing depth and record in the dbSNP database. Since only heterozygous SNVs are informative for calling CNVs, only SNVs with MAF between 0.1 and 0.9 are kept as default. Weighted boxplots and MAF density curves are visualized on chromosomal and arm level, and serve as predictors for CNV calling. The diploid arms are estimated in the first model and used to adjust the expression data for correct prediction of CNVs in the second model. Sex of the sample is estimated based on gene expression on chromosome Y.
Genes with read counts of less than 3 in at least 20% of the analyzed samples were filtered out. Per-gene read counts were normalized with variance stabilizing transformation (VST) from DESeq2 package13 for minimizing the noise of genes with low read counts. Subsequently, gene expression levels were centered by calculating the log2 fold change of the gene expression as to the median expression of the same gene across the cohort. Pseudoautosomal regions on chromosome X interfering with the CNV prediction models were discarded. Each gene was assigned a weight from the in-built or user-provided weight matrix. Weights represent the importance of the gene expression for CNV inference.
To ensure the reliability of SNVs detected from RNA-seq data, only those with adequate sequencing depth and described in the dbSNP database were used. In addition, for CNV evaluation, only the heterozygous SNVs are informative. Thus, only the ones with heterozygous MAF (e.g., between 0.1 and 0.9) were used in the analysis.
Two Random Forest models were used sequentially to automatically improve the interpretability of CNV visualization and provide fast and accurate prediction of CNVs on chromosomal and arm level. The first model classified chromosome arms as either diploid or non-diploid. It improved the accuracy of CNV calling in the second model and enables correction of gene expression level for samples with a high proportion of CNVs. Then, weighted expression level and MAF density curves were used to visualize CNVs on chromosomal and arm levels (Figure 2; Supplementary Figure 1).
Figure 2. Chromosomal level CNVs detected by gene expression and MAF of SNVs.
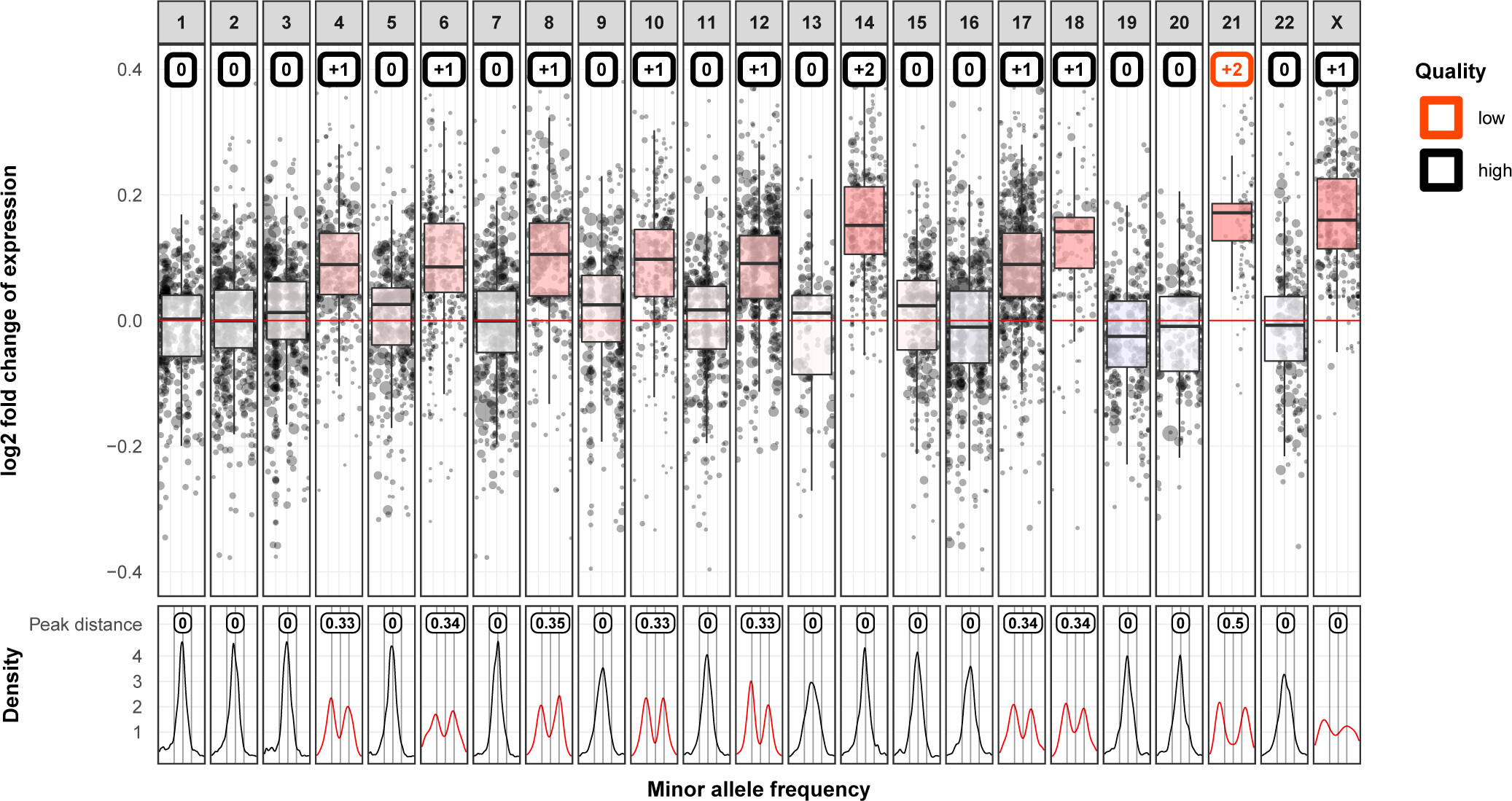
The upper panel shows the chromosomal gene expression level. The Y axis shows log2 fold change of gene expression of the test sample compared to the reference. The X axis shows chromosome 1–22 and X (Y is excluded). The X axis also depicts the position of genes on each chromosome. On each chromosome, a weighted boxplot (¼, ½, ¾, quantile and maximum and minimum are second and third quartile ± 1.5*interquartile range) is drawn based on the distribution of normalized gene expression. Random Forest predictions of chromosome gains or losses are marked in colored boxes at the top of each chromosome panel. The CNV calls voted with less than 85% of trees in the random model are associated with lower confidence (Quality in the figure legend) and thus CNV boxes for such chromosomes are highlighted in red. E.g. the 2-copy gain on chromosome 21 is predicted with low quality because it is not the common form of homozytous 2-copy gain as chromosome 14. The lower panel shows the density curves of MAF for each chromosome. Only the MAF of heterozygous SNVs (e.g, MAF from 0.1 to 0.9) are informative in calling CNVs. Heterozygous SNVs on chromosomes without CNVs are expected to have MAF centered around 0.5 and shown with a single peak at 0.5. Consequently, distortions of MAF density indicates CNVs and are highlighted in red. Peak distance measures the distance between the two highest peaks in the MAF density plot on x axis. This exemplar sample shows 1-copy gain on chromosome 4, 6, 8, 10, 12, 17, 18, X, and ≥2-copy gain on chromosome 14 and 21, which agrees with the results from SNP array data.
Evaluation of RNAseqCNV in an ALL dataset
The ALL dataset consisted of 637 samples (287 high hyperdiploid, 46 low hypodiploid, 28 near haploid and 276 other subtypes). Among them, 272 samples had whole genome sequencing (WGS) data available (https://www.stjude.cloud/)14 that was analyzed by CONSERTING8; 206 samples had Affymetrix SNP Array 6.0 data analyzed by RawCopy12 to provide a reference set of CNVs derived from DNA data. In addition, 159 samples had karyotypic data available. Karyotype reports can be associated with inaccuracies such as selection bias, endoreduplication, or poor chromosome morphology. For this reason, CNVs were validated by our previous CNV visualization method from RNA-seq data, which has been shown with high accuracy compared to SNP array results2. If the copy number of a chromosome was not confirmed, it was excluded from the analysis.
Forty-three samples (28 high hyperdiploid, 3 low hypodiploid, 7 near haploid, 5 other subtypes) with both karyotype report and SNP array data available were set aside to directly compare RNAseqCNV with karyotype with SNP array results serving as a reference. One-fourth of the remaining samples (n=148: 67 high hyperdiploid, 10 low hypodiploid, 4 near haploid, 67 other subtypes) were randomly selected and set aside as a testing dataset.
CNVs were assessed at chromosome arm level. The partial (shorter than chromosome arms) and subclonal CNVs were excluded from the dataset. Unclear CNV calls from WGS or SNP array data due to low tumor purity were discarded as well. Chromosome arms were then assigned into 4 distinct CNV classes: 1 copy deletion, no CNV (two copies), 1 copy gain, and ≥2 copy gain. Because of the inactivation of chromosome X, its MAF density curve is not as informative as autosomal MAFs in calling CNVs, which makes the detection of the exact copy number of chromosome X challenging. In RNAseqCNV, chromosome X is assigned into 3 distinct classes: 1 copy deletion, diploid, and ≥1 copy gain. Lastly, the ≥2 copy gain class and the 1 copy deletion are less common (1.9 percent and 6.7 percent of the training data), which would lead to class imbalance in the training dataset (n=446). Instances of chromosome arms with two copies were randomly under-sampled by 40%, and the two less represented classes were randomly oversampled to contain at least one-fourth of the instances of the major class. (Supplementary Figure 2, Supplementary Table 1–2).
To test the performance of RNAseqCNV in other hematologic malignances, 71 acute myeloid leukemia (AML) samples with both RNA-seq and WGS data were analyzed (https://www.stjude.cloud/) 14. Arm-level CNVs were evaluated by CONSERTING 8 using WGS data. Large-scale CNVs are less frequent in acute myeloid leukemia than in ALL – CNV was detected only in 61 out of the 2,911 chromosome arms. (2,850 two copies, 50 1-copy gains, two ≥2-copy gains, and nine 1-copy deletions).
The performance of RNAseqCNV and alternative tools was evaluated on chromosome or chromosome arm level through accuracy and contingency tables based on predicted copy number states and the reference dataset. Accuracy was calculated as the number of correct predictions of a copy number state divided by the total number of instances evaluated.
Design and testing of prediction method
In the majority of cancer samples, the log2 fold changes of true diploid segments were centered around zero, and the assessment of expression level of chromosomes or arms was straightforward: positive log2 fold change signified gain, and vice versa. However, some samples have a high proportion of chromosomal CNVs of the same type, such as high hyperdiploid and hypodiploid ALL. In such samples, the log2 fold change of diploid segments may be shifted from zero and thus could not be evaluated without the context of gene log2 fold change values on diploid chromosomes. Therefore, an additional prediction model was used to classify chromosome arms as either diploid or non-diploid to identify the baseline expression level on diploid chromosomes. Hence, RNAseqCNV uses two classifier models in sequence to predict CNVs.
To develop optimal CNV prediction models, four classification algorithms (K-Nearest Neighbors (KNN), Support Vector Machine (SVM) with polynomial and radial kernel, and Random Forest (RF)) were tested for both diploid chromosome arm prediction model and CNV prediction model. For each iteration of the algorithm with diploid/CNV classifier, recursive feature elimination (RFE) function (10-fold cross-validation) provided by the caret R package 15 was used for feature selection. Model tuning across parameters was performed by 10-fold cross-validation to maximize accuracy after selecting the best performing features with RFE.
MAF-based features were created to describe the key aspects of the MAF density curve, such as the amplitude of the density curve, the x-axis distance between the amplitude and 0.5 point, the distance between the two highest peaks along the x-axis, and density in the 0.5 point. We also added two MAF-derived features describing the number of chromosome arms with the distance between the two highest peaks in certain ranges (0.2–0.4 and 0.4–0.9) in the sample. The two features were created to provide the model with information about the MAF distribution curves in the whole sample, in addition to the information about the chromosome arm being evaluated. To construct the expression-based features, weighted median, first and third quartile were calculated for each chromosome arm. Sample standard deviation and mean of weighted medians across all of the chromosome arms were subsequently determined. The features themselves were calculated as the arm-specific median, first and the third quartile subtracted by the sample median of weighted medians divided by the sample standard deviation. After the first classification step, additional features describing the expression pattern in relation to diploid chromosome arms were predicted. The standard deviation and the mean of weighted medians on the predicted diploid chromosome arms were used to create three features in relation to the expression level of diploid chromosome arms as described above. The standard deviation and mean of weighted medians overall and based on predicted diploid chromosome arms were included as features as well. Chromosome location was also included as a single variable for the RF model and as 23 dummy variables for SVM and KNN models (Supplementary Table 3).
The RF model had the highest accuracy for both rounds of predictions (Supplementary Table 4–7). Features for the final RF model were selected based on RFE and RF importance score (Supplementary Figure 3). Chromosome arms with an insufficient number of SNVs due to low sequencing depth or short segment length were not used for determining diploid status, and CNV prediction for such arms was made with a separate model using only the expression information.
Develop weighted gene expression in CNV prediction model
Many factors apart from CNV can also affect gene expression level. Therefore, RNAseqCNV assigns a weight value on each gene based on a well-curated CNV dataset to leverage the importance of each gene in predicting CNVs. A weight matrix was precalculated based on the ALL training dataset of samples with curated CNV results.
Multiple methods and parameter iterations were tested to construct weights that can reach the highest accuracy of CNV prediction by the RNAseqCNV model by 10-fold cross-validation on the training dataset. The best-performing weights were based on the correlation of gene expression with CNV divided by expression variance across the cohort. Pearson correlation coefficients were calculated between CNV and normalized gene expression data. Genes with correlation lower than 0.3 and Benjamini-Hochberg adjusted p-value higher than 0.1 were excluded. The accuracy of prediction was highest when correlation was raised to the third power. The gene expression variance was calculated based on 40 diploid samples (confirmed by SNP array data) with the rationale that genes with a high variance of expression are affected by factors other than CNV. However, a higher variance of the raw read counts with higher sequencing depth is a desirable gene trait pointing toward its reliability. Therefore, VST was applied to minimize the correlation between the variance and per gene read count. Only then, the variance can be used as an inverse value for gene weight calculation. Finally, rescaling the exponentiated correlation and inverse variance to a range of 1–100 has increased accuracy compared to no rescaling or different ranges. (Supplementary Table 8).
Assuming that the correlation between gene expression level and DNA copy may vary between different tissues and cancer types, it is recommended to provide a customized reference dataset for each tumor analysis.
Rather than using the in-built weights, RNAseqCNV provides the option to generate a customized weight matrix from user input data. These weights employ a similar strategy, but gene correlation is omitted, and weights are calculated only from the inverse of the rescaled variance.
Diploid adjustment
The premise of DESeq2 normalization is that most genes between samples are not differentially expressed, and when they are, that for one sample, over-expression and under-expression are approximately balanced. However, there are some samples with high numbers of chromosomal CNVs of one type, for which the normalization is biased, and the expression is not comparable across samples.
To solve this issue, the diploid chromosome arm classifier was developed. The median shift from zero on a log2 fold scale was calculated for the classified chromosome arms with two copies, which was then subtracted from all relative gene expression values in the sample (Figure 3).
Figure 3. Diploid adjustment in a sample with a high proportion of CNVs.
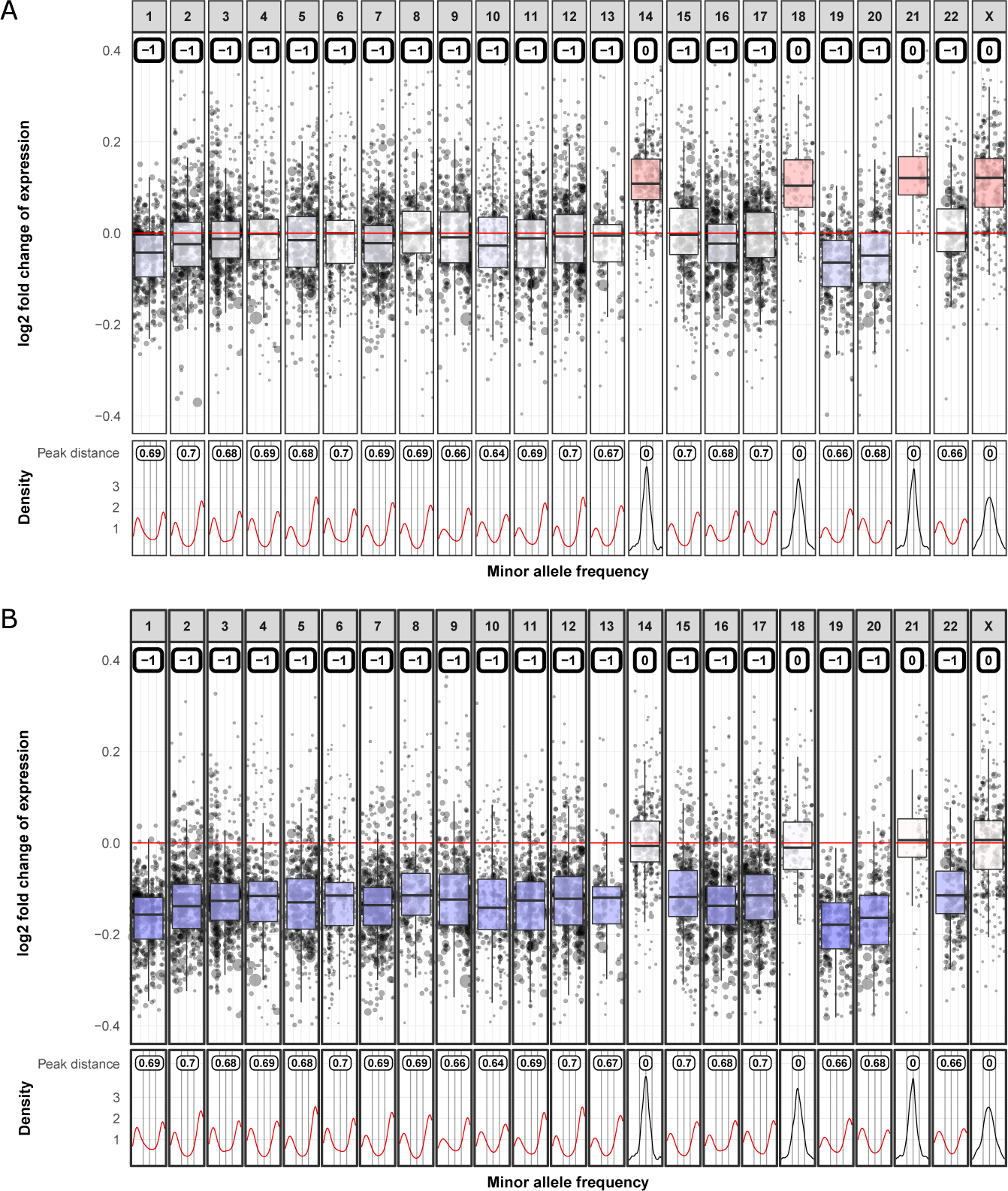
Correct normalization of gene expression levels for samples with large number of copy number gains or losses is challenging. Although gene expression levels are comparable within samples, they cannot be compared across samples. Therefore, when the gene expression is centered using a reference cohort, bias could be introduced. To solve this, a diploid chromosome arm caller was used to determine the diploid chromosome arms. Subsequently, on the estimated diploid arms, median shift from zero is calculated and then subtracted from the expression level of all genes. (A) No diploid adjustment is applied and the expression level of chromosome 14, 18, 21, X suggest copy number gain. (B) After diploid adjustment, it is evident that chromosome 14, 18, 21 and X are actually diploid and the rest carry a 1-copy loss, which agreed with the results acquired from the SNP array data.
Code availability
RNAseqCNV and the tutorial are freely available from https://github.com/honzee/RNAseqCNV. Docker image with RNAseqCNV (v 1.2.1) installation is available at: https://hub.docker.com/repository/docker/honzik1/rnaseqcnv
RESULTS
ALL test data results
Overall accuracy for the testing dataset was 99.1% (5 704 out of 5 758 chromosome arms) and the result was consistent across the cohorts analyzed by SNP array, WGS or karyotype/RNA-seq (98.8%, 99.2%, 99.1%). Most of the chromosome arms were diploid and showed high accuracy (99.3%). For the non-diploid data, RNAseqCNV agreed in 1 257 out of 1 279 (98.3%) CNVs with accuracy of 98.4% for 1-copy gains, 92.9% for ≥2-copy gains and 99.7% for 1-copy deletions. Six out of the 7 incorrectly classified ≥2-copy gains were called as 1-copy gain and 1 as no CNV by the model. The arm-level results were summarized into karytope report and the predicted ALL anueploid subtypes were correctly determined in 97.3% of samples (144 out of 148). In the 4 incorrectly classified samples, the subtypes were missed by maximum 2 chromosome copies (Supplementary table 9).
Comparison with SNP array and karyotyping
SNP array is widely accepted as a robust platform for CNV detection, therefore it was used to generate the gold standard CNV dataset to evaluate RNAseqCNV through the 43 ALL sample dataset. The CNVs estimated by RNAseqCNV were highly consistent with the result by SNP array (1,681 out of the 1,696 chromosome arm CNVs, concordance=99.1%). For non-diploid chromosome arms, the results from RNAseqCNV also achieved high accuracy compared to SNP array (684 out of 691 chromosome arm CNVs, concordance=99.0%). The accuracy for arms with two copies was 99.2%, 100% for 1-copy gains, 85.1% for ≥2-copy gains and 100% for 1-copy deletions. 5 out of the 7 incorrectly classified ≥2-copy gains were called as 1-copy gain and 2 as carrying no CNV by the model. The aneuploid subtype was correctly determined in 100% of samples (Supplementary Table 10).
Karyotyping by cytogenetics is routinely used to identify large-scale CNVs in clinical diagnosis for hematologic malignancies. Since the cytogenetic report can include multiple cell populations with different karyotypes, to determine the concordance between SNP array and karyotype, the copy number was considered as matching if it was identical with SNP array in at least one of the reported clones. This resulted in a concordance of 95.2% (1 615 out of 1 696) for all chromosome arms and 89.4% (618 out of 691) for non-diploid chromosome arms. The accuracy for arms with two copies was 99.2%, 95.4% for 1-copy gains, 76.6% for ≥2-copy gains, and 84.4% for 1-copy deletions. Ten out of the 11 incorrectly classified ≥2-copy gains were called as 1-copy gain and 1 as no CNV. The largest karyotype clone correctly determined the aneuploid subtype in 93% of the samples. In one near haploid sample with 28 chromosomes according to SNP array, karyotype reported 49 chromosomes (Supplementary Table 11).
RNAseqCNV agreed with SNP array in 93.8% (76 out of 81) of the karyotype misclassified chromosome arms, while karyotyping agreed in 66.7% (10 out of 15) of the RNAseqCNV misclassified chromosome arms (Supplementary Figure 4).
Validation in an AML cohort
The arm-level CNVs called by RNAseqCNV showed 99.8% concordance (2,906 out of 2,911) with the WGS-based CNV result, even under the RNA-seq CNV prediction model trained by ALL dataset. High concordance was also observed for the non-diploid chromosome arms (98.4%, 60 out of 61). The accuracy for arms with two copies was 99.9%, 98.0% for 1-copy gains, 100% for ≥2-copy gains, and 100% for 1-copy deletions. The one incorrectly classified 1-copy gain was called as having no CNV. This indicates that the default model within RNAseqCNV is robust for analyzing hematologic malignancies (Supplementary Table 12).
Comparison with alternative algorithms
CNVkit 4 provides an option to analyze CNV from RNA-seq. The main difference between RNAseqCNV and CNVkit is that the latter employs segmentation to detect regions with CNVs. However, RNA-seq data is often insufficient for such operations and results in a high false-positive rate (Supplementary Figure 5). Also, the issue of mis-normalization for samples with high numbers of CNVs is not addressed by CNVkit, making it prone to misinterpreted CNVs. RNAseqCNV and CNVkit (v.0.9.7 default parameters) were run on the same set of 20 testing samples on a standard laptop. Segmentation performed by CNVkit proved to be more time-consuming (74 minutes) than RNAseqCNV (10 minutes; Table 1) in analyzing the testing samples. In addition, without an option to report large-scale CNVs from CNVkit, direct comparison with RNAseqCNV output was not feasible.
TABLE 1.
Capabilities of RNAseq-based DNA copy number calling algorithms
| Per-sample runtime | Arm/Chromosomal CNV | High aneuploid adjustment | Gene-level CNV | |
|---|---|---|---|---|
| RNAseqCNV | 10 minutes | Yes | Yes | No |
| CNVkit | 74 minutes | No | No | Yes |
| CaSpER | 14 minutes | Yes | No | Yes |
| SuperFreq | 2 hours | No | Yes | Yes |
Table notes: Per sample run time refers to average per-sample runtime on a standard PC, except SuperFreq on an high performance computing server
CaSpER R package 5 detects both gene level and large-scale CNVs (Supplementary Figure 5). In the same 20 testing samples analysis, CasSpER (v 0.1.0) took comparable time (14 mins; Table 1) as RNAseqCNV. It also includes a function to estimate arm level CNVs, which was further tested on the ALL testing dataset. The X chromosome was excluded in the comparison since CaSpER omits sex chromosomes in its analysis. In addition, ≥2 copy gain and 1 copy gain classes are not distinguished by CaSpER but merged as copy gain in the output. Compared with the curated CNV result from SNP array, 83.2% overall concordance (4,580 out of 5,503) was reached by CaSpER, and the non-diploid arm level CNVs agreed with 43.6% of the SNP array-based CNVs (501 out of 1,141). The accuracy for arms with two copies was 93.7%, 44.0% for ≥1-copy gains, and 42.7% for 1-copy deletions (Supplementary Table 13).
SuperFreq is a tool that analyses and filters somatic SNVs and short indels, calls copy numbers and tracks clones over multiple samples from the same individual16. This package detects copy number changes from RNA-seq at the gene level and large-scale CNVs, and requires 2–4 CPUs, approximately 10 GB memory per CPU, and a high-performance server to analyze a single sample in around 2 hours (Table 1). Notably, SuperFreq does not automatically report chromosomal copy number changes. Therefore, comparison with RNAseqCNV model was done by manually curating the results from SuperFreq’s CNV visualization (Supplementary Figure 6) on a subset of the testing dataset (n=93, Supplementary table 14). Only whole chromosomes, in which all segments had identical copy number state according to the SuperFreq’s results were selected for the analysis. Both RNAseqCNV and SuperFreq achieved 100% concordance with the reference CNVs curated from SNP array, WGS and/or cytogenetics karyotype. The high concordance with the reference is attributed to the evaluation of only whole chromosome CNVs already reported with high consistence across the chromosomes, which may introduce favorable bias towards SuperFreq. Noticeably, the estimated clonality by SuperFreq is often in contradiction with the log fold changes and MAF in both its own and SNP array-based results, further highlighting the challenges of calling CNVs from RNA-seq data (Supplementary Figure 6).
DISCUSSION
The RNAseqCNV package has a clear focus on hematologic malignancies, such as leukemia for which large-scale CNVs are frequent and important for their initiation and risk-stratification. Although the in-built reference data and RF models are based on ALL samples, users can provide their own training data to customize the analysis.
The RNAseqCNV model had high accuracy overall but somewhat poorer performance for ≥2 copy gains. That said, 86% of the incorrectly classified ≥2 copy gains were still classified as 1-copy gains and there was no deletion classified as a gain and vice versa. This likely reflects the sparser representation of high level gains in the training dataset and multiple possible MAF values for higher copy gains, often not uniquely linked to a certain CNV, making the prediction more challenging.
Results of RNAseqCNV were highly concordant with the CNVs called from SNP array results as well as from WGS. RNAseqCNV is also shown with higher sensitivity than cytogenetics-based karyotyping for detecting large-scale CNVs in ALL. While unable to detect multiple subclones, RNAseqCNV provides extra features such as detecting loss of heterozygosity and it is not as susceptible to clonal outgrowth or selection bias. Poor chromosomal morphology may result in the wrong chromosome being detected as gain or deletion by cytogenetics. In addition, some hypodiploid leukemic cells may undergo endoreduplication, which cannot be distinguished from gain of chromosomes by routine cytogenetics karyotype 17, but can be accurately identified by RNAseqCNV based on the features of MAF distribution. One such sample was even detected in our SNP array validated cohort resulting in a misclassified ALL subtype by karyotyping, but was correctly identified by RNAseqCNV. In addition, the majority of the misclassified CNVs by karyotype were correctly determined by RNAseqCNV.
The package provides a Shiny application for interactive analysis and a robust visualization method to manually infer chromosomal and arm-level CNVs. Manual checking of the graphical output should further improve the accuracy, especially for cases for which there may not have been similar instances in the training data.
RNAseqCNV provides clear and easily interpretable visualization tools and better suited for chromosomal and arm level CNV detection than CNVkit or CasSpER. Most CNV detection tools have difficulties adjusting for samples with high numbers of chromosomal CNVs of one type – high hyperdiploid or near haploid samples. Such shortcomings lower the accuracy of automated detection algorithms and complicate the interpretation of the visualization methods of genome-based methods, including SNP array and WGS data. RNAseqCNV solves this issue by predicting chromosome arms with two copies automatically, avoiding the often-needed manual adjustment. SuperFreq, another alternative tool for calling CNVs from RNA-seq, also performs extremely well even in highly aneuploid samples. In addition, it provides higher resolution of detecting gene-level CNVs from RNA-seq, plus many other functionalities. On the other hand, it is computationally intensive in terms of hardware and runtime, and does not provide an automatic chromosomal and arm level CNV report, which complicates the interpretation of the result and limits its application in leukemia research and disease subtyping.
RNAseqCNV cannot accurately evaluate CNVs on sub-arm level or even large-scale CNVs in samples with low tumor purity. In contrast to cytogenetics-based karyotyping, multiple subclones cannot be detected. Also, usage for solid tumors necessitates providing additional data to customize the package for optimal performance.
In summary, RNAseqCNV is a fast, accurate and user-friendly tool for analyzing CNVs on chromosomal and arm level scale, which provides the ready-to-use result for cancer karyotyping and classification.
Supplementary Material
Acknowledgements
We thank the Biorepository, the Genome Sequencing Facility of the Hartwell Center for Bioinformatics and Biotechnology, and the Cytogenetics core facility of SJCRH. This work was supported by the American Lebanese Syrian Associated Charities of SJCRH, the American Society of Hematology Scholar Award (to Z.G.), the Leukemia & Lymphoma Society’s Career Development Program Special Fellow (to Z.G.), the NIH/NCI K99/R00 Award CA241297 (to Z.G.), NCI Outstanding Investigator Award R35 CA197695 (to C.G.M.), National Institute of General Medical Sciences grant P50 GM115279 (to C.G.M.), NCI grants P30 CA021765 (St. Jude Cancer Center Support Grant).
Footnotes
Competing interests
C.G.M. has received consulting fees from Illumina, speaking fees from Amgen, and research support from Pfizer, Loxo Oncology and Abbvie
References
- 1.Li JF, Dai YT, Lilljebjorn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proceedings of the National Academy of Sciences of the United States of America 2018. Dec 11; 115(50): E11711–E11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nature genetics 2019. Jan 14; 51(2): 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nature genetics 2017. Jul 03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talevich E, Shain AH. CNVkit-RNA: Copy number inference from RNA-Sequencing data. bioRxiv 2018: 408534. [Google Scholar]
- 5.Serin Harmanci A, Harmanci AO, Zhou X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nature communications 2020. Jan 3; 11(1): 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iacobucci I, Mullighan CG. Genetic Basis of Acute Lymphoblastic Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017. Mar 20; 35(9): 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inaba H, Azzato EM, Mullighan CG. Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Front Pediatr 2017; 5: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nature methods 2015. Jun; 12(6): 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012. Feb 01; 28(3): 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G, et al. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome biology 2010; 11(9): R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007. Nov; 17(11): 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayrhofer M, Viklund B, Isaksson A. Rawcopy: Improved copy number analysis with Affymetrix arrays. Scientific reports 2016. Oct 31; 6: 36158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014; 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer discovery 2021. May; 11(5): 1082–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhn M Building Predictive Models in R Using the caret Package. 2008 2008 2008-11-10; 28(5): 26. [Google Scholar]
- 16.Flensburg C, Sargeant T, Oshlack A, Majewski IJ. SuperFreq: Integrated mutation detection and clonal tracking in cancer. PLoS computational biology 2020. Feb; 16(2): e1007603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma SK, Chan GC, Wan TS, Lam CK, Ha SY, Lau YL, et al. Near-haploid common acute lymphoblastic leukaemia of childhood with a second hyperdiploid line: a DNA ploidy and fluorescence in-situ hybridization study. British journal of haematology 1998. Dec; 103(3): 750–755. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.